
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MnTiO3 as a carbon-free cathode for rechargeable Li–O2 batteries†
Doaa Aasef
Ahmed
 *a,
Mustafa
Çelik
bc,
Wernfried
Mayr-Schmölzer
a,
Abdulkadir
Kızılaslan
bc and
Gregor B.
Vonbun-Feldbauer
*ad
*a,
Mustafa
Çelik
bc,
Wernfried
Mayr-Schmölzer
a,
Abdulkadir
Kızılaslan
bc and
Gregor B.
Vonbun-Feldbauer
*ad
aInstitute of Advanced Ceramics, Hamburg University of Technology, 21073 Hamburg, Germany. E-mail: doaa.ahmed@tuhh.de; gregor.feldbauer@tuhh.de
bResearch, Development and Application Center (SARGEM), Sakarya University, Esentepe, Sakarya 54187, Turkey
cDepartment of Metallurgical and Materials Engineering, Engineering Faculty, Sakarya University, Esentepe, Sakarya 54187, Turkey
dInstitute of Soft Matter Modeling, Hamburg University of Technology, 21073 Hamburg, Germany
First published on 17th January 2025
Abstract
Li–O2 batteries (LOBs) are next-generation energy storage systems. However, their main challenges are the sluggish kinetics of oxygen reduction and evolution reactions (ORR/OER) and high charge overpotentials due to the formation of discharge product (Li2O2). To address this challenge, developing a catalyst with a unique structure and exceptional catalytic properties is crucial to enhancing the reversible cycling performance of LOBs, particularly under high current density conditions. Herein, the transition metal-based perovskite MnTiO3 was examined as a carbon-free cathode catalyst using density functional theory (DFT) calculations and experimental techniques. The intrinsic advantages of MnTiO3 stem from the coexistence of Mn and Ti energy levels near the Fermi level, as revealed by our density of states (DOS) analysis. This electronic structure facilitates ORR/OER, thus endowing MnTiO3 with a bifunctional role in promoting battery performance. Our DFT-based investigation elucidates the surface stability and catalytic properties of MnTiO3. Furthermore, Energy Dispersive Spectroscopy (EDS) and X-ray diffraction (XRD) confirm that the electrochemical reaction on MnTiO3 follows a two-electron pathway. Our findings reveal that a LOB with MnTiO3 exhibits a total overpotential of 1.18 V and 1.55 V using DFT and electrochemical measurements, respectively. High current densities up to 1 A g−1 also highlight its potential as a cathode catalyst for LOBs.
1 Introduction
Over the last few years, extensive research and development efforts have resulted in deploying various renewable energy conversion and storage devices, some of which have reached commercialization.1,2 Among energy storage devices, rechargeable lithium–oxygen (Li–O2) batteries (LOBs) are considered highly promising power sources due to their extraordinarily high theoretical energy density of 3500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) W h kg−1, low cost, and environmental friendliness.3,4
W h kg−1, low cost, and environmental friendliness.3,4
A novel rechargeable LOB comprises a Li conductive electrolyte membrane sandwiched by a Li metal foil anode and an electroactive cathode. During discharge oxygen accessed from the environment is reduced on the cathode via the oxygen reduction reaction (ORR, 2Li+ + 2e− + O2 → Li2O2). The charging process involves the decomposition of Li2O2 through the oxygen evolution reaction (OER, Li2O2 → 2Li+ + 2e− + O2).5,6
Although LOBs exhibit significant theoretical energy potential, several challenges presently limit their effectiveness. Namely, the formation of solid and electronically insulating discharge products i.e. LiO2 and Li2O2 during discharge significantly impedes fast reaction kinetics which overall leads to sluggish reactions.6,7
To overcome this problem, carbonaceous materials including porous carbon,8 carbon nanotubes (CNTs), and graphene9 have been widely utilized to elevate the overall conductivity of electrodes. However, the inherent instability of carbon electrodes often leads to reactions with lithium peroxide (Li2O2), resulting in the formation of a non-conductive layer of lithium carbonate (Li2CO3) at the electrode-discharge product interface.10 To address these challenges and increase the overall catalytic performance, various carbon free cathode materials, such as noble metals and their oxides, transition metal carbides, and transition metal compounds, have been explored for LOBs.11 Noble metal materials like Au,12 Pt,13 IrO2,14 and RuO2 (ref. 15) exhibit remarkable electrocatalytic activity, effectively addressing the sluggish kinetics of the ORR or OER. However, their high cost, scarcity, and inferior cycling stability limit their practical application. In this context, it is of great importance to develop a material with a high catalytic effect that will strengthen OER and ORR reactions simultaneously. Recent endeavours in LOB catalysis have shifted focus towards developing mono-component bifunctional catalysts,16–18 aimed at facilitating both the ORR and OER during discharging and charging processes, respectively. Traditionally, these catalysts often involve metals or metal oxides coupled with carbonaceous materials like carbon, CNTs, or graphene.19 This coupling is intended to leverage synergistic effects, enhancing catalytic performance. However, such configurations not only promote the formation of lithium carbonate (LiCO3) but also complicate electrode fabrication processes. Consequently, the exploration of mono-component bifunctional catalysts for LOBs stands as a critical pursuit to address these challenges and streamline electrode development, aiming for enhanced efficiency and simplified fabrication techniques.
Manganese dioxide (MnO2) and titanium dioxide (TiO2) are widely employed as cost-effective catalysts in LOBs. However, when used separately, manganese (Mn)20 and titanium (Ti)21 facilitate redox reactions in battery processes in one way (either OER or ORR) because of their distinct oxidizing and reducing properties and the density of states concentrated at varying energy levels.22 Motivated by the specific electronic structure inherent in a MnTiO3 perovskite, encompassing a broad spectrum of the density of states near the Fermi energy level owing to the simultaneous presence of both Mn and Ti constituents,23 MnTiO3 perovskite is introduced as an electrocatalyst in (LOBs) in this study. In light of its outstanding catalytic performance in diverse applications, including water purification,24 oxidative degradation,25 and photocatalytic activities,26 this study constitutes the first systematic evaluation of the electrochemical performance of MnTiO3 in LOBs. The analysis was conducted through a combination of experimental techniques, including Energy Dispersive Spectroscopy (EDS) and X-ray diffraction (XRD), alongside computational studies based on Density Functional Theory (DFT), providing a comprehensive understanding the electrochemical behaviour of MnTiO3 as a carbon-free electrocatalyst in LOBs.
2 Computational and experimental methods
2.1 First principles calculations
Density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP, version 5.4.4).27–31 The calculations were carried out using the Perdew–Burke–Ernzerhof (PBE) parametrization32 to describe the exchange–correlation functional. The pseudopotentials are obtained by the projector augmented wave (PAW) method.33,34 The states in the electronic configurations 3p4s3d, 3p4s3d, 2s2p, and 1s2s for Ti, Mn, O, and Li, respectively, were treated as valence states.Using generalized gradient approximation (GGA) functionals like PBE often underestimates band gaps for transition metal including compounds. For a better description of the band gap, an effective Hubbard UH,eff parameter following the Dudarev approach35 was used to account for the localization of the Mn d-electrons. In this study, we examined the band gap of MnTiO3 using several UH,eff values, namely 2, 2.5, 3, 3.5, and 4 eV. UH,eff = 3eV yields a band gap of 1.56eV in excellent agreement with our experimentally determined band gap of 1.58eV as illustrated in Fig. S1.†
An energy cutoff of 650eV was used for the plane-wave basis. For slab calculations, a Gamma-centered 5 × 4 × 1 k-point mesh was employed for sampling the Brillouin zone for the slab of the surface stability calculations. For ORR/OER calculations, the slab was doubled in the x-direction, and the k-points were tested to be converged for 3 × 4 × 1. Ionic relaxations were performed until the convergence criterion of 0.01 eV Å−1 in the forces was reached while for electronic self consistency cycles an energy convergence criterion of 10−6 eV was used. To avoid interactions between periodic images of the slab, an 18Å thick vacuum region in the c-direction (perpendicular to the surface) was added to the system.
The adsorption energies (Eadsorption) between the reaction intermediates (adsorbate) and substrate were calculated as;36
| Eadsorption = Esub/ads − Esub − Eads, | (1) |
In the calculations of free energy profiles for ORR/OER reaction, we adopted the Gibbs free energy difference (ΔG) between two consecutive steps for ORR as:
| ΔG = ΔEtotal + ΔEZPE − TΔS − eU, | (2) |
K and pressure p = 1bar in each step. The term eU with the electron charge e has been added to show the effect of a potential U on electrons.37,38 The theoretical overpotential for the ORR ηORR and the OER ηOER was obtained by| ηORR = Ueq − UDC | (3) |
| ηOER = Ueq − UC | (4) |
2.2 Synthesis of MnTiO3
MnTiO3 powders were synthesized by the sol–gel method according to M. Enhessari et al.40 Initially, 0.4mol of stearic acid were liquefied at a temperature of 73 °C. Subsequently, 0.1mol of manganese acetate were dissolved in the liquid stearic acid. The temperature was kept at 73 °C until a dark brown transparent solution was obtained. Following this, 0.1 mol of stoichiometric tetrabutyl titanate was added to the solution and mixed at 73 °C until a uniform brown colloidal solution was formed. The solution was then left to naturally cool down to room temperature and subsequently dried in an oven for 12 hours. Finally, the gel underwent calcination at 1000 °C in four steps, to yield nanocrystallites of MnTiO3. The first step involved heating the dried gel at a rate of 3 °C min−1 up to 470 °C; this was followed by holding the temperature constant at 470 °C for 40 minutes in the second step. Subsequently, the temperature was increased up to 910 °C, and in the final step, the temperature was sustained at 910 °C for 2h under ambient air conditions. Oxidation and crystallization temperatures were determined according to the Differential Thermal Analysis (DTA) measurement as given in ESI Fig. S2.†
2.3 Electrochemical characterization
The electrochemical assessment was conducted utilizing a Swagelok-type battery configuration, comprising a lithium foil anode with a carbon-free composite cathode. The electrolyte employed was 1.5 M LiTFSI (lithium trifluoromethanesulfonate) dissolved in TEGDME (tetraethylene glycol dimethyl ether), while a glassy fiber served as the separator. The fabrication process of the carbon-free cathode involved the preparation of a composite powder consisting of MnTiO3, mixed with polyvinylidene fluoride (PVDF) in 1-methyl-2-pyrrolidone (NMP), without any conducting carbon. The composite was composed of 90 wt% MnTiO3 and 10 wt% binder. Subsequently, the mixture was cast onto a nickel foam current collector and dried at 120 °C for 12h. The mass loading of each cathode, following the drying process, was 0.5 mg cm−2. The batteries were assembled inside an argon-filled glove box (H2O content <0.1 ppm and O2 content <0.1 ppm, manufactured by MBraun, Germany) to ensure an inert atmosphere. The charge–discharge tests were conducted at a current density of 0.1 mA cm−2, facilitated by a continuous flow of pure O2 gas at a rate of 8 ml min−1, maintained at a pressure of 1.0 atm and a temperature of 25 °C in the voltage range of 2.0V to 4.5V. Additionally, cyclic voltammetry (CV) analysis was performed in the same voltage range, employing a scan rate of 0.1 mV s−1 to elucidate the electrochemical reactions and their associated voltage profiles within the Li//MnTiO3 cells. All electrochemical evaluations were executed utilizing a Gamry ref. 1000 potentiostat–galvanostat device under ambient temperature.
3 Results and discussion
3.1 Morphology, crystal, and electronic structure of MnTiO3
A schematic route for the synthesis of MnTiO3 powders is given in Fig. 1a. As shown in Fig. 1b, irregular shaped chunks with different sizes were observed at low magnifications. At higher magnifications, the powders were observed to be in nano-sized irregular shaped particles that were sintered to each others due to high temperature required for the synthesis. | ||
| Fig. 1 (a) Schematic synthesis route for the synthesis of MnTiO3 powders, (b) low magnification, and (c) high magnification FESEM images for the MnTiO3 powders. | ||
Fig. 2a illustrates the optimized crystal structure of MnTiO3. The structure consists of MnO6 and TiO6 octahedrons with corner-sharing oxygen atoms. Within this crystal lattice, Ti and Mn atoms occupy the 6a Wyckoff positions, while O atoms are located at the 18b Wyckoff positions. The DFT-optimized lattice parameters were obtained as a = b = 5.23Å and c = 13.98Å.
 | ||
| Fig. 2 (a) The crystal structure of MnTiO3 with a trigonal unit cell. (b) The XRD patterns of MnTiO3, (c) (αhν)2 − hν curves of the MnTiO3 powders, and the corresponding absorption spectra in the inset obtained from the UV-visible measurements, (d) the calculated band structure, band gap (Eg), and the orbital-projected density of states (PDOS) from DFT. | ||
Synthesized powders were characterized via X-ray diffraction (XRD) analysis to determine their phase composition (Fig. 2b). Our results indicate that the synthesized powders can be indexed within the JCPDS 29-0902 (PDF#29-0902) reference card, exhibiting a trigonal crystal system with the R3c space group. Further analysis was performed by Raman technique to characterize the atomic local environment of the structure as given in Fig. S3.† The UV-vis absorption spectrum was recorded to obtain the band gap Eg, which can be obtained using the following equation;
| (αhν)n = K(hν − Eg) | (5) |
eV as shown in Fig. 2c. A direct band gap of 1.56eV was obtained from DFT calculations with a Hubbard parameter of UH,eff = 3eV which is in well alignment with the experimental measurement. The electronic structure of MnTiO3 was further analyzed using the Projected Density of States (PDOS). The Fermi level was chosen as the reference energy and set to 0eV. The conduction band (CB) primarily derives from the contribution of the Ti-d and Mn-d orbitals, whereas the valence band predominantly stems from the Mn-d and O-p orbitals with a smaller contribution of Ti-d orbitals (Fig. 2d). The existence of electronic states spanning a wide range in energy due to the coexistence of titanium and manganese is expected to promote both OER and ORR reactions simultaneously which take place at different energy levels. Taking into account the influence of orbital states in close proximity to the Fermi level on oxidation-reduction (ORR and OER) activities, it is anticipated that energy levels located on either side of the Fermi level will contribute to the bifunctional ORR and OER capabilities of the resulting material.
3.2 Surface reactivity
The obtained XRD results, depicted in Fig. 2b, reveal distinct peaks denoted by indices (104) and (110) at 38.7° and 39.8°, respectively. Notably, these peaks exhibit a pronounced degree of crystallinity and are indexed to the crystal structure of MnTiO3. Ribeiro et al.41 used DFT calculations to investigate the stoichiometric polar and nonpolar surfaces of MnTiO3, specifically the surfaces indexed as (110), (012), (101), (100), (001), and (111). They determined that the (110) surface exhibited the highest degree of stability. Since our XRD analysis reveals the most pronounced reflections at the (104) plane, we conduct a comparative analysis between the (104) and (110) surfaces, considering their different terminations. For the MnTiO3-(104) surface and the MnTiO3-(110) surface, the atomic layers follow a stacking sequence of (…-O2-O1-Mn-TiO-…) and (…-O2-O1-MnTi-…), respectively, as illustrated in Fig. 3. | ||
| Fig. 3 Top and side views of MnTiO3 slabs showing (a) a (104) surface with O1, O2, Mn and TiO terminations. (b) a (110) surface with O1, O2, and MnTi terminations. | ||
A comprehensive examination of surface stability for the (104) and (110) surfaces is presented in Section S3 of the ESI.† Our investigation (Fig. S4†) reveals that the (104) surface is significantly more stable than the (110) surface. Additionally, (Fig. S5†) elucidates that the (O1 and TiO) terminations exhibit the highest stability among the (104) terminations.
The cathode of a battery operates in an oxygen-rich environment. In order to examine the stability of the MnTiO3-(104) surface under these conditions, we have conducted an analysis of the capacity of the two MnTiO3-(104) terminations, namely O1 and TiO, to interact with oxygen atoms. This investigation was carried out by calculating the adsorption energy of oxygen atoms at multiple sites. The oxygen atoms are placed in high-symmetry points on the top of the two distinct MnTiO3 (104) terminations, O1 and TiO, as shown in Fig. 4. Subsequently, the structures were optimized using DFT calculations to minimize the forces acting on the atoms. Our results for the O1 termination show that regardless of the initial position after relaxation all oxygen atoms were adsorbed at a bridge position between Ti and Mn with an adsorption energy of −4.4eV. This indicates the strong preference of this adsorption site. The adsorption results in the formation of bonds between the oxygen atom and the titanium and manganese atoms, with bond lengths of 1.72 Å and 2.06 Å, respectively. Similarly, for the TiO termination, all initial adsorption structures resulted in the same final geometry after relaxation. On the TiO termination, O atoms prefer the proximity of Ti leading to the formation of O-Ti bonds with a bond length of 1.63Å and with an adsorption energy of −3.6eV. Considering our results, it was found that oxygen adsorption energies are considerably more negative on the O1 termination compared to the TiO termination.
 | ||
| Fig. 4 Top views of (a) O1 and (b) TiO terminations of MnTiO3 (104). In the left, the tested initial adsorption sites for oxygen atoms are shown, while in the right, the geometry after structural optimization is presented. The adsorption energies are given per adsorbed oxygen atom. | ||
3.3 ORR/OER mechanism
The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) processes in a nonaqueous Li–O2 battery can proceed in two reaction paths, namely the 2-electron (2e−) and the 4-electron (4e−) pathways.42 In the 2e− pathway, oxygen is reduced to Li2O2via the reaction 2(Li+ + e−) + O2 → Li2O2 with Ueq = 2.96V. In contrast in the 4e− path, oxygen is fully reduced to Li2O via 4(Li+ + e−) + O2 → 2Li2O with Ueq = 2.91V.
The adsorption of O2 and Li species on the cathode surface can significantly alter the ORR pathway, 2e− or 4e−, and the reaction kinetics.43,44 As illustrated in Fig. 5 and Table S1,† the adsorption energy per adsorbate (calculated according to eqn (1)) for O2 on the investigated cathode surface is −2.88eV, which is significantly more negative than that for Li atoms, which is −1.50eV. This higher energy gain from O2 adsorption indicates that the initial lithiation process (involving the formation of Li2O2) follows the 2e− pathway. In detail, the ORR process starts with the adsorption of an O2 molecule at the clean surface, followed by the reaction of a Li-ion with O2 to form LiO2. A subsequent reaction of another Li-ion with LiO2 yields Li2O2, that is the final product of the first lithiation process. Fig. 5 and Table S1† shows that all intermediates have negative adsorption energies, indicating thermodynamically favorable adsorption processes.
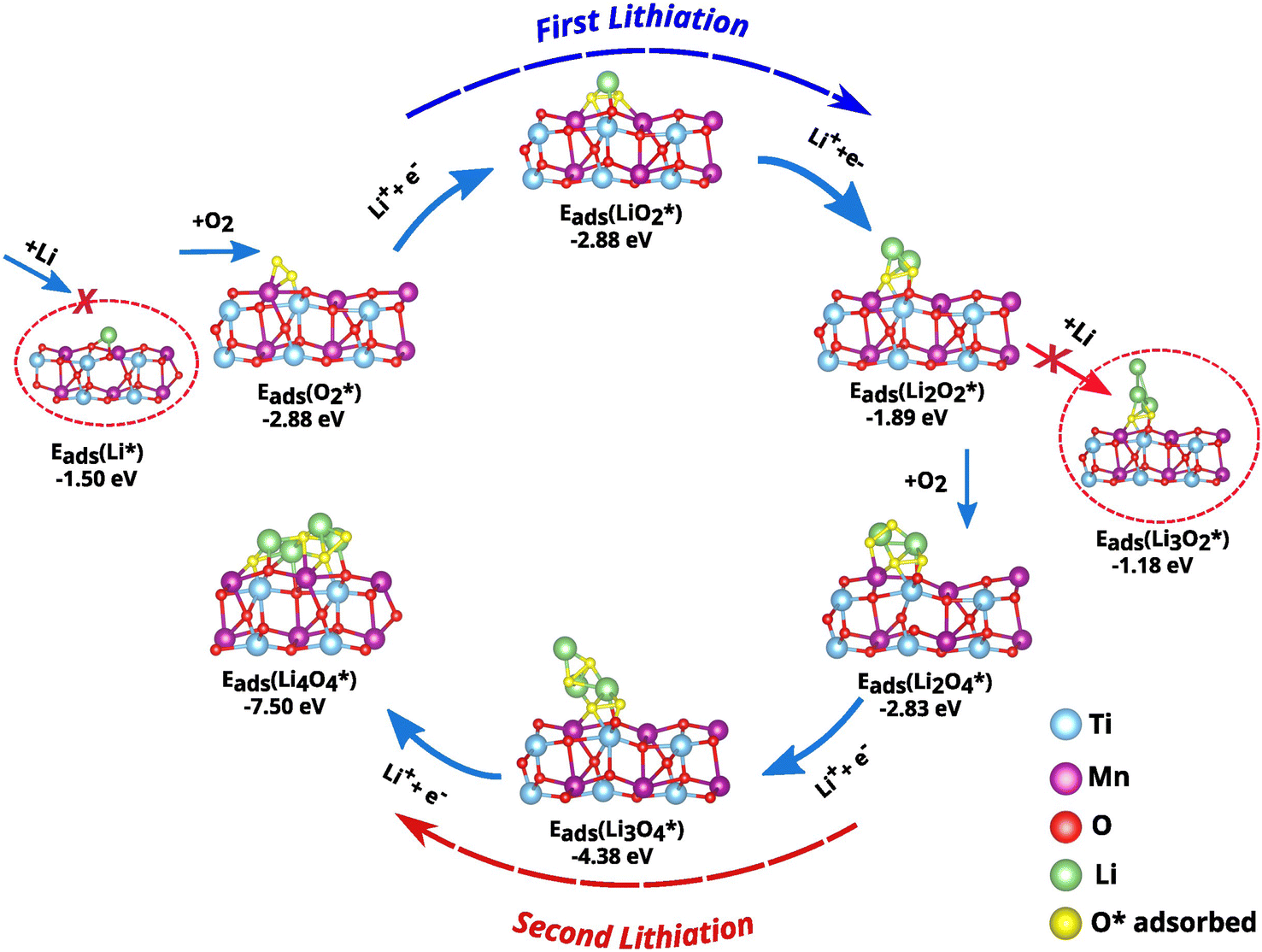 | ||
| Fig. 5 The adsorption energies of ORR intermediates during the first and second lithiation. | ||
To elucidate the second lithiation process, which involves the formation of Li4O4, the analysis was started with the independent adsorption of O2 and Li on the  -covered surface obtained from the first lithiation. The adsorption energy of −2.83eV for O2 is considerably more negative than the −1.18eV for Li atom. These adsorption energies were also calculated using eqn (1).
-covered surface obtained from the first lithiation. The adsorption energy of −2.83eV for O2 is considerably more negative than the −1.18eV for Li atom. These adsorption energies were also calculated using eqn (1).
The significantly higher adsorption energy of O2 suggests that the second lithiation also follows the 2e− pathway. The reaction sequence for the second lithiation can be summarized as follows:  . The high adsorption energy of O2 plays a crucial role in facilitating electron transfer during the electrochemical reaction, thereby accelerating the reaction kinetics. Moreover, throughout the discharge process, the adsorption energy of intermediate LiO2 increases (from −2.88eV in the first lithiation to −4.38eV in the second lithiation), promoting the growth of discharge products through a surface reaction pathway.39
. The high adsorption energy of O2 plays a crucial role in facilitating electron transfer during the electrochemical reaction, thereby accelerating the reaction kinetics. Moreover, throughout the discharge process, the adsorption energy of intermediate LiO2 increases (from −2.88eV in the first lithiation to −4.38eV in the second lithiation), promoting the growth of discharge products through a surface reaction pathway.39
Fig. 6a and b display the simulated Gibbs free energy profiles for ORR/OER corresponding to the first and second lithiation. At zero potential (U = 0V), all the reaction steps are downhill, and exhibit favourable thermodynamics, underscoring the spontaneity of Li2O2 and Li4O4 formation. Utilizing the data from the Gibbs free energy diagrams, we calculated the cathodic discharge (UDC) and charge (UC) potentials for the first and second lithiation as shown in Fig. 6(a) and (b). According to eqn (3) and (4), the total overpotentials η were calculated as 1.66V and 1.18V for the first and second lithiations, respectively.
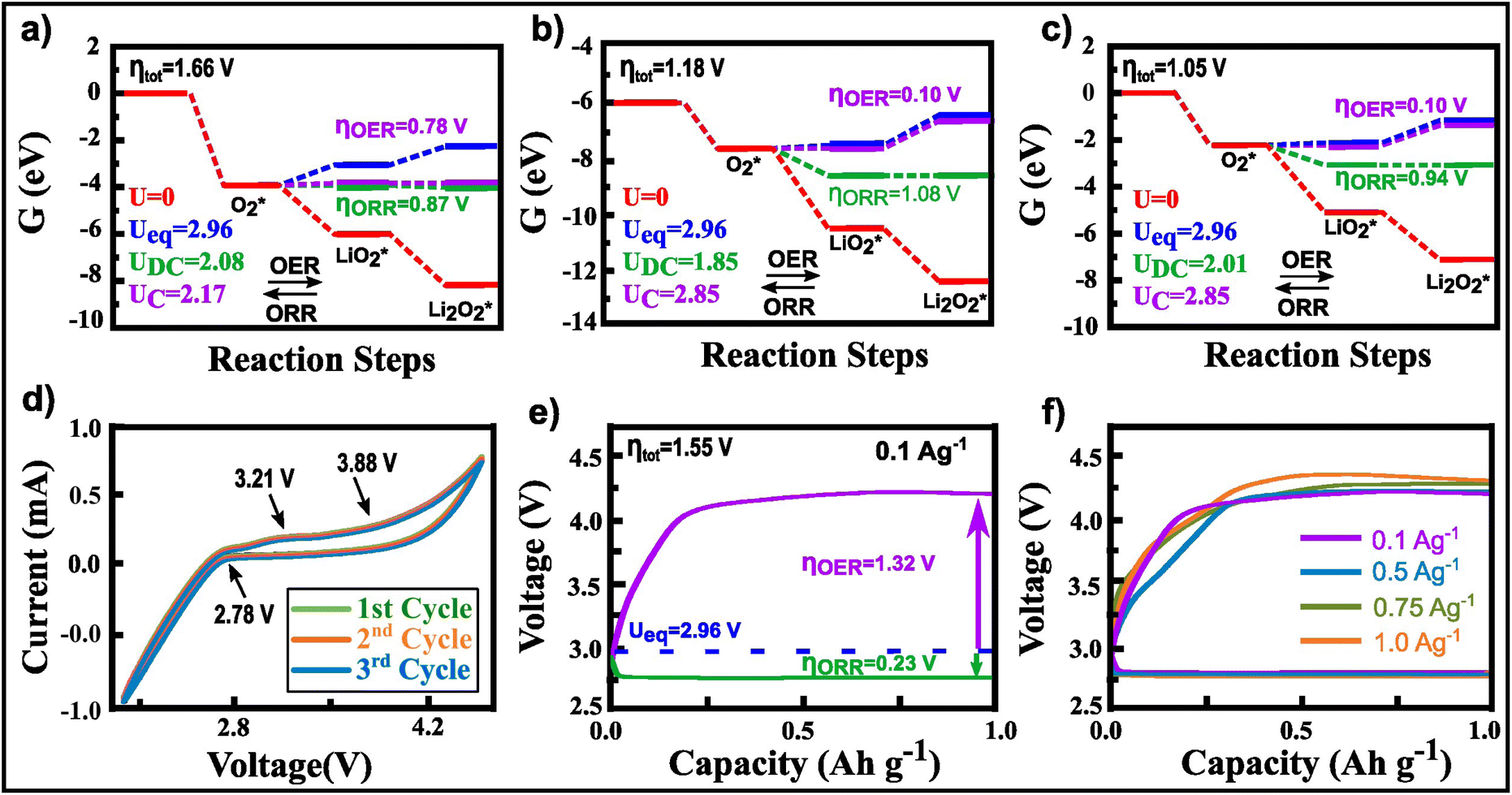 | ||
| Fig. 6 Gibbs free energy diagrams for (a) the first lithiation and (b) the second lithiation for MnTiO3 (104) with O1 termination. (c) Gibbs free energy diagrams after reducing the surface unit cell area of MnTiO3. (d) Cyclic voltammetry curves illustrating electrochemical reactions. (e) Discharging and charging curves with a specific capacity limited to 1 A h g−1 and a current density of 0.1 A g−1. (f) Rate performance at various current densities ranging from 0.1 A g−1 to 1 A g−1. | ||
Furthermore, we examined the first lithiation process on the MnTiO3 surface by reducing the surface supercell along the X-direction until a single Li2O2 molecule fully covered the newly formed surface. The calculated free energy diagram is shown in Fig. 6c. The first lithiation process yields nearly identical values for UDC, UC, and the total overpotential as the second lithiation on the original surface. Additionally, the profiles for ORR/OER closely resemble that of the second lithiation in Fig. 6(b) and (c). Consequently, this observation suggests that, instead of employing a large surface cell area, reducing the surface area can yield equivalent results. Thereby optimizing the computational resources in the analysis of the lithiation process.
The cyclic voltammetry (CV) profile of MnTiO3 shown in Fig. 6d exhibits a prominent cathode peak between 2.0 V and 2.8 V, indicating the formation of discharge products, specifically Li2O2 or LiO2. During the anodic scan, two oxidation peaks are observed: one at 3.21 V and another starting at 3.88V, where the peak at 3.21 V correspond to the oxidation of amorphous Li2O2, particularly when the catalyst has a strong affinity for LiO2 intermediates.45,46 The higher peak starting at 3.88V is associated with the decomposition of bulk Li2O2, which requires a higher dissociation potential.47 The results highlight the importance of considering the role of the MnTiO3 cathode in the formation and decomposition of Li2O2, which can significantly affect charge–discharge characteristics of the battery.
To determine the voltages associated with the OER and ORR, the MnTiO3 cathode underwent charging and discharging cycles with a 1 A h g−1 specific capacity limit, between the potentials from 2 V to 4.5 V, while operating at a current density of 0.1 A g−1 (Fig. 6e). The total overpotential was determined to be 1.55V in close agreement with the predictions from DFT, which yielded values of 1.66V and 1.18V for the first and second lithiation, respectively.
Those results underscore the effectiveness of MnTiO3 in facilitating the oxidation of Li2O2 during the charging phase. Moreover, the charge overpotentials of the MnTiO3 cathode remained relatively constant for different current densities, ranging from 0.1 to 1 A g−1 (Fig. 6f).
In order to gain deeper insights into the superior characteristics exhibited by Li–O2 batteries incorporating the MnTiO3 cathode, the configurations of discharge products were further analyzed through field-emission scanning electron microscopy (FESEM), energy-dispersive X-ray spectroscopy (EDS), and X-ray diffraction (XRD) analyses. Following the initial discharge, the FESEM images revealed the presence of discharge Li2O2 products (Fig. 7), confirming the adherence of the electrochemical reaction to the 2e− pathway. Furthermore, these images demonstrate that the majority of discharge byproducts decomposed after the charging process (Fig. 7). The EDS spectrum of the electrode following discharge indicates a significant increase in oxygen content of 61.79%, resulting from the formation of Li2O2. Nevertheless, the oxygen ratio experienced a notable decline following the decomposition of Li2O2 during the charging phase (Fig. 7a and b). Subsequent XRD analysis corroborated the existence of LiO2 and Li2O2 products, consistent with the reference patterns of JCPD-12-0254 and JCPD-09-0355, respectively (Fig. 7c). After the charging process, those characteristic peaks diminished as the discharge products decomposed, providing further evidence of the reversible nature of this process. Importantly, the XRD pattern did not reveal any additional reactions, emphasizing the effective role of the MnTiO3 perovskite catalyst in preventing the formation of undesired byproducts.
 | ||
| Fig. 7 FESEM images and EDS spectra of MnTiO3 cathode after (a) first discharge and (b) first charge, and the ex situ. (c) XRD patterns after charge and discharge. | ||
4 Conclusions
In this work, a MnTiO3 catalyst was used as a potential oxygen electrode for a Li–O2 battery for the first time. Both ab initio calculations and experimental techniques were employed to investigate the electrochemical behavior. The electronic structure calculations demonstrate that the simultaneous presence of Mn and Ti orbitals near the Fermi level can facilitate the electron transport, thereby promoting ORR and OER reactions. Furthermore, our research elucidated the impact of the reactive surface area on the computational prediction of the electrochemical reactions. DFT adsorption energy and XRD analysis confirm that the electrochemical reaction proceeds following the 2-electron discharge pathway. As a result, the carbon-free MnTiO3 cathode exhibits excellent electrochemical performance and a low overpotential of 1.55V, with a small increase in overpotential at elevated current densities up to 1 A g−1.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Doaa Aasef Ahmed: conceptualization, formal analysis, investigation, methodology, project administration, visualization, writing – original draft, writing – review and editing, data curation. Mustafa Çelik: visualization, investigation, methodology, formal analysis, writing – original draft, writing – review and editing. Wernfried Mayr-Schmölzer: methodology, supervision, writing – review and editing. Abdulkadir Kızılaslan: data curation, conceptualization, supervision, writing – original draft, writing – review and editing. Gregor B. Vonbun-Feldbauer: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 192346071–SFB 986. Doaa Aasef Ahmed was co-funded by the European Union within the Erasmus+ programme. The numerical calculations were partially performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).References
- H. Hou, Y. Cong, Q. Zhu, Z. Geng, X. Wang, Z. Shao, X. Wu, K. Huang and S. Feng, Chem. Eng. J., 2022, 448, 137684 CrossRef CAS.
- X. Mu, C. Xia, B. Gao, S. Guo, X. Zhang, J. He, Y. Wang, H. Dong, P. He and H. Zhou, Energy Storage Mater., 2021, 41, 650–655 CrossRef.
- Q. Qiu, Z.-Z. Pan, P. Yao, J. Yuan, C. Xia, Y. Zhao and Y. Li, Chem. Eng. J., 2023, 452, 139608 CrossRef CAS.
- J. Kang, J.-S. Yu and B. Han, J. Phys. Chem. Lett., 2016, 7, 2803–2808 CrossRef CAS PubMed.
- M.-C. Sung, G.-H. Lee and D.-W. Kim, InfoMat, 2021, 3, 1295–1310 CrossRef CAS.
- J. G. Kim, Y. Kim, Y. Noh, S. Lee, Y. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 5429–5439 CrossRef CAS PubMed.
- C. Xia, M. Waletzko, L. Chen, K. Peppler, P. J. Klar and J. Janek, ACS Appl. Mater. Interfaces, 2014, 6, 12083–12092 CrossRef CAS PubMed.
- D. Zhai, H.-H. Wang, J. Yang, K. C. Lau, K. Li, K. Amine and L. A. Curtiss, J. Am. Chem. Soc., 2013, 135, 15364–15372 CrossRef CAS PubMed.
- Q. Li, P. Xu, W. Gao, S. Ma, G. Zhang, R. Cao, J. Cho, H.-L. Wang and G. Wu, Adv. Mater., 2013, 26, 1378–1386 CrossRef PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- D. Cao, S. Zhang, F. Yu, Y. Wu and Y. Chen, Batteries Supercaps, 2019, 2, 428–439 CrossRef CAS.
- C. Xu, B. M. Gallant, P. U. Wunderlich, T. Lohmann and J. R. Greer, ACS Nano, 2015, 9, 5876–5883 CrossRef CAS PubMed.
- Y.-C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- Y. Zhang, X. Li, M. Zhang, S. Liao, P. Dong, J. Xiao, Y. Zhang and X. Zeng, Ceram. Int., 2017, 43, 14082–14089 CrossRef CAS.
- K. Li, H. Dong, Y. Wang, Y. Yin and S. Yang, J. Colloid Interface Sci., 2020, 579, 448–454 CrossRef CAS PubMed.
- H. Wu, W. Sun, J. Shen, Z. Mao, H. Wang, H. Cai, Z. Wang and K. Sun, ACS Sustainable Chem. Eng., 2018, 6, 15180–15190 CrossRef CAS.
- Y. J. Lee, D. H. Kim, T.-G. Kang, Y. Ko, K. Kang and Y. J. Lee, Chem. Mater., 2017, 29, 10542–10550 CrossRef CAS.
- Y. Xia, X. Yu, Y. Xu, X. Fan, B. Gao, C. Jiang, M. Zhang, X. Huang, H. Gong, J. He and T. Wang, J. Mater. Chem. A, 2023, 11, 24918–24927 RSC.
- S. Zhang, Y. Wang, D. Li, Z. Kang, F. Dong, H. Xie and J. Liu, J. Alloys Compd., 2020, 825, 154054 CrossRef CAS.
- S. H. Kim, Y. J. Lee, D. H. Kim and Y. J. Lee, ACS Appl. Mater. Interfaces, 2017, 10, 660–667 CrossRef PubMed.
- S. Xu, Q. Zhu, J. Long, H. Wang, X. Xie, K. Wang and J. Chen, Adv. Funct. Mater., 2016, 26, 1365–1374 CrossRef CAS.
- T. G. Yun, Y. Heo, H. Bin Bae and S.-Y. Chung, Nat. Commun., 2021, 12, year Search PubMed.
- K. Yoshimatsu, H. Mashiko, N. Umezawa, K. Horiba, H. Kumigashira and A. Ohtomo, J. Phys. Chem. C, 2017, 121, 18717–18724 CrossRef CAS.
- D. Wang, H. Xu, J. Ma, X. Lu, J. Qi and S. Song, ACS Appl. Mater. Interfaces, 2018, 10, 31631–31640 CrossRef CAS PubMed.
- H. Wang, Q. Gao, H. Li, M. Gao, B. Han, K. Xia and C.-G. Zhou, ACS Appl. Nano Mater., 2018, 1, 2727–2738 CrossRef CAS.
- N. Kitchamsetti, P. N. Didwal, S. R. Mulani, M. S. Patil and R. S. Devan, Heliyon, 2021, 7, e07297 CrossRef CAS PubMed.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- T. Zhu, C. Xia, B. Wu, J. Pan, H. Yang, W. Zhang and B. Y. Xia, Appl. Catal., B, 2024, 357, 124315 CrossRef CAS.
- Y. Gong, W. Ding, Z. Li, R. Su, X. Zhang, J. Wang, J. Zhou, Z. Wang, Y. Gao, S. Li, P. Guan, Z. Wei and C. Sun, ACS Catal., 2018, 8, 4082–4090 CrossRef CAS.
- S. Ding, X. Yu, Z.-F. Ma and X. Yuan, J. Mater. Chem. A, 2021, 9, 8160–8194 RSC.
- Y. Cao, Z. Zhu, X. Li, J. Xi, D. J. Singh, L. Xi, J. Yang and W. Zhang, J. Mater. Chem. A, 2022, 10, 11039–11045 RSC.
- M. Enhessari, A. Parviz, E. Karamali and K. Ozaee, J. Exp. Nanosci., 2012, 7, 327–335 CrossRef CAS.
- R. A. Ribeiro, J. Andrés, E. Longo and S. R. Lazaro, Appl. Surf. Sci., 2018, 452, 463–472 CrossRef CAS.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161–1166 CrossRef CAS PubMed.
- F. Zheng, H. Dong, Y. Ji and Y. Li, J. Power Sources, 2019, 436, 226845 CrossRef CAS.
- Y. Jing and Z. Zhou, ACS Catal., 2015, 5, 4309–4317 CrossRef CAS.
- K. R. Yoon, K. Shin, J. Park, S.-H. Cho, C. Kim, J.-W. Jung, J. Y. Cheong, H. R. Byon, H. M. Lee and I.-D. Kim, ACS Nano, 2017, 12, 128–139 CrossRef PubMed.
- Y. Dou, X.-G. Wang, D. Wang, Q. Zhang, C. Wang, G. Chen, Y. Wei and Z. Zhou, Chem. Eng. J., 2021, 409, 128145 CrossRef CAS.
- Y. Dou, R. Lian, Y. Zhang, Y. Zhao, G. Chen, Y. Wei and Z. Peng, J. Mater. Chem. A, 2018, 6, 8595–8603 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05571c |
| This journal is © The Royal Society of Chemistry 2025 |