
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeryllium dinitride monolayer: a multifunctional direct bandgap anisotropic semiconductor containing polymeric nitrogen with oxygen reduction catalysis and potassium-ion storage capability†
Shuang
Ni‡
a,
Jiaxin
Jiang‡
b,
Weiyi
Wang
 c,
Xiaojun
Wu
c,
Zhiwen
Zhuo
*c and
Zhuo
Wang
*a
c,
Xiaojun
Wu
c,
Zhiwen
Zhuo
*c and
Zhuo
Wang
*a
aResearch Center of Laser Fusion, China Academy of Engineering Physics, Mianyang 621900, People's Republic of China. E-mail: wangzhuo_lfrc@yeah.net
bAnhui Province Key Laboratory for Control and Applications of Optoelectronic Information Materials, Key Laboratory of Functional Molecular Solids Ministry of Education, Department of Physics, Anhui Normal University, Wuhu, Anhui 241000, China
cHefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, 96 Jinzhai Rd., Hefei 230026, People's Republic of China. E-mail: zhuozw@ustc.edu.cn
First published on 25th February 2025
Abstract
Searching for two-dimensional multifunctional polynitride materials with novel properties and practical applications presents an attractive challenge. The global energy minimum of the beryllium dinitride monolayer (α-2D-BeN2) was predicted using a global structure search method and first-principles theory. With penta-, hexa-, and hepta-atomic rings and an N4 tetramer in its planar anisotropic structure, α-2D-BeN2 monolayer exhibited lattice dynamic stability, excellent thermal stability, a direct bandgap of 1.82 eV, high carrier mobilities, visible light absorption, a large in-plane Poisson's ratio ranging from 0.228 to 0.368, promising oxygen reduction catalysis, and outstanding potassium storage capability with an ultrahigh specific capacity (2895 mA h g−1), a good voltage range (0.280–0.008 V), and a low migration barrier energy (0.109–0.146 eV). Therefore, the α-2D-BeN2 monolayer is expected to be an anisotropic multifunctional material with potential applications in various fields, such as semiconductors, visible-light detectors, donors in solar cells, ductile materials, iontronic devices, and potassium-ion anode materials, thereby expanding the possibilities for polynitride materials.
1. Introduction
As an extension and frontier in structure and bonding, polynitride materials and polymeric nitrogen allotropes with polymeric Nm substructure units (m > 2) suffer an innate thermodynamic challenge due to the exceptionally high stability of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in nature. Through experimental methods involving conventional high temperature and high pressure, as well as new approaches, various forms have been identified, including an N3 trimer in azides,1,2 an N5 pentatomic ring (CsN5),3 an N6 hexatomic ring (KN3)4 and short chains (ScN3).5 New methods have also led to the discovery of linear N5+ and N8− chains,6,7 infinite [N]n chains (MN4, M = Be, Zn, Mg, Fe)8–12 [N5]n rattan (TiN5),13 polymeric two-dimensional (2D) networks (e.g. supernitride LaN8,14 GaN5,15 ScN5,5 LP-N,16 HLP-N,17 bp-N),18,19 and three-dimensional frameworks (cg-N, t-N).20,21 Owing to theoretical methods, a more diverse range of polynitrides with intricate polymeric structures and extended properties9,22–30 have been identified, such as MnN4 (superconductivity and magnetism),31 ZnN4 (Dirac semimetal),10 and WN6 (superhardness).32 Triggered by the high-energy state of their polymeric substructures, polynitrides often exhibit potential applications in energy storage, propellants, and explosives. However, this high-energy characteristic also poses a challenge for polynitrides in applications requiring high stability, high quality and low spillover. Thus, discovering new materials to expand and develop the polynitride family in terms of structure, properties, and applications remains an attractive challenge, with the aim of overcoming existing limits.
N bond in nature. Through experimental methods involving conventional high temperature and high pressure, as well as new approaches, various forms have been identified, including an N3 trimer in azides,1,2 an N5 pentatomic ring (CsN5),3 an N6 hexatomic ring (KN3)4 and short chains (ScN3).5 New methods have also led to the discovery of linear N5+ and N8− chains,6,7 infinite [N]n chains (MN4, M = Be, Zn, Mg, Fe)8–12 [N5]n rattan (TiN5),13 polymeric two-dimensional (2D) networks (e.g. supernitride LaN8,14 GaN5,15 ScN5,5 LP-N,16 HLP-N,17 bp-N),18,19 and three-dimensional frameworks (cg-N, t-N).20,21 Owing to theoretical methods, a more diverse range of polynitrides with intricate polymeric structures and extended properties9,22–30 have been identified, such as MnN4 (superconductivity and magnetism),31 ZnN4 (Dirac semimetal),10 and WN6 (superhardness).32 Triggered by the high-energy state of their polymeric substructures, polynitrides often exhibit potential applications in energy storage, propellants, and explosives. However, this high-energy characteristic also poses a challenge for polynitrides in applications requiring high stability, high quality and low spillover. Thus, discovering new materials to expand and develop the polynitride family in terms of structure, properties, and applications remains an attractive challenge, with the aim of overcoming existing limits.
Today, a diverse range of 2D materials has been developed both experimentally and theoretically, such as graphene, phosphorene, borophene, h-BN, and MoS2,33–38 possessing novel physical and chemical properties, especially their high specific surface areas with rich active sites for ion and molecular species absorption/loading in ion storage and catalysis. The exploration of 2D polynitrides is still in its early stages but shows great potential for extended properties and applications. Several 2D polynitride materials have been reported, including 2D KN3 with an N3 trimer,39 h-MN2 (M = Be, Mg) with an N4 tetramer,40,41 h-MN3 (M = Be, Ge)42 with N6 hexa-atomic rings, and 2D MN4 (M = Be, Mg, Ir, Rh, Ni, Cu, Au, Pd, and Pt)43,44 with infinite [N]n chains. For instance, the 2D Be–N system, consisting of BeN, Be3N4, Be2N3, BeN2, BeN3, and BeN4, has been explored via the global structure search method.34,42,45 The investigations suggest that the polymerization degree of N atoms increases with the N atom ratio. Among them, the h-BeN2 monolayer with isolated “Y”-shaped N4 tetramers in structure was predicted to exhibit a direct bandgap of 2.23 eV, excellent carrier mobilities, ultrahigh on-state current in transistors,46 ferromagnetic half-metallicity via fluorination,47 oxygen reduction catalytic ability48 and water photocatalytic ability.41 Recently, the experimental progress of the 2D BeN4 monolayer (beryllonitrene),8 which feasibly could have exfoliated from the synthesized new layered material BeN4via decompression technology, raises hope for practical 2D polynitrides with wide-ranging applications. Beryllonitrene is a planar monolayer with parallel armchair-like infinite [N]n chains, possessing anisotropic structures with novel physical and chemical properties, such as anisotropic Dirac cone,8 high lattice thermal conductivity,49 layer-dependent electronic and optical properties,50 CO2 capture,51 hydrogen storage,52 and Li/Na/Ca ion storage for ion batteries.44 To date, practical 2D polynitride materials are still rare, waiting for new discovery and proposed candidates.
Given the light mass of Be and N elements, the high specific surface areas with rich active sites for 2D Be–N structures, and the isoelectronic formula of BeN2 to h-BN (both are 4e per atom) with high strength and stability, in this study, a novel single-atom-thick anisotropic 2D polynitride with 5-, 6-, 7-membered rings and isolated “U”-shaped N4 tetramers in its planar structure, beryllium dinitride monolayer (α-2D-BeN2), is predicted as a global energy minimum by a 2D global structure search method. The thermodynamics, lattice dynamics, and thermal stability results indicate its good synthesizability and high structural stability. The new structure exhibits a direct bandgap, good visible light absorption, a relatively large Poisson's ratio, promising oxygen reduction catalysis ability, and excellent potassium storage ability. The α-2D-BeN2 monolayer is expected to be a highly structurally stable multifunctional material with promising properties for wide-ranging applications in various fields.
2. Computational details
The simulation of structural optimization, energy and electronic structure was based on the first-principles density functional theory (DFT) code with the projector augmented wave (PAW)53 method. An energy cutoff of 520 eV was adopted. The electron exchange–correlation potential was powered by the Perdew–Burke–Ernzerhof (PBE) functional.54 The Brillouin zone was sampled with an 8 × 12 × 1 G-centered K-point grid for geometry optimization and electronic properties. The convergence criteria for energy and forces were set to 10−6 eV and 0.01 eV Å−1, respectively. To gain more accurate results, the Heyd–Scuseria–Ernzerhof (HSE06) functional was adopted for electronic properties.55 The DFT-D3 method56 was applied to describe the adsorption of species during the oxygen reduction reaction (ORR) and potassium atoms. The finite displacement method was used to investigate the lattice dynamic stability via phonon calculation.57 The ab initio molecular dynamics (AIMD) simulations at constant temperature and volume (NVT) were carried out to check the thermal stability via setting the time step at 1 fs and the total simulation time at 5 ps. The climbing image nudged elastic band (CI-NEB) method58 was used to obtain the migration paths and barriers of potassium ions.3. Results and discussion
3.1 Structure and stability
The global structure search was carried out using the artificial bee colony (ABC) algorithm in the 2D structure search mode implemented in the CALYPSO code.59 The structural unit cell with various atom numbers (Be![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N atom = 2:4, 3:6, and 4:8) was considered in computations. The searching population and generation are 20 and 30, respectively. As displayed in Fig. 1a and b, the minimum energy structure for the structural unit cell with an atom number of Be:N = 2:4 is graphene-like β-2D-BeN2 (or h-BeN2), while it is α-2D-BeN2 for the structural unit cell with a larger atom number of Be:N = 4:8. Remarkably, the α-2D-BeN2 monolayer is thermodynamically more stable than the graphene-like h-BeN2 monolayer predicted previously with a decrease of −34 meV per atom in total energy. Based on the energy sequence, the global energy minimum structure in all searches is α-2D-BeN2 (Fig. S1 and S2 in the ESI†). α-2D-BeN2 is a planar monolayer with penta-, hexa-, and hepta-atomic rings (noted as R5, R6, R7, respectively) in structure, different from the h-BeN2 monolayer with only hexa-atomic rings. The α-2D-BeN2 monolayer has a symmetry of PMC21 (space group of No. 26) with lattice constants of a = 7.264 Å and b = 5.134 Å. There are two types of Be atoms with identical positions of Be_1(0.931, 0.360, 0.500) and Be_2(0.389, 0.500, 0.500) in the structure, and four types of N atoms of N_1(0.717, 0.271, 0.500), N_2(0.616, 0.491, 0.500), N_3(0.716, 0.710, 0.500), and N_4(0.896, 0.668, 0.500), respectively. α-2D-BeN2 can be regarded as a framework composed of two units, a ‘U’-shaped chain segment N4 tetramer and a Be atom, as shown in Fig. 1b. The bond length of Be–N (dBe–N) is from 1.589 to 1.672 Å, while that of N–N bonds is dN–N = 1.324–1.346 Å, as shown in Fig. 1f. As comparison, the bond length in the h-BeN2 monolayer is quite similar to dBe–N = 1.616 Å and dN–N = 1.324 Å with the same calculation method (Fig. 1g).
:N atom = 2:4, 3:6, and 4:8) was considered in computations. The searching population and generation are 20 and 30, respectively. As displayed in Fig. 1a and b, the minimum energy structure for the structural unit cell with an atom number of Be:N = 2:4 is graphene-like β-2D-BeN2 (or h-BeN2), while it is α-2D-BeN2 for the structural unit cell with a larger atom number of Be:N = 4:8. Remarkably, the α-2D-BeN2 monolayer is thermodynamically more stable than the graphene-like h-BeN2 monolayer predicted previously with a decrease of −34 meV per atom in total energy. Based on the energy sequence, the global energy minimum structure in all searches is α-2D-BeN2 (Fig. S1 and S2 in the ESI†). α-2D-BeN2 is a planar monolayer with penta-, hexa-, and hepta-atomic rings (noted as R5, R6, R7, respectively) in structure, different from the h-BeN2 monolayer with only hexa-atomic rings. The α-2D-BeN2 monolayer has a symmetry of PMC21 (space group of No. 26) with lattice constants of a = 7.264 Å and b = 5.134 Å. There are two types of Be atoms with identical positions of Be_1(0.931, 0.360, 0.500) and Be_2(0.389, 0.500, 0.500) in the structure, and four types of N atoms of N_1(0.717, 0.271, 0.500), N_2(0.616, 0.491, 0.500), N_3(0.716, 0.710, 0.500), and N_4(0.896, 0.668, 0.500), respectively. α-2D-BeN2 can be regarded as a framework composed of two units, a ‘U’-shaped chain segment N4 tetramer and a Be atom, as shown in Fig. 1b. The bond length of Be–N (dBe–N) is from 1.589 to 1.672 Å, while that of N–N bonds is dN–N = 1.324–1.346 Å, as shown in Fig. 1f. As comparison, the bond length in the h-BeN2 monolayer is quite similar to dBe–N = 1.616 Å and dN–N = 1.324 Å with the same calculation method (Fig. 1g).
 | ||
| Fig. 1 (a) Energy distribution of BeN2 structures determined via global structure searches using the CALYPSO method. The red, green, and black dots represent systems with Be:N atom ratios of 2:4, 3:6, and 4:8, respectively. The pentagrams mark the lowest-energy structures in each system. (b) Structural representations of α-BeN2 and β-BeN2 (h-BeN2). (c) Schematic diagram of α-BeN2, with different colors used to distinguish non-equivalent but identical atoms. (d) Deformation electron density and (e) electron localization function of α-2D-BeN2. (f) Structural information of α-2D-BeN2. (g) Charge transfer analysis of α-BeN2 and β-BeN2. | ||
The stability of the α-2D-BeN2 monolayer structure was examined in different aspects. First, it has a large binding energy of −5.013 eV per atom. Compared with the experimental 2D structures with purely 3-fold atoms, it is lower than that of graphene (−7.974 eV per atom) and h-BN (−7.098 eV per atom), but higher than that of phosphorene (−3.477 eV per atom) and silicene (−3.974 eV per atom), indicating the high-strength Be–N and N–N bonds in α-2D-BeN2. More importantly, the formation energy of α-2D-BeN2 (Ef-BeN2vs. Be metal and N2 molecule, eqn (S1)†) is −0.308 eV per atom, suggesting its good formation feasibility from the Be metal bulk and N2 molecule in thermodynamics. Notably, the interlayer binding energy of bilayer α-2D-BeN2 is merely −20 meV Å−2 (eqn S2†) with a large interlayer distance of 2.92 Å, comparable to that of graphene (−19 meV Å−2) and h-BN (−17 meV Å−2), but significantly smaller than that of borophene (−192 meV Å−2) and silicene (−61 meV Å−2), confirming that the α-2D-BeN2 layers are mainly stabilized by weak vdW interactions without any interlayered chemical bonds. Combining the results of AIMD simulation (Fig. S3†), the α-2D-BeN2 monolayer should not dimerize under ambient condition. Then, according to the calculated phonon dispersion (PhonBand) and density of states (PhonDOS) (Fig. 2a), the α-2D-BeN2 monolayer has no imaginary frequency in the whole Brillouin zone. Therefore, the α-2D-BeN2 monolayer is lattice dynamical stable. Finally, the thermal stability of the new structure was confirmed using AIMD simulations. A 2 × 3 supercell was employed in the simulations at temperatures ranging from 500 to 3000 K with an interval of 500 K. The snapshots of the geometrical structure after 5 ps simulation indicate that the monolayer can maintain its structural integrity up to about 2500 K (Fig. 2b and S4†), and it will melt at 3000 K, suggesting the high thermal stability of α-2D-BeN2 for applications.
 | ||
| Fig. 2 (a) Phonon spectrum and phonon density of states of α-2D-BeN2. (b) Snapshot of the α-2D-BeN2 structure after the AIMD simulation at 2000 K for 5 ps. (c) In-plane Young's stiffness and (d) Poisson's ratio of α-2D-BeN2. | ||
To reveal the chemical bonding nature, the deformation electron density (DED, Fig. 1d), electron localization function (ELF, Fig. 1e), and the Bader charge analysis (Fig. 1g)60 were calculated. The DED result indicates that the electrons are mainly distributed at the bridge site between N atoms and the nearest neighbor Be atoms but with partially between each two neighbor N atoms of N4 chain segment, suggesting significantly covalent hybridizations between Be and N atoms, and inside N4 chain segment. The ELF shows similar distribution characters, that electrons tend to mostly concentrate on the bridge site between N and Be atoms with considerable delocalization in N4 chain segment. The Bader charge analysis demonstrates that there is dominant charge transfer of 1.66/1.68 e− from Be_1/Be_2 atoms to the N4 chain segment, less than 2 e− (the typical valence of Be2+), and the N atoms gain electrons of 0.43–1.22 e− symmetrically but unequally with charge distribution of (N−1.19N−0.43N−0.51N−1.22), different from those in h-BeN2 of (N+0.06)N3−1.13 (Fig. 1g). These results coherently indicate the ionic-covalent bonding feature of Be–N and the covalent bonding of N–N in the α-2D-BeN2 monolayer, in agreement with its isoelectronicity to h-BN (both are 4e and 3 folds per atom on average) with high strength bonds.
3.2 Mechanical, electronic, and optical properties
In mechanical properties, the α-2D-BeN2 monolayer was investigated by examining its elastic constants. The elastic constants were calculated to be C11 = 153.8 N m−1, C22 = 145.4 N m−1, C12 = C21 = 53.5 N m−1, and C66 = 63.8 N m−1, in compliance with the mechanical stability criteria for the 2D structure (C11 > 0, C22 > 0, C11C22–C12C21 > 0, C66 > 0),61 suggesting that the α-2D-BeN2 monolayer is mechanically stable. As shown in Fig. 2c and d, in-plane Young's stiffness (Y(θ), eqn (S3)† and Poisson's ratio (ν(θ), eqn (S4)†) at a rotation angle of θ are directionally varied, showing a certain degree of anisotropy in mechanical properties. For Young's stiffness, the α-2D-BeN2 monolayer has a value of Y(θ) from 127 to 157 N m−1 for all rotation angles, θ, with an anisotropy of Ymax/Ymin = 1.24. The Y are Y11 = 134 and Y22 = 127 N m−1 along the direction-a and -b, respectively. Therefore, the α-2D-Be3N2 monolayer exhibits moderate and anisotropic rigidity for all directions with an observable difference of 30 N m−1. Compared with other 2D structure monolayers, it is comparable to that of MoS2 (121 N m−1),62 and larger than that of silicene (62 N m−1)63 and phosphorene (21–91 N m−1),64 but much smaller than that of graphene (330 N m−1)62 and h-BN (∼279 N m−1).65 Interestingly, the α-2D-BeN2 monolayer has a large in-plane Poisson's ratio, ranging from 0.228 to 0.368 for all rotation angle, θ, with a considerable anisotropy of νmax/νmin = 1.61. Therefore, the α-2D-BeN2 monolayer is a ductile material in most directions (Poisson's ratio > 0.25) with a relatively strong rigidity.As shown in Fig. 3a, α-2D-BeN2 has a direct bandgap of 1.10 eV in the energy band structure for α-2D-BeN2 with the PBE functional. Both the valence band maximum (VBM) and the conduction band minimum (CBM) are positioned at the Y(1/2, 0, 0) point. The calculated projected density of states (PDOS, Fig. 3b) indicated that the VBM was dominantly contributed by N_2pz (especially N_1 and N_4), while the CBM was mainly contributed by the pz orbital of all N atoms and Be_2 atoms (Fig. 3c). The HSE06 method provides that the more accurate bandgap is 1.82 eV with a work function of 4.93 eV. The bandgap of α-2D-Be3N2 is slightly larger than that of phosphorene (1.58 eV),64 but smaller than that of h-BeN2 (2.23 eV) and h-BN (5.66 eV)66 with the HSE06 functional.
 | ||
| Fig. 3 (a) Band structure of α-2D-BeN2 (green: PBE level, gray: HSE06 level) and total density of states (PBE level). (b) Projected density of states (PDOS) of α-2D-BeN2, with the Fermi level set to 0 eV. (c) Partial charge density of the CBM and VBM of α-2D-BeN2. (d) Optical absorption coefficient of α-2D-BeN2. | ||
Furthermore, the carrier mobilities of the α-2D-BeN2 monolayer were calculated using the deformation potential (DP) theory.67 For electron carriers, the effective mass is 0.96/0.61 me with a high mobility of 0.55/6.6 × 104 cm2 V−1 s−1 along the direction-a/-b, respectively. In comparison, the hole carrier has a close effective mass of −0.60/−0.81 me with a relatively small mobility of 1.3/8.3 × 103 cm2 V−1 s−1, along the direction-a/-b, respectively. The highest value is lower than that of graphene (3.4 × 105 cm2 V−1 s−1)68 and h-BeN2 (3.4 × 105 cm2 V−1 s−1),40 but higher than that of the BP monolayer (2.6 × 104 cm2 V−1 s−1),69 offering potential applications of the α-2D-BeN2 monolayer in semiconductor electronics. See more calculation details in the ESI.†
The optical properties of α-2D-BeN2 were computed using the HSE06 method. Due to the anisotropy in structure, the dielectric constant of α-2D-BeN2 is expressed as εxx ≠ εyy ≠ εzz. As shown in Fig. 3d, the absorption coefficients are different along the direction-a (axial a), -b (axial b) and -c (out-of-plane). Possessing a direct bandgap of 1.82 eV, α-2D-Be3N2 displays an anisotropic optical absorption spectrum with abundant absorbance in the visible range. The high intensity region started from about 1.9 eV with peaks up to 2.4 × 105 cm−1 at 4.8, 6.0, and 8.0 eV for the direction-a, at 3.7 and 7.5 eV for the direction-b, and the intensity along the out-of-plane direction is quite slight until about 5.1 eV in the optical absorption spectrum. The in-plane absorption coefficient rapidly stepped up to a high intensity interval of 1.0–2.4 × 105 cm−1 in the visible light region. With high-strength chemical bonds, high thermal stability, and high-density optical absorption in the visible light region, α-2D-BeN2 should be a promising candidate as a visible light detector and a donor in solar cells. See more calculation details in the ESI.†
3.3 Oxygen reduction/evolution reaction catalytic ability
There are rich electronic states around the relatively shallow Fermi level and many bare 3-fold Be atoms in the α-2D-BeN2 structure, which suggests the potential electrocatalytic activity to oxygen reduction reactions. For O2 approach to and adsorption on the surface of α-2D-BeN2, it prefers a side-on configuration on Be_2 with an adsorption free energy of −0.10 eV among the different pathways, and the O–O bond is pronouncedly elongated to 1.24 and 1.36 Å, confirming the O2 activation ability of the α-2D-BeN2 structure. The spatially constrained associative mechanism of ORR catalysis is mainly focused here (Fig. 4a). Specifically, the possible structures of each intermediate ( , OOH*, O*, and OH*) in the four-electron pathway are considered across all active sites, with the assumption that adjacent reactions can only occur on the same or neighboring active sites. The free energy diagrams in Fig. 4a show that all electrochemical steps of the ORR are exothermic at an electrode potential (U) of 0 V. The hydrogenation of
, OOH*, O*, and OH*) in the four-electron pathway are considered across all active sites, with the assumption that adjacent reactions can only occur on the same or neighboring active sites. The free energy diagrams in Fig. 4a show that all electrochemical steps of the ORR are exothermic at an electrode potential (U) of 0 V. The hydrogenation of 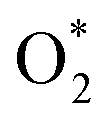 to OOH* at the Be_2 site was determined to be the potential-limiting step, as it possesses the least negative ΔG of −0.43 to −0.53 eV for different pathways. As a result, the limiting potential of the ORR (UL,ORR) was calculated to be 0.43–0.53 V for α-2D-BeN2, higher than that of h-BeN2 (0.40 V)48 and smaller than that of the electrode of 2D biphenylenes (UL,ORR = 0.69–0.73 V)70,71 and Pt metal (0.79 V).72
to OOH* at the Be_2 site was determined to be the potential-limiting step, as it possesses the least negative ΔG of −0.43 to −0.53 eV for different pathways. As a result, the limiting potential of the ORR (UL,ORR) was calculated to be 0.43–0.53 V for α-2D-BeN2, higher than that of h-BeN2 (0.40 V)48 and smaller than that of the electrode of 2D biphenylenes (UL,ORR = 0.69–0.73 V)70,71 and Pt metal (0.79 V).72
 | ||
| Fig. 4 Free energy diagrams with spatial considerations of the (a) ORR and (b) OER processes on α-2D-BeN2. The x-axis represents different adsorption sites, with the assumption that each step of the ORR and OER reactions occurs at the same or adjacent sites. | ||
The reversible process of ORR, the oxygen evolution reaction (OER), was investigated for the electrocatalytic OER. The dehydrogenation of O* to OOH* at the Be_2 site was determined to be the potential-limiting step, as it has the largest positive ΔG of 2.05 eV (Fig. 4b). Therefore, the limiting potential of the OER (UL,OER) was calculated to be 0.82 V for α-2D-BeN2, smaller than that of other 2D structures, such as h-BN (0.93 V),73 and graphene (UL,OER = 1.37 V),74 but higher than that of graphene (0.46 V)75 and RuO2 (0.42 V).76 The calculation details of the ORR and OER are shown in the ESI.†
3.4 K ion adsorption and migration
The adsorption, migration and storage ability of potassium ions on α-2D-BeN2 monolayer were investigated. The adsorption of single K ions on the α-2D-BeN2 monolayer was studied based on a 2 × 3 supercell structure model. As shown in Fig. 5a, three adsorption sites were examined, including R5, R6 and R7 sites. After the structural optimization, the adsorption energy based on eqn (S10)† (Eads, vs. corresponding K metal) is −0.149, −0.050, and −0.268 eV for R5, R6 and R7 sites, respectively. Therefore, the R7 site is the most stable adsorption site. The Bader charge analysis indicates that the K ion on the R7 site loses 0.87 electrons, indicating the high-degree ionic state of K ions (+0.87). | ||
| Fig. 5 (a) Adsorption sites and migration pathways of K on the surface of α-2D-BeN2. (b) Migration energy barriers for K along different pathways, with the red, green, and blue lines representing the initial and final positions of K and their relative energies. (c) Schematic of the K-ion adsorption stage and K-metal slab growth on the surface of α-2D-BeN2. (d) Voltage profiles corresponding to different K adsorption concentrations and side views of the structures. | ||
The migration behavior of K ions is shown in Fig. 5b using the CI-NEB method.58 Eight in-plane migration paths were simulated to cover the periodicity in the structure, including R7 → R7′ (Path-1, labelled as P-1), R7 → R5 (P-2), R7 → R5′ (P-3), R7 → R5'' (P-4), R7 → R6 (P-5), R5 → R6 (P-6), and R5 → R6′ (P-7), R6 → R6′ site (P-8, unstable), as shown in Fig. 5a. The calculated energy barrier (Eb) of all paths ranges from 0.108 to 0.248 eV (Fig. 5b). The combination of “Path-2, -6, -5” along the direction-a is the most favorable migration path with a minimum Eb value of 0.146 eV, while the most favorable migration path is the combination of “Path-6, -7” (Eb = 0.109 eV) along the direction-b to migrate across the whole surface in periodicity, endowing the high K ion conduction ability at room temperature. In general, the K ion possesses the lowest migration energy barrier of only Eb = 0.109–0.146 eV, lower than that in graphite (0.19 eV),77 comparable to that on graphene (0.09 eV),55 but larger than that on MoS2 (0.06 eV).78 Therefore, α-2D-BeN2 monolayer should exhibit excellent K ion adsorption and conduction ability to be applied in iontronic devices via K ion-dominated bandgap change, and anode materials in PIBs through ion capture and transport.
3.5 K ion storage performances
To achieve the reusable K ion storage, the transition between potassiation and depotassation needs to be a reversible process. The half-cell reaction on α-2D-BeN2 associated with K and K+ is as follows: BeN2 + xK+ + xe− ↔ KxBeN2. The thermodynamically stable structures of Kx@α-2D-BeN2 (KxBeN2) at different K ion adsorption concentrations were determined by the formation energy (Ef) of KxBeN2 based on eqn (S11).† As shown in Fig. S5,† the convex hull diagrams of the relationship between the calculated Ef and the K ion proportion in KxBeN2 displays five stable K ion concentration points, which lies on the convex hull, according to thermodynamics stability criteria.The adsorption of K ions includes two stages, namely the ion adsorption stage and the layered potassium metal slab growth stage, which is similar to that on the Ca2Si monolayer.79 The storage of K ion is via ionic interactions between K and the α-2D-BeN2 surface for the first stage (BeN2 + K+ + e− ↔ KBeN2). During the first stage, K ions first adsorbed at the R7 site on the surfaces, but with a placement transition to the R5 sites. For the second stage, the formation of K metal layers via K–K metal bonds dominates the process (KBeN2 + 3K+ + 3e− ↔ K4BeN2, Fig. 5c). It should be noted that, due to the similar storage mechanism to K on the Ca2Si monolayer, only four K layers on each side of α-2D-BeN2 were examined here. For a layer number >5 for K ion adsorption, the process should be an unlimited K metal growth process, which is helpful to form a novel K metal phase and achieve safely ultrahigh capacity storage of K ions, similar to that occurring on the Ca2Si monolayer.79
Based on eqn (S12),† the average stepwise adsorption energy (Estep, vs. K metal bulk) of K ions in the first stage is −0.280, −0.197 eV per K atom for stable concentrations of x = 0.125 and 1 in KxBeN2, and it is −0.033, −0.008, and −0.008 eV per K atom for x = 2, 3, and 4 in KxBeN2 in the second stage, respectively. The maximum specific capacities (CM) of K ions on α-2D-BeN2 were calculated using eqn (S13).† The specific capacity (CM) of K ions is 724 mA h g−1 for the ionic state of K ions, and at least 2895 mA h g−1 for potassium storage with high-degree low valence K ions. Both of these two specific capacities are significantly higher than that of the experimental graphite anode (279 mA h g−1),80 K2C6O6 (218 mA h g−1).81 Considering multilayer adorption of K ions, the specific capacity is higher than most other 2D structures, graphene (558 mA h g−1),70 thgraphene (744 mA h g−1),75 C57 (1117 mA h g−1),82 C36H8 (1278 mA h g−1),83 C18H6 (1449 mA h g−1),84 biphenylene-based carbon allotropes (1116–1489 mA h g−1),70 but smaller than Ca2Si (5459 mA h g−1).79
Based on the formation convex hull result, the open-circuit voltage (VOC) profile was calculated using eqn (S14).† As shown in Fig. 5d, there are four voltage plateaus at different K ion adsorption concentrations, displaying the profile of voltage plateaus with the K concentration range of 0.280 (Kx=0–0.125C6) → 0.197 (x = 0.125–1) → 0.033 (x = 1–2) → 0.008 (x = 2–3) → 0.008 (x = 3–4) V. Therefore, α-2D-BeN2 exhibits a voltage VOC ranging from 0.280 to 0.008 V in K ion storage, indicating its promising voltage performance as a PIB anode material.
4. Conclusions
In summary, we have investigated the structural stability, mechanical properties, electronic properties, optical absorption, oxygen reduction/evolution catalysis and potassium storage ability of the α-2D-BeN2 monolayer (with an isoelectronicity to h-BN, and there are 4e and 3 folds per atom on average for both structures) based on the first-principles theory. The monolayer has good lattice dynamics and excellent thermal stability to maintain its basic structural framework up to about 2500 K. With a moderate direct bandgap of 1.82 eV, it displays a high carrier mobility (up to 6.6 × 104 cm2 V−1 s−1) and high-intensity photon absorption in the visible region. It exhibits a relatively large Poisson's ratio ranging from 0.228 to 0.368, oxygen reduction/evolution catalysis ability, outstanding potassium storage ability with an ultrahigh specific capacity of 2895 mA h g−1, a good voltage ranging from 0.280 to 0.008 V, and a low migration barrier energy of 0.109–0.146 eV. Owing to the novel nature and significant properties, the α-2D-BeN2 monolayer is expected to be an anisotropic multifunctional material for wide-ranging applications in various fields such as semiconductor electronics, visible-light detectors, donors in solar cells, ductile materials, iontronic devices, and potassium ion anode materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. N.: computation executing, methodology, investigation, data curation, writing – original draft. J. J.: computation executing, investigation, formal analysis, data analysis, visualization, writing – original draft. W. W.: discussion of the research scheme, manuscript review, funding acquisition. X. W.: manuscript review, funding acquisition. Z. Z., Z. W.: conceptualization, methodology, data analysis, writing – review & editing, supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Anhui Provincial Natural Science Foundation (No. 2408085MB035), the National Natural Science Foundation for Distinguished Young Scholars (No. 22225301), the Postdoctoral Fellowship Program (No. GZC20232540), the General Program of China Postdoctoral Science Foundation (No. 2024M753088), the Key Research and Development Program of Anhui Province (2022a05020052), the National Natural Science Foundation of China (No. 12104425), and the Hefei Advanced Computing Center, the USTCSCC and the BSCC.References
- H. Xia, T. Jiang, G. Qi, T. Liu, W. Zhang and Q. Zhang, Revisiting Pentazole: An Investigation into the Intriguing Molecule Exhibiting Dual Organic and Inorganic Characteristics, Inorg. Chem., 2024, 63, 13166–13170 CrossRef CAS PubMed.
- M. I. Eremets, M. Y. Popov, I. A. Trojan, V. N. Denisov, R. Boehler and R. J. Hemley, Polymerization of nitrogen in sodium azide, J. Chem. Phys., 2004, 120, 10618–10623 CrossRef CAS PubMed.
- B. A. Steele, E. Stavrou, J. C. Crowhurst, J. M. Zaug, V. B. Prakapenka and I. I. Oleynik, High-Pressure Synthesis of a Pentazolate Salt, Chem. Mater., 2017, 29, 735–741 CrossRef CAS.
- Y. Wang, M. Bykov, I. Chepkasov, A. Samtsevich, E. Bykova, X. Zhang, S.-q. Jiang, E. Greenberg, S. Chariton, V. B. Prakapenka, A. R. Oganov and A. F. Goncharov, Stabilization of hexazine rings in potassium polynitride at high pressure, Nat. Chem., 2022, 14, 794–800 CrossRef CAS PubMed.
- A. Aslandukov, A. Aslandukova, D. Laniel, S. Khandarkhaeva, Y. Yin, F. I. Akbar, S. Chariton, V. Prakapenka, E. L. Bright, C. Giacobbe, J. Wright, D. Comboni, M. Hanfland, N. Dubrovinskaia and L. Dubrovinsky, Stabilization of N6 and N8 anionic units and 2D polynitrogen layers in high-pressure scandium polynitrides, Nat. Commun., 2024, 15, 2244 CrossRef CAS PubMed.
- K. O. Christe, W. W. Wilson, J. A. Sheehy and J. A. Boatz, N5+: A Novel Homoleptic Polynitrogen Ion as a High Energy Density Material, Angew. Chem., Int. Ed., 1999, 38, 2004–2009 CrossRef CAS PubMed.
- Z. Wu, E. M. Benchafia, Z. Iqbal and X. Wang, N8− polynitrogen stabilized on multi-wall carbon nanotubes for oxygen-reduction reactions at ambient conditions, Angew. Chem., 2014, 126, 12763–12767 CrossRef.
- M. Bykov, T. Fedotenko, S. Chariton, D. Laniel, K. Glazyrin, M. Hanfland, J. S. Smith, V. B. Prakapenka, M. F. Mahmood and A. F. Goncharov, High-pressure synthesis of Dirac materials: layered van der Waals bonded BeN4 polymorph, Phys. Rev. Lett., 2021, 126, 175501 CrossRef CAS PubMed.
- D. Laniel, A. A. Aslandukova, A. N. Aslandukov, T. Fedotenko, S. Chariton, K. Glazyrin, V. B. Prakapenka, L. S. Dubrovinsky and N. Dubrovinskaia, High-Pressure Synthesis of the β-Zn3N2 Nitride and the α-ZnN4 and β-ZnN4 Polynitrogen Compounds, Inorg. Chem., 2021, 60, 14594–14601 CrossRef CAS PubMed.
- Y. Zhang, X. Huang, Y. Yao, Z. Zhang, F. Tian, W. Chen, S. Chen, S. Jiang, D. Duan and T. Cui, Dirac nodal-line semimetal zinc polynitride at high pressure, Phys. Rev. B, 2022, 105, 125120 CrossRef CAS.
- D. Laniel, B. Winkler, E. Koemets, T. Fedotenko, M. Bykov, E. Bykova, L. Dubrovinsky and N. Dubrovinskaia, Synthesis of magnesium-nitrogen salts of polynitrogen anions, Nat. Commun., 2019, 10, 4515 CrossRef PubMed.
- M. Bykov, E. Bykova, G. Aprilis, K. Glazyrin, E. Koemets, I. Chuvashova, I. Kupenko, C. McCammon, M. Mezouar, V. Prakapenka, H. P. Liermann, F. Tasnádi, A. V. Ponomareva, I. A. Abrikosov, N. Dubrovinskaia and L. Dubrovinsky, Fe-N system at high pressure reveals a compound featuring polymeric nitrogen chains, Nat. Commun., 2018, 9, 2756 CrossRef CAS PubMed.
- M. Bykov, E. Bykova, A. V. Ponomareva, I. A. Abrikosov, S. Chariton, V. B. Prakapenka, M. F. Mahmood, L. Dubrovinsky and A. F. Goncharov, Stabilization of Polynitrogen Anions in Tantalum–Nitrogen Compounds at High Pressure, Angew. Chem., Int. Ed., 2021, 60, 9003–9008 CrossRef CAS PubMed.
- Y. Zhang, C. Ding, K. Zhang, A. Pakhomova, S. Chen, Y. Ding, S. Jiang, X. Huang, J. Sun and T. Cui, All-Single Bonds Fused N18 Macro-Rings and N8 Cagelike Building Blocks Stabilized in Lanthanum Supernitrides, J. Am. Chem. Soc., 2024, 146, 28174–28181 CAS.
- H. Zhai, R. Xu, J. Dai, X. Ma, X. Yu, Q. Li and Y. Ma, Stabilized Nitrogen Framework Anions in the Ga–N System, J. Am. Chem. Soc., 2022, 144, 21640–21647 CrossRef CAS PubMed.
- D. Tomasino, M. Kim, J. Smith and C.-S. Yoo, Pressure-Induced Symmetry-Lowering Transition in Dense Nitrogen to Layered Polymeric Nitrogen (LP-N) with Colossal Raman Intensity, Phys. Rev. Lett., 2014, 113, 205502 CrossRef PubMed.
- D. Laniel, G. Geneste, G. Weck, M. Mezouar and P. Loubeyre, Hexagonal Layered Polymeric Nitrogen Phase Synthesized near 250 GPa, Phys. Rev. Lett., 2019, 122, 066001 CrossRef CAS PubMed.
- C. Ji, A. A. Adeleke, L. Yang, B. Wan, H. Gou, Y. Yao, B. Li, Y. Meng, J. S. Smith, V. B. Prakapenka, W. Liu, G. Shen, W. L. Mao and H.-k. Mao, Nitrogen in black phosphorus structure, Sci. Adv., 2020, 6, eaba9206 CrossRef CAS PubMed.
- D. Laniel, B. Winkler, T. Fedotenko, A. Pakhomova, S. Chariton, V. Milman, V. Prakapenka, L. Dubrovinsky and N. Dubrovinskaia, High-Pressure Polymeric Nitrogen Allotrope with the Black Phosphorus Structure, Phys. Rev. Lett., 2020, 124, 216001 CrossRef CAS PubMed.
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Single-bonded cubic form of nitrogen, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed.
- Y. Li, X. Feng, H. Liu, J. Hao, S. A. T. Redfern, W. Lei, D. Liu and Y. Ma, Route to high-energy density polymeric nitrogen t-N via He−N compounds, Nat. Commun., 2018, 9, 722 CrossRef PubMed.
- X. Zhang, X. Xie, H. Dong, X. Zhang, F. Wu, Z. Mu and M. Wen, Pressure-Induced High-Energy-Density BeN4 Materials with Nitrogen Chains: First-Principles Study, J. Phys. Chem. C, 2021, 125, 25376–25382 CrossRef CAS.
- Z. Liu, D. Li, F. Tian, D. Duan, H. Li and T. Cui, Moderate Pressure Stabilized Pentazolate Cyclo-N5– Anion in Zn(N5)2 Salt, Inorg. Chem., 2020, 59, 8002–8012 CrossRef CAS PubMed.
- Y. Wang, Z. Li, R. Li, Y. Li, S. Liu, Z. Yao and B. Liu, Two Ultrahigh-Energy-Density Layered Cerium Polynitrides with Molecular Sieve Channel, Inorg. Chem., 2023, 62, 11674–11681 CrossRef CAS PubMed.
- K. Xia, J. Yuan, X. Zheng, C. Liu, H. Gao, Q. Wu and J. Sun, Predictions on High-Power Trivalent Metal Pentazolate Salts, J. Phys. Chem. Lett., 2019, 10, 6166–6173 CrossRef CAS PubMed.
- S. Wei, D. Li, Z. Liu, W. Wang, F. Tian, K. Bao, D. Duan, B. Liu and T. Cui, A Novel Polymerization of Nitrogen in Beryllium Tetranitride at High Pressure, J. Phys. Chem. C, 2017, 121, 9766–9772 CrossRef CAS.
- J. Lin, D. Peng, Q. Wang, J. Li, H. Zhu and X. Wang, Stable nitrogen-rich scandium nitrides and their bonding features under ambient conditions, Phys. Chem. Chem. Phys., 2021, 23, 6863–6870 RSC.
- Z. Liu, D. Li, S. Wei, Y. Liu, F. Tian, D. Duan and T. Cui, Nitrogen-rich GaN5 and GaN6 as high energy density materials with modest synthesis condition, Phys. Lett. A, 2019, 383, 125859 CrossRef CAS.
- B. Jin, S. Liu, K. Hu, Z. Yao and B. Liu, Ambient-Condition Recoverable Polymeric N10 Discovered from the Predicted Zr-N Compounds, Inorg. Chem., 2024, 63, 12615–12623 CrossRef CAS PubMed.
- S. Zhang, Z. Zhao, L. Liu and G. Yang, Pressure-induced stable BeN4 as a high-energy density material, J. Power Sources, 2017, 365, 155–161 CrossRef CAS.
- L. Li, K. Bao, X. Zhao and T. Cui, Bonding Properties of Manganese Nitrides at High Pressure and the Discovery of MnN4 with Planar N4 Rings, J. Phys. Chem. C, 2021, 125, 24605–24612 CrossRef CAS.
- K. Xia, H. Gao, C. Liu, J. Yuan, J. Sun, H.-T. Wang and D. Xing, A novel superhard tungsten nitride predicted by machine-learning accelerated crystal structure search, Sci. Bull., 2018, 63, 817–824 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. Carvalho, M. Wang, X. Zhu, A. S. Rodin, H. Su and A. H. Castro Neto, Phosphorene: from theory to applications, Nat. Rev. Mater., 2016, 1, 16061 CrossRef CAS.
- Z.-Q. Wang, T.-Y. Lü, H.-Q. Wang, Y. P. Feng and J.-C. Zheng, Review of borophene and its potential applications, Front. Phys., 2019, 14, 33403 CrossRef.
- S. K. Tiwari, S. Sahoo, N. Wang and A. Huczko, Graphene research and their outputs: Status and prospect, J. Sci.:Adv. Mater. Devices, 2020, 5, 10–29 Search PubMed.
- L. Song, L. J. Ci, H. Lu, P. B. Sorokin, C. H. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed.
- Y.-H. Lee, X.-Q. Zhang, W. Zhang, M.-T. Chang, C.-T. Lin, K.-D. Chang, Y.-C. Yu, J. T.-W. Wang, C.-S. Chang, L.-J. Li and T.-W. Lin, Synthesis of Large-Area MoS2 Atomic Layers with Chemical Vapor Deposition, Adv. Mater., 2012, 24, 2320–2325 CrossRef CAS PubMed.
- Q. Yang, Y. Zhao, X. Jiang, B. Wang and J. Zhao, Unconventional stoichiometric two-dimensional potassium nitrides with anion-driven metallicity and superconductivity, J. Mater. Chem. C, 2024, 12, 103–109 RSC.
- C. Zhang and Q. Sun, A Honeycomb BeN2 Sheet with a Desirable Direct Band Gap and High Carrier Mobility, J. Phys. Chem. Lett., 2016, 7, 2664–2670 CrossRef CAS PubMed.
- Y. Wei, Y. Ma, W. Wei, M. Li, B. Huang and Y. Dai, Promising Photocatalysts for Water Splitting in BeN2 and MgN2 Monolayers, J. Phys. Chem. C, 2018, 122, 8102–8108 CrossRef CAS.
- X. Li, S. Zhang, C. Zhang and Q. Wang, Stabilizing benzene-like planar N6 rings to form a single atomic honeycomb BeN3 sheet with high carrier mobility, Nanoscale, 2018, 10, 949–957 RSC.
- J. Liu, Y. Shen, X. Gao, L. Lv, Y. Ma, S. Wu, X. Wang and Z. Zhou, GeN3 monolayer: A promising 2D high-efficiency photo- hydrolytic catalyst with High carrier mobility transport anisotropy, Appl. Catal., B, 2020, 279, 119368 CrossRef CAS.
- B. Mortazavi, F. Shojaei and X. Zhuang, Ultrahigh stiffness and anisotropic Dirac cones in BeN4 and MgN4 monolayers: a first-principles study, Mater. Today Nano, 2021, 15, 100125 CrossRef CAS.
- S. Lin, M. Xu, F. Wang, J. Hao and Y. Li, Ultrahigh energy density BeN monolayer: A nodal-line semimetal anode for Li-ion batteries, Phys. Rev. Res., 2024, 6, 013028 CrossRef CAS.
- W. Zhou, S. Guo, H. Zeng and S. Zhang, High-Performance Monolayer BeN2 Transistors With Ultrahigh On-State Current: A DFT Coupled With NEGF Study, IEEE Trans. Electron Devices, 2022, 69, 4501–4506 CAS.
- X. Xin, W. Li, R. Pang, H. Wang, C. Guo, X. Shi and Y. Zhao, Robust ferromagnetism and half-metallicity in fluorinated two-dimensional BeN2 sheets, Appl. Phys. Lett., 2017, 111, 253102 CrossRef.
- Y.-m. Ding, Y. Ji, H. Dong, N. Rujisamphan and Y. Li, Electronic properties and oxygen reduction reaction catalytic activity of h-BeN2 and MgN2 by first-principles calculations, Nanotechnology, 2019, 30, 465202 CrossRef CAS PubMed.
- M. Wang and D. Han, Thermal Properties of 2D Dirac Materials MN4 (M = Be and Mg): A First-Principles Study, ACS Omega, 2022, 7, 10812–10819 CrossRef CAS PubMed.
- A. Bafekry, C. Stampfl, M. Faraji, M. Yagmurcukardes, M. M. Fadlallah, H. R. Jappor, M. Ghergherehchi and S. A. H. Feghhi, A Dirac-semimetal two-dimensional BeN4: Thickness-dependent electronic and optical properties, Appl. Phys. Lett., 2021, 118, 203103 CrossRef CAS.
- M. Mahmoudi, X. Tan and S. C. Smith, Enhanced Charge-Modulated Switchable CO2 Capture on Graphene-like BeN4 with Beryllium Vacancy, J. Phys. Chem. C, 2022, 126, 18189–18197 CrossRef CAS.
- V. Mahamiya, J. Dewangan and B. Chakraborty, Interplay between van der Waals, Kubas, and chemisorption process when hydrogen molecules are adsorbed on pristine and Sc-functionalized BeN4, Int. J. Hydrogen Energy, 2024, 50, 1302–1316 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Togo and I. Tanaka, First principles phonon calculations in materials science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B:Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- R. F. W. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- Y. Ding and Y. Wang, Density Functional Theory Study of the Silicene-like SiX and XSi3 (X = B, C, N, Al, P) Honeycomb Lattices: The Various Buckled Structures and Versatile Electronic Properties, J. Phys. Chem. C, 2013, 117, 18266–18278 CrossRef CAS.
- Y. Li, Y. Liao and Z. Chen, Inside Back Cover: Be2C Monolayer with Quasi-Planar Hexacoordinate Carbons: A Global Minimum Structure, Angew. Chem., Int. Ed., 2014, 53, 7369 CrossRef.
- Z. Zhuo, X. Wu and J. Yang, Two-dimensional silicon crystals with sizable band gaps and ultrahigh carrier mobility, Nanoscale, 2018, 10, 1265–1271 RSC.
- Z. Zhuo, X. Wu and J. Yang, Two-Dimensional Phosphorus Porous Polymorphs with Tunable Band Gaps, J. Am. Chem. Soc., 2016, 138, 7091–7098 CrossRef CAS PubMed.
- S. Roy, X. Zhang, A. B. Puthirath, A. Meiyazhagan, S. Bhattacharyya, M. M. Rahman, G. Babu, S. Susarla, S. K. Saju, M. K. Tran, L. M. Sassi, M. A. S. R. Saadi, J. Lai, O. Sahin, S. M. Sajadi, B. Dharmarajan, D. Salpekar, N. Chakingal, A. Baburaj, X. Shuai, A. Adumbumkulath, K. A. Miller, J. M. Gayle, A. Ajnsztajn, T. Prasankumar, V. V. J. Harikrishnan, V. Ojha, H. Kannan, A. Z. Khater, Z. Zhu, S. A. Iyengar, P. A. d. S. Autreto, E. F. Oliveira, G. Gao, A. G. Birdwell, M. R. Neupane, T. G. Ivanov, J. Taha-Tijerina, R. M. Yadav, S. Arepalli, R. Vajtai and P. M. Ajayan, Structure, Properties and Applications of Two-Dimensional Hexagonal Boron Nitride, Adv. Mater., 2021, 33, 2101589 CrossRef CAS PubMed.
- H. Shan, Q. He, X. Luo and Y. Zheng, First-Principles Calculations of Monolayer h-BN Nanosheets and Nanoribbons with Ultrahigh Phonon-Limited Hole Mobility for Wide Band Gap P-Channel Transistors, J. Phys. Chem. C, 2023, 127, 9278–9286 CrossRef CAS.
- J. Bardeen and W. Shockley, Deformation Potentials and Mobilities in Non-Polar Crystals, Phys. Rev., 1950, 80, 72–80 CrossRef CAS.
- J. Chen, J. Xi, D. Wang and Z. Shuai, Carrier Mobility in Graphyne Should Be Even Larger than That in Graphene: A Theoretical Prediction, J. Phys. Chem. Lett., 2013, 4, 1443–1448 CrossRef CAS PubMed.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed.
- J. Jiang, Y. Chen, H. Guo, X. Wu, N. Lu and Z. Zhuo, Two-Dimensional Biphenylene-Based Carbon Allotrope Family with High Potassium Storage Ability, J. Phys. Chem. Lett., 2023, 14, 9655–9664 CrossRef CAS PubMed.
- T. Liu, Y. Jing and Y. Li, Two-Dimensional Biphenylene: A Graphene Allotrope with Superior Activity toward Electrochemical Oxygen Reduction Reaction, J. Phys. Chem. Lett., 2021, 12, 12230–12234 CrossRef CAS PubMed.
- H. A. Hansen, V. Viswanathan and J. K. Nørskov, Unifying Kinetic and Thermodynamic Analysis of 2 e− and 4 e− Reduction of Oxygen on Metal Surfaces, J. Phys. Chem. C, 2014, 118, 6706–6718 CrossRef CAS.
- D. Perilli, D. Selli, H. Liu and C. Di Valentin, Computational Electrochemistry of Water Oxidation on Metal-Doped and Metal-Supported Defective h-BN, ChemSusChem, 2019, 12, 1995–2007 CrossRef CAS PubMed.
- G. Murdachaew and K. Laasonen, Oxygen Evolution Reaction on Nitrogen-Doped Defective Carbon Nanotubes and Graphene, J. Phys. Chem. C, 2018, 122, 25882–25892 CrossRef CAS PubMed.
- W. Wang, J. Meng, Y. Hu, J. Wang, Q. Li and J. Yang, Thgraphene: a novel two-dimensional carbon allotrope as a potential multifunctional material for electrochemical water splitting and potassium-ion batteries, J. Mater. Chem. A, 2022, 10, 9848–9857 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- C. Yang, X. Sun, X. Zhang, J. Li, J. Ma, Y. Li, L. Xu, S. Liu, J. Yang, S. Fang, Q. Li, X. Yang, F. Pan, J. Lu and D. Yu, Is graphite nanomesh a promising anode for the Na/K-Ions batteries?, Carbon, 2021, 176, 242–252 CrossRef CAS.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Sustainable Recycling Technology for Li-Ion Batteries and Beyond: Challenges and Future Prospects, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- Z. Fang, J. Jiang, H. Guo, X. Lin, X. Wu, Z. Zhuo and N. Lu, Ultrahigh Potassium Storage Capacity of Ca2Si Monolayer with Orderly Multilayered Growth Mechanism, Small, 2024, 20, 2401736 CrossRef CAS PubMed.
- Z. Jian, W. Luo and X. Ji, Carbon Electrodes for K-Ion Batteries, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- R. Lian, C. Zhao, D. Wang, D. Kan, Y. Wang, X. Wang, C. Wang, G. Chen and Y. Wei, Flexible structural changes of the oxocarbon salt K2C6O6 during potassium ion insertion: An in-depth first-principles study, Electrochim. Acta, 2021, 383, 138357 CrossRef CAS.
- Y. Chang, L.-C. Xu, Z. Yang and X. Li, Achieving superior high-capacity K-ion batteries with the C57 carbon monolayer anode by first-principles calculations, Appl. Surf. Sci., 2020, 526, 146638 CrossRef CAS.
- U. Younis, I. Muhammad, Y. Kawazoe and Q. Sun, Design of tetracene-based metallic 2D carbon materials for Na- and K-Ion batteries, Appl. Surf. Sci., 2020, 521, 146456 CrossRef CAS.
- J. Jiang, H. Guo, J. Zhang, G. Z. Zuo, X. Wu, Z. Zhuo and N. Lu, Poly-triphenylene membrane: A multifunctional two-dimensional porous carbon framework with ultra-high carrier mobilities and potassium ion storage capacity, Appl. Surf. Sci., 2023, 631, 157503 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08565e |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2025 |