DOI:
10.1039/D4TA08256G
(Paper)
J. Mater. Chem. A, 2025,
13, 5261-5274
An efficient H2O2-based propylene to propylene oxide (HPPO) reaction catalyzed by ZnO/ZnO2 materials†
Received
20th November 2024
, Accepted 6th January 2025
First published on 8th January 2025
Abstract
Propylene oxide (PO) is an essential feedstock in the plastic industry. Herein, unprecedented, inexpensive and robust zinc oxide (ZnO)-based catalysts were prepared. A ZnO nanorod (ZnO-NR) catalyst was synthesized using a solvothermal method. Another type of ZnO catalyst supported and immobilized on a mesoporous MCM-41 material (ZnO/MCM-41(x)) was also prepared with varied proportions (x = 0.82–9.41 wt%) of Zn content. The catalytic reactions of propylene epoxidation over ZnO-NR and ZnO/MCM-41(x) with H2O2 as an oxidant and acetonitrile as a solvent were studied at 30–70 °C and 5–20 bar. The ZnO catalysts were found capable of catalyzing the reaction with high H2O2 utilization and PO selectivity. Among them, ZnO/MCM-41(7.99) achieved nearly 100% PO selectivity and the highest turnover number of 124.4 (moles of PO per moles of Zn). The fresh catalysts, including ZnO-NR and ZnO/MCM-41(7.99), the spent catalysts, and the ZnO2 nanoparticles were characterized by synchrotron powder X-ray diffraction, transmission electron microscopy, and X-ray absorption spectroscopy. The fractions of ZnO2 in the spent catalysts were quantified to be higher than 50%. Electron paramagnetic resonance and X-ray photoelectron spectroscopy revealed that the ZnO2 phase formed by reacting ZnO with H2O2 in acetonitrile, stored as green redox oxide materials, contained hydroperoxide, peroxide and superoxide species, which can be essential for catalytic production of PO. The observed 18O enrichment in PO using H218O2 suggested that the reactive oxygenated species are generated from H2O2 and anchored on/in ZnO2 for electrophilic epoxidation, with the assistance of acetonitrile.
1 Introduction
Propylene oxide (PO) is an essential feedstock in the plastic industry, as it is utilized in the production of chemicals and polymers.1,2 According to forecasts, the yearly demand for PO is expected to surpass 20 Mt by 2025.2 PO can be generated by the epoxidation of propylene. The three industrial techniques for PO production are the chlorohydrin process, organic hydroperoxide-based processes, and hydrogen peroxide (H2O2)-to-propylene oxide (HPPO) processes.2–7 The chlorohydrin process is known to have significant environmental drawbacks as it generates halogen and alkali salt wastes.3,5 The processes based on organic hydroperoxides (e.g., cumene hydroperoxide, tert-butyl hydroperoxide and ethylbenzene hydroperoxide) usually need additional purification steps to separate PO from co-products (e.g., styrene and tert-butanol).3,8 With the high reactivity of cumene with O2, however, the Sumitomo process using Ti-containing silica catalysts can efficiently recycle cumene for PO production.6 In comparison, the direct use of H2O2 for epoxidizing propylene with water as the only co-product is much more appealing from both environmental and economic points of view. The developed commercial HPPO plants produce H2O2 by the oxidation and reduction of anthraquinones or related molecules9,10 and use titanosilicalite-1 (TS-1)11,12 catalysts for the epoxidation reaction.13–16 Alternatively, H2O2 can be generated in situ by gas-phase reactions of H2 and O2,13 but temperature control is crucial for the gas-phase HPPO process because over-oxidation of PO may take place at high temperatures. In addition to the three industrial processes, direct epoxidation of propylene with O2 over copper and silver-based catalysts has also been studied.4,17 However, the reaction typically occurs at high temperatures, thermodynamically favoring C–H bond cleavage rather than double bond epoxidation,18 and therefore PO's selectivity and formation rate are typically low.4
Reports have shown that the reaction conditions and catalyst type greatly affect the selectivity of PO (SPO), conversion of H2O2 (XH2O2), and the utilization efficiency of H2O2 (UH2O2) in the HPPO process. The reaction is typically carried out under mild conditions (around 40 °C and 1–10 bar) with protic (e.g., methanol, water, and other alcohols) or aprotic (e.g., acetonitrile and acetone) solvents.19,20 For the reaction catalyzed by TS-1, Liu et al. compared the catalytic performance in methanol and acetonitrile. They found that lower PO selectivity (SPO = 95%) and higher conversion of H2O2 (XH2O2 = 97%) were obtained for the reaction conducted in methanol than that in acetonitrile (SPO = 100% and XH2O2 = 66%).21 As a protic solvent, methanol may coordinate with the Ti centers and employ hydrogen bonding to stabilize a five-membered Ti-hydroperoxide (Ti–OOH) intermediate with high efficiency,22,23 but solvolysis (or hydrolysis in the case of water) of PO may take place, lowering the PO selectivity.6,24 In an aprotic solvent like acetonitrile, on the other hand, apart from the higher solubility of propylene than that in methanol, the Ti–OOH intermediate is not stabilized by hydrogen bonding,25,26 but possible solvolysis of PO may be prevented.27 Moreover, acetonitrile may react with H2O2 to form a peroxyacetimidic acid intermediate (CH3–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)–O–O–H) to create an additional organic hydroperoxide-like route of epoxidation.7,28,29 In addition, the coordination state of Ti species has been shown to influence the catalytic activity of TS-1 and related Ti-based catalysts for the HPPO reaction and epoxidation of other alkenes.30–33 For example, Wang et al. prepared a TS-1 material with penta-coordinated (Ti(OH)2(OSi)3) and hexa-coordinated (Ti(OH)4(OSi)2) Ti sites. They found that the hexa-coordinated Ti sites exhibit significantly higher catalytic activity than the penta- and tetra-coordinated sites in TS-1.32 Other Ti-containing catalysts, including Ti-substituted BEA zeolites,11,34 Ti-substituted MWW zeolites,12,25 and even zeolites or oxide materials containing Nb, Ta or other metals,35,36 have also been investigated for the HPPO reaction and alkene epoxidation.
NH)–O–O–H) to create an additional organic hydroperoxide-like route of epoxidation.7,28,29 In addition, the coordination state of Ti species has been shown to influence the catalytic activity of TS-1 and related Ti-based catalysts for the HPPO reaction and epoxidation of other alkenes.30–33 For example, Wang et al. prepared a TS-1 material with penta-coordinated (Ti(OH)2(OSi)3) and hexa-coordinated (Ti(OH)4(OSi)2) Ti sites. They found that the hexa-coordinated Ti sites exhibit significantly higher catalytic activity than the penta- and tetra-coordinated sites in TS-1.32 Other Ti-containing catalysts, including Ti-substituted BEA zeolites,11,34 Ti-substituted MWW zeolites,12,25 and even zeolites or oxide materials containing Nb, Ta or other metals,35,36 have also been investigated for the HPPO reaction and alkene epoxidation.
Herein, we report the study on the HPPO reaction over zinc oxide (ZnO) with acetonitrile as a solvent. ZnO is a cheap, nontoxic material with unique physical and chemical properties and shows attractive applications in optoelectronics, electronics, laser technology, and photocatalysis.37,38 It has been demonstrated that ZnO with a hexagonal wurtzite structure could interact with H2O2 and convert to cubic zinc peroxide (ZnO2)39 and that ZnO2 may partially decompose to produce O2 and hexagonal ZnO with a significant number of defects as the temperature increases, with a complete decomposition taking place at around ∼250 °C.39 ZnO2 may react with water to produce paramagnetic superoxide species40,41 and has been applied for reactions including the oxidation of aromatic alcohols to the corresponding carbonyl compounds,42 decomposition of dye molecules in wastewater,43 and detoxification of mustard gas.44 As for the HPPO or related reactions, rather limited studies on ZnO-based catalysts, mainly in photocatalysis, e.g., photocatalytic olefin epoxidation with O2 over Pd/ZnO45 and cyclohexane oxidation/epoxidation with H2O2 over Fe2O3–ZnO46 have been reported. It is noted that Arca and coworkers reported the preparation of zinc salt pre-treated TS-1 for the HPPO reaction47 and found that the coordination of Zn to the Ti site (through bridging oxygen) resulted in the increase in the solvent donor properties to the Ti site as well as the reduction of the Ti–OOH electrophilicity, thereby giving rise to high SPO and UH2O2. However, the contribution from ZnO2 possibly formed during the reaction was not discussed. In this study, ZnO nanorods (NRs; the sample is designated as ZnO-NR) and the composites of ZnO-NR and mesoporous MCM-41 materials (designated as ZnO/MCM-41) were prepared and applied to catalyze the reaction of propylene epoxidation with aqueous H2O2 in acetonitrile. As compared to ZnO-NR, ZnO/MCM-41 with optimum composition exhibited superior epoxidation performance with an SPO of 100% and a UH2O2 of over 98%. The fresh and spent catalysts of ZnO-NR and the best ZnO/MCM-41 composite, as well as the ZnO2 nanoparticles (NPs), were characterized by multiple techniques, including synchrotron powder X-ray diffraction (SXRD), transmission electron microscopy (TEM), and X-ray absorption spectroscopy (XAS). Electron paramagnetic resonance (EPR) spectroscopy and X-ray photoelectron spectroscopy (XPS) were applied to investigate the possible reactive species formed in the working catalysts. Based on the results, a possible reaction mechanism of the HPPO process over ZnO/ZnO2 catalysts was proposed.
2 Materials and methods
2.1. Chemicals and materials
All chemicals, including zinc acetate dihydrate (Zn(CH3COO)2·2H2O, 99.8%), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 98%), ethanol (EtOH, 99.9%), sodium hydroxide pellets (NaOH, 99.9%), hydrogen peroxide (H2O2, 35 wt% aqueous solution), and acetonitrile (CH3CN, 98%), were of analytical grade purchased from Sigma-Aldrich and were used without further purification. The propylene oxide standard (C3H6O, 99%) and methanol (CH3OH, 99%) were purchased from Sigma-Aldrich, whereas 7 N ammonia (NH3) solution was purchased from Acros Organics. The propylene gas was purchased from Huei Chyi Gas Co. Ltd, Taiwan. The mesoporous MCM-41 material (CAS no. 1318-02-1) was also purchased from Sigma-Aldrich. Isotope-labeled H218O2 (90 atom%, 2.0–2.4% aqueous solution) and 18O2 (97 atom%) were purchased from ICON Isotopes and Sigma-Aldrich, respectively.
2.2. Catalyst preparation
The ZnO-NR sample was prepared by a reported solvothermal method.48 Zn(CH3COO)2·2H2O (2.195 g) and NaOH (0.8 g) were first dissolved in ethanol (80 mL). The solution was poured into a 15 mL Teflon-sealed autoclave and was heated at 180 °C for 12 h. The solid product was collected by centrifugation and was repeatedly washed with ethanol and water. The dried solid was further heated at 500 °C for 3 h with a temperature ramp of 10 °C min−1. The composites of ZnO-NR and MCM-41 were prepared by the immobilization method.49,50 A total of 25 mg of ZnO-NR was added into 75 mg of MCM-41 and stirred in a mixture of CH3CN (40 mL) and 10 mL of EtOH (4/1 v/v) at 30 °C, and 7 N ammonia solution in methanol (2 mL) was added dropwise into the mixture to reach a pH value of 9.0. After the mixture was further stirred for 12 h, the solid was collected by centrifugation, washed with ethanol, and dried at 60 °C in a vacuum. The composites are designated as ZnO/MCM-41(x), where x denotes the content of Zn in the composites (wt%; x = 0.82–9.41). A reference sample of ZnO2 NPs was also prepared by mixing an aqueous solution of Zn(CH3COO)2·2H2O (1.0 g in 50 mL of water) with an aqueous solution of H2O2 (35 wt%, 2 mL).51 The mixture was stirred at room temperature for 12 h, and the solid product of ZnO2 NPs was collected by centrifugation, washed with water, and dried at 60 °C under vacuum.
2.3. Catalyst characterization
SXRD patterns were recorded using the TPS 19A1 beamline, with an X-ray wavelength of 0.77489 Å, at the National Synchrotron Radiation Research Center (NSRRC), Taiwan. TEM images were obtained using a JEOL JEM-F200 microscope operating at 200 kV. The compositions of the ZnO-based materials were determined using an inductively coupled plasma-mass spectrometer (ICP-MS, Thermo-element XR) and energy dispersive X-ray spectroscopy (EDX) on a field-emission scanning electron microscope (SEM, FEI Quanta 200F) with an EDX detector (ULTRA PLUS, Carl Zeiss). The SEM images were obtained using the microscope operated at 10 kV. Nitrogen physisorption isotherms were obtained at 77 K using a Micromeritics 3Flex instrument. Each sample was evacuated at 120 °C for 12 h prior to the measurement. The Brunauer–Emmett–Teller (BET) surface area was calculated from the adsorption branch in the relative pressure range of 0.05–0.30, and the average pore diameter was calculated from the desorption branch using the Barrett–Joyner–Halenda (BJH) method. The total pore volume was evaluated at a relative pressure of 0.95. XAS measurements at the Zn K-edge were conducted in transmission mode using the TLS 17C1 and TPS 44A beamlines at the NSRRC, Taiwan. Zinc foil was measured simultaneously as a reference sample for energy calibration. Multiple scans were performed and averaged to enhance the signal-to-noise ratio. For a measured spectrum, the extended X-ray absorption fine structure (EXAFS) function χ was obtained by subtracting the post-edge background from the initial spectrum and then normalizing to the edge jump step. The energy space spectrum of the normalized χ(E) was then transformed into the k-space spectrum of χ(k). The χ(k) was multiplied by k3 to compensate for the oscillation dampening in the high-k region, and the k3-weighted EXAFS data in the range of 3.0–14 Å−1 were converted to the R-space data by Fourier transform (FT). Athena software was used to process the XAS spectra, including background subtraction, normalization of the edge jump step, and FT of the k3-weighted EXAFS data. Artemis software was used to fit the FT profiles. Wavelet transform analysis was further conducted to gain more insights into the k-space resolution. Athena software was also used for linear combination fitting (LCF) of XANES and k3-weighted EXAFS data of the spent catalysts. EPR spectra were recorded on a Bruker EMX spectrometer (Bruker ST4102 cavity) operating at X-band frequency and 100 kHz field modulation. To insert the sample (10 mg), a cylindrical quartz tube container was used, which was maintained at a temperature of 77 K. Thermogravimetric analysis combined with gas chromatography and mass spectrometry (TGA-GC/MS) was conducted on a Mettler-Toledo 2-HT analyzer coupled with an Agilent 7890 GC/5975 MS system. XPS measurements were performed on an ultrahigh vacuum system using an Omicron DAR400 Al Kα X-ray source and an Omicron EA125 energy analyzer. The C 1s core level peak (284.8 eV) was used as an energy reference, and the XPS spectra were analyzed using CasaXPS software.
2.4. Epoxidation of propylene with hydrogen peroxide
The catalytic HPPO reactions were carried out in a 50 mL stainless steel Parr autoclave reactor equipped with a Teflon liner. The autoclave was loaded with a catalyst (12.5–62.5 mg), H2O2 (35 wt% aqueous solution, 5.87–14.10 mmol), and CH3CN (5 mL). The reactor was sealed, pressurized with propylene (5–20 bar), and then heated to the designated temperature (30–70 °C). The reaction mixture was stirred at 880 rpm and allowed to react for 1–8 h (starting from the time when the reaction mixture reached the designated temperature). After the reaction was complete, the stirring was stopped, and to avoid loss of PO (with a boiling point of ∼35 °C), the reactor was cooled to 10 °C using iced water before being slowly depressurized and opened. The reaction products were analyzed by gas chromatography (GC, Shimadzu GC-2010plus) or GC/MS (Agilent GC/MS-5977B/MSD) with methyl tert-butyl ether (MTBE) as an internal standard. The amount of H2O2 remaining after the reaction was determined by UV-vis spectroscopy with a potassium titanium oxalate solution.52,53 For the catalytic test of ethylene epoxidation, the products were analyzed by 1H-NMR on a Bruker AVA-AV400 (400 MHz) spectrometer. All resonances were referenced to the deuterated solvent CD3CN (δ = 1.93 ppm). The ZnO-NR and ZnO/MCM-41(7.99) catalysts after the catalytic tests under optimum reaction conditions were collected by centrifugation for further characterization, and the spent catalysts are designated by adding “sp” to the designation of the original catalysts (e.g., ZnO-NRsp refers to the ZnO-NR collected after the catalytic reaction). Small losses of the spent catalyst during collection by centrifugation were accounted for to keep the mass of the applied catalyst constant in the recyclability test. The values of conversion of H2O2 (XH2O2), the utilization efficiency of H2O2 (UH2O2), the yield of PO (YPO, based on the initial molar amount of H2O2), the selectivity of PO (SPO), the productivity of PO (PPO), and the turnover number (TON) were calculated using these criteria:| |  | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
| | 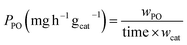 | (5) |
| |  | (6) |
where  and nH2O2 denote the molar amounts of H2O2 before and after the reactions, nPO denotes the molar amount of PO formed (calculated from the volume of the reaction mixture and the value of molar concentration determined by GC or GC/MS), nothers denotes the molar amount of other byproducts detected, wPO and wcat denote the weights of PO formed and the catalyst and nZn denotes the molar amount of Zn in a catalyst. Some catalysts also produced acetic acid (AC) or acetaldehyde (AA), and the criteria for the yields and selectivity of these reaction products were the same as those for PO.
and nH2O2 denote the molar amounts of H2O2 before and after the reactions, nPO denotes the molar amount of PO formed (calculated from the volume of the reaction mixture and the value of molar concentration determined by GC or GC/MS), nothers denotes the molar amount of other byproducts detected, wPO and wcat denote the weights of PO formed and the catalyst and nZn denotes the molar amount of Zn in a catalyst. Some catalysts also produced acetic acid (AC) or acetaldehyde (AA), and the criteria for the yields and selectivity of these reaction products were the same as those for PO.
3 Results and discussion
3.1. Catalytic study of propylene epoxidation
The structure and morphology of all the ZnO-NR and ZnO/MCM-41(x) materials were well characterized by SXRD, TEM, and SEM. The compositions of the ZnO/MCM-41(x) composites were determined by ICP-MS and EDX. The catalytic performance of ZnO-NR for propylene epoxidation with H2O2 in CH3CN was first examined with varied amounts of H2O2 (Entries 1–5, Table 1). While no PO was produced in the absence of H2O2 (Entry 1, Table 1), efficient conversion of propylene to PO with nearly 100% selectivity was observed in the presence of H2O2 and ZnO-NR. With an amount of 5.87 mmol H2O2 for a reaction of 6 h, the conversion of H2O2 (XH2O2) and the utilization efficiency of H2O2 (UH2O2) reached 97.98% and 97.53%, respectively. Under the reaction conditions, the yield of PO (YPO) based on the initial molar amount of H2O2 reached 95.56% and the productivity of PO (PPO) was 1448 mg h−1 gcat−1. The value of TON based on the molar amount of Zn was 14.3. As the amount of H2O2 increased up to 14.10 mmol, the reactions gave a higher TON and PPO (23.3 and 2398 mg h−1 gcat−1 with 14.10 mmol H2O2, Table 1), but at the cost of reduced conversion and utilization efficiency of H2O2. The amount of 5.87 mmol H2O2 with the highest values of XH2O2, UH2O2, and YPO was thus chosen to study other reaction parameters. Next, catalytic reactions with varied amounts (12.5–62.5 mg) of ZnO-NR were conducted (Entries 2, 6–9, Table 1). Basically, volcano relationships were observed between the amount of ZnO-NR, with an optimum value of 37.5 mg, and the parameters of XH2O2, UH2O2, and YPO. No PO was formed in the blank test with no ZnO-NR. It was noted that acetic acid (AC) as an over-oxidized minor product was detected with smaller (12.5 and 25.0 mg) amounts of ZnO-NR, and an AC selectivity of 10.41% was measured with 12.5 mg of ZnO-NR. Finally, the choice of solvent was examined and the mixtures of acetonitrile and water, water, and methanol were applied to the reaction. Under conditions similar to those for Entry 2 of Table 1 (5.87 mmol of H2O2, 37.5 mg of a catalyst, 5 mL of solvent, and 20 bar propylene pressure at 50 °C for 6 h), we found worse catalytic performance of ZnO-NR in the acetonitrile/water mixtures than in pure acetonitrile and observed no conversion of propylene in water or methanol (Table S1†). The results clearly demonstrated the crucial role of acetonitrile as a solvent in the catalytic reaction over ZnO-based catalysts.
Table 1 Results of propylene epoxidation over ZnO-NR with H2O2 using varied amounts of ZnO-NR and H2O2a
| No |
ZnO-NR (mg) |
H2O2 (mmol) |
X
H2O2 (%) |
U
H2O2 (%) |
Y
PO (%) |
S
PO (%) |
P
PO (mg h−1 gcat−1) |
Y
AC (%) |
S
AC (%) |
TON |
|
Reactions were conducted with 5 mL of CH3CN and a propylene pressure of 20 bar at 50 °C for 6 h.
Not determined.
|
| 1 |
37.5 |
0 |
0 |
0 |
0 |
NDb |
0 |
— |
— |
0 |
| 2 |
37.5 |
5.87 |
97.98 |
97.53 |
95.56 |
100 |
1448 |
— |
— |
14.3 |
| 3 |
37.5 |
9.34 |
74.43 |
85.82 |
64.11 |
100 |
1546 |
— |
— |
15.1 |
| 4 |
37.5 |
11.75 |
74.70 |
85.26 |
63.69 |
100 |
1932 |
— |
— |
18.8 |
| 5 |
37.5 |
14.10 |
77.19 |
85.33 |
65.87 |
100 |
2398 |
— |
— |
23.3 |
| 6 |
12.5 |
5.87 |
72.07 |
48.04 |
34.62 |
89.59 |
1574 |
4.02 |
10.41 |
15.3 |
| 7 |
25.0 |
5.87 |
86.71 |
75.91 |
65.82 |
97.23 |
1496 |
1.88 |
2.77 |
14.6 |
| 8 |
50.0 |
5.87 |
86.39 |
93.61 |
80.87 |
100 |
919 |
— |
— |
9.0 |
| 9 |
62.5 |
5.87 |
88.95 |
96.28 |
85.64 |
100 |
779 |
— |
— |
9.0 |
Since ZnO-NR contained aggregates of nanorods and exhibited a relatively low surface area (31 m2 g−1, cf. Fig. S1 and Table S2†), an attempt was made to disperse and immobilize varied amounts of ZnO-NR on mesoporous MCM-41 aluminosilicate (with a BET surface area of 856 m2 g−1 and an average pore diameter of 3.1 nm, cf. Fig. S1 and Table S2†) for the catalytic reaction. We first examined the catalytic performance of the composite catalysts ZnO/MCM-41(x) with varying Zn content x (in wt%) (Table 2). While MCM-41 alone did not exhibit any catalytic activity, the composites with a Zn content higher than 1.45% were capable of epoxidizing propylene (Table 2). Except for ZnO/MCM-41(1.45), for which acetaldehyde (AA) appeared as the major product, other composite catalysts with higher Zn contents showed 100% selectivity of PO. The values of XH2O2 and UH2O2, as well as YPO, increased with increasing the Zn content of the composites. The best catalyst among the composites prepared and studied was ZnO/MCM-41(7.99), showing nearly 100% PO selectivity (SPO), H2O2 utilization efficiency (UH2O2 = 98.33%) and PO yield (YPO = 97.44%) under the reaction conditions at 50 °C for 6 h. The catalytic performance of ZnO/MCM-41(7.99) (Entry 7, Table 2) and ZnO-NR under otherwise the same reaction conditions (Entry 2, Table 1) was compared. With the same catalyst amount of 37.5 mg, the Zn (or ZnO) content in ZnO/MCM-41(7.99) was much lower than that for ZnO-NR. The fact that both catalysts produced PO as the only product and exhibited similar or comparable values of XH2O2, UH2O2, YPO, and PPO clearly indicated that ZnO-NR was successfully dispersed and immobilized on MCM-41 to expose a larger catalytically active surface area to convert propylene to PO with H2O2 in acetonitrile. As a result of low Zn (or ZnO) content, the calculated value of TON for ZnO/MCM-41(7.99) (124.4) was nearly an order of magnitude higher than that for ZnO-NR (14.3). The reactions with varied amounts of H2O2 and ZnO/MCM-41(7.99) were further conducted (cf. Table S3†). In these reactions (except the one without adding H2O2), PO was the only product (i.e., SPO = 100%) and acetic acid was not detected. Similar to the trends observed for ZnO-NR, an increase in the amount of H2O2 resulted in lower values of H2O2 conversion, H2O2 utilization efficiency, and PO yield but better TON and PO productivity. The highest values of TON (225.0) and PPO (2691 mg h−1 gcat−1) were observed for the reaction with 14.1 mmol H2O2. Besides, similar volcano relationships between the catalyst amount and parameters of XH2O2, UH2O2, and YPO were also observed for ZnO/MCM-41(7.99).
Table 2 Results of propylene epoxidation over ZnO/MCM-41(x) with H2O2 using varying Zn loadingsa
| No |
Zn content x (wt%) |
X
H2O2 (%) |
U
H2O2 (%) |
Y
PO (%) |
S
PO (%) |
P
PO (mg h−1 gcat−1) |
Y
AA (%) |
S
AA (%) |
TON |
|
Reactions were conducted with 5.87 mmol of H2O2, 37.5 mg of catalyst, 5 mL of solvent, and a propylene pressure of 20 bar at 50 °C for 6 h.
Not determined.
|
| 1 |
0 |
NDb |
ND |
0 |
ND |
0 |
— |
— |
0 |
| 2 |
0.82 |
ND |
ND |
0 |
ND |
0 |
— |
— |
0 |
| 3 |
1.45 |
10.36 |
24.52 |
2.54 |
15.84 |
39 |
5.12 |
84.16 |
20.5 |
| 4 |
4.23 |
44.06 |
80.79 |
35.60 |
100 |
538 |
— |
— |
90.7 |
| 5 |
4.34 |
49.91 |
80.54 |
40.20 |
100 |
609 |
— |
— |
94.5 |
| 6 |
4.83 |
60.83 |
94.38 |
57.41 |
100 |
871 |
— |
— |
120.5 |
| 7 |
7.99 |
99.09 |
98.33 |
97.44 |
100 |
1476 |
— |
— |
124.4 |
| 8 |
9.41 |
95.46 |
97.35 |
92.93 |
100 |
1408 |
— |
— |
101.0 |
The influences of propylene pressure and reaction temperature on the catalytic performance of ZnO-NR and ZnO/MCM-41(7.99) with fixed amounts of catalyst (37.5 mg), H2O2 (5.87 mmol), and acetonitrile (5 mL) were then studied. The results of the reactions with propylene pressures of 5–20 bar at 50 °C for 6 h are shown in Fig. 1a and Tables S4 and S5.† For both catalysts, PO was the only detected product, and the conversion and utilization efficiency of H2O2, the yield of PO, and TON all increased with increasing propylene pressure. The results suggest that the mass transfer of the gaseous reactant is a limiting factor for the catalytic process. Next, catalytic reactions with a propylene pressure of 20 bar over the two catalysts at 30–70 °C were conducted for 6 h. As shown in Fig. 1b and Tables S4 and S5,† while all the reactions exhibited 100% selectivity for PO, volcano relationships were observed between the reaction temperature and XH2O2, UH2O2, YPO, and TON for both ZnO-NR and ZnO/MCM-41(7.99). The poorer catalytic performance at a temperature higher than 50 °C may be attributed to the accelerated decomposition of H2O2.54
 |
| | Fig. 1 Effects of (a) propylene pressure (at 50 °C) and (b) reaction temperature (with 20 bar propylene pressure) on PO yield (YPO) and TON for the reactions over ZnO-NR and ZnO/MCM-41(7.99) with fixed amounts of H2O2 (5.87 mmol), catalyst (37.5 mg), and acetonitrile (5 mL) for 6 h. | |
Changes in catalytic performance with time for the reactions over ZnO-NR and ZnO/MCM-41(7.99) were then compared. As shown in Fig. 2a, for both catalysts, no induction time was observed for the catalytic reactions to start, and the evolutions of H2O2 conversion and PO yield are very close to each other, implying a one-to-one equivalence of the limiting reactant H2O2 and the produced PO. The values of XH2O2 and YPO (and UH2O2, Table S6†) for the two catalysts are similar, which should be, as aforementioned, associated with the dispersion of ZnO-NR on MCM-41. The 6 h and 8 h TON values are very close (Table S6†), indicating that the reactions over the two catalysts approached completion within 6 h. The time courses shown in Fig. 2a and b could be well fitted with the pseudo-first-order kinetic model, and the derived rate constants (k) from PO yield are 0.43 h−1 for ZnO-NR (with a correlation coefficient (R2) of 0.941) and 0.50 h−1 for ZnO/MCM-41(7.99) (R2 = 0.948). Poorer fittings were found for the data of H2O2 conversion, and the rate constant values derived from H2O2 conversion are slightly higher than those derived from YPO (k = 0.54 h−1 and R2 = 0.929 for ZnO-NR; k = 0.58 h−1 and R2 = 0.875 for ZnO/MCM-41(7.99)). This may be attributed to the spontaneous decomposition of H2O2 under the reaction conditions.
 |
| | Fig. 2 (a) Changes in H2O2 conversion (XH2O2) and PO yield (YPO) with reaction time for propylene epoxidation over ZnO-NR and ZnO/MCM-41(7.99) with 5.87 mmol of H2O2, 37.5 mg of catalyst, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C. (b) Time courses fitted with the pseudo-first-order kinetic model. (c) YPO and TON for the two catalysts in cycle tests of propylene epoxidation with 5.87 mmol of H2O2, 37.5 mg of catalyst, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C for 6 h. | |
The stability of ZnO-NR and ZnO/MCM-41(7.99) was further examined. After the reactions with 5.87 mmol of H2O2, 37.5 mg of catalyst, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C for 6 h, the spent catalysts were collected and reused to catalyze the reactions under the same conditions for four cycles. The results of cycle tests are shown in Fig. 2c and Table S7.† Slight decreases in XH2O2, UH2O2, YPO, PPO, and TON were found for both catalysts, and around a 10% drop in TON was observed after the fourth cycle. The decreases may be mainly associated with the loss of catalysts during the collection of spent catalysts by centrifugation. For ZnO/MCM-41(7.99), a slight degree of leaching of Zn species was another reason, and a Zn leaching of ∼5.7% was detected by ICP-MS after the fourth cycle. With factors of catalyst loss and Zn leaching being taken into account, the calculated values of TON and other parameters are very close to those for the first catalytic cycle, suggesting high stability of the catalysts. A comparison of the catalytic performance of ZnO-NR and ZnO/MCM-41(7.99) with reported HPPO catalysts was made. As shown in Table S8,† the values of PO yield, PO selectivity, H2O2 conversion, and H2O2 utilization for ZnO-NR and ZnO/MCM-41(7.99) are comparable to those for industrially applicable TS-1 catalysts25,55 for the epoxidation of propylene with H2O2(aq). The two ZnO-based catalysts outperform the Nb-EISA catalyst35,36 in terms of PO yield and H2O2 conversion.
An attempt was made to test the catalytic activity of the best catalyst, ZnO/MCM-41(7.99), for the epoxidation of ethylene. The reaction was conducted with 2.94 mmol of H2O2, 37.5 mg of catalyst, 2.5 mL of deuterated acetonitrile (CD3CN), and an ethylene pressure of 20 bar at 50 °C for 6 h, and the solution collected after the reaction by centrifugation was analyzed by 1H-NMR. Ethylene oxide (EO), identified by the signal at 2.64 ppm (Fig. S2†), was the major product of the reaction with a selectivity of 92.73%, with ethylene glycol as the only minor product. The conversion and utilization of H2O2 were lower than the values for the reaction of propylene epoxidation (Table S9†), despite a lower initial amount of the oxidant. Nevertheless, the results show that ZnO/MCM-41(7.99) could catalyze the epoxidation of ethylene, giving an EO yield of 67.26%, an EO productivity of 387 mg h−1 gcat−1, and a TON of 46.3 under the reaction conditions.
3.2. Structural analysis of fresh and spent catalysts
The synchrotron powder X-ray diffraction (SXRD) patterns (X-ray wavelength of 0.77489 Å) of ZnO-NR and ZnO/MCM-41(x) all showed sharp peaks at 15.96°, 17.15°, 18.16°, 23.52°, 27.87°, and 30.53° (Fig. 3 and S3†), which correspond to the (100), (002), (101), (102), (210), and (103) reflections of crystalline ZnO with a hexagonal wurtzite structure (space group: P63mc; lattice constants: c = 5.21 Å and a = 3.25 Å).56 For ZnO/MCM-41(x), the peaks in the small-angle region correspond well to the reflections of MCM-41 exhibiting a two-dimensional hexagonal mesostructure (with a unit cell parameter of ∼3.9 nm).57 The nanorod morphology of ZnO was confirmed by SEM (Fig. S4†). The nanorods in ZnO-NR were mostly assembled or bundled, while those in ZnO/MCM-41(x) were mainly segregated, presumably by the mixed MCM-41 particles. The MCM-41 particles could be observed in the samples with a relatively higher amount of the material (e.g. ZnO/MCM-41(4.23), Fig. S4e†), but they could not be clearly discerned in ZnO/MCM-41(7.99) due to the low contrast and low relative amount of MCM-41. The SEM observation confirmed the role of MCM-41 in dispersing ZnO nanorods to expose the surface of segregated nanorods for catalysis. TEM was further performed to analyze ZnO-NR and ZnO/MCM-41(7.99). For ZnO-NR, the bundled nanorods were again found (Fig. 4a), and fringe patterns were clearly observed in the ZnO nanorods in the high-resolution TEM (HRTEM) images (Fig. 4b). In ZnO/MCM-41(7.99), on the other hand, individual ZnO nanorods with high crystallinity were observed among irregularly shaped MCM-41 particles (Fig. 5a and b). Basically, the results of TEM are consistent with the SEM observations.
 |
| | Fig. 3 SXRD patterns of ZnO-NR, ZnO-NRsp, ZnO/MCM-41(7.99), ZnO/MCM-41(7.99)sp, and ZnO2 NPs (X-ray wavelength of 0.77489 Å). The peaks marked by asterisks are attributed to cubic ZnO2. | |
 |
| | Fig. 4 TEM (a and c) and HRTEM (b and d) images of ZnO-NR (a and b) and ZnO-NRsp (c and d). | |
 |
| | Fig. 5 TEM (a and c) and HRTEM (b and d) images of ZnO/MCM-41(7.99) (a and b) and ZnO/MCM-41(7.99)sp (c and d). | |
The ZnO-NR and ZnO/MCM-41(7.99) catalysts after the catalytic reactions under optimum conditions (i.e., 5.87 mmol of H2O2, 37.5 mg of catalyst, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C for 6 h) were collected by centrifugation, and the spent catalysts ZnO-NRsp and ZnO/MCM-41(7.99)sp were further characterized in order to identify the active species for the reaction. ZnO-NRsp gave an SXRD pattern with some broad and weak reflections, in addition to those attributed to hexagonal ZnO (Fig. 3), suggesting the presence of some minor phase with relatively low crystallinity in the spent catalyst. For ZnO/MCM-41(7.99)sp, the SXRD pattern is completely different from that of ZnO/MCM-41(7.99) and contains relatively broad reflections that could be indexed to ZnO2 with a cubic-pyrite structure (space group: Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) lattice constants: a = 4.87 Å).39 A reference sample of phase-pure cubic ZnO2 NPs was synthesized by following a reported procedure.51 Its SXRD pattern is also shown in Fig. 3, and analyses of TEM, HRTEM, and EDX revealed that the sample comprised particles with tens of nanometers in size, each of which contained nanosized domains of cubic ZnO2 (Fig. S5†). Based on SXRD, the minor phase in ZnO-NRsp was identified to be ZnO2 and the three additional peaks were indexed to be the (200), (220), and (331) reflections of cubic ZnO2. The spent catalysts were further analyzed by TEM. Most ZnO nanorods in ZnO-NRsp were much shorter than those in the as-prepared catalyst, and some nanoparticles with irregular shapes (several to tens of nanometers in size) were also observed (Fig. 4c). The nanoparticles showed a lattice fringe spacing of 0.282 nm, corresponding to the (111) plane of cubic ZnO2 (Fig. 4d). For ZnO/MCM-41(7.99)sp, no rod-shaped particles were observed (Fig. 5c). EDX elemental mapping images confirmed that the Zn-containing species were dispersed in the composite (Fig. S6†), and the particles with darker contrast also showed lattice fringes of ZnO2 (Fig. 5d). The results of SXRD and TEM/EDX indicated that ZnO nanorods interacted with H2O2 and converted to ZnO2 NPs during the catalytic reaction. Moreover, while only part of the ZnO nanorods in ZnO-NR were converted, most ZnO nanorods in ZnO/MCM-41(7.99) were transformed into ZnO2. Such a dramatic transformation may be attributed to the much better accessibility of the dispersed ZnO nanorods in the catalyst.
lattice constants: a = 4.87 Å).39 A reference sample of phase-pure cubic ZnO2 NPs was synthesized by following a reported procedure.51 Its SXRD pattern is also shown in Fig. 3, and analyses of TEM, HRTEM, and EDX revealed that the sample comprised particles with tens of nanometers in size, each of which contained nanosized domains of cubic ZnO2 (Fig. S5†). Based on SXRD, the minor phase in ZnO-NRsp was identified to be ZnO2 and the three additional peaks were indexed to be the (200), (220), and (331) reflections of cubic ZnO2. The spent catalysts were further analyzed by TEM. Most ZnO nanorods in ZnO-NRsp were much shorter than those in the as-prepared catalyst, and some nanoparticles with irregular shapes (several to tens of nanometers in size) were also observed (Fig. 4c). The nanoparticles showed a lattice fringe spacing of 0.282 nm, corresponding to the (111) plane of cubic ZnO2 (Fig. 4d). For ZnO/MCM-41(7.99)sp, no rod-shaped particles were observed (Fig. 5c). EDX elemental mapping images confirmed that the Zn-containing species were dispersed in the composite (Fig. S6†), and the particles with darker contrast also showed lattice fringes of ZnO2 (Fig. 5d). The results of SXRD and TEM/EDX indicated that ZnO nanorods interacted with H2O2 and converted to ZnO2 NPs during the catalytic reaction. Moreover, while only part of the ZnO nanorods in ZnO-NR were converted, most ZnO nanorods in ZnO/MCM-41(7.99) were transformed into ZnO2. Such a dramatic transformation may be attributed to the much better accessibility of the dispersed ZnO nanorods in the catalyst.
XAS measurements at the Zn K-edge were further conducted for bulk and quantitative analysis of the catalysts. ZnO-NR and ZnO/MCM-41(7.99) exhibited identical absorption edges (9661.30 eV) in their XANES spectra, while the edge for ZnO2 NPs is slightly blue shifted to 9664.70 eV (Fig. 6a and b). The measured values of the absorption edge for the samples are almost identical to the reported values of bulk hexagonal ZnO58 and cubic ZnO2.39 The slight difference in absorption edges for ZnO and ZnO2 may be associated with the coordination geometry of Zn2+ in the two structures (four-coordinated tetrahedral geometry in hexagonal ZnO and six-coordinated octahedral geometry in cubic ZnO2)58,59 and the oxidation state of the coordinating oxygen anions. Moreover, the normalized white line absorption of ZnO-NRsp is more intense than that of ZnO-NR, possibly also associated with the change in the coordination state of Zn.59 The fitted results of Zn K-edge k3-weighted EXAFS data and their FT profiles as well as the wavelet transforms for EXAFS signals further confirmed the crystalline structure of hexagonal ZnO and cubic ZnO2 in the samples (Table S10 and Fig. S7†). Next, the spent catalysts were analyzed. The absorption edges for ZnO-NRsp and ZnO/MCM-41(7.99)sp were in between those for ZnO-NR and ZnO2 NPs (Fig. 6a and b), suggesting that the spent catalysts contained both ZnO and ZnO2. The XANES spectra were fitted with those of ZnO-NR and ZnO2 NPs by linear combination, and the results of LCF (Fig. 6c and d) suggested that around 50% (R factor = 0.001) and 77% (R factor = 0.003) of ZnO in ZnO-NRsp and ZnO/MCM-41(7.99)sp, respectively, were converted to ZnO2. The relative amounts of ZnO2 in spent catalysts were also estimated by fitting the k3-weighted EXAFS data (Fig. 6e and f), and the values of 57% (R factor = 0.034) for ZnO-NRsp and 84% (R factor = 0.034) for ZnO/MCM-41(7.99)sp were obtained. The quantitative analysis confirmed a higher degree of conversion of ZnO nanorods in ZnO/MCM-41(7.99) during the catalytic reaction than those in the ZnO-NR catalyst.
 |
| | Fig. 6 (a and b) Normalized Zn K-edge XANES spectra of Zn foil, ZnO-NR, ZnO-NRsp, ZnO/MCM-41(7.99), ZnO/MCM-41(7.99)sp, and ZnO2 NPs. (c and d) LCF results for the Zn K-edge XANES of the spent catalysts. (e and f) LCF results for the k3-weighted EXAFS data of the spent catalysts. | |
3.3. Study on active sites and the mechanism of the HPPO process over ZnO-based catalysts
The fact that a significant amount of ZnO converted to ZnO2 during a catalytic reaction points to the possibility that ZnO2 might be the actual active species for the epoxidation of propylene. The catalytic performance of ZnO2 NPs for the reaction was therefore studied. Under identical reaction conditions (i.e., 37.5 mg of ZnO2 NPs, 5.87 mmol of H2O2, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C for 6 h), it gave a PO yield of 85.12% (100% PO selectivity) and a PO productivity of 1290 mg h−1 gcat−1, with a TON of 14.4, a H2O2 conversion of 91.66%, and a H2O2 utilization efficiency of 92.86%. No conversion of propylene was observed when CH3CN was replaced by water. Overall, the catalytic performance of ZnO2 NPs is very similar to that of ZnO-NR (Entry 2 of Table 1 and Entry 1 of Table S1†). This strongly suggests that ZnO2, formed from ZnO under reaction conditions, is the main catalytically active species for the epoxidation reaction.
The key role of H2O2 in the epoxidation of propylene over ZnO-based catalysts was further confirmed by reactions with 18O-enriched oxidants. The catalytic reactions with H218O2 (conditions: 37.5 mg of ZnO-NR, 300 μL of the aqueous solution of 2.0–2.4% H218O2, 5 mL of CH3CN, and a propylene pressure of 20 bar at 50 °C for 6 h) or with H216O2 and 18O2 (conditions: 37.5 mg of ZnO-NR, 5.87 mmol of H2O2, 5 mL of CH3CN, 20 bar of propylene pressure with ∼5% 18O2 at 50 °C for 6 h) were conducted and the resulting product of PO was analyzed by GC/MS. As shown in Fig. 7, no 18O-enriched PO was detected for the reaction with H216O2 and 18O2, while an enrichment of 72% was observed for the reaction with H218O2, with roughly one-to-one stoichiometry of H2O2 and PO. The results indicated that the ZnO-based catalysts reacted with H2O2 but not with O2 to generate reactive oxygenated species to further react with propylene to produce PO.
 |
| | Fig. 7 The MS data of propylene epoxidation over ZnO-NR with (a) H216O2, (b) 18O2 (97 atom%) and H216O2, and (c) H218O2 (90 atom%, 2.0–2.4% aqueous solution). | |
The HPPO process over ZnO-based catalysts should be associated with the reactive species in the ZnO2 NPs formed upon the reaction of ZnO with H2O2. Selected catalysts were further analyzed by EPR and XPS to probe and investigate the possible reactive species. The EPR spectra of fresh and spent catalysts of ZnO-NR and ZnO/MCM-41(7.99) as well as the spectrum of ZnO2 NPs were recorded at 77 K. In principle, both ZnO and ZnO2 are EPR silent, but the peroxide (O22−) ions on ZnO2 may react with water to form EPR-active superoxide (O2−) species.40 As shown in Fig. 8a, while the fresh catalysts did not show any signals in their spectra, the spectrum of ZnO2 NPs displayed an axial peak at g‖ ∼ 2.030 and g⊥ ∼ 2.000, where the perpendicular absorption linewidth is large so that the difference between gyy and gxx cannot be significantly resolved. The corresponding results further supported the presence of superoxide species adsorbed to Zn2+ in the sample.40,41,60 The signals were also observed for the two spent catalysts. The integration intensity ratio for ZnO-NRsp, ZnO/MCM-41(7.99)sp, and ZnO2 NP was determined to be 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0:2.0 (Fig. S8†). Interestingly, the Zn species with a relatively small amount (7.99%) in ZnO/MCM-41(7.99)sp resulted in comparable spin intensity (no signal of the MCM-41-only material) for paramagnetic superoxide species, implicating the role of the mesoporous materials in assisting the dispersion and storage of the reactive species in the composite. The comparable values of PO productivity (PPO, per gram of catalyst) for ZnO-NR and ZnO/MCM-41(7.99) further support this point (Table S1†). For ZnO-NRsp and ZnO2 NPs, the additional signal at g ∼ 1.95, which could be attributed to the oxygen vacancy with trapped electrons,40,61,62 was also observed. Since water was present in the reaction media for all the catalytic tests, one could conclude that both peroxide and superoxide species should be present in the ZnO-based catalysts in the HPPO process. The peroxide and superoxide species were thermally stable up to 200 °C and were converted to O2 at 210–250 °C, as revealed by analysis of TGA-GC/MS (Fig. S9†).
:1.0:2.0 (Fig. S8†). Interestingly, the Zn species with a relatively small amount (7.99%) in ZnO/MCM-41(7.99)sp resulted in comparable spin intensity (no signal of the MCM-41-only material) for paramagnetic superoxide species, implicating the role of the mesoporous materials in assisting the dispersion and storage of the reactive species in the composite. The comparable values of PO productivity (PPO, per gram of catalyst) for ZnO-NR and ZnO/MCM-41(7.99) further support this point (Table S1†). For ZnO-NRsp and ZnO2 NPs, the additional signal at g ∼ 1.95, which could be attributed to the oxygen vacancy with trapped electrons,40,61,62 was also observed. Since water was present in the reaction media for all the catalytic tests, one could conclude that both peroxide and superoxide species should be present in the ZnO-based catalysts in the HPPO process. The peroxide and superoxide species were thermally stable up to 200 °C and were converted to O2 at 210–250 °C, as revealed by analysis of TGA-GC/MS (Fig. S9†).
 |
| | Fig. 8 (a) EPR spectra of MCM-41, ZnO-NR, ZnO-NRsp, ZnO/MCM-41(7.99), ZnO/MCM-41(7.99)sp, and ZnO2 NPs measured at 77 K. (b) O 1s XPS spectra and deconvoluted peaks of ZnO-NR, ZnO-NRsp, and ZnO2 NPs. | |
XPS was further applied to probe the binding energy (BE) and chemical state of oxygen-containing species on the surface of these catalysts. The O 1s spectra of the fresh and spent catalysts and ZnO2 NPs are shown in Fig. 8b and S10.† The spectrum of ZnO-NR can be deconvoluted into three peaks at ∼530.6, 531.5, and 532.5 eV, corresponding to the O2− anions in the lattice (OL), the oxygen anions (Ox−) in the vicinity of oxygen vacancies (VO), and the chemisorbed O2 or H2O (OC), respectively.63,64 The values of BE for VO and OC are characteristic of defect-rich oxides.65 Similarly, the spectrum of ZnO/MCM-41(7.99) was fitted with the contributions of the three species (Fig. S10†), with a much more intense peak at 532.6 eV mainly attributed to the framework oxygen (Si–O–Si/Al) of MCM-41 as well as the surface OH groups and the OC on ZnO-NR.53,66 For ZnO2 NPs, on the other hand, its spectrum can be well fitted with three components (Fig. 8b). The most intense peak at 531.7 eV should be mainly attributed to both the O22− anions (and related hydroperoxide, OOH−) of ZnO2 as well as the EPR-active superoxide species on the surface.63,64 The presence of VO species, as suggested by EPR, might also contribute to the peak. The other two peaks at 530.3 and 532.7 eV are relatively weak and they are assigned to the O2− anions of ZnO, possibly formed by the decomposition of ZnO259 and the chemisorbed species (OC), respectively. The spectrum of the spent catalyst ZnO-NRsp resembles that of ZnO2, with the values of BE of the three fitted peaks being identical to those for ZnO2. Comparing the relative intensities of the three peaks suggests that ZnO-NRsp possessed fewer peroxide (and hydroperoxide), superoxide, and VO species, yet more OC on its surface than ZnO2 did. Similar changes in the surface species were also observed for ZnO/MCM-41(7.99)sp after catalytic reaction (Fig. S10†).
Based on our findings, we propose a catalytic cycle for the HPPO process over ZnO-based catalysts with acetonitrile as a solvent. As shown in Scheme 1, under the reaction conditions, the hexagonal ZnO-NR first reacts with H2O2 to transform the four-coordinated Zn2+ cations with tetrahedral geometry to the six-coordinated octahedral Zn centers of cubic ZnO2. The process can store additional oxygen atoms from H2O2 with the reactive species anchored at the Zn centers. The Zn-peroxide species may react with water to form EPR-active Zn-superoxide species and may also generate related Zn-hydroperoxide species (suggested by XPS) on ZnO2. All these species may be capable of epoxidizing propylene via a direct O-atom transfer reaction6 with the assistance of acetonitrile and a nearby aqueous hydrogen bonding network. The acetonitrile molecule may serve as a decent ligand33,67 to coordinate and interact with Zn2+ through the nitrogen atom and open up the Zn–O coordination during the catalytic cycle for O-atom transfer to the olefin to selectively produce PO. The coordination and geometry of the proposed intermediate states might resemble those in TS-1 catalysts.5,6 Moreover, the presence of a nearby water molecule can be essential for forming the H-bonding network to activate the peroxide (and other reactive species) and enable epoxidation. In our study, acetonitrile was the solvent and the water molecules needed for the proposed catalytic cycle came from the aqueous solution of H2O2. Such reaction conditions seemed to be necessary, noting that the corresponding chemistry cannot be conducted in a protic solvent of methanol or an aqueous environment. After forming PO, the Zn center within a six-membered ring of (ZnO)3, resembling the structure of ZnO, may further react with another molecule of H2O2 to regenerate the active site on ZnO2, forming one molecule of water as a byproduct. It is noted that similar species of peroxide, hydroperoxide and superoxide were also detected for olefinic epoxidation and aromatic hydroxylation with H2O2 over TS-1 and TiMCM-41.68–70 In our study, the results of XAS and nitrogen physisorption reveal that the dispersion of ZnO by MCM-41 is beneficial for forming and accumulating more (up to 84%) reactive ZnO2 in the spent ZnO/MCM-41(7.99) catalyst. With the assistance of acetonitrile, an aprotic polar organic solvent, the hydrophobic propylene substrate may diffuse into the mesopores of MCM-41 and may also approach the active sites on ZnO2 more easily with reduced activation free energy due to enthalpy–entropy compensation.20 We could thus bridge the microscopic properties and the macroscopic catalytic performance for the HPPO process over highly active ZnO/ZnO2 catalysts.71–73 Finally, the fact that no amide or organic peroxide was detected in our catalytic studies strongly suggests that an organic peroxide process was not involved in the HPPO process over the ZnO-based catalysts.
 |
| | Scheme 1 A proposed mechanism for the HPPO process over ZnO-NR with H2O2 in CH3CN. | |
4 Conclusions
ZnO-based catalysts, including ZnO-NR and ZnO/MCM-41(x) composites, have been prepared and applied for the epoxidation of propylene with H2O2 in acetonitrile under low temperatures (30–70 °C) and high pressures (5–20 bar). The ZnO catalysts exhibited high epoxidation activity with high H2O2 utilization and PO selectivity. The amount of ZnO in ZnO/MCM-41(x) composites has been optimized to give ≥98% H2O2 utilization and nearly 100% PO selectivity. The catalytic performance of the ZnO-based catalysts is comparable to that of TS-1 catalysts under similar reaction conditions. Moreover, ZnO2 has been identified as forming from the reaction of ZnO with H2O2 and served as a novel green oxygen-storage oxide material. The reactive species on ZnO2, including peroxide, hydroperoxide, and superoxide, are proposed to epoxidize propylene through a direct O-atom transfer reaction with the assistance of acetonitrile and a nearby aqueous hydrogen bonding network. The ZnO-based catalytic system with H2O2 in acetonitrile shows high efficiency and recyclability and may be promising for practical applications.
Data availability
The authors confirm that the data supporting the findings of this study are within the article and ESI.†
Author contributions
Gebretinsae Yeabyo Nigussie: data collection, methodology, conceptualization, data analysis, writing – original draft, review, and editing. Yi-Fang Tsai: data collection, conceptualization, and methodology. Tsung-Cheng Yang: data collection and methodology. Chia-Min Yang: funding acquisition, supervision, conceptualization, methodology, data analysis, investigation, writing – original draft, review and editing. Steve S.-F. Yu: funding acquisition, supervision, conceptualization, methodology, data analysis, investigation, writing – original draft, review and editing.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
This work was supported by Academia Sinica, Taiwan, and grants from the Innovative Materials and Analysis Technology Exploration Program (AS-iMATE-112-22) and the National Science and Technology Council, Taiwan (NSTC 111-2113-M-001-040, 112-2113-M-001-038- and 112-2113-M-007-019-MY3). We are grateful to Drs Jyh-Fu Lee, Yu-Chun Chuang, Chih-Wen Pao, Jeng-Lung Chen and the staff of the National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan, for their kind assistance with XAS and high-resolution SXRD studies. We also thank Miss Ting-Yin Cheng and Yi-Jen Yu at the Instrumentation Center of National Tsing Hua University for TGA-GC/MS and TEM measurements and acknowledge the Instrument Center of National Chung Hsing University (NSTC 112-2740-M-005-001-/111-2740-M-005-001-) and the Core Facility Center of National Cheng Kung University (NSTC 113-2740-M-006-002-/NSTC 112-2740-M-006-001-) for their assistance with XPS and ICP-MS measurements, respectively.
References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- J. Teržan, M. Huš, B. Likozar and P. Djinović, ACS Catal., 2020, 10, 13415–13436 CrossRef.
- J. Tsuji, J. Yamamoto, M. Ishino and N. Oku, Sumitomo Kagaku, 2006, 1, 4–10 Search PubMed.
- Q. Hua, T. Cao, X. K. Gu, J. Lu, Z. Jiang, X. Pan, L. Luo, W. X. Li and W. Huang, Angew. Chem., Int. Ed., 2014, 126, 4956–4961 CrossRef.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2013, 52, 1168–1178 CrossRef CAS.
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, ChemCatChem, 2022, 14, e202101132 CrossRef CAS.
- A. S. Sharma, V. S. Sharma, H. Kaur and R. S. Varma, Green Chem., 2020, 22, 5902–5936 RSC.
- J. Yamamoto, C. Works, H. Koike and S. Yoshida, Sumitomo Kagaku Report, 2019, 1–9 Search PubMed.
- J. Wei, S. Wang, J. Wu, D. Cao and D. Cheng, Ind. Chem. Mater., 2024, 2, 7–29 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- J. Z. Tan, D. T. Bregante, C. Torres and D. W. Flaherty, J. Catal., 2022, 405, 91–104 Search PubMed.
- M. Alvear, C. Schmidt, O. Reinsdorf, E. Lebron-Rodrigez, A. Al Abdulghani, I. Hermans, M. Peurla, M. Lastusaari, K. Eränen and D. Y. Murzin, Catal. Lett., 2023, 1–12 Search PubMed.
- W. Li, L. Chen, M. Qiu, W. Li, Y. Zhang, Y. Zhu, J. Li and X. Chen, ACS Catal., 2023, 13, 10487–10499 CrossRef CAS.
- C. Miao, N. He, Q. Zhu, Y. Yi, Z. Feng and H. Guo, Catal. Lett., 2020, 150, 281–290 CrossRef CAS.
- N. Sheng, Z. Liu, Z. Song, D. Lin, X. Feng, Y. Liu, X. Chen, D. Chen, X. Zhou and C. Yang, Chem. Eng. J., 2019, 377, 119954 Search PubMed.
- Z. Song, X. Feng, N. Sheng, D. Lin, Y. Li, Y. Liu, X. Chen, X. Zhou, D. Chen and C. Yang, Catal. Today, 2020, 347, 102–109 CrossRef CAS.
- A. Pulido, P. Concepción, M. Boronat and A. Corma, J. Catal., 2012, 292, 138–147 CrossRef CAS.
- E. J. Lee, J. Lee, M. Lee, H.-K. Min, S. Park and D. H. Kim, Mol. Catal., 2019, 467, 111–119 CrossRef CAS.
- Y. Yu, N. Fang, Z. Chen, D. Liu, Y. Liu and M. He, ACS Sustainable Chem. Eng., 2022, 10, 11641–11654 CrossRef CAS.
- D. S. Potts, D. T. Bregante, J. S. Adams, C. Torres and D. W. Flaherty, Chem. Soc. Rev., 2021, 50, 12308–12337 RSC.
- X. Liu, X. Wang, X. Guo and G. Li, Catal. Today, 2004, 93, 505–509 CrossRef.
- X. Nie, X. Ji, Y. Chen, X. Guo and C. Song, Mol. Catal., 2017, 441, 150–167 CrossRef CAS.
- G. Bellussi, A. Carati, M. G. Clerici, G. Maddinelli and R. Millini, J. Catal., 1992, 133, 220–230 CrossRef CAS.
- Q. Qin, H. Liu, Y. Guo, B. Wang, J. Zhu and J. Ma, Phys. Chem. Chem. Phys., 2023, 25, 21358–21375 RSC.
- F. Song, Y. Liu, L. Wang, H. Zhang, M. He and P. Wu, Stud. Surf. Sci. Catal., 2007, 170, 1236–1243 CrossRef.
- X. Lu, H. Wu, J. Jiang, M. He and P. Wu, J. Catal., 2016, 342, 173–183 CrossRef CAS.
- J. Yin, X. Jin, H. Xu, Y. Guan, R. Peng, L. Chen, J. Jiang and P. Wu, Chin. J. Catal., 2021, 42, 1561–1575 Search PubMed.
- G. Laus, J. Chem. Soc., Perkin Trans. 2, 2001, 864–868 RSC.
- T. Mallat and A. Baiker, Catal. Sci. Technol., 2011, 1, 1572–1583 RSC.
- L. Wu, X. Deng, S. Zhao, H. Yin, Z. Zhuo, X. Fang, Y. Liu and M. He, Chem. Commun., 2016, 52, 8679–8682 RSC.
- D. Pan, L. Kong, H. Zhang, Y. Zhang and Y. Tang, ACS Appl. Mater. Interfaces, 2023, 15, 28125–28134 CrossRef CAS.
- Y. Wang, H. Yang, Y. Zuo, D. Tian, G. Hou, Y. Su, Z. Feng, X. Guo and C. Li, Appl. Catal., B, 2023, 325, 122396 CrossRef CAS.
- Y.-J. Lu, D. Janmanchi, T. Natarajan, Z.-H. Lin, W. H. Wanna, I.-J. Hsu, D.-L. M. Tzou, T. Ayalew Abay and S. S.-F. Yu, ChemCatChem, 2022, 14, e202200030 CrossRef CAS.
- D. T. Bregante, A. M. Johnson, A. Y. Patel, E. Z. Ayla, M. J. Cordon, B. C. Bukowski, J. Greeley, R. Gounder and D. W. Flaherty, J. Am. Chem. Soc., 2019, 141, 7302–7319 CrossRef CAS PubMed.
- S. K. Maiti, A. Ramanathan and B. Subramaniam, Ind. Eng. Chem. Res., 2019, 58, 17727–17735 CrossRef CAS.
- D. T. Bregante, N. E. Thornburg, J. M. Notestein and D. W. Flaherty, ACS Catal., 2018, 8, 2995–3010 CrossRef CAS.
- A. Kołodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef.
- Z. Mirzaeifard, Z. Shariatinia, M. Jourshabani and S. M. Rezaei Darvishi, Ind. Eng. Chem. Res., 2020, 59, 15894–15911 CrossRef CAS.
- D. Bocharov, A. Chesnokov, G. Chikvaidze, J. Gabrusenoks, R. Ignatans, R. Kalendarev, M. Krack, K. Kundzins, A. Kuzmin and N. Mironova-Ulmane, J. Phys. Chem. Solids, 2022, 160, 110318 CrossRef CAS.
- R. Iyengar and V. S. Rao, J. Phys. Chem. C, 1971, 75, 3089–3092 CrossRef CAS.
- A. I. Shames, O. Lev, A. A. Mikhaylov, A. G. Medvedev, J. Gun and P. V. Prikhodchenko, J. Phys. Chem. C, 2019, 123, 20884–20892 CrossRef CAS.
- S. Verma and S. L. Jain, Inorg. Chem. Front., 2014, 1, 534–539 RSC.
- S. Chawla, H. Uppal, M. Yadav, N. Bahadur and N. Singh, Ecotoxicol. Environ. Saf., 2017, 135, 68–74 CrossRef CAS PubMed.
- D. A. Giannakoudakis, M. Florent, R. Wallace, J. Secor, C. Karwacki and T. J. Bandosz, Appl. Catal., B, 2018, 226, 429–440 CrossRef CAS.
- M. Hosseini-Sarvari and Z. Bazyar, ChemistrySelect, 2020, 5, 8853–8857 CrossRef CAS.
- D. G. Montjoy, E. A. Wilson, H. Hou, J. D. Graves and N. A. Kotov, Nat. Commun., 2023, 14, 857 CrossRef CAS.
- V. Arca, A. B. Boscoletto, N. Fracasso, L. Meda and G. Ranghino, J. Mol. Catal. A: Chem., 2006, 243, 264–277 CrossRef CAS.
- P. Ghasemipour, M. Fattahi, B. Rasekh and F. Yazdian, Sci. Rep., 2020, 10, 4414 Search PubMed.
- D. Liu, L. Cao, G. Zhang, L. Zhao, J. Gao and C. Xu, Fuel Process. Technol., 2021, 216, 106770 CrossRef CAS.
- R. Al-Gaashani, S. Radiman, A. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef CAS.
- V. Alvarado-Pérez, L. I. Cabrera-Lara, G. López-Téllez, D. Mendoza-Anaya, S. Hernández-López and M. Camacho-López, Mater. Chem. Phys., 2019, 233, 180–184 CrossRef.
- R. M. Sellers, Analyst, 1980, 105, 950–954 RSC.
- Z. Zhou, Y. Wu, H. Ling, J. Guo, S. Wang and M. Li, J. Ind. Eng. Chem., 2023, 119, 218–225 CrossRef CAS.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2014, 53, 6274–6287 CrossRef CAS.
- W. Cheng, X. Wang, G. Li, X. Guo and S. Zhang, J. Catal., 2008, 255, 343–346 CrossRef CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 4430–4431 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. Kresge, K. Schmitt, C. Chu, D. H. Olson, E. Sheppard and S. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- K. M. McPeak, M. A. Becker, N. G. Britton, H. Majidi, B. A. Bunker and J. B. Baxter, Chem. Mater., 2010, 22, 6162–6170 CrossRef CAS.
- T. Daley, K. B. Opuni, E. Raj, A. J. Dent, G. Cibin, T. I. Hyde and G. Sankar, J. Phys.: Condens. Matter, 2021, 33, 264002 CrossRef CAS.
- M. Che and A. J. Tench, Adv. Catal., 1983, 32, 1–148 CAS.
- M. Buryi, V. Babin, Y. Y. Chang, Z. Remeš, J. Mičová and D. Šimek, Appl. Surf. Sci., 2020, 525, 146448 CrossRef CAS.
- A. B. Djurišić, Y. H. Leung, W. C. H. Choy, K. W. Cheah and W. K. Chan, Appl. Phys. Lett., 2004, 84, 2635–2637 CrossRef.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 130, 6162–6167 CrossRef.
- H. Yuan, S. A. A. A. Aljneibi, J. Yuan, Y. Wang, H. Liu, J. Fang, C. Tang, X. Yan, H. Cai and Y. Gu, Adv. Mater., 2019, 31, 1807161 CrossRef PubMed.
- K. Banger, Y. Yamashita, K. Mori, R. Peterson, T. Leedham, J. Rickard and H. Sirringhaus, Nat. Mater., 2011, 10, 45–50 CrossRef CAS PubMed.
- C. Miao, K. Shang, L. Liang, S. Chen and J. Ouyang, ACS Sustain. Chem. Eng., 2022, 10, 12771–12782 CrossRef CAS.
- S. F. Rach and F. E. Kühn, Chem. Rev., 2009, 109, 2061–2080 CrossRef CAS.
- Q. Zhao, X.-H. Bao, Y. Wang, L.-W. Lin, G. Li, X.-W. Guo and X.-S. Wang, J. Mol. Catal. A: Chem., 2000, 157, 265–268 CrossRef CAS.
- K. Chaudhari, D. Srinivas and P. Ratnasamy, J. Catal., 2001, 203, 25–32 CrossRef CAS.
- D. Srinivas, P. Manikandan, S. C. Laha, R. Kumar and P. Ratnasamy, J. Catal., 2003, 217, 160–171 CAS.
- W. Chen, G. Qian, Y. Wan, D. Chen, X. Zhou, W. Yuan and X. Duan, Acc. Chem. Res., 2022, 55, 3230–3241 CrossRef CAS.
- X. Zhang, N. Fei, Q. Wang, A. R. Khan, W. Chen, G. Qian, J. Zhang, D. Chen, X. Duan, X. Zhou and W. Yuan, J. Mater. Chem. A, 2024, 12, 21884–21894 RSC.
- W. Chen, W. Fu, X. Duan, B. Chen, G. Qian, R. Si, X. Zhou, W. Yuan and D. Chen, Engineering, 2022, 14, 124–133 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Yi-Fang
Tsai
a,
Tsung-Cheng
Yang
d,
Chia-Min
Yang
abc,
Yi-Fang
Tsai
a,
Tsung-Cheng
Yang
d,
Chia-Min
Yang






 and nH2O2 denote the molar amounts of H2O2 before and after the reactions, nPO denotes the molar amount of PO formed (calculated from the volume of the reaction mixture and the value of molar concentration determined by GC or GC/MS), nothers denotes the molar amount of other byproducts detected, wPO and wcat denote the weights of PO formed and the catalyst and nZn denotes the molar amount of Zn in a catalyst. Some catalysts also produced acetic acid (AC) or acetaldehyde (AA), and the criteria for the yields and selectivity of these reaction products were the same as those for PO.
and nH2O2 denote the molar amounts of H2O2 before and after the reactions, nPO denotes the molar amount of PO formed (calculated from the volume of the reaction mixture and the value of molar concentration determined by GC or GC/MS), nothers denotes the molar amount of other byproducts detected, wPO and wcat denote the weights of PO formed and the catalyst and nZn denotes the molar amount of Zn in a catalyst. Some catalysts also produced acetic acid (AC) or acetaldehyde (AA), and the criteria for the yields and selectivity of these reaction products were the same as those for PO.








