DOI:
10.1039/D5TA00347D
(Paper)
J. Mater. Chem. A, 2025,
13, 13976-13987
A single lithium-ion conducting monomer as a SEI-forming additive for lithium-ion batteries†
Received
14th January 2025
, Accepted 27th March 2025
First published on 28th March 2025
Abstract
A novel single lithium-ion conducting (SLIC) monomer, lithium((4-(methacryloyloxy)phenyl)sulfonyl)((trifluoromethyl)sulfonyl)imide (LiMPTFSI) was designed and synthesized as a solid-electrolyte interphase (SEI)-forming additive. The formation of a single lithium-ion conducting polymeric SEI on the surface of a graphite anode was confirmed by nuclear magnetic resonance (NMR) spectroscopy and X-ray photoelectron spectroscopy (XPS). The thin and stable SLIC polymer layer formed by LiMPTFSI on the graphite anode effectively suppressed the reductive decomposition of the liquid electrolyte. Based on the excellent electrochemical properties of the polymeric SEI, the graphite/LiNi0.8Co0.1Mn0.1O2 (NCM) full cells employing LiMPTFSI exhibited a high capacity retention of 71.0% after 1000 cycles at a rate of 1.0C and 25 °C. The cell containing LiMPTFSI also exhibited excellent rate capability, which was approximately 2-fold higher than the discharge capacity of the lithium-ion cell without the additive at 5.0C rate.
1. Introduction
Lithium-ion batteries (LIBs) have been widely used in portable electronic devices since the 1990s. These batteries are now an essential part of daily life owing to their high energy density, long cycle life, and affordability.1–3 Because of the recent focus on carbon neutrality, these batteries have been incorporated into electric vehicles (EVs) and large-scale energy storage systems (ESS).4,5 Graphite anodes are indispensable components of LIBs due to the layered structure that facilitates the highly reversible intercalation/deintercalation of lithium ions, which supports the long cycle life of LIBs.6–8 However, despite the superiority of graphite anodes, some technical challenges still remain unaddressed. The formation of an unstable solid electrolyte interphase (SEI) and the sluggish diffusion of lithium ions through the SEI result in performance deterioration, such as the degradation of the active material, gas evolution, disruption of the SEI and lithium plating, which consume limited lithium resources and cause significant capacity loss.9–11 In addition, the cycle life of LIBs remains insufficient to meet the demands of certain applications, such as EVs and ESS.12,13 Numerous strategies, such as surface modification of active materials, separator functionalization, tuning of the solvation structure of lithium ions, and introduction of functional additives, have been actively studied to address these challenges.14–21 The addition of low quantities of electrolyte additives is an effective strategy that results in significant improvement of cell performance.22–24 These additives perform various functions, such as scavenging corrosive substances (for example, HF), providing overcharge protection, forming a protective surface film on the electrode, and increasing the flame retardancy. Among them, the film-forming additives have been extensively studied. They enhance the cycling performance of LIBs by forming an electrochemically stable and ion-conductive film on the surface of active materials. Vinylene carbonate (VC) has been used as a SEI-forming additive for graphite anodes in LIBs. VC is known to form a SEI through preferential reduction on the graphite surface during the charging process. The mechanism of film formation by the preferential decomposition of additives is explained by the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) according to the molecular orbital theory.25–29 VC has a lower LUMO energy level than other electrolyte components such as ethylene carbonate (EC), ethyl methyl carbonate (EMC), and dimethyl carbonate (DMC). In previous studies, the SEI derived from VC was described as poly(VC).30–32 This polymeric SEI enhances the cycling stability by suppressing the reductive decomposition of electrolyte components.33–35 Similarly, many electrolyte additives can form the polymeric SEI (for example, 4,5-dimethyl-1,3-dioxol-2-one, allyl methyl carbonate, and vinyl acetate); however, the polymerization of these electrolyte additives has not been clearly confirmed.36–39 The polymerization of most additives has been investigated using electrochemical decomposition via potentiostatic methods, including linear sweep voltammetry (LSV) and cyclic voltammetry (CV). Previous studies have shown that the interfacial resistance increased after the first few cycles due to the formation of a polymeric SEI; however, these validations cannot prove that the additives undergo polymerization to form the SEI.40–45
In this study, lithium((4-(methacryloyloxy)phenyl)sulfonyl)((trifluoromethyl)sulfonyl) imide (LiMPTFSI) was designed and synthesized as a SEI-forming additive, which is a single lithium-ion conducting (SLIC) monomer with electron-withdrawing groups, such as (trifluoromethyl sulfonyl)imide and phenyl.46,47 These moieties weaken the binding energy of lithium ions by delocalizing the negative charge of the molecules, which is beneficial for the dissociation of lithium ions. Nuclear magnetic resonance (NMR) and X-ray photoelectron spectroscopy (XPS) were used to examine the formation of poly(LiMPTFSI) based on the charging voltage on the graphite anode in the lithium-ion full cells. A SLIC polymer layer was formed on the graphite anode, resulting in improved transport of lithium ions and mitigation of the side reactions of the liquid electrolyte. Therefore, the graphite/LiNi0.8Co0.1Mn0.1O2 (Gr/NCM) cells with 0.5 wt% LiMPTFSI exhibited significantly enhanced cycle life and excellent rate.
2. Experimental section
2.1. Materials
Sodium 4-hydroxybenzenesulfonate (>98.0%, TCI), lithium hydroxide (>98.0%, Sigma-Aldrich), magnesium sulfate (Daejung), and trifluoromethane sulfonamide (>98.0%, TCI) were used after vacuum drying at room temperature for 12 h. Thionyl chloride (SOCl2, 97%, Sigma-Aldrich) was distilled over linseed oil before use. Methacrylic acid (99%, Sigma-Aldrich), trifluoroacetic acid (TFA, 99%, Daejung), trifluoroacetic anhydride (TFAA, >99%, Sigma-Aldrich), acetone (>99.5%, Sigma-Aldrich), tetrahydrofuran (THF, >99.5%, Sigma-Aldrich), N,N-dimethylformamide (DMF, 99.8%, Sigma-Aldrich), dichloromethane (DCM, >99.8%, Sigma-Aldrich), and acetonitrile (ACN, 99.8%, Sigma-Aldrich) were used as received.
2.2. Synthesis of LiMPTFSI
In the first step, sodium 4-hydroxybenzenesulfonate (20.0 g, 102.0 mmol) and methacrylic acid (9.7 g, 112.2 mmol) were added to trifluoroacetic acid (TFA) (130 mL) at 0 °C under a dry argon atmosphere; thereafter, the mixture was stirred for 1 h. Trifluoroacetic anhydride (TFAA) (60 mL) was slowly added to the solution, and the mixture was stirred for 14 h at room temperature. The light-yellow solution was concentrated via reduced-pressure rotary evaporation. To remove impurities and unreacted materials, the product was washed several times with acetone. Sodium 4-(methacryloyloxy)benzenesulfonate was obtained as a white powder after vacuum drying for 24 h at 25 °C (yield, 89.8%). In the second step, sodium 4-(methacryloyloxy)benzenesulfonate (16.1 g, 60.9 mmol) and N,N-dimethylformamide (DMF) (1.7 mL) were added to tetrahydrofuran (THF) (25.0 mL) under a dry argon atmosphere. Subsequently, the mixture was cooled to 0 °C, and an excess amount of SOCl2 (24.3 mL, 335.2 mmol) was added in a dropwise manner while stirring. The reaction proceeded for 1 h at 0 °C and for 12 h at 25 °C. The resulting solution was cautiously poured into excess water at 0 °C to perform quenching. The lower organic layer was diluted with dichloromethane (80.0 mL) and washed six times with water. The mixture was then dried over magnesium sulfate. The precipitate was filtered to yield a light-yellow oil, which was subsequently dried under vacuum at 25 °C for 24 h. 4-(Chlorosulfonyl)phenyl 2-methylpropanoate was obtained as light purple crystals (yield: 86.0%). Finally, 4-(chlorosulfonyl)phenyl 2-methylpropanoate (11.8 g, 45.3 mmol) and lithium hydroxide (2.0 g, 83.5 mmol) were dissolved in ACN at 0 °C under a dry argon atmosphere. Trifluoromethane sulfonamide (6.1 g, 41.1 mmol) was added to the mixture, which was then heated to 25 °C and stirred for 24 h. The precipitate was filtered, and the filtrate was dried in a vacuum oven for 24 h at 25 °C. LiMPTFSI was obtained as a white powder (yield: 78.5%) and purified by silica gel column chromatography using a gradient of hexane to hexane/ethyl acetate (1/1 by volume).
2.3. Characterization
The 1H NMR, 7Li NMR, and 19F NMR spectra of the chemicals were obtained using NMR spectroscopy (VNMRS 600 MHz, Varian). Fourier transform infrared (FT-IR) spectroscopy was conducted between 400 and 4000 cm−1 using a Nicolet iS50 spectrometer (Thermo Fisher Scientific Inc.). The chemical composition of the surface formed on the graphite anode was investigated by X-ray photoelectron spectroscopy (XPS; K-Alpha+, Thermo Fisher) using Al-Kα as the X-ray source under ultrahigh vacuum. The surface and cross-sectional images of the electrodes were obtained using field-emission scanning electron microscopy (FE-SEM; Verios G4UC, FEI). The morphologies of the anodes processed using a Focused Ion Beam (FIB, Scios, FEI) were examined using high-resolution TEM (HR-TEM; JEOL 2100F).
2.4. Computational details
Density functional theory (DFT) calculations were performed using the Gaussian 16 program. Geometry optimizations and energy profile calculations were carried out using the B3LYP functional with the 6-311++G(d,p) basis set. Vibrational frequency and Intrinsic Reaction Coordinate (IRC) calculations were conducted to identify the minimum energy structure and transition state. The energy diagram for the polymerization of LiMPTFSI was examined. The activation energy (Ea) and reaction energy (ΔE) for the initiation and propagation steps were calculated using DFT. Polymerization of LiMPTFSI was initiated by the formation of a radical from ethylene carbonate (EC) in the electrolyte. Additionally, polymerization occurred at the methyl methacrylate moiety of LiMPTFSI. Ea and ΔE were calculated using the following equations.
where EIS, ETS, and EFS are the energies of the initial, transition, and final states, respectively.
2.5. Preparation of electrodes and cell assembly
The cathode was fabricated by slurry casting on an aluminium current collector. The slurry was composed of 95 wt% LiNi0.8Co0.1Mn0.1O2 (NCM, L&F Co. Ltd), 3 wt% poly(vinylidene fluoride) (PVdF, Solef 5130, Solvay) and 2 wt% Super P (TIMCAL) in N-methyl-3-pyrrolidone (NMP). The anode was fabricated using the same procedure on a copper current collector with a slurry containing 91 wt% artificial graphite (S360, BTR), 1 wt% Super P, and 8 wt% PVdF in NMP. The mass loadings of NCM and artificial graphite in the electrodes were 20.5 and 9.8 mg cm−2, respectively. The base electrolyte (1.0 M LiPF6 in EC/EMC = 3/7 by volume) was purchased from the Dongwha Electrolyte Company Limited. A 2032 coin-type cell was assembled by sandwiching a polyethylene separator between the graphite anode and the NCM cathode. The liquid electrolyte was then injected into the cell. All cell assembly procedures were conducted in a glove box (MBRAUN) filled with high purity argon gas at room temperature.
2.6. Electrochemical measurements
Electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 1 mHz to 1 × 106 Hz using an impedance analyzer (ZIVE MP1, Wonatech). Linear sweep voltammetry (LSV) was performed at a scan rate of 1 mV s−1 using a CHI 660 analyzer (CH Instruments, Inc.). To electrochemically polymerize LiMPTFSI on Cu foil, the cell was fabricated using a liquid electrolyte containing a low quantity of LiMPTFSI with Cu foil and stainless steel (SS). The cell was charged to 5.0 V for 24 h. Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were conducted using a Li metal (RE)/electrolyte/SS (WE) cell with scan rates of 1 mV s−1 and 0.1 mV s−1, respectively. The surface of the lithium metal was similarly modified to measure the lithium-ion transference number (t+). The cells assembled with lithium metal as the WE and SS as the RE were charged at 5 V for 24 h using a base electrolyte with and without 0.5 wt% LiMPTFSI. The cells were disassembled to obtain surface-modified lithium, and symmetric Li/electrolyte/Li cells were fabricated for t+ measurements. The t+ value was measured with a diluted electrolyte (0.01 M LiPF6 in EC/EMC = 3/7 by volume) by a combination of AC impedance and DC polarization measurements at room temperature. A galvanostatic cycling test of the Gr/NCM full cells was conducted in a voltage range of 2.5–4.3 V at 25 °C with a battery tester (WBCS 3000, WonAtech). In the cycling test, the charging process was conducted in constant-current/constant-voltage mode and discharging was conducted in constant-current mode.
3. Results and discussion
The three-step synthetic pathway for LiMPTFSI is shown in Fig. 1a. LiMPTFSI was characterized using NMR, Fourier transform infrared (FT-IR) spectroscopy, and liquid chromatography-mass spectrometry (LC-MS). The 1H NMR peaks of the methacrylate group were observed at 2.01, 5.93, and 6.31 ppm, whereas those of the phenyl group were observed at 7.30 and 7.81 ppm (Fig. S1a†). The 19F NMR and 7Li NMR peaks of the CF3 group (Fig. S1b†) and Li (Fig. S1c†) were observed at −78.67 and −1.24 ppm, respectively. Fig. S2† shows the FT-IR peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, aromatic C–C, SO2, CF3, and S–N. The molecular weight of the LiMPTFSI anion was consistent with the expected value in the LC-MS spectrum (Fig. S3†). Because the LUMO energy level is related to the electron-accepting properties of an electrolyte component, it can be used to predict the reducibility of electrolyte additives.48–51Fig. 1b shows the HOMO and LUMO energy levels of electrolyte components, which are obtained using DFT calculations. Compared with other electrolyte components (EC, EMC, VC, and LiPF6), LiMPTFSI exhibited the lowest LUMO energy level (−2.22 eV). This implies that LiMPTFSI can be preferentially reduced and polymerized on the anode surface and form a SLIC polymeric layer during the first charging process.28 In general, EC is used as an electrolyte solvent for LIBs, and its reductive decomposition is known to form a polymeric SEI, as demonstrated in Fig. S4a.†52–55 Based on this scheme, a polymerization mechanism was proposed for EC with LiMPTFSI, as shown in Fig. S4b.† To verify this hypothesis, the energetics of polymerization of one EC was compared with that of another EC and LiMPTFSI (Fig. 1c). During the initiation step of EC polymerization, the Ea was lower with LiMPTFSI (0.21 eV) than with the other EC (2.21 eV), indicating that EC was preferentially initiated with LiMPTFSI. Moreover, the Ea value of the propagation of LiMPTFSI polymerization (0.29 eV) was significantly lower than that of EC initiation (2.21 eV). Therefore, LiMPTFSI can be polymerized on the anode surface, leading to the formation of poly(LiMPTFSI). In addition to EC-derived reactions, poly(LiMPTFSI) can also be formed through the polymerization of the methacrylate moiety in the LiMPTFSI monomer.
C, aromatic C–C, SO2, CF3, and S–N. The molecular weight of the LiMPTFSI anion was consistent with the expected value in the LC-MS spectrum (Fig. S3†). Because the LUMO energy level is related to the electron-accepting properties of an electrolyte component, it can be used to predict the reducibility of electrolyte additives.48–51Fig. 1b shows the HOMO and LUMO energy levels of electrolyte components, which are obtained using DFT calculations. Compared with other electrolyte components (EC, EMC, VC, and LiPF6), LiMPTFSI exhibited the lowest LUMO energy level (−2.22 eV). This implies that LiMPTFSI can be preferentially reduced and polymerized on the anode surface and form a SLIC polymeric layer during the first charging process.28 In general, EC is used as an electrolyte solvent for LIBs, and its reductive decomposition is known to form a polymeric SEI, as demonstrated in Fig. S4a.†52–55 Based on this scheme, a polymerization mechanism was proposed for EC with LiMPTFSI, as shown in Fig. S4b.† To verify this hypothesis, the energetics of polymerization of one EC was compared with that of another EC and LiMPTFSI (Fig. 1c). During the initiation step of EC polymerization, the Ea was lower with LiMPTFSI (0.21 eV) than with the other EC (2.21 eV), indicating that EC was preferentially initiated with LiMPTFSI. Moreover, the Ea value of the propagation of LiMPTFSI polymerization (0.29 eV) was significantly lower than that of EC initiation (2.21 eV). Therefore, LiMPTFSI can be polymerized on the anode surface, leading to the formation of poly(LiMPTFSI). In addition to EC-derived reactions, poly(LiMPTFSI) can also be formed through the polymerization of the methacrylate moiety in the LiMPTFSI monomer.
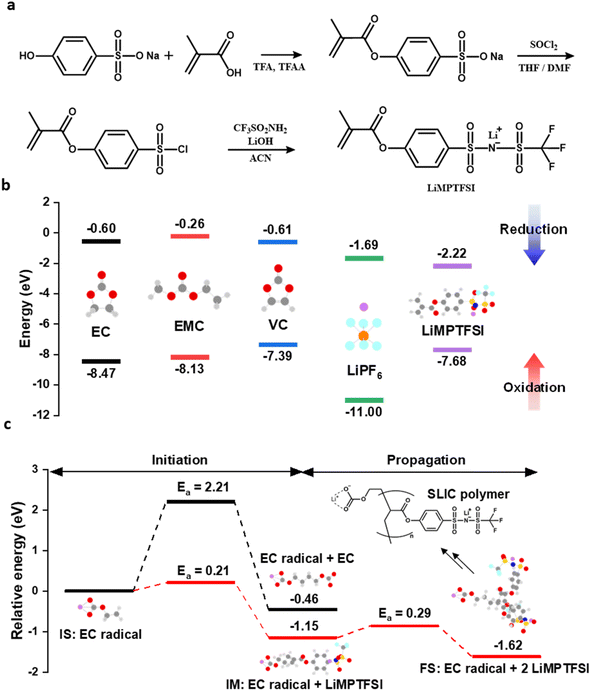 |
| | Fig. 1 (a) Synthetic route of LiMPTFSI. (b) HOMO and LUMO energy levels of EC, EMC, VC, LiPF6 and LiMPTFSI. (c) Relative energies for dimerization of EC and LiMPTFSI by the EC radical. Initial state (IS): EC radical, intermediate state (IM): IS + LiMPTFSI, final state (FS): IM + LiMPTFSI. | |
Cu foil/stainless steel (SS) cells were fabricated using 1.0 M LiPF6 in EC/EMC = 3/7 (v/v) (base electrolyte) without and with 0.5 wt% LiMPTFSI to assess the polymerization of LiMPTFSI on the surface of the electrode. The two-electrode cell was charged at 5.0 V for 24 h and then disassembled to obtain surface-modified Cu foil.56 The FT-IR spectra of bare Cu, LiMPTFSI powder, and surface-modified Cu foil (using the base electrolyte or electrolyte with 0.5 wt% LiMPTFSI) are shown in Fig. 2a. The spectrum of bare Cu foil was used as the reference (background) for all the FT-IR spectra. The peaks associated with the decomposition of carbonate solvents (CO at 1664 cm−1 and C–O at 1104 cm−1) and salt (P–O at 846 cm−1) were observed on the Cu surface modified with the base electrolyte. When comparing the FT-IR spectra of LiMPTFSI and modified Cu using an electrolyte with LiMPTFSI, the vibration peak at 1644 cm−1 corresponding to CC disappeared. However, the aromatic C–C peaks at 1586 and 1490 cm−1, SO2 peaks at 1327 and 1130 cm−1, CF3 peak at 1204 cm−1, and S–N–S peaks at 786 and 757 cm−1 were observed in both spectra, indicating that LiMPTFSI was polymerized on the surface of the Cu foil. The difference in the peak intensities of specific functional groups, such as CF3 and asymmetric SO2, is due to the physical state of samples and presence or absence of a substrate. This poly(LiMPTFSI) is expected to mitigate the decomposition of electrolyte components at the electrolyte–electrode interface and facilitate a uniform lithium-ion flux and high selectivity of lithium ions, which enhance the cycle life and rate capability of LIBs.52–54,57–60 The poly(LiMPTFSI) layer was formed on the surface of the lithium metal in the same manner, as described above (lithium metal as the WE instead of Cu foil), and its electrochemical properties were examined. To measure the t+, the symmetrical Li cells were assembled with modified lithium metal (by the base electrolyte or electrolyte with 0.5 wt% LiMPTFSI) using a dilute electrolyte (0.01 M LiPF6 in EC/EMC = 3/7 by volume).61 The t+ was then measured using a combination method of AC impedance and DC polarization measurements (Bruce–Vincent method). The Li modified by the base electrolyte exhibited a value of t+ = 0.37 (Fig. S5†). A low value of t+ may result in a concentration gradient in the cell, which increases the overpotential of the cell and ultimately disrupts its long-term cycling.62–64 After the formation of poly(LiMPTFSI) on the surface of the lithium metal (Fig. 2b), t+ increased to 0.64 owing to the single lithium-ion conducting behavior of poly(LiMPTFSI), which implied that the electrode covered with the SLIC poly(LiMPTFSI) facilitated a high current with a low overpotential.63–66 This was confirmed by measuring the exchange current density using Tafel plots for the symmetric Li cells.67 As shown in Fig. 2c, the exchange current density of the electrolyte with LiMPTFSI was approximately 1.5 times higher than that of the base electrolyte. In addition, the activation energy of the lithium-ion diffusion during the formation of different types of SEIs on graphite anodes was measured. Regarding this process, the graphite/NCM full cells were subjected to five charge–discharge cycles at 25 °C and 0.1C rate. The symmetric graphite cells were fabricated by removing the anode from the cycled full cells. The interfacial resistance was then measured using electrochemical impedance spectroscopy (EIS) at temperatures ranging from −20 to 40 °C. The SEI formed on the cycled graphite anode reflects the layer produced under actual operating conditions of the LIBs.
 |
| | Fig. 2 (a) FT-IR spectra of bare Cu, LiMPTFSI powder, and surface modified Cu foils using the base electrolyte or electrolyte with 0.5 wt% LiMPTFSI. (b) t+ measurement and (c) Tafel plots of the surface modified Li symmetric cells using the base electrolyte with and without 0.5 wt% LiMPTFSI. (d) Temperature dependence of the interfacial resistance of the graphite anode after pre-cycling. | |
The activation energy for the interfacial resistance (Ea,int) was calculated using an Arrhenius plot (Fig. 2d).68–71 The calculated Ea,int of the base electrolyte was 48.7 kJ mol−1, whereas the estimated Ea,int of LiMPTFSI was 38.1 kJ mol−1. The lower Ea,int value for LiMPTFSI indicates that poly(LiMPTFSI) facilitates a more facile interfacial electrochemical reaction, which is consistent with its higher exchange current density. The above electrochemical analyses support the formation of poly(LiMPTFSI) at the electrolyte–electrode interface, which increases the value of t+ and enhances the charge transfer kinetics of the lithium ions.
The dQ/dV plot and cyclic voltammogram of the lithiation process in the lithium/graphite half-cell also confirmed the polymerization of LiMPTFSI on the graphite surface (Fig. 3a and S6†).72 The electrolyte containing LiMPTFSI exhibited a reductive peak at 1.4 V vs. Li/Li+, which was not observed in the base electrolyte. Additionally, the cathodic current increased with increasing LiMPTFSI content. Accordingly, the reductive peak observed at 1.4 V vs. Li/Li+ can be attributed to SEI formation resulting from the reductive polymerization of LiMPTFSI. The cathodic peaks observed at 0.6–0.9 V (Fig. 3a) corresponded to the reductive decomposition of EC and EMC solvents.73 The electrochemical reactions associated with these reductive peaks contributed to the formation of the SEI layer. The SEI resistance of the graphite/NCM full cell containing 0.5 wt% LiMPTFSI was higher than that of the cell containing the base electrolyte after pre-cycling, as given in Table S1.† However, the charge transfer resistance of the cell containing LiMPTFSI was lower than that of the base electrolyte (Fig. 3b). The reduced charge transfer resistance indicated that the SEI derived from LiMPTFSI improved the interfacial reaction kinetics of lithium ions at the electrode–electrolyte interface. The CV curves of graphite/NCM full cells are presented in Fig. S7.† In the electrolyte containing 0.5 wt% LiMPTFSI, an increased current density was observed above 3.5 V during the first anodic scan, which may be attributed to the additional current generated by the decomposition of LiMPTFSI. The time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profiles of the surface of the precycled graphite (Fig. 3c) showed that poly(LiMPTFSI) was present on the graphite anode surface. The poly(LiMPTFSI) layer was identified by its S− signal, which was distinguishable from that of other electrolyte components, whereas the P−, PO−, and LiF− signals represented the SEI components derived from LiPF6. The S− signal was strong at the beginning and decreased gradually with depth, whereas the C− signal increased and plateaued gradually. Because the plateau of the C− signal corresponds to the bulk of pristine graphite, the top layer of the SEI was likely composed of poly(LiMPTFSI).74,75 The longer sputtering time for the SEI region occurred because of the formation of poly(LiMPTFSI) compared with the depth profiles of the precycled graphite with the base electrolyte (Fig. S8†). This was consistent with the higher SEI resistance values shown in Fig. 3b. In addition, a 3D rendering image of the ToF-SIMS signal (Fig. 3d) showed that poly(LiMPTFSI) was formed on the graphite anode. Different quantities of LiMPTFSI were added to the electrolyte to determine the adequate amount of LiMPTFSI in the electrolyte for graphite/NCM full cells, and the cycling performance of the cells was compared. 0.5 wt% LiMPTFSI was confirmed as optimal for the best capacity retention after 200 cycles (Fig. S9†). Low LiMPTFSI contents caused an unstable SEI, whereas excessive quantities of LiMPTFSI formed a thick layer. Hence, the optimal quantity of LiMPTFSI for improving the cycling performance of the graphite/NCM cell was determined to be 0.5 wt%.
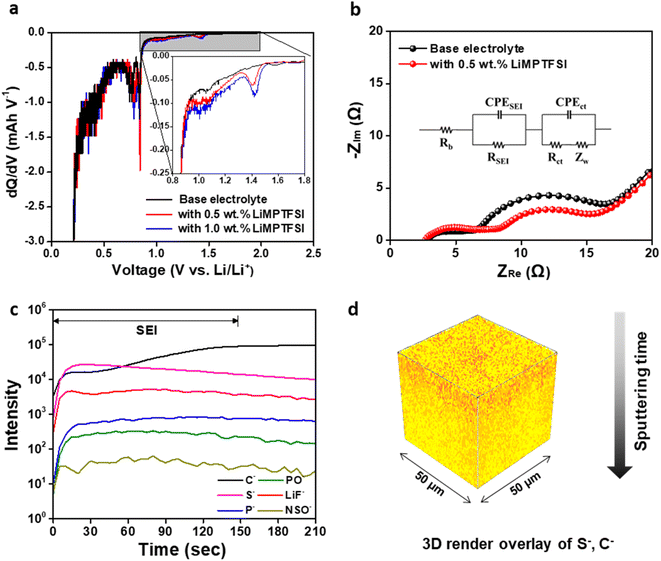 |
| | Fig. 3 (a) dQ/dV plot of the graphite electrodes containing different electrolytes. (b) Electrochemical impedance spectra of the Gr/NCM full cell after pre-cycling. (c) ToF-SIMS depth profile and (d) 3D rendered image of the graphite anode using the base electrolyte with 0.5 wt% LiMPTFSI after pre-cycling (S−: orange, C−: yellow). | |
The cycling performance of graphite/NCM cells with different electrolytes was evaluated at 25 °C and a rate of 1.0C. The capacity retention at the 1000th cycle significantly increased from 56.9% to 71.0% when adding 0.5 wt% LiMPTFSI into the base electrolyte (Fig. 4a–c). Although the cells initially exhibited somewhat low efficiencies, they achieved high efficiencies exceeding 99.9% once a stable SEI was formed (Fig. 4c). Notably, the cell with the electrolyte containing 0.5 wt% LiMPTFSI demonstrated the highest efficiency during cycling. The large capacity fading during the early cycles of cells containing the base electrolyte likely occurred because of the excessive formation of the SEI layer. This can be inferred from the relatively high rate of the increase in interfacial resistances (including the SEI and charge transfer resistance) with cycling (Fig. 4d).76,77 While the interfacial resistances of the cell employing the base electrolyte significantly increased after the 100th cycle, no significant increase in the interfacial resistances of the cell with the electrolyte containing 0.5 wt% LiMPTFSI was observed (Fig. 4e). Furthermore, the cell containing 0.5 wt% LiMPTFSI exhibited outstanding rate capability, as shown in Fig. 4f and S10.† As the C rate increased, the cell containing 0.5 wt% LiMPTFSI exhibited superior performance than that of the cell without LiMPTFSI. At a rate of 5.0C, the cell with the base electrolyte delivered a discharge capacity of 47 mA h g−1, whereas the cell with 0.5 wt% LiMPTFSI exhibited a discharge capacity of 90 mA h g−1. The cell containing 0.5 wt% LiMPTFSI exhibited improved cycle life and rate capability compared with those of the cell containing the same quantity of VC as a SEI-forming additive. This result can be attributed to the decrease of the continuous decomposition of the electrolyte by the presence of a poly(LiMPTFSI) layer (similar to VC), coupled with the effect of enhanced surface kinetics due to its high Li+ ion-conducting behavior. The graphite/NCM full cells were cycled at a charge/discharge rate of 2C to assess long-term cycling at a high current rate (Fig. S11†). The cell containing the base electrolyte exhibited a large capacity decline, delivering a discharge capacity of 47.6 mA h g−1 at the 600th cycle, whereas the cell using the electrolyte that contains 0.5 wt% LiMPTFSI delivered a high discharge capacity of 101.5 mA h g−1 at the 600th cycle. DFT calculations were performed to compare the polymerization of the VC radical with that of another VC and LiMPTFSI (Fig. S12a†). The initiation of VC polymerization was more favorable with LiMPTFSI (Ea = 0.51 eV) than with the other VC (Ea = 0.62 eV) (Fig. S12b†). Therefore, the polymerization of VC was facilitated using LiMPTFSI. This result suggests that LiMPTFSI can be copolymerized with VC and that this preferred reaction forms a robust lithium-ion conductive polymer layer. Additionally, the increase in capacity retention of the dual addition of LiMPTFSI and VC was greater than that observed when a single additive was applied to the graphite/NCM cell (Fig. S12†). As mentioned earlier, the SEI formed by the copolymerization of VC and LiMPTFSI is more robust and ion-conductive than when either LiMPTFSI or VC is added individually, leading to improved cycling stability.
 |
| | Fig. 4 Voltage profiles of the Gr/NCM full cells using the base electrolyte (a) without and (b) with 0.5 wt% LiMPTFSI. (c) Cycling performance of the Gr/NCM full cells containing different electrolytes at 1.0C and 25 °C. AC impedance spectra of the cells using the base electrolyte (d) without and (e) with 0.5 wt% LiMPTFSI with cycling. (f) Rate capability of the Gr/NCM full cells containing different electrolytes at 25 °C. | |
To examine the consumption behavior of LiMPTFSI during the first charge of the full cell, 1H NMR analysis was conducted on the electrolyte and SEI components formed by LiMPTFSI on the graphite anode of graphite/NCM cells with and without 0.5 wt% LiMPTFSI after charging to various voltages (interval of 0.2 V within 2.5–4.3 V). During the charging process, LiMPTFSI was decomposed before the graphite anode began to lithiate, as shown in Fig. 3a. The 1H NMR spectra in Fig. 5a show that some of the monomers were converted into polymers. During the polymerization of LiMPTFSI, the two proton peaks in the phenyl group experienced an up-field shift from 7.81 and 7.30 ppm to 7.57 and 6.79 ppm, respectively. The two peaks of CC at 6.30 and 5.93 ppm shifted to approximately 1.98 ppm. In addition, a significant up-field shift of methyl groups from 2.0 ppm to a broad range of 0.7–1.1 ppm was observed.78–80 These shifts were most pronounced between 3.7 and 3.9 V. Some LiMPTFSI remained after the cell voltage attained 4.3 V. Based on the integral values of peak intensities at 4.3 V, 70 mol% of the initial LiMPTFSI was consumed, half of which was polymerized to poly(LiMPTFSI) (Fig. 5b). The F 1s, S 2p, and N 1s XPS profiles of the graphite anode after pre-cycling with 0.5 wt% LiMPTFSI (Fig. S14†) confirmed that LiMPTFSI formed the SEI composed of organic components containing poly(LiMPTFSI) and inorganic components such as LiF, Li2S, and Li3N. The inorganic components in the SEI layer are known to exhibit relatively high ionic conductivity and good mechanical properties.81–83 However, the remaining 30 mol% LiMPTFSI remained in the electrolyte because it did not decompose. XPS analysis was conducted to investigate the chemical composition of the SEI layer formed on the surface of the graphite anode. The C 1s spectra of the surface of the graphite anode obtained at different charging voltages could be divided into five characteristic peaks (Fig. 5c): C–C (284.6 eV), polymer species (285.6 eV), C–O (286.7 eV), O–CO (288.1 eV), and C–F (291.3 eV).84 The intensities of the C–C and C–F peaks corresponding to the PVdF binder decreased as charging proceeded. In contrast, the dominance of the peaks corresponding to the polymer species and the C–O signal from poly(LiMPTFSI) increased as the charging voltage increased. These results were consistent with the consumption tendency of LiMPTFSI observed in the 1H NMR spectra (Fig. 5a). The 1H NMR and C 1s XPS results presented in Fig. 5 provide evidence for the polymerization of LiMPTFSI on the surface of the graphite anode. Fig. S15† shows the surface SEM images of the pristine NCM and cycled NCM cathodes obtained after 500 cycles. No noticeable differences were observed in the morphologies of the cathodes cycled in different electrolytes, indicating that LiMPTFSI did not significantly affect the cathode. Fig. 6b and c show the surface SEM images of the graphite anodes cycled in different electrolytes. Compared with pristine graphite (Fig. 6a), the surface of the graphite cycled using the base electrolyte was covered with by-products, indicating severe decomposition of the electrolyte components.85,86 A distinct morphological difference between the two graphite anodes was observed using low-magnification SEM images (Fig. S16†). After the addition of 0.5 wt% LiMPTFSI to the electrolyte, a smooth and uniform surface was observed. The SEI layer was visible at the graphite anode using cross-sectional TEM images and EDS mapping (Fig. 6d and e). The thickness of the SEI layer of the graphite cycled in the base electrolyte was 3-fold greater than that of the graphite cycled in the electrolyte containing LiMPTFSI. The thick SEI layer formed on the graphite cycled in the base electrolyte was the main factor behind the increased interfacial resistance.87,88 However, the mitigation of concentration polarization by poly(LiMPTFSI) with high t+ facilitated the formation of a thin SEI. The XPS spectra in Fig. 6f–h also support the decomposition of the electrolyte components in the absence of LiMPTFSI and the mitigation of reductive decomposition by poly(LiMPTFSI) after the addition of LiMPTFSI. The lithium carbonate observed at 290.1 eV, which resulted from the decomposition of carbonate solvents, was predominantly observed in the C 1s spectra of the base electrolyte.89 In contrast, the polymer species assigned to poly(LiMPTFSI) produced from the electrolyte containing 0.5 wt% LiMPTFSI occupied a large proportion even after 500 cycles (Fig. 6f). The overall distribution of the SEI components formed on graphite cycled in the electrolyte containing 0.5 wt% LiMPTFSI did not exhibit significant change with cycling (Fig. S17†). In contrast, the P–F (687.0 eV), LiF (685.1 eV), LiPxFy (136.9 eV), and LixPOyFz (133.4 eV) in the SEI layer formed on the graphite cycled in the base electrolyte significantly increased after 500 cycles compared to those of the SEI layer formed on the graphite surface after pre-cycling (Fig. S18†).90–92 These results suggest that the SEI layer formed by LiMPTFSI effectively mitigated the reductive decomposition of the electrolyte during cycling.
 |
| | Fig. 5 Consumption pattern of LiMPTFSI in the graphite/NCM cell at different voltages during the first charging process. (a) 1H NMR spectra of LiMPTFSI, (b) relative integration values of monomer and polymer at 2.5 and 4.3 V, and (c) C 1s XPS spectra of the graphite anode surface during the initial charge process. | |
 |
| | Fig. 6 Surface SEM images of (a) a pristine graphite anode and graphite anodes cycled (b) without and (c) with 0.5 wt% LiMPTFSI. Cross-sectional TEM and EDS mapping images of graphite anodes cycled (d) without and (e) with 0.5 wt% LiMPTFSI. (f) C 1s, (g) P 2p and (h) F 1s XPS spectra of the surface of the graphite anode after 500 cycles. | |
4. Conclusions
LiMPTFSI, a SLIC monomer used as a SEI-forming additive, was designed and synthesized. DFT calculations and FT-IR, LSV, CV, ToF-SIMS, 1H NMR, and XPS analyses were used to investigate its polymerization on the electrode. 1H NMR and XPS analyses at different charging voltages showed that LiMPTFSI was consumed in the first charging step, forming poly(LiMPTFSI) on the surface of the graphite anode. Compared with the SEI layer formed on the graphite cycled in the base electrolyte, poly(LiMPTFSI) exhibited a high rate of electrochemical kinetics at the electrode–electrolyte interface owing to the low activation energy required for the transport of lithium ions. When 0.5 wt% LiMPTFSI was added into the electrolyte as a SEI-forming additive in the graphite/NCM full cell, the capacity retention after the 1000th cycle was significantly higher than that of the cell using the base electrolyte without LiMPTFSI. Moreover, the addition of 0.5 wt% LiMPTFSI supported a high current density, exhibiting outstanding performance under high C rate conditions. Surface and cross-sectional analyses showed that the thin SEI layer formed by LiMPTFSI effectively mitigated electrolyte decomposition during long-term cycling. The LIB performance of the cells containing LiMPTFSI surpassed that of cells containing the same quantity of VC. These results provide new insights into SEI-forming additives and demonstrate the exclusive formation of SLIC poly(LiMPTFSI) as a SEI layer on graphite anodes that improves the cycling performance of LIBs.
Data availability
All the data have been presented in the article or the ESI.†
Author contributions
Jin-Hong Seok contributed to the conceptualization, formal analysis, validation and writing – original draft. Kyeounghak Kim and Seongjae Lee contributed to the software and validation. Da-Ae Lim contributed to the conceptualization, data curation, formal analysis and methodology. Kyoung Ho Ahn and Chul Haeng Lee contributed to the validation. Dong-Won Kim contributed to the conceptualization, funding acquisition, project administration and writing – review & editing. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by LG Energy Solutions and the National Research Foundation of Korea (RS-2024-00454354).
Notes and references
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CAS.
- M. S. Ziegler and J. E. Trancik, Energy Environ. Sci., 2021, 14, 1635–1651 Search PubMed.
- W. Liu, T. Placke and K. Chau, Energy Rep., 2022, 8, 4058–4084 Search PubMed.
- J. Lin, X. Zhang, E. Fan, R. Chen, F. Wu and L. Li, Energy Environ. Sci., 2023, 16, 745–791 Search PubMed.
- J. Dahn, R. Fong and M. Spoon, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 6424 CAS.
- J. Dahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 9170 CAS.
- L. Zhao, B. Ding, X. Y. Qin, Z. Wang, W. Lv, Y. B. He, Q. H. Yang and F. Kang, Adv. Mater., 2022, 34, 2106704 CAS.
- J. C. Garcia, I. Bloom, C. Johnson, D. Dees and H. Iddir, J. Phys. Chem. C, 2020, 124, 8162–8169 CAS.
- C. Mao, R. E. Ruther, J. Li, Z. Du and I. Belharouak, Electrochem. Commun., 2018, 97, 37–41 CAS.
- W. Cai, C. Yan, Y. X. Yao, L. Xu, X. R. Chen, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13007–13012 CAS.
- X. Wang, L. Liu and Z. Niu, Mater. Chem. Front., 2019, 3, 1265–1279 RSC.
- S. Heng, X. Shan, W. Wang, Y. Wang, G. Zhu, Q. Qu and H. Zheng, Carbon, 2020, 159, 390–400 CrossRef CAS.
- L. Fu, H. Liu, C. Li, Y. Wu, E. Rahm, R. Holze and H. Wu, Solid State Sci., 2006, 8, 113–128 CrossRef CAS.
- B. Boateng, X. Zhang, C. Zhen, D. Chen, Y. Han, C. Feng, N. Chen and W. He, Nano Sel., 2021, 2, 993–1010 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2007, 164, 351–364 CrossRef CAS.
- Y. X. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed.
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, 2201207 CrossRef CAS.
- Q. Li, G. Liu, H. Cheng, Q. Sun, J. Zhang and J. Ming, Chem.–Eur. J., 2021, 27, 15842–15865 CrossRef CAS.
- W. Zhao, Y. Ji, Z. Zhang, M. Lin, Z. Wu, X. Zheng, Q. Li and Y. Yang, Curr. Opin. Electrochem., 2017, 6, 84–91 CrossRef CAS.
- X. Zhuang, S. Zhang, Z. Cui, B. Xie, T. Gong, X. Zhang, J. Li, R. Wu, S. Wang and L. Qiao, Angew. Chem., Int. Ed., 2024, 63, e202315710 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- Y. Li, G. M. Veith, K. L. Browning, J. Chen, D. K. Hensley, M. P. Paranthaman, S. Dai and X.-G. Sun, Nano Energy, 2017, 40, 9–19 CrossRef CAS.
- R. Chen, F. Wu, L. Li, Y. Guan, X. Qiu, S. Chen, Y. Li and S. Wu, J. Power Sources, 2007, 172, 395–403 CrossRef CAS.
- W. Zhang, S. Zhang, L. Fan, L. Gao, X. Kong, S. Li, J. Li, X. Hong and Y. Lu, ACS Energy Lett., 2019, 4, 644–650 CrossRef CAS.
- M. C. Madhusudhanan, S. A. Kumar, S. Nair, N. Srinivasan, M. Buragohain and S. Kunnikuruvan, Batteries Supercaps, 2023, 6, e202200430 CrossRef CAS.
- F.-M. Wang, H.-M. Cheng, H.-C. Wu, S.-Y. Chu, C.-S. Cheng and C.-R. Yang, Electrochim. Acta, 2009, 54, 3344–3351 CrossRef CAS.
- N. Yao, S. Y. Sun, X. Chen, X. Q. Zhang, X. Shen, Z. H. Fu, R. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210859 CrossRef CAS PubMed.
- Z.-Z. Dong, J.-H. Zhang, L. Zhu, X.-Z. Fan, Z.-G. Liu, Y.-B. Yan and L. Kong, Chin. J. Chem., 2025, 36, 109773 CAS.
- H.-H. Lee, Y.-Y. Wang, C.-C. Wan, M.-H. Yang, H.-C. Wu and D.-T. Shieh, J. Appl. Electrochem., 2005, 35, 615–623 CrossRef CAS.
- H. Ota, K. Shima, M. Ue and J.-i. Yamaki, Electrochim. Acta, 2004, 49, 565–572 CrossRef CAS.
- H. Zhao, X. Yu, J. Li, B. Li, H. Shao, L. Li and Y. Deng, J. Mater. Chem. A, 2019, 7, 8700–8722 CAS.
- L. Chen, K. Wang, X. Xie and J. Xie, J. Power Sources, 2007, 174, 538–543 CAS.
- L. El Ouatani, R. Dedryvere, C. Siret, P. Biensan, S. Reynaud, P. Iratçabal and D. Gonbeau, J. Electrochem. Soc., 2008, 156, A103 Search PubMed.
- D. Pritzl, S. Solchenbach, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2625 CrossRef CAS.
- Y. Liu, K. Xie, Y. Pan, Y. Li, H. Wang, Z. Jin and C. Zheng, J. Electrochem. Soc., 2017, 164, A3949 CrossRef CAS.
- K. Abe, H. Yoshitake, T. Kitakura, T. Hattori, H. Wang and M. Yoshio, Electrochim. Acta, 2004, 49, 4613–4622 CrossRef CAS.
- M. Xu, L. Zhou, L. Xing, W. Li and B. L. Lucht, Electrochim. Acta, 2010, 55, 6743–6748 CrossRef CAS.
- S. H. Beheshti, M. Javanbakht, H. Omidvar, M. S. Hosen, A. Hubin, J. Van Mierlo and M. Berecibar, iScience, 2022, 25, 103862 CrossRef CAS.
- X. Wang, X. Zheng, Y. Liao, Q. Huang, L. Xing, M. Xu and W. Li, J. Power Sources, 2017, 338, 108–116 Search PubMed.
- C. Korepp, W. Kern, E. Lanzer, P. Raimann, J. Besenhard, M. Yang, K.-C. Möller, D.-T. Shieh and M. Winter, J. Power Sources, 2007, 174, 637–642 Search PubMed.
- K.-C. Möller, H. Santner, W. Kern, S. Yamaguchi, J. Besenhard and M. Winter, J. Power Sources, 2003, 119, 561–566 CrossRef.
- Y. Ha, T. R. Martin, S. Frisco, L. Rynearson, M. C. Schulze, S.-D. Han, S. E. Trask, B. L. Lucht, G. Teeter and N. R. Neale, J. Electrochem. Soc., 2022, 169, 070515 Search PubMed.
- W. Wang, H. Hu, X. Zeng, W. Fan, T. Yang, X. Zhao, C. Fan, X. Zuo and J. Nan, ACS Appl. Energy Mater., 2021, 4, 7101–7111 CrossRef CAS.
- Y. Li, S. Wan, G. M. Veith, R. R. Unocic, M. P. Paranthaman, S. Dai and X. G. Sun, Adv. Energy Mater., 2017, 7, 1601397 CrossRef.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS PubMed.
- B. Hoge, J. Bader and J. Fluor, Chem, 2007, 128, 857–861 CAS.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J.-G. Zhang and W. Xu, Nat. Energy, 2017, 2, 1–8 Search PubMed.
- Q. Zheng, Y. Yamada, R. Shang, S. Ko, Y.-Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291–298 CrossRef CAS.
- H. Zhang, L. Qiao, H. Kühnle, E. Figgemeier, M. Armand and G. G. Eshetu, Energy Environ. Sci., 2023, 16, 11–52 RSC.
- Q. Fan, J. Zhang, S. Fan, B. Xi, Z. Gao, X. Guo, Z. Duan, X. Zheng, Y. Liu and S. Xiong, Adv. Mater., 2024, 36, 2409521 CrossRef CAS.
- R. Yazami, Electrochim. Acta, 1999, 45, 87–97 CrossRef CAS.
- H. Tavassol, J. W. Buthker, G. A. Ferguson, L. A. Curtiss and A. A. Gewirth, J. Electrochem. Soc., 2012, 159, A730 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, Carbon, 2016, 105, 52–76 CrossRef CAS.
- R. Hu, H. Qiu, H. Zhang, P. Wang, X. Du, J. Ma, T. Wu, C. Lu, X. Zhou and G. Cui, Small, 2020, 16, 1907163 CrossRef CAS.
- M. Gu, A. M. Rao, J. Zhou and B. Lu, Energy Environ. Sci., 2023, 16, 1166–1175 RSC.
- M. Martinez-Ibañez, E. Sanchez-Diez, L. Qiao, Y. Zhang, X. Judez, A. Santiago, I. Aldalur, J. Carrasco, H. Zhu and M. Forsyth, Adv. Funct. Mater., 2020, 30, 2000455 CrossRef.
- D. M. Shin, J. E. Bachman, M. K. Taylor, J. Kamcev, J. G. Park, M. E. Ziebel, E. Velasquez, N. N. Jarenwattananon, G. K. Sethi and Y. Cui, Adv. Mater., 2020, 32, 1905771 CrossRef CAS PubMed.
- S. Gao, F. Sun, N. Liu, H. Yang and P.-F. Cao, Mater. Today, 2020, 40, 140–159 Search PubMed.
- H. Dai, X. Gu, J. Dong, C. Wang, C. Lai and S. Sun, Nat. Commun., 2020, 11, 643 CrossRef CAS PubMed.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CAS.
- H. G. Buss, S. Y. Chan, N. A. Lynd and B. D. McCloskey, ACS Energy Lett., 2017, 2, 481–487 CAS.
- L. Gao, S. Luo, J. Li, B. Cheng, W. Kang and N. Deng, Energy Storage Mater., 2021, 43, 266–274 Search PubMed.
- Y. Zhao, L. Wang, Y. Zhou, Z. Liang, N. Tavajohi, B. Li and T. Li, Adv. Sci., 2021, 8, 2003675 CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- X. Liu, J. Liu, B. Lin, F. Chu and Y. Ren, ACS Appl. Energy Mater., 2021, 5, 1031–1040 Search PubMed.
- Y. Yin, T. Zheng, J. Chen, Y. Peng, Z. Fang, Y. Mo, C. Wang, Y. Wang, Y. Xia and X. Dong, Adv. Funct. Mater., 2023, 33, 2215151 Search PubMed.
- K. Huang, S. Bi, B. Kurt, C. Xu, L. Wu, Z. Li, G. Feng and X. Zhang, Angew. Chem., 2021, 133, 19381–19389 Search PubMed.
- M. Smart and B. Ratnakumar, J. Electrochem. Soc., 2011, 158, A379 CAS.
- Z. Wang, F. Qi, L. Yin, Y. Shi, C. Sun, B. An, H. M. Cheng and F. Li, Adv. Energy Mater., 2020, 10, 1903843 CrossRef CAS.
- M. Weiling, C. Lechtenfeld, F. Pfeiffer, L. Frankenstein, D. Diddens, J. F. Wang, S. Nowak and M. Baghernejad, Adv. Energy Mater., 2024, 14, 2303568 CAS.
- X. Z. Fan, J. H. Zhang, N. Yao, J. X. Chen, X. Chen and L. Kong, Adv. Energy Mater., 2024, 14, 2303336 Search PubMed.
- L. Hu, Z. Zhang and K. Amine, J. Power Sources, 2013, 236, 175–180 CAS.
- M. Klett, P. Svens, C. Tengstedt, A. Seyeux, J. Swiatowska, G. Lindbergh and R. W. Lindström, J. Phys. Chem. C, 2015, 119, 90–100 CrossRef CAS.
- H. Xu, Z. Li, T. Liu, C. Han, C. Guo, H. Zhao, Q. Li, J. Lu, K. Amine and X. Qiu, Angew. Chem., 2022, 134, e202202984 Search PubMed.
- X. Lin, J. Park, L. Liu, Y. Lee, A. Sastry and W. Lu, J. Electrochem. Soc., 2013, 160, A1701 CrossRef CAS.
- Y.-X. Lin, Z. Liu, K. Leung, L.-Q. Chen, P. Lu and Y. Qi, J. Power Sources, 2016, 309, 221–230 CrossRef CAS.
- Y. Tanaka, R. Shinohe, S. Yuki, T. Ohashi and H. Mori, Polym. Chem., 2023, 14, 4538–4546 RSC.
- X. Shan, S. Jin, S. Zhao, H. Yang and P.-F. Cao, MRS Commun., 2023, 13, 848–853 CrossRef CAS.
- E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, P. Grysan, G. Lingua, C. Gerbaldi, A. S. Shaplov and Y. S. Vygodskii, Electrochim. Acta, 2022, 413, 140126 CrossRef CAS.
- S. Ni, M. Zhang, C. Li, R. Gao, J. Sheng, X. Wu and G. Zhou, Adv. Mater., 2023, 35, 2209028 CrossRef CAS.
- S. Kim, K. Y. Cho, J. Kwon, K. Sim, D. Seok, H. Tak, J. Jo and K. Eom, Small, 2023, 19, 2207222 CrossRef CAS PubMed.
- H.-H. Sun, A. Dolocan, J. A. Weeks, R. Rodriguez, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2019, 7, 17782–17789 RSC.
- Z. Lin, O. Sheng, X. Cai, D. Duan, K. Yue, J. Nai, Y. Wang, T. Liu, X. Tao and Y. Liu, J. Energy Chem., 2023, 81, 358–378 CrossRef CAS.
- Z. Li, X. Lin, H. Zhou, L. Xing, G. Lan, W. Zhang, J. Chen, M. Liu, Q. Huang and W. Li, J. Power Sources, 2020, 467, 228343 CrossRef CAS.
- H. L. Andersen, L. Djuandhi, U. Mittal and N. Sharma, Adv. Energy Mater., 2021, 11, 2102693 CrossRef CAS.
- D. Hu, Y. Su, L. Chen, N. Li, L. Bao, Y. Lu, Q. Zhang, J. Wang, S. Chen and F. Wu, J. Energy Chem., 2021, 58, 1–8 CrossRef CAS.
- H. Da, S. Pan, J. Li, J. Huang, X. Yuan, H. Dong, J. Liu and H. Zhang, Energy Storage Mater., 2023, 56, 457–467 CrossRef.
- F. Buchner, M. Fingerle, J. Kim, T. Späth, R. Hausbrand and R. J. Behm, Adv. Mater. Interfaces, 2019, 6, 1801650 Search PubMed.
- W. Deng, W. Dai, X. Zhou, Q. Han, W. Fang, N. Dong, B. He and Z. Liu, ACS Energy Lett., 2020, 6, 115–123 CrossRef.
- P. Vanaphuti, Z. Cui and A. Manthiram, Adv. Funct. Mater., 2024, 34, 2308619 CAS.
- R. Guo, D. Wang, L. Zuin and B. M. Gallant, ACS Energy Lett., 2021, 6, 877–885 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Seongjae
Lee
a,
Da-Ae
Lim
a,
Kyoung Ho
Ahn
b,
Chul Haeng
Lee
b,
Kyeounghak
Kim
a,
Seongjae
Lee
a,
Da-Ae
Lim
a,
Kyoung Ho
Ahn
b,
Chul Haeng
Lee
b,
Kyeounghak
Kim





