
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and characterization of an adaptive polymer electrolyte for lithium metal solid-state battery applications†
Matthew
Newman
 ,
Jian
Liu
,
Hoyeon
Jang
,
Rinky
Ghosh
,
Sriloy
Dey
,
Hanna
Cho
,
Yael
Vodovotz
,
Jay
Sayre
* and
Marcello
Canova
*
,
Jian
Liu
,
Hoyeon
Jang
,
Rinky
Ghosh
,
Sriloy
Dey
,
Hanna
Cho
,
Yael
Vodovotz
,
Jay
Sayre
* and
Marcello
Canova
*
The Ohio State University, Columbus, USA. E-mail: canova.1@osu.edu; sayre.17@osu.edu
First published on 11th February 2025
Abstract
A major challenge for Li-metal solid-state batteries (LIMSSBs) lies in the mechanical degradation of the solid interface between Li-metal and the solid electrolyte. This work focuses on the synthesis and electrochemomechanical characterization of an adaptive polymer electrolyte (A-PE) that could potentially be applied as an interlayer to stabilize the interfacial contact between the lithium metal anode and the solid electrolyte, mitigating the effects of void formation leading to contact loss. The A-PE operates based on the principle of utilizing the polarization of conducting polymer particles, polypyrrole doped with dodecylbenzenesulfonate (PPy(DBS)), to impart adaptive properties to a polymer electrolyte matrix. The A-PE was synthesized via hot pressing and subsequent UV polymerization resulting in free-standing films with various amounts of PPy(DBS) and ionic conductivities in the range of 0.11–0.16 mS cm−1 at 25 °C. Film characterizations included insoluble fraction, mechanical response under electric field, and Li symmetric cycling with intermittent electrochemical impedance spectroscopy (EIS). The measured mechanical responses of the film were expansion with atomic force microscopy (AFM) and block force response by exciting the film with electric field of variable strength. The results obtained suggest that the addition of PPy(DBS) particles provides adaptive capability in polymer electrolytes at room temperature (20–25 °C), with an expansion response of up to 6% strain with 1 wt% PPy(DBS) at an electric field strength of 0.3 V μm−1. The results indicate that the A-PE shows promise for application as an interlayer in LIMSSBs, with the potential to reduce mechanical degradation at the lithium metal–solid electrolyte interface and enhance durability by expanding to maintain contact with the lithium metal anode.
1. Introduction
With the rising demand for high energy density Li-ion batteries for electric vehicles and grid storage applications, solid-state electrolytes offer improved safety due to their non-flammability and the opportunity to enable the use of a lithium anode in Li-metal solid-state batteries (LIMSSBs). This technology could result in gravimetric and volumetric energy density gains of up to 40% and 70%, respectively, compared to conventional Li-ion batteries.1 On the other hand, the solid–solid anode–electrolyte interface in LIMSSBs has been recognized as one of the main limiting factors for the performance and durability, due to various mechanical and electrochemical challenges.1–6 Mechanically, a rigid lithium metal–solid electrolyte interface cannot maintain sufficient contact due to the large volume and morphology change of the Li-metal anode from repeated Li stripping and plating during cell operation. Contact loss and Li filament growth has been shown to ultimately short the cell, except in cases of unrealistically high stack pressure forcing the interface to maintain contact.3,6–10 Electrochemically, many ceramic solid electrolytes are not inherently stable at the low potential conditions that occur when direct contact with Li-metal is established, which can necessitate the use of a protective layer or interlayer.4,5To address these challenges, solutions such as engineered interfaces, polymer/ceramic composite solid electrolytes, or polymer interlayers have been proposed to improve the initial interface, provide flexibility to accommodate some of the volume change of the Li-metal anode, and increase the electrochemical stability.11–20 Composite solid electrolytes may compromise on ionic conductivity through the entire separator thickness as the polymer electrolyte component typically has lower ionic conductivity than ceramic solid electrolytes.14–17 Polymer interlayers can offer similar benefits of flexibility and electrochemical stability without compromising on ionic conductivity through the whole separator thickness but instead through a thin interlayer thickness.18–20 However, these solutions all have limited ability to maintain the interfacial contact in the event of non-uniform Li stripping and plating, and long-term stability is not guaranteed as contact loss can cause the cell to eventually fail.
This paper introduces a novel concept of an adaptive polymer electrolyte (A-PE) designed to offer self-adaptive capabilities at room temperature (20–25 °C). The A-PE is envisioned as an interlayer between the Li-metal anode and the solid electrolyte, where it could help maintain interfacial contact in the event of partial contact loss. A conceptual representation of the A-PE working principles and its potential future role as an interlayer is shown in Fig. 1. It is hypothesized that the A-PE could expand under an electric field inherently present in a battery to maintain interfacial contact with the anode. As long as some contact is maintained, the electric field across the interlayer would induce expansion to restore interfacial integrity. Although this approach may result in a slight reduction in energy density due to the interlayer's thickness, the trade-off could be improved durability and safety of the battery. This adaptive behavior is imparted by the polarization of conducting polymer (CP) particles, which exhibit electrorheological (ER) properties under an electric field. The mobility of electrons in CPs allow the particles to polarize and align along the field at relatively low electric field strengths.21–25 While ER properties are well studied in fluids, most research on ER polymers or elastomers has focused on the change in storage modulus under an electric field rather than on deformation or expansion responses.26–29 This study presents a comprehensive electrochemomechanical characterization of an A-PE envisioned for application in Li-metal solid-state batteries. Unlike traditional approaches to enhancing lithium metal solid-state batteries, the A-PE uniquely leverages the polarization of CP particles in a solid polymer matrix, providing a mechanical response under relatively low electric field strengths.
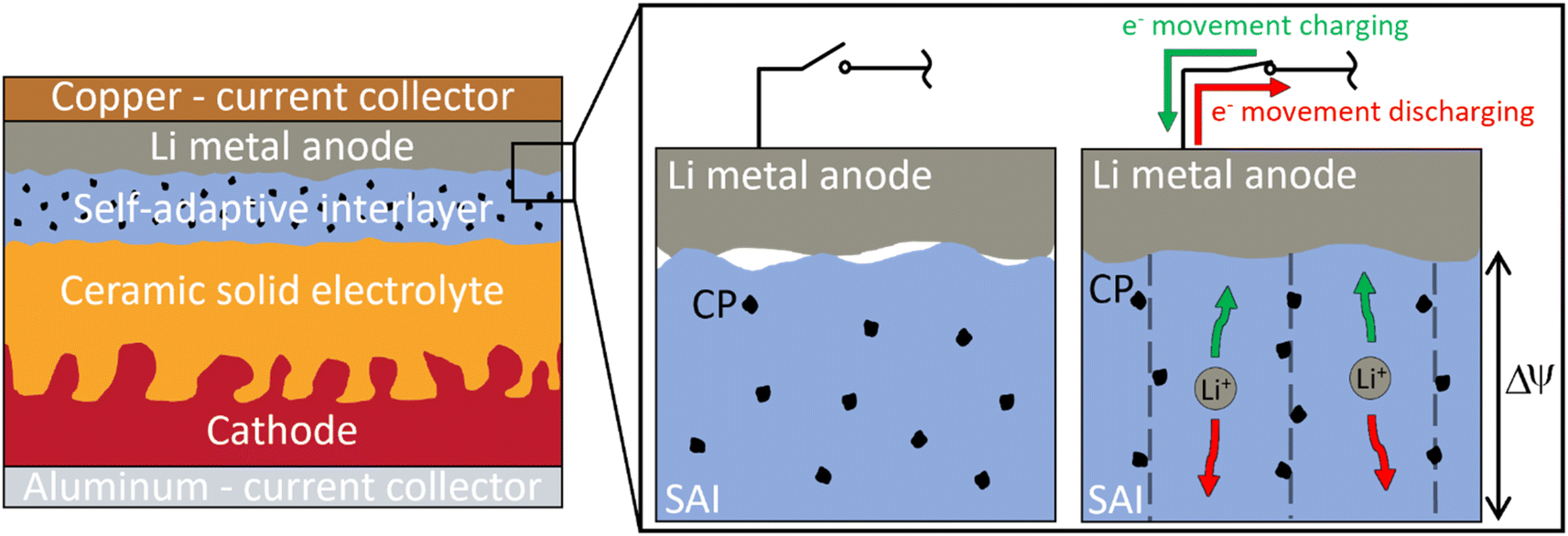 | ||
| Fig. 1 Concept of a self-adaptive interlayer applied between a Li-metal anode and ceramic solid electrolyte. The self-adaptive interlayer (SAI) is shown as a polymer electrolyte matrix in blue with black CP particles dispersed in the matrix, which would try to align along an electric field (ΔΨ) to expand and re-establish interface contact. Li-ion transfer is shown to occur primarily through the polymer electrolyte matrix. | ||
This work conducts an experimental evaluation of the A-PE's physical, electrochemical, and mechanical properties. Physical characterizations include insoluble fraction and electrical conductivity measurements, X-ray diffraction (XRD), differential scanning calorimetry (DSC), and dynamic mechanical analysis (DMA). Mechanical response characterizations are conducted using atomic force microscopy (AFM) and block force measurements to demonstrate the adaptive property through expansion and force response under applied electric fields. Electrochemical characterizations encompass electrochemical impedance spectroscopy (EIS), Li–Li symmetric cycling, and electrochemical stability window (ESW) measurements. These results establish the A-PE's potential as a promising interlayer material for LIMSSB, with the possibility of mitigating interfacial degradation and enhancing battery performance.
This paper is organized as follows: Section 2 explains the synthesis process for the polymer electrolyte investigated. Section 3 presents the physical, mechanical response, and electrochemical characterizations used. Section 4 presents the results of the polymer electrolyte characterizations.
2. Adaptive polymer electrolyte synthesis process
Polypyrrole doped with dodecylbenzenesulfonate (PPy(DBS)) is the CP used in this work for its polarization under an electric field in the polymer electrolyte. While other types of CPs can exhibit similar properties, polypyrrole (PPy) is chosen for this work because PPy-based materials have been reported to respond to low electric field strengths of 0.1 V μm−1 or less.22,24,26 Doping PPy can increase the mobility of electrons for polarization of the particles. Dodecylbenzenesulfonate (DBS−) is used as a large, immobile dopant (compared to smaller dopants that can migrate from the CP) and has also been shown to improve stability of PPy.30,31 However, a limitation with the addition of CPs is the electrical conductivity that provides CPs (such as PPy(DBS)) this property can make a composite (with an insulative matrix) electrically conductive at a high enough CP filler loading, known as the percolation threshold. The percolation threshold widely varies depending on the properties of the electrically conductive filler (such as size, morphology, aspect ratio), insulative matrix, and the mixing method.32–34 Thus, the electrical conductivity of composites should be measured for any new compositions including the compositions in this work. The amount of PPy(DBS) added in a polymer electrolyte or polymer interlayer for LIMSSBs must stay below the percolation threshold, otherwise, the cell could electrically short, Li could plate on the opposite side of the interlayer, and no appreciable electric field strength could be maintained across the polymer interlayer. With the amount of PPy(DBS) below the percolation threshold, the ionic conduction mechanism of the polymer electrolyte, through the segmental motion of polymer chains and Li-ions in the polymer electrolyte matrix, will remain relatively unchanged.A polymer electrolyte that can operate near room temperature (25 °C) is desirable for more equivalent comparisons between the characterization techniques used in this work. Thus, a polyethylene oxide (PEO) based polymer electrolyte adapted primarily from Zhang and coworkers35 is used as the polymer electrolyte matrix in which the CP particles are dispersed. PEO based polymer electrolytes are widely studied and show adequate room temperature ionic conductivity (≥0.1 mS cm−1 at 25 °C) if the crystallinity can be inhibited. PEO (M.W. 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) as the main long chain polymer gives a balance between mechanical properties and potential for lower crystallinity.35,36 Tetraethylene glycol dimethacrylate (TEGDMA) as an additional cross-linking short chain monomer can inhibit crystallinity and improve interfacial contact.35 Tetraethylene glycol dimethyl ether (TEGDME) as a cross-linking plasticizer can further reduce crystallinity and reduce the coordination of Li-ions with EO in long chain PEO. Lithium bis(trifluoromethane)sulfonimide (LiTFSI) as the main Li salt is the salt of choice in most polymer electrolytes, showing relatively high ionic conductivity and the TFSI− anion can have a plasticizing effect.35,36 Lithium bis(oxalato)borate (LiBoB) as an additional Li salt has lower ionic conductivity than LiTFSI but used in small amounts can improve the interface stability with Li-metal.37 4-Methylbenzophenone (MBP) is a hydrogen abstraction photoinitiator used to generate free radical chains from the methylene groups in PEO, TEGDMA, and TEGDME for UV-polymerization. Photopolymerization is a rapid process and does not require volatile solvents as compared to thermal polymerization or solvent evaporation synthesis methods.35,36
000) as the main long chain polymer gives a balance between mechanical properties and potential for lower crystallinity.35,36 Tetraethylene glycol dimethacrylate (TEGDMA) as an additional cross-linking short chain monomer can inhibit crystallinity and improve interfacial contact.35 Tetraethylene glycol dimethyl ether (TEGDME) as a cross-linking plasticizer can further reduce crystallinity and reduce the coordination of Li-ions with EO in long chain PEO. Lithium bis(trifluoromethane)sulfonimide (LiTFSI) as the main Li salt is the salt of choice in most polymer electrolytes, showing relatively high ionic conductivity and the TFSI− anion can have a plasticizing effect.35,36 Lithium bis(oxalato)borate (LiBoB) as an additional Li salt has lower ionic conductivity than LiTFSI but used in small amounts can improve the interface stability with Li-metal.37 4-Methylbenzophenone (MBP) is a hydrogen abstraction photoinitiator used to generate free radical chains from the methylene groups in PEO, TEGDMA, and TEGDME for UV-polymerization. Photopolymerization is a rapid process and does not require volatile solvents as compared to thermal polymerization or solvent evaporation synthesis methods.35,36
2.1. Materials
All chemicals were used as received. Iron(III) sulfate (Fe2(SO4)3) pentahydrate (97%) and PEO (M.W. 100000) were purchased from Thermo Scientific. Pyrrole (reagent grade 98%), sodium dodecylbenzenesulfonate (NaDBS, technical grade), TEGDMA (≥90% technical grade), TEGDME (≥99%), LiTFSI (99.95% trace metals basis), LiBoB, MBP (99%), and indium tin oxide (ITO) coated polyethylene terephthalate (PET) sheets (ITO-PET, 130 nm thick ITO coating on one side of 178 μm thick PET, surface resistivity 60 Ω sq−1) were purchased from Sigma-Aldrich. Clear PET film (127 μm thick) and copper tape with conductive adhesive were purchased from McMaster-Carr. Li-metal chips (0.6 mm thick) were purchased from MTI.
2.2. Chemical polymerization of CP particles
PPy(DBS) was chemically polymerized as described by Kudoh, reported to have an electrical conductivity of approximately 21.7 S cm−1 and molecular weight per residue of approximately 138.4 g mol−1.31 In brief, a polymerization solution of 0.375 M pyrrole monomer, 0.030 M NaDBS for DBS− dopant, and 0.100 M Fe2(SO4)3 chemical oxidant was prepared in 200 mL of deionized (DI) water. One half solution of pyrrole and NaDBS was stirred in 100 mL of DI water, and the other half solution of Fe2(SO4)3 was stirred in a separate 100 mL of DI water. The solution with Fe2(SO4)3 was slowly poured into the solution with pyrrole and NaDBS until fully combined, and the polymerization reaction was allowed to proceed for 16 h at room temperature (20–25 °C) under slow magnetic stirring. The solution was then poured through 0.8 μm filter paper, rinsed with DI water until the filtrate is neutral pH, rinsed with ethanol several times, dried at 40 °C under vacuum (gauge pressure −0.95 bar) for 24 h, and manually grinded with a mortar and pestle. The reaction yielded 2.1 g of PPy(DBS) particles (approximately 13.6 mL loosely packed).2.3. Polymer electrolyte preparation
The synthesis process of the polymer electrolyte film is summarized in Fig. 2. The polymer electrolyte was composed of 30.3 wt% PEO (approximate 100000 M.W.) as the main long chain polymer, 15.1 wt% TEGDMA as an additional cross-linking short chain monomer, 30.3 wt% TEGDME as cross-linking plasticizer, 17.8 wt% LiTFSI as the main Li salt, 3.5 wt% LiBoB as additional Li salt (20% of LiTFSI amount), and 3.0 wt% MBP as photoinitiator. PPy(DBS) CP particles were added as an additional 0–2 wt% of PEO and TEGDMA amounts (0–0.9 wt% of total components, ∼0–15 vol% of final polymer electrolyte film) for their properties of polarizing and trying to align under an electric field. The PPy(DBS) particles are insoluble in the polymer electrolyte solution and will remain as particles dispersed in the cured polymer electrolyte matrix. PPy(DBS) added as up to 0.9 wt% of the total components is well below the percolation threshold reported for roughly spherical PPy particles of at least 3–5 wt%.33,34 The components were combined in an argon-filled glovebox, magnetic stirred overnight (at room temperature, 20–25 °C, in a sealed vial) to form a viscous liquid mixture, then ultrasonicated for 10 min prior to hot-pressing. In ambient air, the mixture was hot-pressed between PET sheets at 80 °C under 4.4 bar of pressure for 10 min (film thickness set to around 125 μm with a spacer). The film was polymerized for 10–20 min under UV light with 150 mW cm−2 intensity at the film surface (main wavenumber 365 nm), flipping the film over halfway through the UV curing process, to obtain the polymer electrolyte. The polymer electrolyte film was vacuum dried (gauge pressure −0.95 bar) overnight at 40 °C to try to remove residual moisture prior to further characterizations. Hereafter, the amount of PPy(DBS) is referred to as the wt% of PEO and TEGDMA amounts in the compositions (0–2 wt% of the polymer and monomer).
 | ||
| Fig. 2 Summary of the polymer electrolyte components and the synthesis process used. | ||
3. Procedures for characterization of adaptive polymer electrolyte
The polymer electrolyte films were characterized with a combination of physical, mechanical, and electrochemical tests. Physical characterizations consisted of insoluble fraction, electrical conductivity, XRD, DSC, and DMA. Mechanical response characterizations included block force response and AFM. Electrochemical characterizations consisted of EIS, Li–Li symmetric cycling, and ESW.3.1. Physical characterizations
Insoluble fraction measurements were performed by taking the initial dry weight of the film (14.3 mm dia.), sandwiching the film between 304 stainless steel mesh, soaking in acetonitrile overnight at room temperature (20–25 °C) to dissolve the soluble (non-cross-linked) components,35,36 vacuum drying (gauge pressure −0.95 bar) overnight at 40 °C, and taking the final dry weight of the film. Electrical conductivity measurements were performed with the polymer film between copper foil contacts, using an Extech EX330 multimeter to obtain the electrical resistance.XRD was collected at room temperature (20–25 °C) from 10° to 40° 2θ with a Rigaku MiniFlex 600 with a Cu K-α radiation source (λ = 1.5406 Å) at 40 kV voltage and 15 mA current. DSC was performed with a TA Instruments DSC 2500 (TA Instruments-Waters LLC, New Castle, DE). Samples between 2.5 mg to 7.5 mg were used in Tzero hermetic aluminum pans. Samples were cooled from room temperature to −90 °C at 10 °C min−1, then heated to 120 °C at 5 °C min−1. This process was repeated for a second scan to remove any thermal history. In the DSC analysis, the glass transition temperature (Tg) was determined from the inflection point in the slope towards the middle of the glass transition step. The DSC slope of heat flow after the glass transition was determined by performing a linear regression of the slope from −20 °C to 120 °C. DMA was performed with a TA Instruments Q800 (TA Instruments-Waters LLC, New Castle, DE) equipped with a tension clamp and Thermal Advantage Q Series version 5.5.22 software. The DMA was operated under tension analysis (0.1 N preload) on polymer films with sample dimensions of 20 mm L (±1 mm) × 10.5 mm W (±0.6 mm) × 0.28 mm T (±0.2 mm) from −120 °C to 150 °C at a 3 °C min−1 heating rate and a 1 Hz frequency. Thicker films for DMA were obtained by using thicker spacers in the synthesis process.
3.2. Mechanical response characterizations
AFM was used to measure the expansion response of polymer electrolyte films under an applied electric field at room temperature (20–25 °C) in air. A schematic of the AFM sample setup and picture of a sample are shown in Fig. 3. Small alterations were made to the polymer synthesis process for producing ∼10 μm thick polymer film samples. Polymer solution was poured onto an ITO-PET sheet (ITO side facing up) to form a 15 mm dia. pool of solution, then hot-pressed between another ITO-PET sheet (ITO side facing polymer solution) without any spacer. After curing, one of the ITO-PET sheets is peeled off and 1 cm × 2 cm samples of 10 μm thick polymer on ITO-PET are cut out. A 50 nm thick layer of gold is sputtered on top of the polymer using a 20 mA sputter current with an EMS150R ES sputter coater (Electron Microscopy Sciences/Quorum). The outer edges of the sample are cut off then a section of polymer film is removed on one end, and copper tape attached to the ITO-PET for the bottom electrical contact. Another copper tape is attached to the gold coating as the top electrical contact, with insulative tape between the top and bottom copper tapes to prevent electrical shorting. The sample is attached to a glass slide using double-sided tape then loaded into an MFP-3D Infinity AFM system (Oxford Instrument, Asylum Research). The cross-section and gold-coated surface morphology were investigated with a Thermo Scientific Apreo scanning electron microscope (SEM). The thicknesses of the films were confirmed to be approximately 10 μm thick by measuring with a micrometer and with cross-sectional SEM (Fig. S1†). | ||
| Fig. 3 (a) Schematic of AFM sample setup for the A-PE and (b) picture of a sample with the area used for AFM characterizations indicated by the dotted line box (grid size 5 mm). | ||
Continuous point measurement with the AFM probe in contact with the sample surface was used to measure the displacement of the film over time. The displacement of the film surface (expansion and contraction) was obtained from the Z-piezo movement required to maintain a constant cantilever deflection. A commercial AFM cantilever was used for continuous point measurements (Olympus AC160-R3, nominal spring constant 4 N m−1, nominal tip radius 7 nm, and nominal resonant frequency 300 kHz). An NI USB-6008 multifunction I/O data acquisition module (National Instruments) was used with LabVIEW to apply a voltage for different electric field strengths across the polymer film for periods of 10 min excitations (voltage applied) followed by 10 min relaxations (no voltage applied). Additionally, topography maps were obtained using tapping mode using a commercial AFM cantilever (Olympus AC240-R3, nominal spring constant 2 N m−1, nominal tip radius 7 nm, and nominal resonant frequency 70 kHz).
A block force setup was utilized to measure the force response of polymer electrolyte films under an applied electric field at room temperature (20–25 °C) in air. The film (14.3 mm dia., 125 μm thick) was sandwiched between 304 stainless steel (304SS) spacers (15.5 mm dia.) with conductive adhesive copper tape attached to each 304SS spacer for electrical connection. A calibrated MF01A-N-221-A04 membrane force sensor (Alpha Taiwan) was used to measure the force. A polytetrafluoroethylene (PTFE) spacer (9.5 mm dia., 1.0 mm thick) was used between the membrane force sensor and the 304 SS/polymer electrolyte/304SS stack to ensure the force was applied in the membrane force sensor active area (10.2 mm dia.) and for electrical isolation. This stack was clamped between insulated surfaces of a small vice mounted on a vibration isolation table. A programmable DC power supply (Tektronix PWS4305) connected with a linear amplifier (Piezo Systems, Inc. model EPA-104) was used to apply a voltage for different electric field strengths across the polymer electrolyte film. Deviations in the membrane force sensor readings from a stable ∼1 N clamp force were monitored to determine the force response of the polymer electrolyte.
3.3. Electrochemical characterizations
CR2032 coin cells were used for all electrochemical testing at 25 °C in a Thermo Scientific Heratherm Refrigerated Incubator. Li-metal chips were polished with a nylon brush to remove any oxide layer prior to coin cell assembly in an argon-filled glovebox. For EIS and Li–Li symmetric cycling, cells with 304SS spacer (15.5 mm dia.)/Li-metal (14.3 mm dia.)/polymer electrolyte (14.3 mm dia.)/Li-metal (11.1 mm dia.)/304SS spacer (15.5 mm dia.) were used (as shown in Fig. S2†). EIS was performed with a Gamry Instruments Reference 600+ potentiostat/galvanostat/ZRA at 25 °C from 5 MHz to 0.1 Hz with a 10 mV AC amplitude applied around the open-circuit voltage (OCV). Gamry Echem Analyst was used for fitting EIS spectra with equivalent circuits using the Simplex method and excluding data points after the end of the semicircle on the low frequency side (right side of the Nyquist plots). Li symmetric cell cycling was performed with an Arbin LBT21084 at 25 °C and a current density of ±0.1 mA cm−2 (based on the area set by the 11.1 mm dia. Li-metal) for 30 min each direction with 30 min rests.The ESW was determined by linear sweep voltammetry (LSV) and cyclic voltammetry (CV) at 25 °C with an Arbin LBT20084. Separate cells were used for anodic LSV and cathodic CV scans. For anodic LSV scans (OCV to 7 V vs. Li/Li+ at 0.1 mV s−1 scan rate), cells with the polymer electrolyte film (14.3 mm dia.) sandwiched between 304SS (15.5 mm dia.) as the working electrode (WE) and Li-metal (11.1 mm dia.) as the counter/reference electrode (CE/RE) were used (shown in Fig. S3a†). For cathodic CV scans (OCV to −0.2 V vs. Li/Li+ at 0.1 mV s−1 scan rate), cells with the polymer electrolyte film (14.3 mm dia.) sandwiched between Cu foil (14.3 mm dia.) as the WE and Li-metal (11.1 mm dia.) as the CE/RE were used (shown in Fig. S3b†).
4. Results and discussion
4.1. Physical characterizations
As produced, the polymer electrolytes were free-standing films (shown for 0 wt% PPy(DBS) composition in Fig. S4†) that could be peeled off the PET sheet and were non-tacky. The cross-linked fraction of the polymer electrolytes was determined through insoluble fraction measurements, as the cross-linked components of the film remain after soaking in acetonitrile.35,36 The results in Fig. 4a show the insoluble fractions of compositions with various amounts of PPy(DBS). The target insoluble fraction of ≥50 wt% was selected based on the similar polymer electrolyte composition from Zhang and coworkers35 and above this threshold the films were free-standing. For a given UV curing time, as the amount of PPy(DBS) in the composition increased the insoluble fraction typically decreased and the variability in insoluble fraction between samples from the same film increased. With longer UV curing times for each composition, the insoluble fraction increased and the variability decreased. The PPy(DBS) particles are opaque, black particles that reduced UV transmittance through the thickness of the film (inhibiting UV curing), resulting in lower insoluble fractions and higher variability. The optical appearance of the films with increasing amounts of PPy(DBS) are shown in the inset in Fig. 4a. The 0 wt% PPy(DBS) film was optically clear, becoming darker with more PPy(DBS) until appearing almost completely opaque at 2.0 wt% PPy(DBS). The 2.0 wt% PPy(DBS) film eventually reached the target insoluble fraction after 20 min UV curing time but resulted in a very brittle film (likely due to overcuring in areas of the film the UV transmittance was less obstructed by PPy(DBS) particles). The 2.0 wt% PPy(DBS) films did not reach full solidification below this curing time and the insoluble fraction could not be measured. | ||
| Fig. 4 Physical characterizations of the polymer electrolytes. (a) Insoluble fraction measurements with markers indicating average values and error bars indicating standard deviations. Markers are offset horizontally for visibility. Inset shows appearance of films with different amounts of PPy(DBS) (grid size 5 mm). (b) XRD patterns (c) DSC profiles, and (d) DMA tan delta thermograms a.u. denotes arbitrary units. XRD, DSC, and DMA curves are offset vertically for visibility. | ||
Electrical conductivities obtained from 5 samples of each composition are shown in Fig. S5.† The electrical conductivities (σelec) were calculated using the resistance values measured with the multimeter (R), thickness of the film (t) and contact area (A) in the following equation:
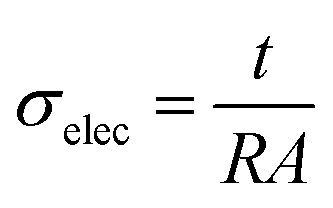 | (1) |
Materials with an electrical conductivity below 10−8 S cm−1 are typically considered electrical insulators, with the transition to electrical semiconductors for conductivities above 10−8 S cm−1.38,39 Films with 0 wt% through 2.0 wt% PPy(DBS) only show a minor increase in electrical conductivity from 4.6–6.3 × 10−9 S cm−1, confirming the compositions are below the percolation threshold (where a sharp increase in electrical conductivity of multiple orders of magnitude would be observed). Thus, the upper limit on the amount of PPy(DBS) that can be added to these compositions is due to the UV curing process, not the percolation threshold. For subsequent experiments, only 0 wt% through 1.0 wt% PPy(DBS) films from 10 min UV curing time were used. These films were selected to avoid potential effects from overcuring at longer UV cure times. The average insoluble fractions were above the target fraction for these films, even though the films with increasing amounts of PPy(DBS) had lower insoluble fractions.
XRD scans of polymer electrolyte films containing 0 wt% through 1.0 wt% PPy(DBS) particles (with background signal from the glass slide removed) are shown in Fig. 4b. There are no sharp crystalline peaks present in the XRD pattern – only a broad amorphous peak centered around 2θ of 19.5° is present, indicating highly amorphous films. The amorphous peak appears to broaden with increasing amounts of PPy(DBS), suggesting the addition of PPy(DBS) may lead to increasingly disordered films.
DSC profiles from the second heating scan of the different polymer electrolyte compositions are shown in Fig. 4c with results summarized in Table S1.† During DSC heating scans, thermal transitions such as the glass transition, melting, and crystallization can be observed. The glass transition occurs within the amorphous phase of PEO as the material changes from a rigid glassy state to a more mobile rubbery state due to heating. It presents itself as an endothermic step change in heat flow within the DSC heat scan. The polymer samples in this study showed Tg values between −37 °C and −41 °C, in order of increasing PPy(DBS). With Tg values well below room temperature, the polymer will exhibit a highly mobile rubbery structure around room temperature. The polymer samples do not display any distinct melting peaks in the DSC data up to 120 °C. This suggests the films are highly amorphous, as confirmed by XRD data, compared to the semi-crystalline behavior typically observed in similar PEO based polymer electrolytes.35,36 Li-ion transport in PEO based polymer electrolytes is typically considered to occur through segmental motion of polymer chains in amorphous regions and the subsequent Li-ion hopping along and between the polymer chains.40 Thus, a highly amorphous structure enhances segmental motion mechanisms for increasing ionic conductivity because there are no crystalline lattice structures to inhibit the movement of the polymer chains and lithium ions. As expected from literature on other filler particles incorporated in PEO lowering Tg (such as SiO2, SnO2, and TiO2),41–43 incorporating increasing amounts of PPy(DBS) shifts the Tg to slightly lower temperatures. This could be caused by PPy(DBS) particles physically disrupting and interfering with the packing of PEO chains allowing the glass transition to occur at lower temperatures. The slope of the heat flow after the glass transition in Fig. 4c increased with the addition of PPy(DBS) from 20.1 mW (g °C)−1 to 85.9–115.0 mW (g °C)−1, summarized in Table S1.† This increase in slope of the heat flow suggests an increase in specific heat capacity (requiring additional heat flow to maintain the same heating rate). This difference is not expected from the heat capacity of PPy(DBS), which is typically lower than the heat capacity of PEO. Instead, the increasingly mobile rubbery structures (as indicated by decreasing Tg) may cause the increase in specific heat capacity.44–47
DMA tan delta thermograms are shown in Fig. 4d, with storage and loss modulus profiles shown in Fig. S6.† Results are only shown to 20 °C as the films became too soft above this temperature and broke or the measurement signal became unstable. The peak of the tan delta curve (when the material demonstrates the most viscous response to deformations) is used to determine Tg of the films from DMA in this analysis with the results summarized in Table S1.† There are multiple methods to determine Tg from DMA, with the tan delta peak typically giving the highest value of Tg.48 This is a conservative determination with respect to enhanced segmental motion of polymer chains for increased ionic conductivity in PEO based electrolytes. DMA results show Tg values between −43 °C and −57 °C in good agreement with the trend of Tg generally decreasing with increasing amounts of PPy(DBS), as observed with DSC. The obtained values of Tg differed between DSC and DMA, which is not uncommon as glass transition is a region of behavior with values often differing by 10–25 °C between the techniques.48 The difference in Tg between techniques can be attributed to the different property changes related to the glass transition region observed with DSC and DMA (measurement parameters can also affect the values obtained). Changes in heat flow due to the change in heat capacity of the polymer during its transition from a rigid glassy state to a mobile rubbery state are observed with DSC, whereas the change in mechanical properties are observed with DMA. These Tg values from DSC and DMA are within the range of similar PEO based polymer electrolytes with reported Tg values between −34 °C and −78 °C.35,36 The storage and loss moduli did not show a consistent trend with increasing amounts of PPy(DBS). If the addition of PPy(DBS) particles affected the storage and loss moduli, increasing or decreasing values with increasing amounts of PPy(DBS) would be expected but no such trend is observed. The variability between the storage and loss moduli for the different compositions could obscure any effect from PPy(DBS).
4.2. Mechanical response characterizations
Fig. 5 shows the AFM continuous point measurements for the films under an electric field strength of 0.3 V μm−1 for 10 min followed by 0 V μm−1 for 10 min for 5 representative excitations and relaxations. The 0 wt% PPy(DBS) film (Fig. 5a) does not show any appreciable response beyond the measurement uncertainty and noise. The 0.25–1 wt% PPy(DBS) films (Fig. 5b–d) show a clear expansion response up to 0.6 μm (corresponding to 6% strain for the approximately 10 μm thick films) under an electric field strength of 0.3 V μm−1 followed by a relaxation when the electric field is removed (0 V μm−1). This response is primarily attributed to the polarization of the PPy(DBS) particles in the polymer film. The PPy(DBS) particles are polarized under an electric field and would try to align, causing the film to expand. The expansion response increases from an average expansion of 0.44 μm for 0.25 wt% PPy(DBS) to 0.56 μm for 1 wt% PPy(DBS) and appears to be repeatable over several cycles. The expansion response decreases with decreasing electric field strength as shown in Fig. S7,† where the film does not produce an appreciable response below 0.2 V μm−1 regardless of the concentration of PPy(DBS). | ||
| Fig. 5 AFM continuous point measurement of polymer film under 0.3 V μm−1 followed by 0 V μm−1 electric field strength for (a) 0 wt% PPy(DBS), (b) 0.25 wt% PPy(DBS), (c) 0.5 wt% PPy(DBS), and (d) 1 wt% PPy(DBS). (e) Film strain at the end of each excitation and relaxation and (f) excitation and relaxation time constants with markers indicating median values and error bars indicating standard deviations. | ||
The films not returning to zero displacement at the end of the relaxations suggests there is a residual expansion left in the films. This could be due to an irreversibility such as plastic deformation or an irreversible rearrangement of PPy(DBS) particles and polymer matrix chains during the initial excitation. This behavior is also seen to some extent in the storage modulus response for PPy-based and other ER elastomers.26–28 The strain of the films at the end of excitations and relaxations (εexcitation and εrelaxation, corresponding to expansion and residual expansion, respectively) were calculated from the displacement compared to the beginning of the excitation (taken as zero) divided by the film thickness (approximately 10 μm). The results are shown in Fig. 5e with 0 wt% PPy(DBS) included for comparison, even though it did not display a clear expansion and relaxation response. For the 0.25–1 wt% PPy(DBS) films, εexcitation increased from ∼4–6% with increasing PPy(DBS) whereas εrelaxation did not display a clear trend. Further characterization will be needed to fully elucidate the relaxation behavior.
The temporal characteristics of the response was investigated with the time constant for expansion and relaxation (τexcitation and τrelaxation, respectively). τexcitation was determined from the time taken for the excitation to reach 1 − 1/e (63.2%) of the final value. τrelaxation was determined from the time taken for the relaxation to decay to 1/e (36.8%) of the final excitation value compared to the final relaxation value. The results are shown in Fig. 5f, with the response times a similar magnitude to those of storage modulus for a PPy-based ER elastomer.26 There was no significant trend for the time constants with different amounts of PPy(DBS). However, τrelaxation was lower than τexcitation by about 1 min for each case. This suggests the polymer starts to return to its relaxed state faster than the excitation. This is seen in the storage modulus response for a PPy-based ER elastomer as well.26 The storage modulus quantifies the elastic response of the polymer when it is subjected to a deformation. The energy is stored elastically when undergoing deformation and can be released once the stress is removed. This energy is required to rearrange the polymer chain entanglements and cross links in response to the stress (in this case, in response to the PPy(DBS) particles polarizing and trying to align in the polymer matrix under an electric field). However, this elastic energy can allow the polymer chains in the matrix to return to the relaxed state very fast, which could result in τrelaxation being lower than τexcitation. Also, the films do not return to zero displacement during the relaxation, which may contribute to the lower time constant.
Note that, while the AFM measurements were generally reliable, they were subject to uncertainties such as thermal drift over the long timescale of the polymer response. Some sharp increases or decreases were noted during the transitions between excited and relaxed states. These variations are likely attributable to measurement artifacts, as the direction of these changes was inconsistent across similar transitions. Despite these uncertainties, the results indicate the 0.25–1 wt% PPy(DBS) films produce a significant expansion strain response during excitation (4.2–5.9%) and residual strain following relaxation (1.1–2.5%) compared to the 0 wt% PPy(DBS) control (with strains of 0.2% ± 0.3% and 0.1% ± 0.4%, respectively).
The surface morphology of gold coated 1 wt% PPy(DBS) film is shown in Fig. 6. SEM micrographs (Fig. 6a and e) of the pristine film show relatively flat surfaces with the exception of a few raised areas. AFM topography maps were used to investigate the uniformity of the polymer film expansion in local regions. The AFM topography maps of the film at rest (Fig. 6b and f), after the first excitation at 0.3 V μm−1 (Fig. 6c and g), and after the first relaxation (Fig. 6d and h) do not show any significant changes in the surface locally, suggesting the expansion response is relatively uniform across the surface of the film.
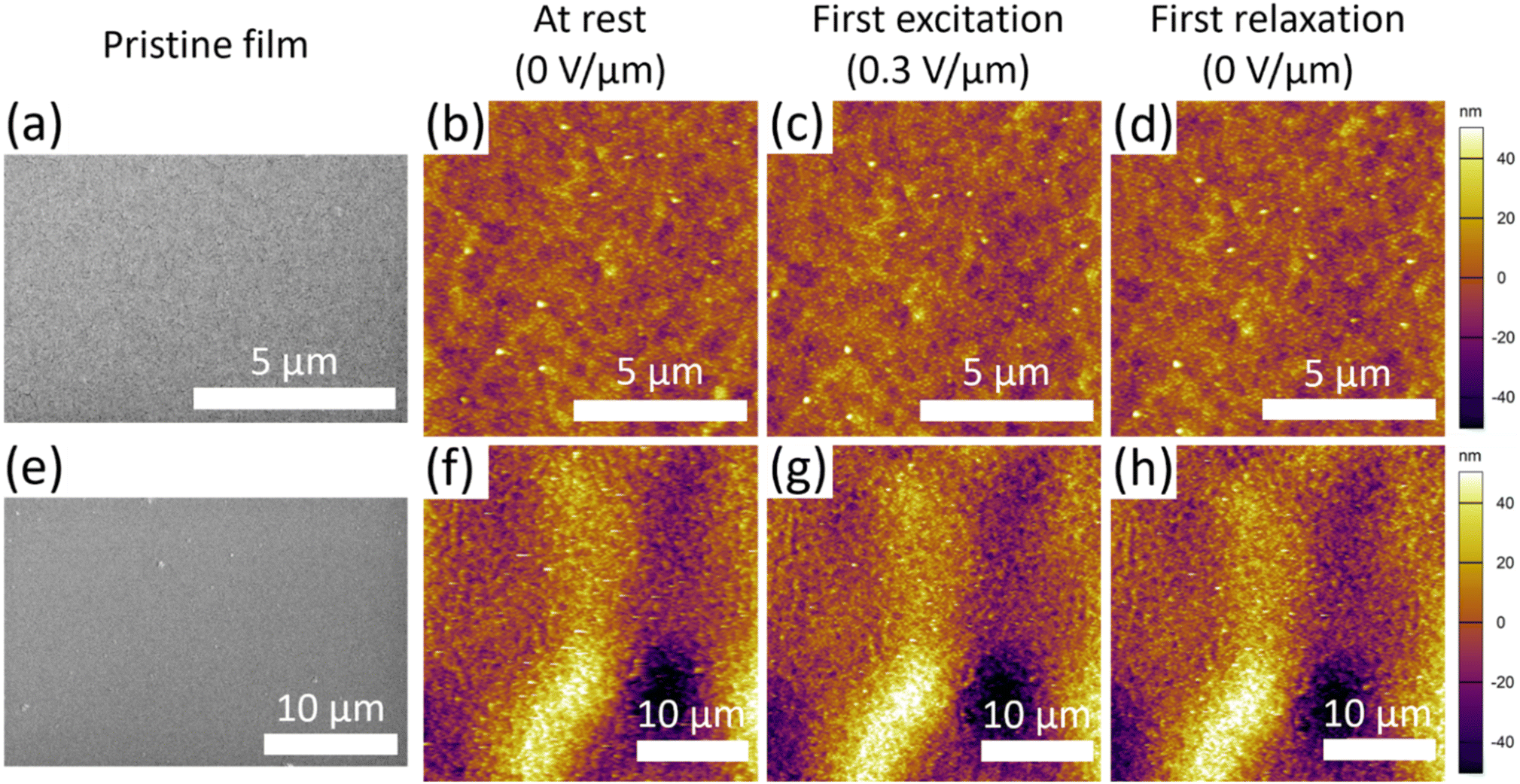 | ||
| Fig. 6 Surface of 50 nm gold coated 1 wt% PPy(DBS) film (a) SEM micrograph, (b–d) AFM topography maps of 10 μm × 10 μm area (b) at initial rest, (c) after the first excitation for 10 min, and (d) after the first relaxation for 10 min. Zoomed-out (e) SEM micrograph, (f–h) AFM topography maps of 30 μm × 30 μm area (f) at initial rest, (g) after the first excitation for 10 min, and (h) after the first relaxation for 10 min. SEM micrographs were obtained with secondary electron mode at an accelerating voltage of 5 kV and beam current of 0.20 nA. | ||
A simplified schematic of the block force setup (insulative layers between electrically conductive components not shown) used for measuring the force response of the polymer electrolyte compositions under an electric field is shown in Fig. 7a. The polymer electrolyte film was sandwiched between electrical contacts that were connected to a voltage source to apply an electric field across the film. A membrane force sensor was used in-line to measure force applied from the film (as deviations from the ∼1 N clamp force) with the entire stack clamped in a small vise. This setup roughly corresponds to the polymer electrolyte as an interlayer sandwiched between a Li-metal anode and ceramic solid electrolyte, however, the clamp force used here is lower than the stack pressure would be in a solid-state battery due to resolution limitations of force sensors with higher load capacities.
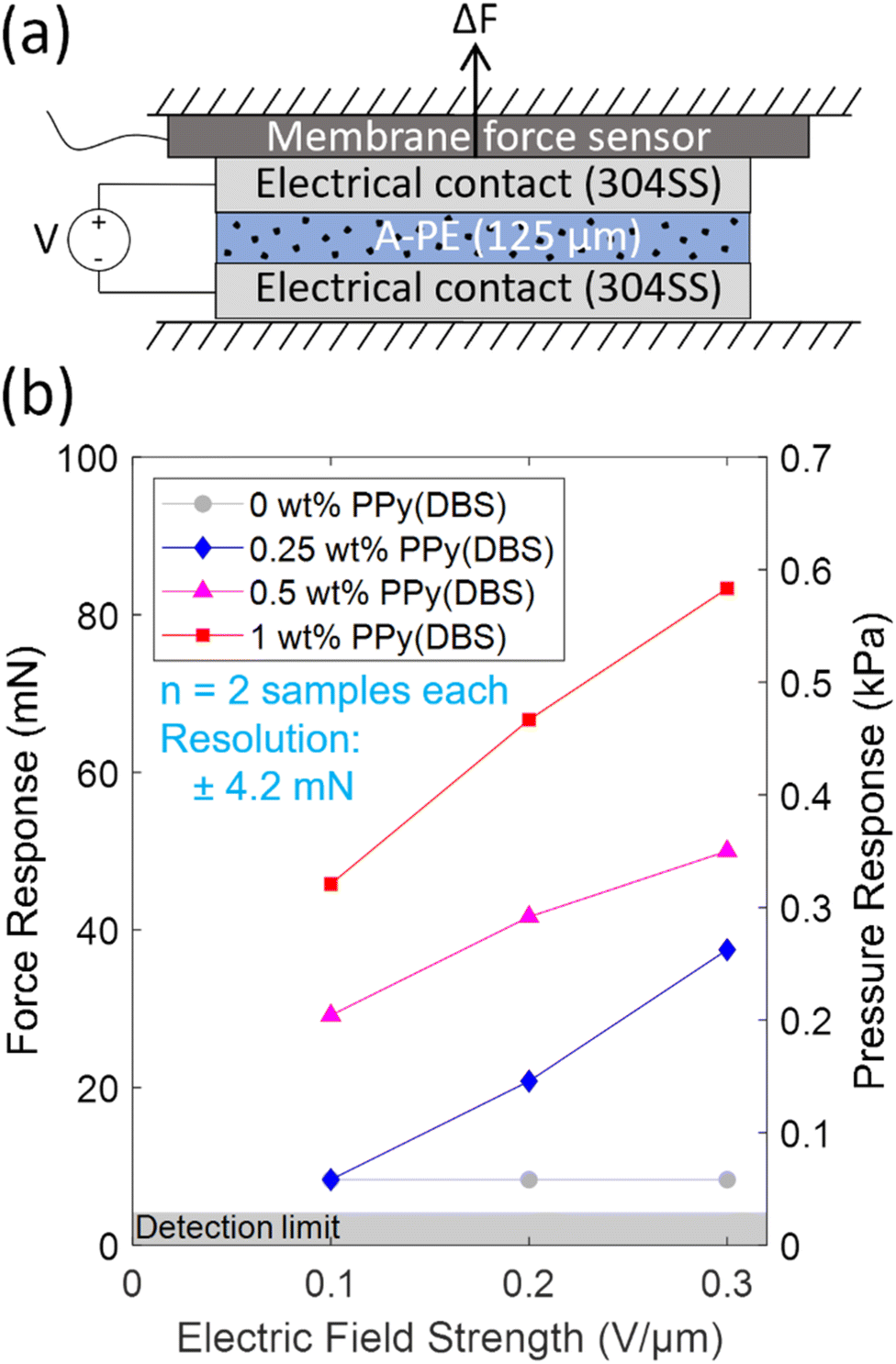 | ||
| Fig. 7 (a) Schematic of block force setup for the A-PE and (b) force response measurements under different electric field strengths. Markers indicate average values, and the shaded area indicates the region below the detection limit of the force sensor. | ||
The resulting force response measurements are shown in Fig. 7b. For films with PPy(DBS), the force response increased with increasing wt% of PPy(DBS) (from 0.25 wt% to 1 wt%) and increasing electric field strength (from 0.1 V μm−1 to 0.3 V μm−1). This force response trend is associated primarily to the polarization of PPy(DBS) under an electric field as the particles may try to align, since this response of CP particles such as PPy(DBS) has typically been shown to increase with increased CP loading and electric field strength.21–29 Since the film is mechanically compressed under the clamp, changes in stiffness of the film under electric field may partially contribute to the measured response, implying that the force required to maintain the same level of compression would increase.26–29
It is worth noting that the 0 wt% PPy(DBS) films showed a non-zero response (Fig. 7b) that, however, did not indicate any appreciable relationship with the applied electric field strength. Polarization of ions within the film under an electric field could contribute to the response. The film responses in Fig. 7b are only reported for the first actuation to mitigate possible effects on the response from decomposition of the films (as the applied voltages are above the ESW of around 4.5 V in Fig. 8d). However, these results still suggest a desirable force response due to the polarization of the PPy(DBS) particles as the responses of films with PPy(DBS) are significantly larger than the responses in the 0 wt% PPy(DBS) films, increasing with increasing electric field strength and amount of PPy(DBS) (and the ESW is similar between all compositions).
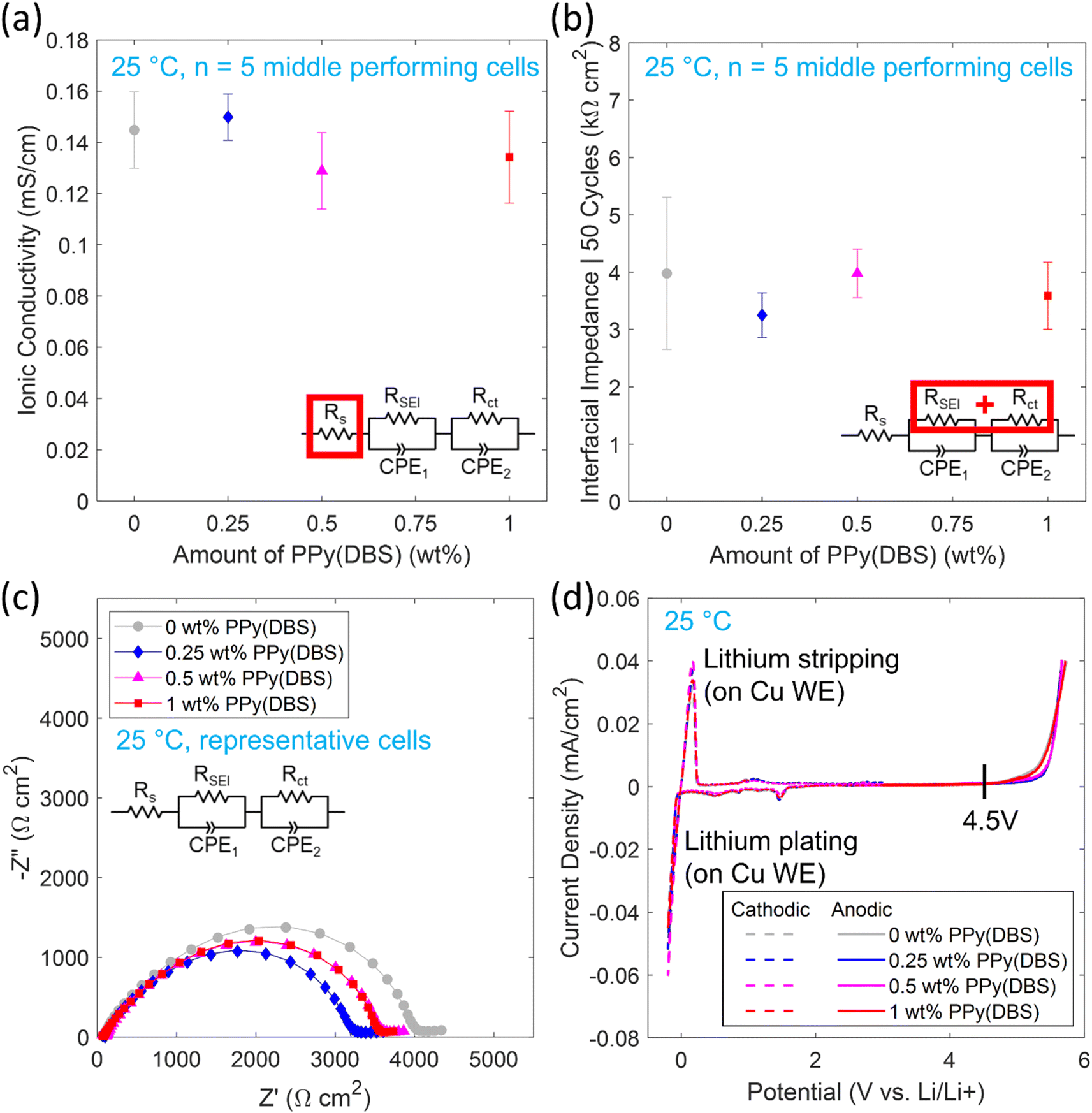 | ||
| Fig. 8 Summary of polymer electrolyte electrochemical characterizations at 25 °C. (a) Ionic conductivity from EIS and (b) interfacial impedance from EIS after 50 Li symmetric cycles with markers indicating median values (corresponding to an actual coin cell) and error bars indicating standard deviations. (c) EIS spectra of representative Li/polymer/Li coin cells after 50 Li symmetric cycles for 0–1 wt% PPy(DBS) with markers indicating measured values and solid lines representing the equivalent circuit fit. Equivalent circuit elements consist of series resistance (Rs), solid electrolyte interphase resistance (RSEI), charge transfer resistance (Rct), and constant phase elements (CPE1, CPE2). (d) ESW from CV and LSV. | ||
4.3. Electrochemical characterizations
The electrochemical characterizations of the polymer electrolyte compositions were all performed at 25 °C to investigate the performance around room temperature. Symmetric Li/polymer/Li coin cells were used to compare the polymer electrolyte performance of the compositions with PPy(DBS) to the 0 wt% PPy(DBS) composition. For symmetric cycling (30 min cycles at ±0.1 mA cm−2) and EIS, ≥10 coin cells were tested for each composition. Results based on the 5 middle performing coin cells are reported in this work.A summary of the measured ionic conductivities for the 5 middle performing coin cells at 25 °C are shown in Fig. 8a. The ionic conductivities (σion) were calculated using Rs from the equivalent circuit as the bulk polymer electrolyte impedance (Rb), thickness of the electrolyte (t) and contact area (A) in the following equation:
 | (2) |
The results indicate no significant trend for ionic conductivity between the compositions when considering the variability, with ionic conductivities between 0.11–0.16 mS cm−1.
Out of the 5 middle performing coin cells for each composition the Li symmetric cycling overpotentials are shown in Fig. S8† and the Nyquist plots obtained from EIS are shown in Fig. S9† for 0–50 cycles of the representative cells. Equivalent circuit model parameter fitting was performed with a series resistor (Rs) to model the bulk impedance of the polymer electrolyte and two sets of parallel resistors (RSEI, Rct) with constant phase elements (CPE1, CPE2) to model the solid electrolyte interphase and charge transfer of the Li/polymer interfaces, respectively. The total interfacial impedance is taken as the combination of RSEI and Rct of both Li/polymer interfaces, assuming the interfaces are nearly identical. CPEs are used to account for non-ideal capacitive behavior. The equivalent circuit model parameters for the representative cells are summarized in Table S2.† The bulk impedance (which can be approximated as the left-side intersection of semicircle with the real impedance Z′ axis) remained relatively stable over 50 cycles. The interfacial impedance (can be approximated as the right-side intersection of semicircle with the real impedance Z′ axis) varied between the cells over 50 cycles with interfacial impedance decreasing (suggesting the interface is still stabilizing), increasing, a combination of the two, or remaining stable. Thus, the cells are compared after 50 cycles when there was less variability between the 5 cells with the interfacial impedances shown in Fig. 8b and Nyquist plots from EIS for representative cells shown in Fig. 8c.
As with the ionic conductivity, no significant trend in interfacial impedances was observed among the compositions with increasing amounts of PPy(DBS) when considering the variability of the 5 cells of each composition in Fig. 8b. One notable difference was the variability between the 0 wt% PPy(DBS) composition cells was significantly higher than each of the compositions with PPy(DBS). The use of wave springs in the coin cells could contribute to increased variability in interfacial impedance, due to uneven pressure applied by wave springs.49–51 Overall, the total interfacial impedances of the cells were on the order of 4 kΩ cm2. This value is higher than comparable PEO based polymer electrolytes35,36 of ∼500–700 Ω cm2 which could be due to some experimental processing limitations in this work such as: hot-pressing and UV-curing the films in ambient air (possibility of moisture contamination that vacuum drying may not fully remove), hot-pressing under lower pressure, greater amount of photoinitiator used (evidenced by reaching target insoluble fraction in a shorter curing time compared to Zhang and coworkers35 where photoinitiator amount was not specified), and differences in coin cell configuration (total spacer thickness and type of spring will affect stack pressure and thus interfacial impedance, which were not specified35,36). Excess photoinitiator in particular could cause increased interfacial impedance due to increased storage modulus52 or premature termination and incomplete cure of the polymer matrix.53 During polymerization, excess photoinitiator could cause more polymer chains to become crosslinked leading to a higher crosslink density. An increased storage modulus could result from the higher crosslink density due to less mobility within the polymer chains and the polymer would not be able to conform as easily to the Li-metal surface, increasing the interfacial impedance. An incomplete cure could lead to less crosslinks in the matrix than expected or inhomogeneous crosslinking. Regions of lower crosslinking could have higher mobility and leaching of non-crosslinked moieties and regions of higher crosslinking could have more defects resulting in cracks or voids in the interface.
Although some polymer electrolytes have achieved sufficient ionic conductivities at 25 °C, high interfacial impedances >100 Ω cm2 limit the commercial viability of polymer electrolytes in LIMSSBs for applications such as electric vehicles and consumer electronics (apart from semi-solid gel polymer electrolytes that can pose safety concerns). Further advancements are needed to lower interfacial impedance at 25 °C such as developments with in situ polymerization or single ion conducting polymer electrolytes.54–57 The incorporation of PPy(DBS) particles and resulting properties presented in this work are expected to be applicable for other polymer electrolyte compositions.
The ESW at 25 °C with Li-metal of each polymer electrolyte composition was determined with cathodic CVs and anodic LSV shown in Fig. 8d (with zoomed-in plots shown in Fig. S10a and b,† respectively). For the cathodic sweep, a small peak is initially seen around 1.5 V vs. Li/Li+ that may be attributed to the decomposition of some polymer electrolyte components to form a passivation layer with Li-metal,58 as many polymer electrolytes are not inherently stable with Li-metal but form a stable passivation layer or interphase.6,20,35,36 This peak is present in all compositions, suggesting components in the 0 wt% PPy(DBS) composition are responsible, not PPy(DBS). Reversible Li plating and stripping on the Cu surface is seen from −0.2 V to 0.3 V vs. Li/Li+, as Li will plate on the Cu WE when its potential goes below 0 V vs. Li/Li+ then strip after the potential increases above 0 V vs. Li/Li+ again. From the anodic sweep, the oxidative stability is conservatively determined as when the current density surpasses 1 μA cm−2 giving a stability up to 4.5 V vs. Li/Li+ for all compositions.
5. Conclusions
This study demonstrates the adaptive behavior of a polymer electrolyte at room temperature (20–25 °C) by incorporating conducting polymer particles, with the aim of investigating its potential for application as an interlayer to enhance the performance and durability of lithium metal solid-state batteries (LIMSSBs). The adaptive polymer electrolyte (A-PE) in this study consisted of a polyethylene oxide based polymer electrolyte containing polypyrrole doped with dodecylbenzene sulfonate (PPy(DBS)) conducting polymer particles. The incorporation of PPy(DBS) particles imparted adaptive properties to a polymer electrolyte matrix, enabling force and expansion responses under electric fields through the polarization of PPy(DBS) particles.Experimental results confirmed the A-PE's adaptive behavior. Atomic force microscopy (AFM) measurements revealed an expansion response of films of up to 6% strain under a field of 0.3 V μm−1, repeatable over several cycles, with reduced expansion observed at lower electric field strengths. Block force response measurements showed a corresponding increase in force with higher electric field (starting from 0.1 V μm−1) and greater PPy(DBS) loading. These findings are consistent with the hypothesis that the adaptive response is driven by the polarization of PPy(DBS) particles, a behavior commonly observed in electrorheological fluids and elastomers.21–29 However, the direct alignment of PPy(DBS) particles in solid films was not observed and remains an area for further exploration.
The incorporation of PPy(DBS) particles affected the polymer matrix by inhibiting UV curing, lowering the insoluble fraction of the film and restricting the PPy(DBS) particle amount to ∼1 wt% of polymer and monomer content (equivalent to ∼0.45 wt% or ∼8 vol% of final film). Despite these constraints during the synthesis process, the films remained electrical insulators with no significant increase in electrical conductivity, indicating that the percolation threshold was not reached. Furthermore, no substantial trends were observed in the electrochemical performance of the polymer electrolytes at 25 °C with increasing amounts of PPy(DBS). The amorphous structure of the films showed only minor changes with the amounts of PPy(DBS) incorporated in this study.
Beyond LIMSSBs, this adaptive polymer concept shows promise for other applications, such as low voltage soft actuators. Further research should focus on characterizing the reversibility of the adaptive response, residual expansion upon relaxation, and direct observation of PPy(DBS) particle alignment in solid films. Exploring synthesis methods that allow for the addition of more PPy(DBS) is also of interest to determine if there is an optimal amount before the films become electrically conductive. Additionally, evaluating the performance of the A-PE as a thin interlayer (ideally <10 μm thick) between the lithium metal anode and ceramic solid electrolyte in full LIMSSBs will be critical to understanding its potential to mitigate interfacial changes upon lithiation/delithiation, and its effects on battery durability.
Data availability
Data for this article (insoluble fraction, electrical conductivity, XRD, DSC, DMA, AFM, block force, EIS, Li symmetric cell cycling, ESW) are available at Harvard Dataverse at https://doi.org/10.7910/DVN/FJ4VZW. The following data supporting this article have been included in the ESI:† SEM cross-section of thin polymer film for AFM experiments, diagrams of coin cell configurations used for electrochemical tests, photographs of free-standing polymer electrolyte film, electrical conductivity results, additional DMA results, additional AFM results, Li symmetric cycling overpotentials, EIS equivalent circuit fitting parameters.Author contributions
M. N., H. C., J. S., and M. C. designed the experiments. M. N. fabricated the samples, performed XRD, block force, and electrochemical characterizations, and analyzed all data. J. L. and H. J. performed AFM characterizations. R. G. performed DSC characterizations and contributed to interpreting the data. S. D. performed DMA characterizations and contributed to interpreting the data. H. C. contributed to interpreting the data and provided resources for AFM characterizations. Y. V. contributed to interpreting the data and provided resources for DSC and DMA characterizations. J. S. and M. C. contributed to interpreting all data and provided supervision. M. N. drafted the manuscript and all authors contributed to reviewing and editing the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge Dr Vishnu Baba Sundaresan for initiating the project and securing initial funding. We thank Dr Vicky Doan-Nguyen, Dr Jung-Hyun Kim, Lakshmi Surag Singavarapu, and Hans Herath for valuable discussions. We are grateful to Dr Jung-Hyun Kim for use of equipment in the Energy Innovation Lab. We additionally wish to acknowledge Dr Christopher Brooks, Honda Research Institute USA, Inc. for mentorship provided during the project. Electron microscopy was performed at the Center for Electron Microscopy and Analysis (CEMAS) at The Ohio State University. Financial support was supplied by the member organizations of the Smart Vehicle Concepts Center (https://www.SmartVehicleCenter.org), a graduated National Science Foundation Industry-University Cooperative Research Center formerly operating under Grant NSF IIP 1738723.References
- J. Janek and W. G. Zeier, A solid future for battery development, Nat. Energy, 2016, 1, 16141 CrossRef.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Solid-State Li–Metal Batteries: Challenges and Horizons of Oxide and Sulfide Solid Electrolytes and Their Interfaces, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, From nanoscale interface characterization to sustainable energy storage using all-solid-state batteries, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, Origin of Outstanding Stability in the Lithium Solid Electrolyte Materials: Insights from Thermodynamic Analyses Based on First-Principles Calculations, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Electrochemical Stability of Li10GeP2S12 and Li7La3Zr2O12 Solid Electrolytes, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- S. Sen, E. Trevisanello, E. Niemöller, B.-X. Shi, F. J. Simon and F. H. Richter, The role of polymers in lithium solid-state batteries with inorganic solid electrolytes, J. Mater. Chem. A, 2021, 9, 18701–18732 RSC.
- M. Nagao, A. Hayashi, M. Tatsumisago, T. Kanetsuku, T. Tsuda and S. Kuwabata, In situ SEM study of a lithium deposition and dissolution mechanism in a bulk-type solid-state cell with a Li2S–P2S5 solid electrolyte, Phys. Chem. Chem. Phys., 2013, 15, 18600 RSC.
- Y. Ren, Y. Shen, Y. Lin and C.-W. Nan, Direct observation of lithium dendrites inside garnet-type lithium-ion solid electrolyte, Electrochem. Commun., 2015, 57, 27–30 CrossRef CAS.
- T. Krauskopf, H. Hartmann, W. G. Zeier and J. Janek, Toward a Fundamental Understanding of the Lithium Metal Anode in Solid-State Batteries – An Electrochemo-Mechanical Study on the Garnet-Type Solid Electrolyte Li6.25Al0.25La3Zr2O12, ACS Appl. Mater. Interfaces, 2019, 11, 14463–14477 CrossRef CAS PubMed.
- X. Zhang, Q. J. Wang, K. L. Harrison, S. A. Roberts and S. J. Harris, Pressure-Driven Interface Evolution in Solid-State Lithium Metal Batteries, Cell Rep. Phys. Sci., 2020, 1, 100012 CrossRef.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Negating interfacial impedance in garnet-based solid-state Li metal batteries, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- Y. Ruan, Y. Lu, Y. Li, C. Zheng, J. Su, J. Jin, T. Xiu, Z. Song, M. E. Badding and Z. Wen, A 3D Cross-Linking Lithiophilic and Electronically Insulating Interfacial Engineering for Garnet-Type Solid-State Lithium Batteries, Adv. Funct. Mater., 2021, 31, 2007815 CrossRef CAS.
- W. Zhang, X. Wang, Q. Zhang, L. Wang, Z. Xu, Y. Li and S. Huang, Li7La3Zr2O12 Ceramic Nanofiber-Incorporated Solid Polymer Electrolytes for Flexible Lithium Batteries, ACS Appl. Energy Mater., 2020, 3, 5238–5246 CrossRef CAS.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Flexible and ion-conducting membrane electrolytes for solid-state lithium batteries: Dispersion of garnet nanoparticles in insulating polyethylene oxide, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- K. Liu, R. Zhang, J. Sun, M. Wu and T. Zhao, Polyoxyethylene (PEO)|PEO-Perovskite|PEO Composite Electrolyte for All-Solid-State Lithium Metal Batteries, ACS Appl. Mater. Interfaces, 2019, 11, 46930–46937 CrossRef CAS PubMed.
- J. Zheng, P. Wang, H. Liu and Y.-Y. Hu, Interface-Enabled Ion Conduction in Li10GeP2S12–Poly(ethylene Oxide) Hybrid Electrolytes, ACS Appl. Energy Mater., 2019, 2, 1452–1459 CrossRef CAS.
- S. Wang, X. Zhang, S. Liu, C. Xin, C. Xue, F. Richter, L. Li, L. Fan, Y. Lin, Y. Shen, J. Janek and C.-W. Nan, High-conductivity free-standing Li6PS5Cl/poly(vinylidene difluoride) composite solid electrolyte membranes for lithium-ion batteries, Journal of Materiomics, 2020, 6, 70–76 CrossRef.
- Z. Yang, H. Yuan, C. Zhou, Y. Wu, W. Tang, S. Sang and H. Liu, Facile interfacial adhesion enabled LATP-based solid-state lithium metal battery, Chem. Eng. J., 2020, 392, 123650 CrossRef CAS.
- H. Duan, Y. X. Yin, Y. Shi, P. F. Wang, X. D. Zhang, C. P. Yang, J. L. Shi, R. Wen, Y. G. Guo and L. J. Wan, Dendrite-Free Li-Metal Battery Enabled by a Thin Asymmetric Solid Electrolyte with Engineered Layers, J. Am. Chem. Soc., 2018, 140, 82–85 CrossRef CAS PubMed.
- F. J. Simon, M. Hanauer, A. Henss, F. H. Richter and J. Janek, Properties of the Interphase Formed between Argyrodite-Type Li6PS5Cl and Polymer-Based PEO10:LiTFSI, ACS Appl. Mater. Interfaces, 2019, 11, 42186–42196 CrossRef CAS PubMed.
- D. S. Jang, W. L. Zhang and H. J. Choi, Polypyrrole-wrapped halloysite nanocomposite and its rheological response under electric fields, J. Mater. Sci., 2014, 49, 7309–7316 CrossRef CAS.
- G. C. Teare and N. M. Ratcliffe, Electrorheological properties of polypyrrole prepared by the action of mineral acids on pyrrole, J. Mater. Chem., 1996, 6, 301 RSC.
- D.-H. Kim and Y. D. Kim, Electrorheological Properties of Polypyrrole and its Composite ER Fluids, J. Ind. Eng. Chem., 2007, 13, 879–894 CAS.
- F. F. Fang, Y. D. Liu and H. J. Choi, Electrorheological and magnetorheological response of polypyrrole/magnetite nanocomposite particles, Colloid Polym. Sci., 2013, 291, 1781–1786 CrossRef CAS.
- P. Chen, Q. Cheng, L. M. Wang, Y. D. Liu and H. J. Choi, Fabrication of dual-coated graphene oxide nanosheets by polypyrrole and poly(ionic liquid) and their enhanced electrorheological responses, J. Ind. Eng. Chem., 2019, 69, 106–115 CrossRef CAS.
- P. Ludeelerd, S. Niamlang, R. Kunaruksapong and A. Sirivat, Effect of elastomer matrix type on electromechanical response of conductive polypyrrole/elastomer blends, J. Phys. Chem. Solids, 2010, 71, 1243–1250 CrossRef CAS.
- W. Wichiansee and A. Sirivat, Electrorheological properties of poly(dimethylsiloxane) and poly(3,4-ethylenedioxy thiophene)/poly(stylene sulfonic acid)/ethylene glycol blends, Mater. Sci. Eng. C, 2009, 29, 78–84 CrossRef CAS.
- X. Yuan, X. Zhou, Y. Liang, L. Wang, R. Chen, M. Zhang, H. Pu, S. Xuan, J. Wu and W. Wen, A stable high-performance isotropic electrorheological elastomer towards controllable and reversible circular motion, Composites, Part B, 2020, 193, 107988 CrossRef CAS.
- N. Ma, Y. Yao, Q. Wang, C. Niu and X. Dong, Properties and mechanical model of a stiffness tunable viscoelastic damper based on electrorheological elastomers, Smart Mater. Struct., 2020, 29, 045041 CrossRef.
- N. Aydemir, P. A. Kilmartin, J. Travas-Sejdic, A. Kesküla, A. L. Peikolainen, J. Parcell, M. Harjo, A. Aabloo and R. Kiefer, Electrolyte and solvent effects in PPy/DBS linear actuators, Sens. Actuators, B, 2015, 216, 24–32 CrossRef CAS.
- Y. Kudoh, Properties of polypyrrole prepared by chemical polymerization using aqueous solution containing Fe2(SO4)3 and anionic surfactant, Synth. Met., 1996, 79, 17–22 CrossRef CAS.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, Electrically conductive polymers and composites for biomedical applications, RSC Adv., 2015, 5, 37553–37567 RSC.
- C. Cassignol, M. Cavarero, A. Boudet and A. Ricard, Microstructure–conductivity relationship in conducting polypyrrole/epoxy composites, Polymer, 1999, 40, 1139–1151 CrossRef CAS.
- P. C. Vargas, C. Merlini, S. D. A. da Silva Ramôa, R. Arenhart, G. M. de Oliveira Barra and B. G. Soares, Conductive Composites Based on Polyurethane and Nanostructured Conductive Filler of Montmorillonite/Polypyrrole for Electromagnetic Shielding Applications, Mat. Res., 2018, 21, e20180014 CAS.
- Y. Zhang, W. Lu, L. Cong, J. Liu, L. Sun, A. Mauger, C. M. Julien, H. Xie and J. Liu, Cross-linking network based on poly(ethylene oxide): solid polymer electrolyte for room temperature lithium battery, J. Power Sources, 2019, 420, 63–72 CrossRef CAS.
- L. Porcarelli, C. Gerbaldi, F. Bella and J. R. Nair, Super Soft All-Ethylene Oxide Polymer Electrolyte for Safe All-Solid Lithium Batteries, Sci. Rep., 2016, 6, 19892 CrossRef CAS PubMed.
- R. He, F. Peng, W. E. Dunn and T. Kyu, Chemical and electrochemical stability enhancement of lithium bis(oxalato)borate (LiBOB)-modified solid polymer electrolyte membrane in lithium ion half-cells, Electrochim. Acta, 2017, 246, 123–134 CrossRef CAS.
- M. Chanda, in Science of Engineering Materials, Macmillan Education UK, London, 1980, pp. 61–126 Search PubMed.
- L. A. Fielding, J. K. Hillier, M. J. Burchell and S. P. Armes, Space science applications for conducting polymer particles: synthetic mimics for cosmic dust and micrometeorites, Chem. Commun., 2015, 51, 16886–16899 RSC.
- Z. Xue, D. He and X. Xie, Poly(ethylene oxide)-based electrolytes for lithium-ion batteries, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- K.-S. Ji, H.-S. Moon, J.-W. Kim and J.-W. Park, Role of functional nano-sized inorganic fillers in poly(ethylene) oxide-based polymer electrolytes, J. Power Sources, 2003, 117, 124–130 CrossRef CAS.
- A. K. Thakur and S. A. Hashmi, Polymer matrix–filler interaction mechanism for modified ion transport and glass transition temperature in the polymer electrolyte composites, Solid State Ionics, 2010, 181, 1270–1278 CrossRef CAS.
- K. Vignarooban, M. A. K. L. Dissanayake, I. Albinsson and B.-E. Mellander, Effect of TiO2 nano-filler and EC plasticizer on electrical and thermal properties of poly(ethylene oxide) (PEO) based solid polymer electrolytes, Solid State Ionics, 2014, 266, 25–28 CrossRef CAS.
- B. A. Lunn, J. Unsworth, N. G. Booth and P. C. Innis, Determination of the thermal conductivity of polypyrrole over the temperature range 280–335 K, J. Mater. Sci., 1993, 28, 5092–5098 CrossRef CAS.
- T. H. Gilani, Evidence for Metal-like Electronic Contribution in Low-Temperature Heat Capacity of Polypyrrole, J. Phys. Chem. B, 2005, 109, 19204–19207 CrossRef CAS PubMed.
- T. Albrecht, S. Armbruster, S. Keller and G. Strobl, Dynamics of Surface Crystallization and Melting in Polyethylene and Poly(ethylene oxide) Studied by Temperature-Modulated DSC and Heat Wave Spectroscopy, Macromolecules, 2001, 34, 8456–8467 CrossRef CAS.
- K. Pielichowski and K. Flejtuch, Phase Behavior of Poly(Ethylene Oxide) Studied by Modulated-Temperature DSC—Influence of the Molecular Weight, J. Macromol. Sci., Part B:Phys., 2004, 43, 459–470 CrossRef.
- K. P. Menard and N. Menard, Dynamic Mechanical Analysis, Taylor and Francis, Boca Raton, 3rd edn, 2020 Search PubMed.
- J. S. Okasinski, I. A. Shkrob, A. Chuang, M.-T. F. Rodrigues, A. Raj, D. W. Dees and D. P. Abraham, In situ X-ray spatial profiling reveals uneven compression of electrode assemblies and steep lateral gradients in lithium-ion coin cells, Phys. Chem. Chem. Phys., 2020, 22, 21977–21987 RSC.
- F. C. Strobridge, B. Orvananos, M. Croft, H.-C. Yu, R. Robert, H. Liu, Z. Zhong, T. Connolley, M. Drakopoulos, K. Thornton and C. P. Grey, Mapping the Inhomogeneous Electrochemical Reaction Through Porous LiFePO4-Electrodes in a Standard Coin Cell Battery, Chem. Mater., 2015, 27, 2374–2386 CrossRef CAS.
- F. Dai and M. Cai, Best practices in lithium battery cell preparation and evaluation, Commun. Mater., 2022, 3, 64 CrossRef.
- B. Steyrer, P. Neubauer, R. Liska and J. Stampfl, Visible Light Photoinitiator for 3D-Printing of Tough Methacrylate Resins, Materials, 2017, 10, 1445 CrossRef PubMed.
- K. Wang, B. Li, K. Ni, B. Li and Z. Wang, Optimal photoinitiator concentration for light-cured dental resins, Polym. Test., 2021, 94, 107039 CrossRef CAS.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na, A. K. Nanjundan, S. Wang, J. Yu and Y. Yamauchi, Progress in Solid Polymer Electrolytes for Lithium-Ion Batteries and Beyond, Small, 2022, 18, 2103617 CrossRef CAS PubMed.
- P. Lennartz, B. A. Paren, A. Herzog-Arbeitman, X. C. Chen, J. A. Johnson, M. Winter, Y. Shao-Horn and G. Brunklaus, Practical considerations for enabling Li|polymer electrolyte batteries, Joule, 2023, 7, 1471–1495 CrossRef CAS.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Solid-state polymer electrolytes with in-built fast interfacial transport for secondary lithium batteries, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- H. Zhang, F. Chen, O. Lakuntza, U. Oteo, L. Qiao, M. Martinez-Ibañez, H. Zhu, J. Carrasco, M. Forsyth and M. Armand, Suppressed Mobility of Negative Charges in Polymer Electrolytes with an Ether-Functionalized Anion, Angew. Chem., Int. Ed., 2019, 58, 12070–12075 CrossRef CAS PubMed.
- G. Lingua, P. Grysan, P. S. Vlasov, P. Verge, A. S. Shaplov and C. Gerbaldi, Unique Carbonate-Based Single Ion Conducting Block Copolymers Enabling High-Voltage, All-Solid-State Lithium Metal Batteries, Macromolecules, 2021, 54, 6911–6924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08556f |
| This journal is © The Royal Society of Chemistry 2025 |