DOI:
10.1039/D4TA08701A
(Paper)
J. Mater. Chem. A, 2025,
13, 7215-7227
Synergistically enhanced electronic modulation in trimetallic Cu–Co–Mo-based heterostructured nanomaterials for green H2 production via efficient alkaline electrolysis†
Received
7th December 2024
, Accepted 26th January 2025
First published on 27th January 2025
Abstract
Trimetallic Cu, Co, and Mo-based bifunctional electrocatalysts have been developed for H2 production through water electrolysis at a low synthesis cost of $0.051, $0.053 and $0.052 cm−2 for CuCoMo-LDH, CuCoMo-P and CuCoMo-S, respectively. CuCoMo-LDH, CuCoMo-P and CuCoMo-S required 307, 331 and 311 mV overpotentials for oxygen evolution and 215, 117 and 265 mV for the hydrogen evolution reaction at 50 mA cm−2, respectively. Density functional theory calculations show that intermediate species migrating to Cu atoms neighboring Co and Mo on the electrocatalyst surface enhance water adsorption. Electronic modulations among the metals boost the density of electronic states near the Fermi level, resulting in higher electrical conductivity. Among the alkaline electrolyzers, CuCoMo-P (±) exhibited the highest overall water splitting performance, requiring the lowest recorded cell potential of 1.393 V at 10 mA cm−2 followed by CuCoMo-LDH (±) (1.427 V) and CuCoMo-S (±) (1.540 V), all surpassing RuO2 (+)‖Pt/C (−) (1.554 V). To our knowledge, the use of CuCoMo-P in water electrolysis has not been previously reported. CuCoMo-P (±) achieves 98.7% faradaic efficiency with an H2 generation rate of 3.4 mL min−1 costing $0.12255 per 1000 L H2(g). Consequently, the synthesized electrocatalysts are anticipated to significantly advance water electrolysis for large-scale H2 production.
1. Introduction
Large-scale green hydrogen (GH2) production via electrochemical water splitting powered by renewable resources is important for sustainable living, owing to the high energy density (120–142 MJ kg−1), renewability and carbon-zero properties of H2.1–4 Hence, the main focus of the research is to synthesize economically viable and highly competent non-noble transition metal-based electrocatalysts to accelerate the sluggish proton-coupled electron transfer steps of the oxygen evolution reaction (OER) at the anode.1,3 The sluggish kinetics of the OER eventually hinder the hydrogen evolution reaction (HER) at the cathode, necessitating improved bifunctional electrocatalysts for efficient overall water splitting (OWS).3,5–7 The use of platinum-based electrocatalysts for the HER and RuO2 and IrO2 for the OER is discouraged due to the exorbitant cost and scarcity of noble metals.5,6,8–12 Instead, non-noble transition metal-based materials are being explored to develop highly efficient, cost-effective, and durable electrocatalysts for water electrolysis. This approach aims to reduce electrolyzer fabrication costs and the overpotential (η),5,6,8–10 considering that the theoretical thermodynamic potential is 1.23 V at normal temperature and pressure.4,13 Among these, trimetallic electrocatalysts have garnered significant interest due to their advantages of strong synergistic effects among the metals to modulate the electronic structure favoring the OER and HER.14–22 Besides metals, considering the importance of counter ions in enhancing the water electrolysis activity of an electrocatalyst,23 numerous reports on mono- and multi-non-noble transition metal-based hydroxides,12,24,25 phosphides,14,26–28 sulfides,29,30 and oxides,28,29 are reported. From density functional theory (DFT) studies on Ni2P, Liu et al. showed that the presence of phosphorus (P) atoms in an electrocatalyst facilitates the adsorption/desorption of hydrogen atoms, unveiling its potential applicability as an efficient HER electrocatalyst following the Sabatier principle.31,32 Considering the advancement with P atoms in electrocatalysts, Yu et al. in 2019 developed N-doped carbon shell-coated CoP(MoP)–CoMoO3 as an OER and HER electrocatalyst requiring overpotentials of 296 and 198 mV at 10 mA cm−2, respectively.33 Later, Viswanathan et al. in 2023 developed a carbon cloth-supported Ni-doped CoMo ternary phosphate for the OER (272 mV@10 mA cm−2) and the HER (96 mV@10 mA cm−2).22 Similarly, Hou et al. reported carbon fiber cloth-supported oxygenated-CoS2-MoS2 heteronanosheets requiring an overpotential of 272 and 97 mV for the OER and HER at 10 mA cm−2, respectively.34 Considering the bifunctionality of layered double hydroxides (LDHs) and the advancement from P in an electrocatalyst, Mai et al. reported a cell voltage of 1.68 V at 50 mA cm−2 for the symmetrical electrolyzer constructed using Co5.0Mo1P/NiFe-LDH/NF.26 In addition, Hu et al. reported a Cu2+-guided electrodeposition strategy to synthesize trimetallic amorphous-CoMoO3/Cu for water electrolysis, benefiting from synergistic effects among the metals, achieving 10 mA cm−2 at a cell voltage of 1.5 V with the fabricated symmetrical electrolyzer.35 DFT studies reveal that Cu facilitates water electrolysis by augmenting the adsorption of H2O molecules on the electrocatalyst surface.35
Despite extensive research on developing electrocatalysts for water electrolysis, the technology is yet in the developmental stage without widespread adoption. The demerits include a lack of sufficiently efficient electrocatalysts that can withstand high current densities, along with issues of short durability and poor bifunctionality. In most cases, the cell voltages of the fabricated electrolyzers exceed 1.5 V to achieve the benchmark current density of 10 mA cm−2.14,36–38 The complex synthesis process involved in producing electrode materials results in higher production costs, hindering widespread adoption and reducing overall efficiency. Additionally, reporting OER and HER data in iR-corrected form prevents understanding the actual scenario of an electrocatalyst efficiency in a real-world application.14,25,33,34 During the fabrication of electrodes, the use of a binder also reduces the efficiency of an electrocatalyst by covering the active sites and reducing the conductivity. Therefore, it is essential to develop a cost-effective binder-free bifunctional electrocatalyst for water electrolysis that can withstand high current densities. In addressing these concerns, the current article outlines the design and in situ synthesis of trimetallic Cu, Co, and Mo-based bifunctional electrocatalysts on Ni foam (NF) for water electrolysis. This approach leverages the enhanced water adsorption ability and the excellent electronic conductivity of Cu along with the presence of high-valence Mo metals. The simple synthesis process of the electrocatalysts results in low material costs of $0.051, $0.053, and $0.052 cm−2 for CuCoMo-LDH, CuCoMo-P, and CuCoMo-S, respectively, as shown by the calculations in the ESI.† Furthermore, the anticipated reduction in electrocatalyst costs during bulk synthesis is expected to be substantial, while the cost of electricity consumption is projected to remain constant. The evaluated water electrolysis activities of Cu, Co, and Mo-based mono- and bimetallic materials indicate that the presence of all three metals in the electrocatalysts is crucial for achieving efficient water electrolysis. DFT studies of CuCoMo-P reveal that the electronic structure of the electrocatalyst is modulated by the synergistic effects among Cu, Co, and Mo metals. This modulation increases the density of electronic states near the Fermi level, resulting in enhanced electrical conductivity, resembling metallic-like properties. CuCoMo-P exhibited an overpotential of merely 246 mV at 10 mA cm−2 for the OER and 46 mV at −15 mA cm−2 for the HER without any iR-correction. The symmetrical alkaline electrolyzer CuCoMo-P (±) demonstrated the highest OWS in 1 M KOH, achieving an ultra-low cell voltage of 1.393 V at 10 mA cm−2. This performance surpasses that of CuCoMo-LDH (±) (1.427 V), CuCoMo-S (±) (1.540 V), and RuO2(+)‖Pt/C(−) (1.554 V). The high activity of CuCoMo-P for OWS shows its excellent bifunctionality for both the OER and the HER. Furthermore, all the electrocatalysts showed robust durability over 100 h at high applied current density for the OER, HER, and OWS. To our knowledge, the use of CuCoMo-P in water electrolysis represents a novel contribution to the field, as the fabricated symmetrical electrolyzer has demonstrated the lowest recorded cell potential at 10 mA cm−2 to date, surpassing previously reported results in the literature. The faradaic efficiency (FE) of the CuCoMo-P (±) electrolyzer is 98.7% for both the OER and the HER with an H2 generation rate of 3.4 mL min−1 and an estimated H2 production cost of $0.12255 per 1000 L, as shown by the calculation in the ESI.† This research work is expected to significantly advance sustainable energy technology and pave the way for further exploration of trimetallic phosphides in the field of efficient water electrolysis.
2. Results and discussion
2.1. Physicochemical analysis
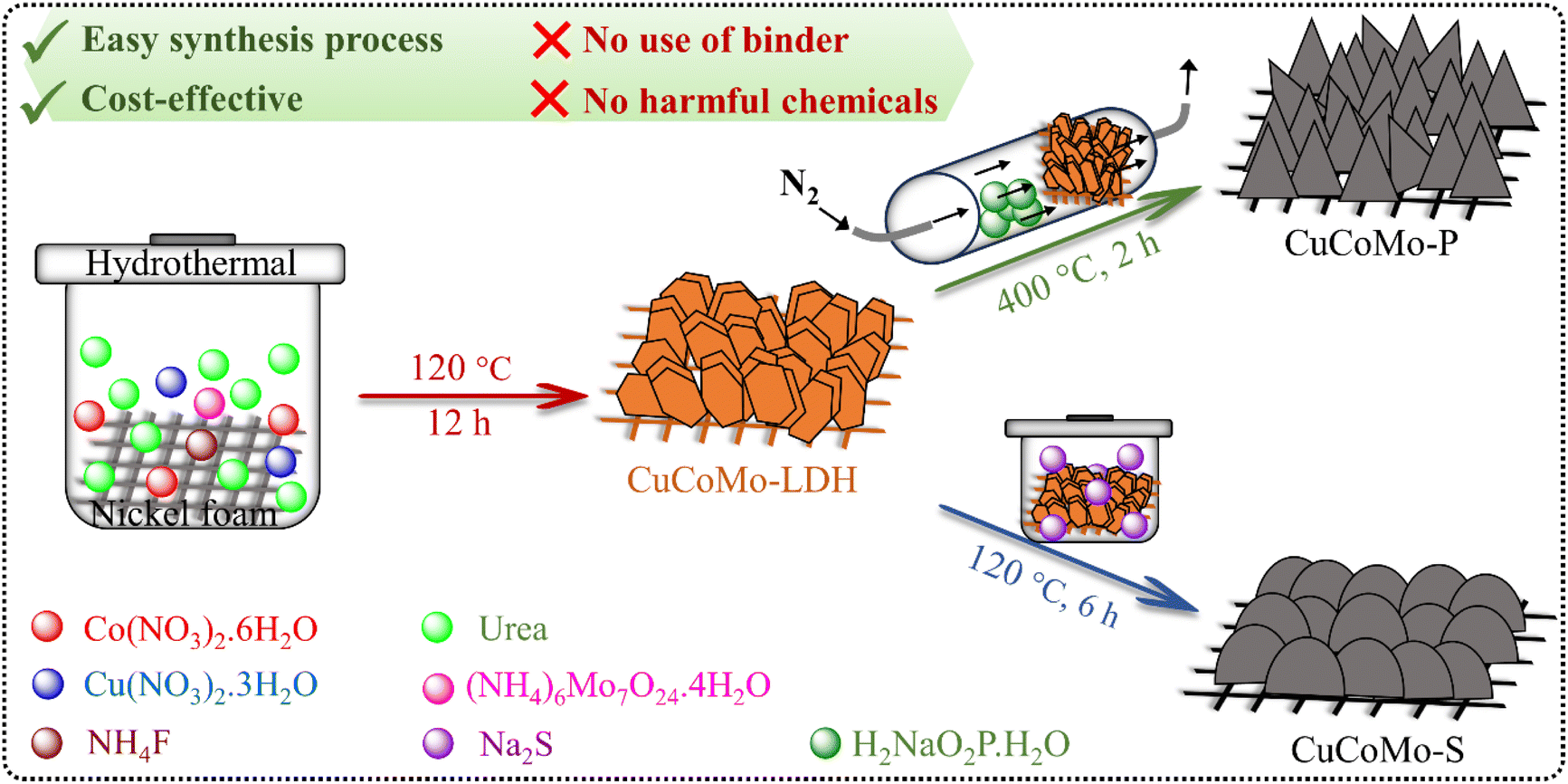
The detailed synthesis process of the electrocatalyst materials is provided in the synthesis of electrocatalyst materials section in the ESI.† For better comprehension, a schematic illustration of the synthesis process is provided in Scheme 1. Urea is employed in the synthesis process due to its slow and gradual hydrolysis within a confined vessel at temperatures nearing 100 °C. This controlled process enables the gradual release of OH− ions (eqn (1) and (2)). These ions react with metal ions to form a brucite-like layered M(OH)2 structure, as depicted in eqn (3).4 The use of NH4F in the synthesis process has demonstrated effectiveness in maintaining the structural integrity of the electrocatalyst, advancing by the adhesive nature of F− ions.4 However, no phase change of the materials was observed because of NH4F.| | | NH2CONH2 → NH4+ + NCO− | (1) |
| | | NCO− + 3H2O → HCO3− + NH4+ + OH− | (2) |
| | | M + 2OH− → M(OH)2 (M = metal ion) | (3) |
 |
| | Scheme 1 Schematic illustration of the synthesis process of CuCoMo-LDH, CuCoMo-P, and CuCoMo-S. | |
Initially, powder X-ray diffraction (PXRD) was performed on the synthesized CuCoMo-LDH, CuCoMo-P, and CuCoMo-S materials to examine their basic crystallographic structure and phase. The PXRD patterns show that the materials consist of multiple phases (Fig. S1a–c, ESI†). Specifically, CuCoMo-LDH is composed of Cu(OH)2 (ICDD no. 01-80-0656), CoMo-LDH (ICDD no. 46-0605), and MoO2.5(OH)0.5 (ICDD no. 014-0041) (Fig. S1a, ESI†).39–41 For CuCoMo-LDH, the sharp peaks at lower 2θ values of 11.76°, 23.06°, and 33.98° are ascribed to the characteristic (003), (006) and (009) basal planes, respectively. Broad asymmetric peaks observed at higher 2θ values further confirm the formation of the LDH structure.42 The PXRD pattern of CuCoMo-P (Fig. S1b, ESI†) shows peaks indexed to CuP2 (ICDD no. 01-076-1190),28 MoP (ICDD no. 24-0771),14 CoMoP (ICDD no. 32-0300),27 and CoMoP2 (ICDD no. 33-0428),14 evidencing the presence of multiple phases. Similarly, the PXRD pattern of CuCoMo-S (Fig. S1c, ESI†) comprises peaks corresponding to CuS (ICDD no. 01-075-2235),43 CoS2 (ICDD no. 03-0772),30 CoS (ICDD no. 19-0366),44 and MoS2 (ICDD no. 86-2308) phases.44 Additionally, the PXRD patterns of the mono- and bimetallic electrocatalysts (Fig. S2, ESI†) confirm that the monometallic electrocatalyst is Co(OH)2 (ICDD no. 30-0443).45 The bimetallic electrocatalyst containing Co and Mo metals, named CoMo–OH, is confirmed to have CoMo-LDH and MoO2.5(OH)0.5 phases.40,41 The Cu and Co-containing electrocatalyst, named CuCo–OH, consists of Cu(OH)2 and Co(OH)2 phases.39,45 Fourier-transform infrared (FT-IR) spectra of the electrocatalysts (Fig. S3, ESI†) were recorded to provide basic information on the chemical composition of the materials. The FT-IR spectrum of CuCoMo-LDH (Fig. S3, ESI†) shows a peak at a wavenumber of 521 cm−1, ascribed to the stretching vibration of the Co–O bond.4 The two peaks at 1280 and 1410 cm−1 are due to the carboxylate anion's symmetric and asymmetric stretching vibrations, respectively.4 The broad band observed between 3000 and 3500 cm−1 is ascribed to the stretching vibration of the O–H bond from the layered –OH groups and adsorbed interlamellar H2O molecules, and the weak peak at 1606 cm−1 is attributed to the bending vibration.4,46 The band ranging from 600 to 930 cm−1 is ascribed to the M–M, M–O, and M–OH bonds (where M = Cu, Co and Mo).46 The peak at 709 cm−1 in the FT-IR spectrum of CuCoMo-P (Fig. S3, ESI†) is attributed to the stretching vibrational mode of P–O–P linkages in PO43−.47 The broad peak at 1038 cm−1 is attributed to the stretching vibration of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in PO43−, while the band at 1307 cm−1 is attributed to the asymmetric stretching mode.48,49 The FT-IR spectrum of CuCoMo-S (Fig. S3, ESI†) shows a broad peak at around 620 cm−1, arising from the overlap of the vibrational modes of Cu–S, Co–S and Mo–S bonds.50–52 The band ranging from 1000 to 1200 cm−1 is attributed to the collective vibrations of S–O, SO, MO and Co–S bonds, where the oxides result from partial surface oxidation.50,52 The band across 3000 to 3500 cm−1 arises from the stretching vibration of the O–H bond in adsorbed and hydrated H2O molecules during the synthesis process, while the bending vibration of the O–H bond is observed at 1630 cm−1.50–52 Interestingly, the stretching and bending vibrations of the O–H bond are relatively less in the case of CuCoMo-P, likely owing to the heat treatment involved in its synthesis process.
O bond in PO43−, while the band at 1307 cm−1 is attributed to the asymmetric stretching mode.48,49 The FT-IR spectrum of CuCoMo-S (Fig. S3, ESI†) shows a broad peak at around 620 cm−1, arising from the overlap of the vibrational modes of Cu–S, Co–S and Mo–S bonds.50–52 The band ranging from 1000 to 1200 cm−1 is attributed to the collective vibrations of S–O, SO, MO and Co–S bonds, where the oxides result from partial surface oxidation.50,52 The band across 3000 to 3500 cm−1 arises from the stretching vibration of the O–H bond in adsorbed and hydrated H2O molecules during the synthesis process, while the bending vibration of the O–H bond is observed at 1630 cm−1.50–52 Interestingly, the stretching and bending vibrations of the O–H bond are relatively less in the case of CuCoMo-P, likely owing to the heat treatment involved in its synthesis process.
Field emission scanning electron microscopy (FE-SEM) images were recorded to comprehend the surface morphology and microstructure of the nickel foam-supported synthesized materials, as presented in Fig. 1. The surface morphology of CuCoMo-LDH (Fig. 1a and b) has several trapezoidal prisms with an average length of 1 μm and width of 259 nm, which are assembled to form half spheres with a diameter of 2.98 μm. The elemental mapping shown in Fig. 1c reveals the existence and distribution of Cu, Co, Mo, and O elements on the surface of CuCoMo-LDH. During the conversion of CuCoMo-LDH to CuCoMo-P at high temperatures, the trapezoidal prisms in CuCoMo-LDH transformed into pyramidal shapes after phosphorization, as shown in Fig. 1d and e. This transformation resulted in the creation of pores and increased surface roughness. The edge lengths of the pyramids range from 740 nm to 2.29 μm (Fig. 1e). The elemental mapping of CuCoMo-P shows the presence and distribution of Cu, Co, Mo and P elements on its surface (Fig. 1f). Besides, during the conversion of CuCoMo-LDH to CuCoMo-S under hydrothermal conditions, the surface morphology transformed into two-dimensional (2D) interconnected nanosheets measuring 2.24 μm in length and 260 nm in width, as shown in Fig. 1g and h. The elemental mapping of CuCoMo-S shows the presence of Cu, Co, Mo and S elements and their distribution on the surface in Fig. 1i. Additionally, high-resolution transmission electron microscopy (HR-TEM) images of the electrocatalysts were recorded to gain deep insight into their microstructures (Fig. 2). The HR-TEM image of CuCoMo-LDH (Fig. 2a) shows long-range lattice fringes corresponding to the (110) plane of Cu(OH)2, indicating a highly ordered structure. The selected area electron diffraction (SAED) pattern (Fig. 2b) also shows the diffraction rings corresponding to the (110) and (020) planes of Cu(OH)2 with interplanar spacings of 0.28 and 0.52 nm, respectively. The additional diffraction rings (200) and (110) corresponding to the planes of MoO2.5(OH)0.5 and CoMo-LDH, respectively, demonstrate interplanar spacings of 0.19 and 0.15 nm, respectively, consistent with the PXRD results. Fig. 2c shows the presence and distribution of Cu, Co, Mo, and O elements in CuCoMo-LDH material, and the comprehensive quantitative analysis of the elements is furnished in the energy-dispersive X-ray (EDX) spectroscopy spectrum (Fig. S4, ESI†). The HR-TEM image of CuCoMo-P (Fig. 2d) reveals the presence of the (004) plane related to the CoMoP2 phase. The lattice fringes appear relatively more disordered in CuCoMo-P, demonstrating its partial amorphous nature, which is further supported by the observed amorphous region in the HR-TEM image (Fig. 2d) and broad humps present in the PXRD pattern (Fig. S1b, ESI†). The SAED pattern of CuCoMo-P (Fig. 2e) shows diffraction rings corresponding to the (011) and (120) planes, which relate to the interplanar spacings of 0.44 and 0.22 nm for the CuP2 phase, whereas the rings indexed to (011) and (301) with an interplanar spacing of 0.32 and 0.18 nm are ascribed to the CoMoP phase. The ring indicated as (102) with an interplanar spacing of 0.25 nm is ascribed to the CoMoP2 phase. The elemental mapping images in Fig. 2f show the presence and distribution of Cu, Co, Mo and P elements in CuCoMo-P, and the corresponding quantitative analysis data are provided in the EDX spectrum (Fig. S5, ESI†). The HR-TEM image of CuCoMo-S (Fig. 2g) shows interplanar spacings of 0.32 and 0.28 nm, corresponding to the (101) and (103) planes of the CuS phase. The SAED pattern of CuCoMo-S (Fig. 2h) shows diffraction rings ascribed to the (204) and (330) planes of the CoS phase, while (104) and (008) planes correspond to the MoS2 and CuS phases, respectively. The elemental mapping images in Fig. 2i show the distribution of Cu, Co, Mo, and S elements on the electrocatalyst, with the corresponding quantitative analysis data provided in the EDX spectrum (Fig. S6, ESI†). To determine the specific surface area (SSA), all samples underwent characterization using the Brunauer–Emmett–Teller (BET) method. The results show that all samples exhibit type-IV BET isotherms (Fig. S7a, ESI†), with notable mesoporosity evident in the cumulative pore volume distribution curves (Fig. S7b, ESI†). Among them, CuCoMo-P exhibited the highest pore volume and the most favorable hysteresis loop, leading to the largest SSA of 21.35 m2 g−1. This was followed by CuCoMo-LDH with an SSA of 7.436 m2 g−1 and CuCoMo-S with 2.261 m2 g−1, as depicted in the inset of Fig. S7a, ESI.†
 |
| | Fig. 1 FE-SEM images of (a and b) CuCoMo-LDH, (d and e) CuCoMo-P and (g and h) CuCoMo-S; and elemental mapping of (c) CuCoMo-LDH, (f) CuCoMo-P and (i) CuCoMo-S. | |
 |
| | Fig. 2 HR-TEM images of (a) CuCoMo-LDH, (d) CuCoMo-P and (g) CuCoMo-S; SAED patterns of (b) CuCoMo-LDH, (e) CuCoMo-P and (h) CuCoMo-S; and EDX elemental mapping images of (c) CuCoMo-LDH, (f) CuCoMo-P and (i) CuCoMo-S. | |
To analyze the surface elemental composition and chemical valence states of the electrocatalysts, X-ray photoelectron spectroscopy (XPS) was employed. The survey spectrum of CuCoMo-P (Fig. S8a, ESI†) shows the presence of Cu, Co, Mo, P, and O elements, whereas the survey spectrum of CuCoMo-LDH (Fig. S8b, ESI†) shows the presence of Cu, Co, Mo, and O. The presence of O in the CuCoMo-P material originates from the physisorbed H2O molecules and partial oxidation of the surface. The deconvoluted Co 2p spectra of CuCoMo-P (Fig. 3a) and CuCoMo-LDH (Fig. S9a, ESI†) show a pair of spin–orbit doublets belonging to Co 2p3/2 and Co 2p1/2.23,53 The Co 2p peaks at binding energies (BEs) of 781.2 and 797.6 eV for CuCoMo-P are ascribed to the +3, whereas the peaks at BEs of 782.5 and 798.9 eV are ascribed to the +2 oxidation state of Co.4,53 Interestingly, the spin–orbit doublets of Co 2p for CuCoMo-LDH show a negative shift, indicating redistribution of electron density around Co atoms. The peaks corresponding to the +3 oxidation state of Co shift to BEs of 775.8 and 792.2 eV, whereas those corresponding to the +2 oxidation state shift to BEs of 778.4 and 794.8 eV.53 The deconvoluted Cu 2p spectra of CuCoMo-P (Fig. 3b) and CuCoMo-LDH (Fig. S9b, ESI†) reveal a pair of spin–orbit doublets corresponding to Cu 2p3/2 and Cu 2p1/2.12 The Cu 2p peaks at BEs of 932.3 and 952.3 eV for CuCoMo-P correspond to the +1 oxidation state, while the peaks at BEs of 933.6 and 953.6 eV correspond to the +2 oxidation state of Cu.12 In contrast, the Cu 2p peaks in CuCoMo-LDH show a shift to lower binding energies with peaks at 926 and 946 eV corresponding to the +1 oxidation state and peaks at 929 and 949 eV corresponding to the +2 oxidation state of Cu (Fig. S9b, ESI†).12,54 The spin–orbit doublets are also observed in the deconvoluted Mo 3d spectra of CuCoMo-P (Fig. 3c) and CuCoMo-LDH (Fig. S9c, ESI†), corresponding to Mo 3d5/2 and Mo 3d3/2. The Mo 3d peaks of CuCoMo-P at 231 and 234.3 eV correspond to the +4 oxidation state, whereas the peaks at 232.8 and 236.1 eV correspond to the +6 oxidation state of Mo.14,55 A negative shift in BE is observed in CuCoMo-LDH compared to CuCoMo-P. This negative shift in BE in CuCoMo-LDH compared to CuCoMo-P is attributed to the increased electron density around the Cu, Co and Mo atoms in CuCoMo-LDH, suggesting unique electron density modulation in both electrocatalyst systems.53 The peaks at 132.9 and 134.7 eV in the deconvoluted P 2p spectrum of CuCoMo-P (Fig. 3d) are attributed to P 2p3/2 and P2p1/2 from the metal-phosphorus (M–P) bond.23,53 Another peak at 135.3 eV is from the phosphorus–oxygen (P–O) bond of the phosphates, resulting from the surface oxidation of phosphide samples.27,53 The deconvoluted O 1 s spectrum of CuCoMo-LDH shows three peaks at 524.3, 526.3 and 527.65 eV corresponding to the metal–oxide (M–O), metal–hydroxide (M–OH) and adsorbed H2O molecules, respectively (Fig. S9d, ESI†).4,35 Thus, the synthesized materials have been thoroughly characterized through the aforementioned physicochemical analysis, revealing the presence of Cu, Co, and Mo constituents along with their corresponding oxidation states. This indicates that the developed materials are likely to enhance electrocatalytic efficiency for water electrolysis by modulating both the crystal and electronic structures.
 |
| | Fig. 3 XPS deconvoluted spectra of (a) Co 2p, (b) Cu 2p, (c) Mo 3d, and (d) P 2p of CuCoMo-P. | |
2.2. Oxygen evolution reaction study
Initially, the electrocatalytic OER performance of the electrocatalysts was evaluated by recording linear sweep voltammetry (LSV) curves in an aqueous 1 M KOH solution. Fig. 4a shows the LSV curves of the synthesized materials, showing that CuCoMo-P achieves an overpotential of 331 mV at a current density of 50 mA cm−2, with a Tafel slope of 84 mV dec−1 (Fig. 4b). In comparison, CuCoMo-LDH exhibits an overpotential of 307 mV and a Tafel slope of 73 mV dec−1, while CuCoMo-S shows 311 mV and 85 mV dec−1). Nevertheless, CuCoMo-P shows the lowest overpotential in the high current density region (above 300 mA cm−2). Impressively, all the reported electrocatalysts exhibit superior OER performance compared to the benchmark RuO2 (369 mV, 99 mV dec−1) and the bare Ni foam (440 mV and 134 mV dec−1). Unlike many recently developed materials, these electrocatalysts achieve exceptionally high current densities at 2 V (Fig. 4a), with CuCoMo-P reaching 644 mA cm−2 and CuCoMo-LDH achieving 575 mA cm−2, an essential factor for their practical application in real electrolyzer systems. A comparison of overpotentials and current densities with those of recently reported materials is shown in Table S1, ESI,† highlighting the excellent electrocatalytic activity of these materials. This enhanced OER performance of the electrocatalysts is mainly due to the synergistically enhanced electronic modulation among the Cu, Co, and Mo metals. To evaluate the synergistic effect, monometallic Co(OH)2, bimetallic CoMo–OH and CuCo–OH were synthesized and tested by recording LSV curves (Fig. S10a, ESI†). The results show that Co(OH)2, CoMo–OH and CuCo–OH exhibit higher overpotentials of 379, 353 and 399 mV at 50 mA cm−2, respectively, demonstrating the presence of synergistic effects on the trimetallic CuCoMo-LDH, CuCoMo-P and CuCoMo-S materials contributing to their superior performance. Another important factor influencing the catalytic activity of the materials is the electrochemically active surface area (ECSA). To evaluate the ECSAs of the synthesized materials, the double-layer capacitance (Cdl) method was employed, as shown in Fig. S11 and S12 of the ESI.† Among the developed materials, CuCoMo-P exhibits the highest ECSA value of 440 cm2, followed by CuCoMo-LDH (162.5 cm2) and CuCoMo-S (80 cm2), as shown in Table S2, ESI,† along with all other electrocatalysts. The results shown in Table S2, ESI,† reveal that the trimetallic materials exhibit significantly higher ECSA values compared to their mono- and bimetallic counterparts. The enhanced ECSA is mainly due to the improved active sites available for electrochemical reactions, further supporting synergistic behavior among the three metal constituents. The highest ECSA value of CuCoMo-P is further supported by its highest specific surface area as determined from BET analysis. The roughness factor (RF) of the electrocatalysts follows the same trend as the ECSA (Table S2, ESI†), showing the highest RF value for CuCoMo-P, followed by CuCoMo-LDH. The higher RF value of CuCoMo-P is consistent with its high OER activity as the potential scans toward higher values. To further evaluate the conductivity of the materials, electrochemical impedance spectroscopy (EIS) was performed at 1.53 V (vs. RHE), and the resulting Nyquist plots are shown in Fig. 4c. All three trimetallic electrocatalysts displayed lower charge transfer resistance (Rct) compared to both the benchmark RuO2 and bare NF, signifying faster reaction kinetics and more efficient charge transfer, as shown in the Nyquist plots in Fig. 4c. The equivalent circuit model for the Nyquist plots of the electrocatalysts, as shown in the inset of Fig. 4c, includes solution resistance (Rs), Rct, mass transfer resistance (Rm), and constant phase elements (CPEs). The presence of CPEs in the circuit indicates the non-ideal capacitive behavior of the electrocatalyst systems. Among the electrocatalysts, CuCoMo-P exhibited the lowest Rs of 0.5563 Ω and Rct of 0.2503 Ω, unveiling its potential as a highly active OER electrocatalyst (Table S3, ESI†). These low Rs and Rct values are in good agreement with its high OER activity, particularly as the potential scans extend to higher values. Additionally, the highest exchange current density (j0) of CuCoMo-P (25.65 mA cm−2) further supports its superior OER performance compared to CuCoMo-S and CuCoMo-LDH (Table S4, ESI†). Furthermore, to gain deeper insights into the intrinsic activity of the electrocatalysts for the OER, turnover frequencies (TOFs) were calculated from the areas of the oxidation curves (Fig. S13a–c, ESI†). CuCoMo-P exhibited the highest TOF of 1.977 s−1 at 1.7 V, 6.5 and 13.7 times higher than those of CuCoMo-LDH and CuCoMo-S, respectively (Fig. S13d, ESI†). The active site counts and TOF values for the electrocatalysts are given in Table S5, ESI.† In addition to catalytic activity, stability is a crucial aspect of an electrocatalyst for practical applications. To evaluate durability, the electrocatalysts were tested using the chronopotentiometry (CP) technique, and the corresponding CP profiles are shown in Fig. 4d. All electrocatalysts exhibited strong stability over 100 h at a current density of 50 mA cm−2. After observing the encouraging stability results of the CuCoMo-P, CuCoMo-LDH and CuCoMo-S catalysts, the used electrocatalysts underwent subsequent durability testing at significantly high current densities of 100 and 200 mA cm−2 for an additional 9 hours, as shown in the inset of Fig. 4d. The LSVs recorded after the CP test are shown in Fig. S14a, ESI.† As anticipated, all three catalysts demonstrated excellent stability (Fig. 4d), although a slight decrease in stability was observed for CuCoMo-LDH, while CuCoMo-S exhibited a significant improvement. Though CuCoMo-S showed lower catalytic activity initially, its performance noticeably increased after the stability test.
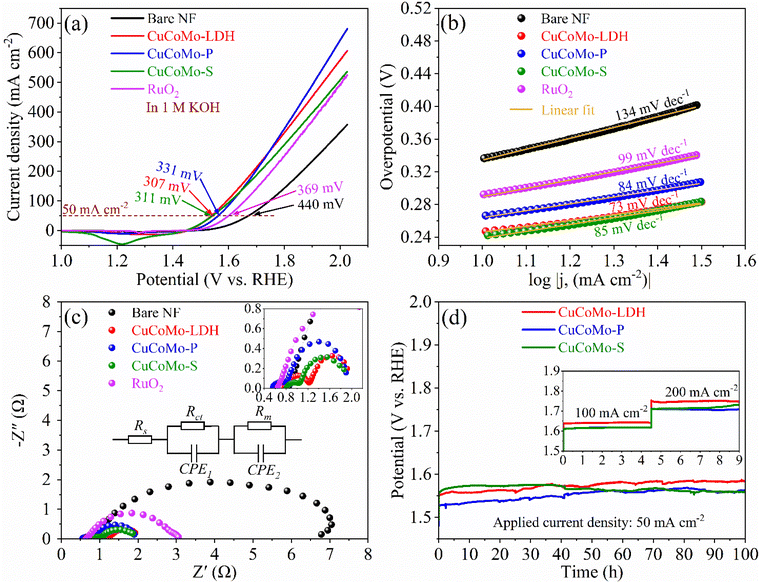 |
| | Fig. 4 OER studies: (a) LSV curves at a 2 mV s−1 scan rate, (b) Tafel plots, (c) Nyquist plots, and (d) chronopotentiometric stability test profiles. | |
The OER analysis shows that CuCoMo-P exhibited superior OER activity compared to CuCoMo-LDH and CuCoMo-S. This enhancement can be attributed to several factors: (i) metal phosphides exhibit higher electronic conductivity compared to metal LDHs and metal sulfides, enabling improved charge transport rates during the OER, as demonstrated by the experimental EIS data, (ii) the electronic modulations resulting from the interaction between the metals and phosphide counter ions enhance the active sites in CuCoMo-P and maximize their catalytic capabilities, as evidenced by the experimental ECSA data, (iii) the unique bonding characteristics between the metal and phosphorus can stabilize the reaction intermediates, lowering the energy barrier for the OER, which is further discussed in the later section of DFT calculations, and (iv) during the OER process in an alkaline medium, the surface of the metal phosphide is expected to undergo oxidation, resulting in the formation of an in situ oxidized metal hydroxide or (oxy)hydroxide. This phase may exhibit unusual amorphous or metastable characteristics, which can lead to enhanced catalytic activity.23 These factors collectively enhance the catalytic activity and stability of CuCoMo-P, offering advantages over CuCoMo-LDH and CuCoMo-S.
2.3. Hydrogen evolution reaction study
The HER LSV curves of the electrocatalysts are shown in Fig. 5a, with a detailed comparison of overpotentials at various current densities shown in Table S6, ESI.† CuCoMo-P exhibits the highest current density and the lowest overpotential among the developed materials, requiring overpotentials of only 45 and 117 mV at current densities of −15 and −50 mA cm−2, respectively. This performance surpasses that of CuCoMo-LDH, which demands 79 mV at −10 mA cm−2 and 215 mV at −50 mA cm−2, and CuCoMo-S, which demands 167 mV at −10 mA cm−2 and 265 mV at −50 mA cm−2. The HER catalytic activity of CuCoMo-P is close to that of Pt/C/NF (22 mV at −15 mA cm−2 and 83 mV at −50 mA cm−2) and much higher than that of bare Ni foam (230 mV at −10 mA cm−2). In addition, to gain insights into the HER kinetics and underlying mechanisms, the Tafel plots were derived from the respective LSV curves, as shown in Fig. 5b. Among the studied electrocatalysts, CuCoMo-P exhibited the lowest Tafel slope of 132 mV dec−1 compared to CuCoMo-LDH (152 mV dec−1) and CuCoMo-S (184 mV dec−1), indicative of its superior HER kinetics, while Pt/C/NF exhibited 49 mV dec−1 and the bare Ni foam substrate exhibited a Tafel slope of 271 mV dec−1 in the low potential region and 157 mV dec−1 in the high potential region, respectively. The higher Tafel slope of bare NF in the low potential region shows its sluggish kinetics toward the HER. The consistent LSV curves of the electrocatalysts across all potentials signify their reliable HER activity. Furthermore, comparing the HER activity of the electrocatalysts with recent results (Table S6, ESI†) shows better activity in most cases, highlighting their superior performance for the HER. The admirable HER activity of the electrocatalysts is attributed to the synergistically enhanced electronic modulations among Cu, Co and Mo metals. The existence of these electronic modulations is evidenced by XPS analysis and DFT calculations, which are discussed in the following section. In addition, as observed from Fig. S10b, ESI,† the lower HER activity of monometallic Co(OH)2 (350 mV at −50 mA cm−2), bimetallic CoMo–OH (325 mV at −50 mA cm−2), and CuCo–OH (345 mV at −50 mA cm−2) compared to CuCoMo-LDH, CuCoMo-P and CuCoMo-S indicates the importance of synergistic effects among Cu, Co, and Mo metals in enhancing HER activity. To assess the charge transfer resistance of the developed materials, Nyquist plots were obtained from EIS analysis, as shown in Fig. 5c. The equivalent circuit model for Nyquist plots of the electrocatalysts, presented in the inset of Fig. 5c, includes Rs, Rct, and a CPE, where the CPE corresponds to the non-linear capacitor behavior of the electrocatalysts. CuCoMo-P exhibits the lowest Rct value of 1.61 Ω, followed by CuCoMo-LDH (10.04 Ω) and CuCoMo-S (47.66 Ω) (Table S7, ESI†). The lowest Rct value of CuCoMo-P accounts for its faster charge transfer kinetics at the electrode–electrolyte interface, resulting in faster HER kinetics. This is consistent with the trend observed from the Tafel slope values. The highest exchange current density (j0) value of CuCoMo-P (7.97 mA cm−2) further indicates its excellent HER activity among the examined electrocatalysts (Table S4, ESI†).
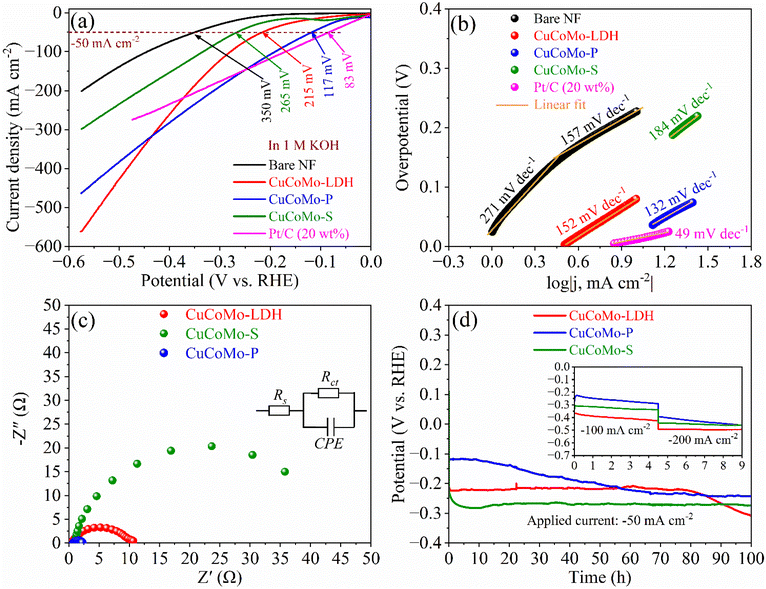 |
| | Fig. 5 HER studies: (a) LSV curves at a 2 mV s−1 scan rate, (b) Tafel plots, (c) Nyquist plots, and (d) chronopotentiometric stability test profiles. | |
In addition to the reduced overpotentials and Tafel slopes that indicate superior HER catalytic activity, long-term durability is another essential consideration for real-world applications. Therefore, the HER durability of the electrocatalysts was evaluated by performing a chronopotentiometry stability test at −50 mA cm−2 for 100 h, with the corresponding CP profiles presented in Fig. 5d. Among all the examined electrocatalysts, CuCoMo-P retained the highest HER activity even after 100 h of CP testing, as observed from the post-CP LSV curves in Fig. S14b, ESI.† In addition, similar to the OER stability tests, the used electrocatalysts underwent rigorous stability testing at high current densities of −100 and −200 mA cm−2 for an additional 9 hours, as shown in the inset of Fig. 5d. The results showed encouraging durability, with CuCoMo-P maintaining superior performance among the electrocatalysts tested.
2.4. Density functional theory calculations
DFT calculations were conducted to explicitly investigate and gain a deeper understanding of the HER and OER processes in CuCoMo-P. Fig. 6a–e provide the pictorial representations of the catalyst studied, including key intermediates involved in the HER and OER on the electrocatalyst surfaces. The general pathway of the HER can be depicted using a three-state diagram: beginning with a primary state of H+ + e−, followed by adsorption of the H* intermediate, and culminating in the formation of H2 as the final product. The HER activity of the catalyst was evaluated using hydrogen adsorption free energy (ΔGH*), where an ideal value of |ΔGH*| should be close to zero following the Sabatier principle. Fig. 6f shows the optimal ΔGH* value (−0.11 eV) for the adsorption of H* intermediates onto CuCoMo-P, signifying an appropriate adsorption strength of H* intermediates, which is close enough to zero, proving an ideal electrocatalyst for the HER. Interestingly, the ΔGH* value of CuCoMo-P is much closer to zero than those recently reported for Co2P@Fe2P (−0.17 eV) and CoMoP2-PV (−0.29 eV).14,23 The Tafel slope in the HER indicated that the rate-determining step (RDS) is 2H* → 2* + H2, further supported by the calculated ΔGH*. Additionally, the distances between hydrogen and metal atoms Cu and Mo were 1.66 and 1.83 Å, respectively. This low energy barrier and minimal distance between hydrogen and Cu atoms resulted in a favorable ΔGH* and faster HER kinetics, as shown in Fig. 6f.
 |
| | Fig. 6 Schematic representations are shown for (a) the electrocatalyst, intermediate structures of the electrocatalyst (b) with H for the HER, (c) with OH, (d) with O, and (e) with OOH for the OER, (f) the free energy profile for the HER, and (g) the free energy profile for the OER. The red, green, and blue arrows represent the a, b, and c vectors, respectively. | |
For the OER, the conventional absorbate evolution mechanism was applied to simulate the oxygen evolution process, widely recognized as the primary mechanism in this system. Fig. 6g illustrates the geometrical arrangements of the stable adsorbed intermediates. Transition metal atoms in CuCoMo-P exhibit high electroactivity and play a crucial role in alleviating these stable intermediates, enabling efficient variation during the OER. Fig. 6g shows the energetic progression of the OER pathway. Initially, the OH molecule adsorbs on the catalyst, resulting in OH*. Subsequently, O*, OOH*, and finally O2 are sequentially evolved. The OER exhibits a constant uphill drift at the standard potential (U = 1.23 V). The calculated energy barrier for the reaction [3OH− + OH] to [2OH− + H2O + O] is 0.945 eV. In addition, the highest barrier in the OER occurs during the transformation from [OOH] to [O2], with a value of 3.00 eV, indicating the RDS. Therefore, the calculated energy profiles (Fig. 6g) of CuCoMo-P indicate that O2 formation is the RDS. Moreover, the investigation explores the structural alterations of the electrocatalyst and adsorbed molecules and intermediates at each step of the OER. At each reaction step, the intermediate species migrates from its initial position to the catalyst surface, binding to a Cu atom adjacent to Co and Mo. The increase in the bond distance from OH* to the OOH* intermediate underscores a strong interaction between the intermediate and the catalyst. However, a slight increase in the bond distance is observed between the adsorbed moieties and the catalyst, from OH* to O*, which decreases further in the case of OOH* (Fig. S15, ESI†). This alternation in the bond distance highlights the strong adsorption of the OER intermediate onto the catalyst.
An electronic property analysis was conducted to investigate the intrinsic relationship between the electronic structure and catalytic activity of the electrocatalyst toward the HER and OER by calculating the density of states (DOS) and the projected density of states (PDOS) of CuCoMo-P and intermediates involved in the HER and OER (Fig. S16, ESI†). The DOS of CuCoMo-P shows a higher number of electronic states at energy levels close to the Fermi level (EF), resembling metallic properties, suggesting higher electrical conductivity and improved electron transport during electrochemical reactions. For effective comparison, the PDOS within the energy range of −24 to 4 eV is considered. Furthermore, the electron densities at EF highlight the electroactive region. For the metal, a robust perturbation to the electronic distribution of the lattice is observed, resulting in a more electron-rich catalyst surface. Analysis of the PDOS shows that the metal-d and P-P states are primarily localized between −7.7 and 5 eV. The metal sites dominate the main electronic contribution near the EF. The O-p state shows electron-rich characteristics at a lower energy site. Additionally, metal-d orbitals exhibit good overlap with O-p orbitals, supporting efficient site-to-site electron transfer and stable bonding. The O-sites also exhibit a notable upward trend from −3.93 eV to −4.03 eV, indicating a significant electron transfer from the O sites to neighboring metal sites, implying high electroactivity. Moreover, a clear linear correlation exists between OH* and OOH*, indicating significant electron transfer and transformation of intermediates during the OER.
2.5. Overall water splitting study
After conducting rigorous studies on the OER and HER activities of the electrocatalysts, the impressive electrocatalytic activities facilitates the development of three symmetrical alkaline electrolyzers: CuCoMo-LDH (±), CuCoMo-P (±) and CuCoMo-S (±). Fig. 7a shows the LSV curves of the fabricated electrolyzers, recorded in aqueous 1 M KOH solution at a scan rate of 2 mV s−1. Among the three, the CuCoMo-P (±) electrolyzer exhibited the lowest recorded cell potential of 1.393 V to reach the benchmark current density of 10 mA cm−2, followed by CuCoMo-LDH (±) (1.427 V at 10 mA cm−2) and CuCoMo-S (±) (1.599 V at 15 mA cm−2). Impressively, to achieve a high current density of 50 mA cm−2, the CuCoMo-P (±) electrolyzer demands only 1.633 V, while CuCoMo-LDH (±) and CuCoMo-S (±) required 1.732 V and 1.818 V, respectively. Remarkably, the CuCoMo-P (±) electrolyzer, with a 1 cm2 active area, exhibited the highest current density of 250 mA cm−2 at 2 V, surpassing most of the reported results. This performance of the electrolyzer is superior to the majority of the findings that have been recently reported.26,38,56 Hence, we anticipate that the alkaline or anion exchange membrane electrolyzer stack fabricated with the CuCoMo-P (±) material is expected to show high efficiency. These results are also compared with those of the electrolyzer fabricated using the benchmark RuO2 as the anode and Pt/C (20 wt%) as the cathode, where RuO2(+)‖Pt/C(−) required 1.554 and 1.741 V cell potentials at 10 and 50 mA cm−2, respectively. The electrolyzer constructed using bare NF as both the anode and the cathode (bare NF(±)) required a high cell potential of 1.772 V at 10 mA cm−2, revealing low OWS capability compared to deposited materials. Furthermore, the OWS performance of symmetrical alkaline electrolyzers constructed using monometallic Co(OH)2, bimetallic CoMo–OH and CuCo–OH was also analyzed (Fig. S10c, ESI†). All three electrolyzers, Co(OH)2(±), CoMo–OH(±) and CuCo–OH(±), underperformed compared to the trimetallic CuCoMo-LDH (±), CuCoMo-P (±) and CuCoMo-S (±) electrolyzers, requiring cell potentials of 1.928, 1.881 and 1.926 V at 50 mA cm−2, respectively. The obtained results, summarized in Table S8, ESI,† are compared with those of recently reported materials, showing the superior OWS performance of the CuCoMo-P (±) electrolyzer. To check the durability of CuCoMo-LDH (±), CuCoMo-P (±) and CuCoMo-S (±) electrolyzers, a CP stability test was conducted using fresh electrode materials, as shown in Fig. 7b. After 100 h of continuous electrolysis at a current density of 50 mA cm−2, negligible decay was noticed for CuCoMo-P (±), maintaining a current density of 228 mA cm−2 at 2 V (Fig. S14c, ESI†). Meanwhile, a noticeable enhancement in activity was observed for CuCoMo-S (±) (Fig. S14c, ESI†). In addition, the used electrolyzers were further subjected to rigorous durability tests at even higher current densities of 100 and 200 mA cm−2 for an additional 9 h, as shown in the inset of Fig. 7b. Notably, no significant decay was observed, even under these high current conditions, indicating that the electrode materials did not undergo deterioration. The slight increase in potential in the curves can be due to a decrease in KOH concentration near the electrode surface and the partial blockage of active sites by the formed gas bubbles. Fig. 7c shows the potential difference between the half-cell anodic OER and cathodic HER potentials of the developed materials at 50 mA cm−2, indicating that these observed potentials closely match those in the fabricated electrolyzers (Fig. 7a). This alignment shows the reliability of the material testing. The electrolysis performance of CuCoMo-P (±) was further investigated using a 1.5 V AA battery, as shown in Fig. 7d and Video S1,† where the electrolyzer successfully initiated the splitting of alkaline water into H2 and O2 gases. According to the LSV data for the CuCoMo-P (±) electrolyzer in Fig. 7a, a cell potential of 1.5 V produces a current density of 21 mA cm−2, accompanied by the visual evolution of H2 and O2 bubbles. Finally, the faradaic efficiency (FE) of the CuCoMo-P (±) electrolyzer was calculated and is presented in Fig. S17, ESI and Video S2.† The FE for H2 and O2 generation was found to be 98.7%.
 |
| | Fig. 7 OWS studies: (a) LSV curves of the fabricated electrolyzers at a 2 mV s−1 scan rate, (b) chronopotentiometric stability test of the fabricated electrolyzers, (c) half-cell HER and OER LSV curves plotted together, and (d) OWS performed with the CuCoMo-P(±) electrolyzer powered by a 1.5 V AA battery. | |
These rigorous studies show that CuCoMo-P (±) is highly suited to use in both alkaline and anion exchange membrane electrolyzers, supporting large-scale H2 production at high operating currents. The H2 production cost using the CuCoMo-P (±) electrolyzer is approximately $0.12255 per 1000 L, with a generation rate of 3.4 mL min−1, as shown in the calculations in the ESI.†
3. Conclusions
In this study, a series of trimetallic Cu, Co and Mo-based bifunctional electrocatalysts, specifically CuCoMo-LDH, CuCoMo-P, and CuCoMo-S, were synthesized via a simple and cost-effective hydrothermal method, followed by heat treatment for efficient overall water electrolysis to produce H2. Electrochemical studies show that the CuCoMo-P material exhibits exceptional bifunctional catalytic activity for both the OER and the HER, exceeding the catalytic activity of benchmark RuO2 for the OER and Pt/C for the HER, as well as most recently reported materials. Especially, the symmetrical alkaline electrolyzer fabricated with CuCoMo-P (±) demonstrated exceptional OWS performance, requiring a record low cell potential of 1.393 V at a current density of 10 mA cm−2. Following closely, CuCoMo-LDH (±) exhibited a cell potential of 1.427 V under the same conditions. Both the electrolyzers' performance exceeds that of the benchmark RuO2(+)‖Pt/C(−), which requires a cell potential of 1.554 V at the same current density. Additionally, all three developed trimetallic electrolyzers showed exceptional stability, maintaining performance over 109 hours even at varied and high current densities. The exceptional bifunctional catalytic performance of CuCoMo-P is explained by density functional theory (DFT) calculations. DFT studies indicate that electronic modulations among the metal atoms enhance the density of electronic states near the Fermi level, resulting in improved electrical conductivity and favorable reaction pathways for both the OER and the HER. The estimated H2 production cost of the CuCoMo-P (±) electrolyzer with 98.7% faradaic efficiency is approximately $0.12255 per 1000 L, with a generation rate of 3.4 mL min−1. This suggests that an alkaline or anion exchange membrane electrolyzer stack fabricated with the developed CuCoMo-P material is expected to achieve high efficiency for large-scale GH2 production at minimal cost.
Data availability
The data will be made available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr Gaddam Rajeshkhanna would like to acknowledge the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India (sanction order no: SRG/2021/001028), for providing the funding to carry out the research work.
Notes and references
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174 CrossRef CAS PubMed.
- H. Shi, T. Y. Dai, W. B. Wan, Z. Wen, X. Y. Lang and Q. Jiang, Adv. Funct. Mater., 2021, 31, 2102285 CrossRef CAS.
- J. M. Ibrahim and S. Xiaoming, J. Energy Chem., 2019, 34, 111 CrossRef.
- A. Borah, Sumit, S. Palaniyappan and G. Rajeshkhanna, Sustainable Energy Fuels, 2024, 8, 2265 RSC.
- H. Wang, T. Zhou, P. Li, Z. Cao, W. Xi, Y. Zhao and Y. Ding, ACS Sustain. Chem. Eng., 2018, 6, 380 CrossRef CAS.
- Q. Zhou, T. T. Li, J. Qian, Y. Hu, F. Guo and Y. Q. Zheng, J. Mater. Chem. A, 2018, 6, 14431 RSC.
- C. Panda, P. W. Menezes, M. Zheng, S. Orthmann and M. Driess, ACS Energy Lett., 2019, 4, 747 CrossRef CAS.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305 CrossRef CAS PubMed.
- G. Rajeshkhanna, T. I. Singh, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 42453 CrossRef CAS PubMed.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- T. I. Singh, G. Rajeshkhanna, U. N. Pan, T. Kshetri, H. Lin, N. H. Kim and J. H. Lee, Small, 2021, 17, 2101312 CrossRef CAS PubMed.
- L. Zhang, W. Cai and N. Bao, Adv. Mater., 2021, 33, 2100745 CrossRef CAS PubMed.
- A. B. Sumit, S. Palaniyappan and G. Rajeshkhanna, Nanoscale, 2024, 16, 14020 RSC.
- L. Zheng, C. Liu, W. Zhang, B. Gao, T. Yan, Y. Zhang, X. Cao, Q. Gao and Y. Tang, J. Mater. Chem. A, 2024, 12, 1243 RSC.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196 CrossRef CAS.
- E. Loni, A. Shokuhfar and M. H. Siadati, Catal. Surv. Asia, 2021, 25, 114 CrossRef CAS.
- N. Zaman, T. Noor and N. Iqbal, RSC Adv., 2021, 11, 21904 RSC.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- X. P. Li, C. Huang, W. K. Han, T. Ouyang and Z. Q. Liu, Chin. Chem. Lett., 2021, 32, 2597 CrossRef CAS.
- X. Cao, T. Wang and L. Jiao, Adv. Fiber Mater., 2021, 3, 210 CrossRef CAS.
- L. Zhang, H. Zhao, S. Xu, Q. Liu, T. Li, Y. Luo, S. Gao, X. Shi, A. M. Asiri and X. Sun, Small Struct., 2020, 2, 2000048 CrossRef.
- P. Viswanathan and K. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 16571 CrossRef CAS PubMed.
- G. Rajeshkhanna, A. Borah, T. I. Singh, T. H. Nguyen, V. A. Dinh, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2024, 12, 24656 RSC.
- Y. Hu, T. Shen, Z. Song, Z. Wu, S. Bai, G. Liu, X. Sun, Y. Wang, S. Hu, L. Zheng and Y. F. Song, ACS Catal., 2023, 13, 11195 CrossRef CAS.
- M. A. U. Din, S. Irfan, H. M. A. Sharif, S. Jamil, M. Idrees, Q. U. Khan, G. Nazir and N. Cheng, Int. J. Hydrogen Energy, 2023, 48, 5755 CrossRef.
- W. Mai, Q. Cui, Z. Zhang, K. Zhang, G. Li, L. Tian and W. Hu, ACS Appl. Energy Mater., 2020, 3, 8075 CrossRef CAS.
- Y. Lu, X. Zheng, Y. Liu, J. Zhu, D. Li and D. Jiang, Inorg. Chem., 2022, 61, 8328 CrossRef CAS PubMed.
- S. Riyajuddin, K. Azmi, M. Pahuja, S. Kumar, T. Maruyama, C. Bera and K. Ghosh, ACS Nano, 2021, 15, 5586 CrossRef CAS PubMed.
- J. Choi, K. Morey, A. Kumar, D. Neupane, S. R. Mishra, F. Perez and R. K. Gupta, Mater. Today Chem., 2022, 24, 100848 CrossRef CAS.
- A. A. Yadav, Y. M. Hunge and S. W. Kang, Surf. Interfaces, 2021, 23, 101020 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871 CrossRef CAS PubMed.
- Y. Yu, Q. Chen, J. Li, P. Rao, R. Li, Y. Du, C. Jia, W. Huang, J. Luo, P. Deng, Y. Shen and X. Tian, J. Colloid Interface Sci., 2022, 607, 1091 CrossRef CAS PubMed.
- L. Yu, Y. Xiao, C. Luan, J. Yang, H. Qiao, Y. Wang, X. Zhang, X. Dai, Y. Yang and H. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 6890 CrossRef CAS PubMed.
- J. Hou, B. Zhang, Z. Li, S. Cao, Y. Sun, Y. Wu, Z. Gao and L. Sun, ACS Catal., 2018, 8, 4612 CrossRef CAS.
- E. Hu, Y. Yao, Y. Chen, Y. Cui, Z. Wang and G. Qian, ACS Appl. Energy Mater., 2021, 4, 6740 CrossRef CAS.
- Z. Pan, Z. Tang, M. Yaseen and Y. Zhan, ACS Appl. Nano Mater., 2022, 5, 16793 CrossRef CAS.
- Q. Sun, Y. Tong, P. Chen, B. Zhou and X. Dong, ACS Sustain. Chem. Eng., 2021, 9, 4206 CrossRef CAS.
- S. Yuan, M. Xia, Z. Liu, K. Wang, L. Xiang, G. Huang, J. Zhang and N. Li, Chem. Eng. J., 2022, 430, 132697 CrossRef CAS.
- Z. Li, Y. Xin, Z. Zhang, H. Wu and P. Wang, Sci. Rep., 2015, 5, 10617 CrossRef CAS PubMed.
- J. Bao, Z. Wang, J. Xie, L. Xu, F. Lei, M. Guan, Y. Huang, Y. Zhao, J. Xia and H. Li, Inorg. Chem. Front., 2018, 5, 2964 RSC.
- A. Wang, M. Zhang, Z. Huang, H. Liu, Z. Wang, Z. Song, W. Zhou, G. Zhu and S. Shao, J. Solid State Chem., 2022, 307, 122760 CrossRef CAS.
- K. Klemkaite, I. Prosycevas, R. Taraskevicius, A. Khinsky and A. Kareiva, Cent. Eur. J. Chem., 2011, 9, 275 CAS.
- G. Heidari, M. Rabani and B. Ramezanzadeh, Int. J. Hydrogen Energy, 2017, 42, 9545 CrossRef CAS.
- S. Mathew, J. H. Sim, R. Rajmohan, O. L. Li and Y. R. Cho, Electrochim. Acta, 2022, 403, 139586 CrossRef CAS.
- Y. S. He, D. W. Bai, X. Yang, J. Chen, X. Z. Liao and Z. F. Ma, Electrochem. Commun., 2010, 12, 570 CrossRef CAS.
- M. S. Mostafa and N. H. Mohamed, Egypt. J. Pet., 2016, 25, 221 CrossRef.
- I. Ahmed, R. Biswas, R. Sharma, V. Burman and K. K. Haldar, Front. Chem., 2023, 11, 1129133 CrossRef CAS PubMed.
- K. H. Kim, J. M. Jeong, S. J. Lee, B. G. Choi and K. G. Lee, J. Colloid Interface Sci., 2016, 484, 44 CrossRef CAS PubMed.
- P. Arunachalam, M. N. Shaddad, A. S. Alamoudi, M. A. Ghanem and A. M. Al-Mayouf, Catalysts, 2017, 7, 119 CrossRef.
- S. Nandhini and G. Muralidharan, J. Mater. Sci., 2022, 57, 5933 CrossRef CAS.
- P. Naveenkumar, P. G. Kalaignan, S. Arulmani and S. Anandan, J. Mater. Sci.: Mater. Electron., 2018, 29, 16853 CrossRef CAS.
- Y. Wang, M. Zhen, H. Liu and C. Wang, J. Solid State Electrochem., 2018, 22, 3069 CrossRef CAS.
- R. Na, K. Min, M. Kim, S. Min and S. H. Baeck, Adv. Sustainable Syst., 2023, 7, 2300130 CrossRef CAS.
- M. Chandra, K. Bhunia and D. Pradhan, Inorg. Chem., 2018, 57, 4524 CrossRef CAS PubMed.
- W. Xin, B. Liu, Y. Zhao, G. Chen, P. Chen, Y. Zhou, W. Li, Y. Xu, Y. Zhong and Y. A. Nikolaevich, Electrochim. Acta, 2022, 404, 139748 CrossRef CAS.
- S. J. Patil, N. R. Chodankar, S. K. Hwang, P. A. Shinde, G. S. R. Raju, K. S. Ranjith, Y. S. Huh and Y. K. Han, Chem. Eng. J., 2022, 429, 132379 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Gaddam
Rajeshkhanna
and
Gaddam
Rajeshkhanna