
A local proton-transport promoter for industrial CO2 electroreduction to multicarbon products†
Haiyi
Guo
a,
Qi
Huang
a,
Di
Li
a,
Shiyu
Dai
b,
Kang
Yang
a,
Sheng
Chen
 a,
Wei
Ma
*b,
Qiang
Li
*a and
Jingjing
Duan
*a
a,
Wei
Ma
*b,
Qiang
Li
*a and
Jingjing
Duan
*a
aMIIT Key Lab Thermal Control Electronic Equipment, School of Energy and Power Engineering, School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, China. E-mail: jingjing.duan@njust.edu.cn
bKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China
First published on 16th November 2024
Abstract
The industrial electrochemical carbon dioxide reduction reaction (eCO2RR) is of wide interest; however, it is a great challenge to ensure sufficient and fast mass supply to achieve industrial-level current densities. Herein, a local proton-transport promoter was developed by hybridizing Cu catalytic sites with proton hopping sites from dual-conductive polymers to tackle the mass-diffusion limitation. The as-prepared Cu/polypyrrole composite exhibits an extraordinary eCO2RR to C2+ performance with a high FEC2+ of 80.0% under an industrial current density of 700 mA cm−2. Experimentally and theoretically, it was found that protons transfer via the Grotthuss mechanism, and proton conductivity is determined by the hydrogen bond formation and breakage (“–HN1⋯H N2H–” to “–HN1 H⋯N2H–”) at the hopping site in dual-conductive polypyrrole, rather than the diffusion coefficient of the proton source and hydrous/anhydrous protons. Significantly, the advantageous proton transport of Cu/PPy was further confirmed using in situ scanning electrochemical microscopy based on the proton change in the diffusion layer and local catalytic sites.
Introduction
To tackle current environment and energy crisis, the electrochemical carbon dioxide reduction reaction (eCO2RR) has attracted tremendous attentions since it could eliminate CO2 pollution, generate valuable carbon-based fuels/chemicals and provide a pathway to store/utilize intermittent energy if the eCO2RR is driven by renewable electricity.1 Theoretically, the eCO2RR is a multi-proton coupled electron transfer (PCET) process,2 which is complicated since it involves 2–18 or even more electrons and protons in a one-step reaction.3 In particular, to reach an industrial-level current density (usually above 150 or 200 mA cm−2) for multicarbon (C2+) products and thus promote its commercialization, the eCO2RR requires quick supply of CO2, electrons, and protons simultaneously.Gas-diffusion electrodes (GDEs) are usually used to facilitate the transport of CO2 reactants from the gas flow channel to a catalyst layer (CL) through a gas-diffusion layer (GDL) and are highly porous, hydrophobic and thus aerophilic substrates.4 An improved overpotential could facilitate electron transport but compromise the efficiency of energy conversion from electricity to chemical energy.5 Besides, protons should be properly managed since a low amount unavoidably leads to the current density-limit of the eCO2RR,6 while a large quantity would result in severe side-reactions such as the hydrogen evolution reaction (HER), which is the main competitive reaction consuming electrons that are supposed to be used in the eCO2RR. Currently, alkaline electrolytes are utilized in the eCO2RR to suppress the side HER7 by decreasing proton concentration in the bulk and local environment through a competitive adsorption between protons and other alkaline cations such as K+ and Na+.8–10 Therefore, strategies to efficiently regulate proton transport in the local environment11 are needed to realize an industrial-scale eCO2R-to-C2+ process.12,13
Electron-conductive polymers, such as polyaniline (PANI), polypyrrole (PPy), polythiophene (PTh) and their derivatives, are a class of polymeric materials that can quickly transfer electrons because their π-conjugated configuration creates a unique off-domain structure that enables electrons to transport along π-bonds in the molecular chain.14,15 Meanwhile, nitrogen, sulfur, or oxygen-containing groups in the molecular structure of such polymers could also modulate protons by interacting with anhydrous/hydrous protons (H+, H3O+, and H5O2+) or water molecules. Therefore, electrocatalysts hybridized with such polymers are expected to promote the local electron and proton transport simultaneously during eCO2RR, decreasing the ohmic and reaction kinetics loss.16 In addition, conductive polymers could also act as electrocatalyst substrates to stabilize the metal nanoparticles of the CL.17
Herein, a dual-conductive strategy has been developed by hybridizing Cu electrocatalysts with electron and proton-conductive polymers through an in situ polymerization method, to manipulate the local electron and proton transport effectively. As compared with Cu/PTh, Cu/PPy exhibits higher electron and proton conductivity, with a slightly higher activation energy for electron transport but a much lower activation energy for proton transport, which is further confirmed by theoretical molecular dynamics (MD) simulation and in situ scanning electrochemical microscopy (SECM) analysis. As a result, Cu/PPy displays an extraordinary eCO2RR performance with an optimal faradaic efficiency of C2+ products (FEC2+) of 80.0% under a large current density up to 700 mA cm−2, much higher than that of Cu/PTh in a wide working range. Importantly, the C2+ product selectivity has been interpreted by density functional theory (DFT) computation and thus a selectivity Pourbaix diagram has been proposed based on variable potentials and pH of electrolytes. This multi-scale study clarifies the determinative factors and proton transport mechanism in the widely investigated Cu-promoted-eCO2RR system, providing a possible pathway for the industrialization of the eCO2RR → C2+ process.
Results and discussion
Characterization of dual-conductive Cu–polymer composites
The Cu/conductive polymer composites have been prepared by a two-step strategy. In detail, granular PPy has been synthesized by polymerization of pyrrole monomers, followed by in situ growth of Cu nanoparticles on PPy through chemical reduction, forming the Cu/PPy hybrid. The Cu/PTh composite is prepared by the same method using thiophene monomers instead of pyrrole. From scanning and transmission electron microscopy (SEM and TEM, Fig. 1a and S1†), PPy particles show diameters ranging between 200 and 500 nanometers (nm) and a wrinkled and rough structure, which encapsulates Cu nanoparticles of around 20–50 nm. At a larger magnification of TEM, a lattice spacing of 2.099 nm has been spotted, corresponding to the Cu (111) facet (Fig. 1b), which is also evidenced by selected area electron diffraction (SAED, Fig. 1c). Furthermore, SEM and TEM EDS mapping of Cu/PPy show a two-phase distribution of the Cu and PPy phases including C and N (Fig. 1d and S1†). The Cu/PTh composite displays similar results (Fig. S2†). In addition, PPy and PTh display a broad peak at 12.3° in the X-ray diffraction (XRD) spectra, which originates from the C of polymers, while for the composites, the carbon peak is hardly observed possibly due to the strong XRD peaks of Cu nanocrystals at 43.19°, 50.30° and 73.89°, corresponding to the Cu (111), (200) and (220) planes, respectively (Fig. 1e).18 The XRD results are consistent with the aforementioned SEM and TEM mapping. Furthermore, the loading amount of Cu in the composite is varied and the resultant sample is denoted as Cux/PPy, where x is the Cu percentage (Table S1;† the Cu/PPy sample is actually Cu40/PPy). As the Cu amount increases, the intensity of the dominant (111) peak of the Cu/PPy composite increases until the maximum value at Cu40/PPy (Fig. S3†), consistent with the SEM characterization (Fig. S4†). The Cu amount of Cu/PTh is also varied (Fig. S5†).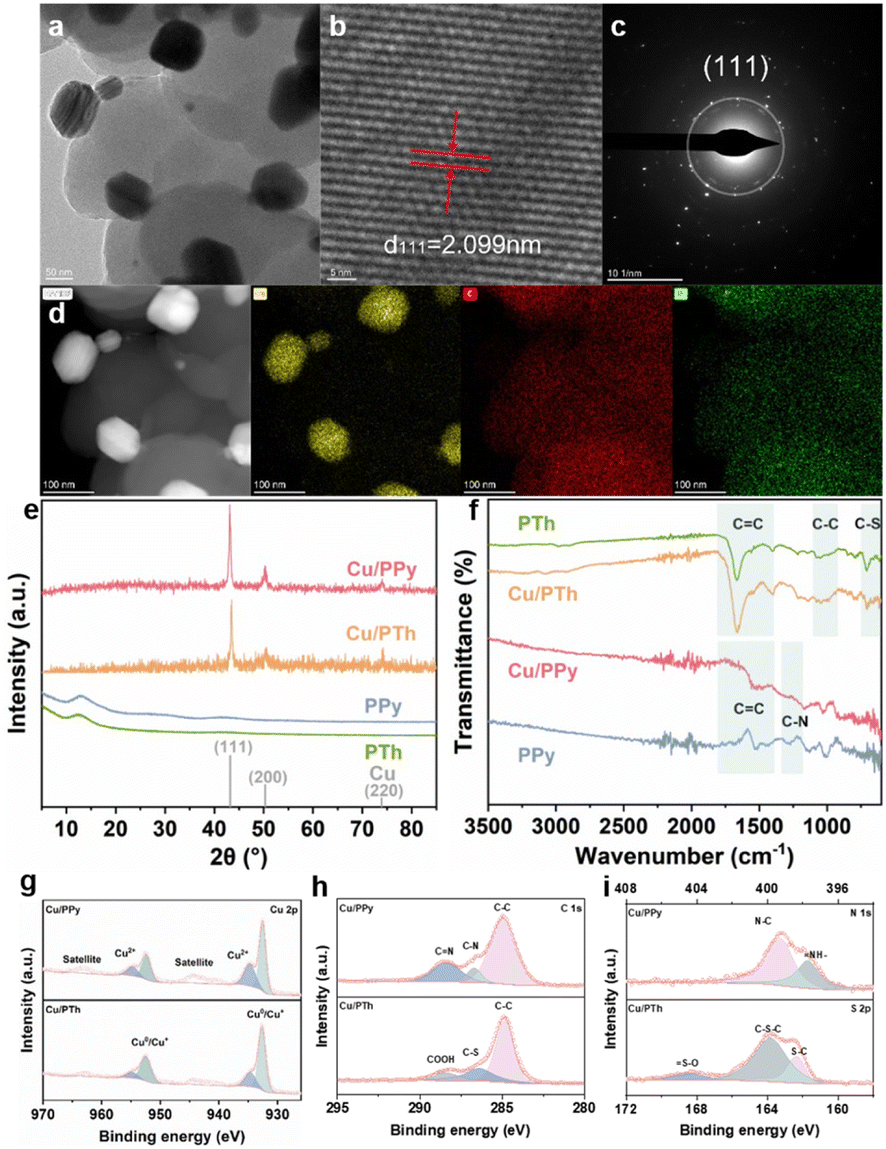 | ||
| Fig. 1 (a) TEM, (b) high-resolution TEM, (c) SAED, and (d) STEM and its EDS mapping image of the Cu, C, and N elements of Cu/PPy; (e and f) XRD and FTIR spectra of PPy, PTh and their composites with Cu; (g) Cu 2p, (h) C 1s, and (i) N 1s XPS spectra of Cu/PPy and Cu/PTh. | ||
Besides, Fourier transform infrared spectroscopy (FTIR, Fig. 1f) has also been conducted to confirm the successful hybridization of conductive polymers and Cu catalysts. For pure PPy, characteristic peaks at 1556 cm−1 and 1299 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration and C–N tensile vibration of the pyrrole ring,19,20 respectively, while the weak characteristic peak at 1044 cm−1 corresponds to plane deformation vibration of the N–H.21 These characteristic peaks are also observed in the FTIR spectrum of Cu/PPy, suggesting that the hybridization does not change the molecular structure of PPy.22,23 Raman spectra display similar results, where peaks at 1365 cm−1 and 1580 cm−1 assigned to C–C and CC vibration are both observed before and after Cu loading (Fig. S6a†).24 For the PTh and Cu/PTh samples, FTIR peaks at 1651 cm−1 and 1456 cm−1 correspond to the CC stretching vibration of the thiophene ring,14 and that at 625 cm−1 corresponds to the C–S bending vibration.19 Raman spectra show the characteristic peaks of CC planar expansion and contraction vibration at 1511 cm−1 and two modes of C–C expansion and contraction vibration at 1175 cm−1 and 1216 cm−1, confirming that the original structure of PTh is also kept after Cu hybridization (Fig. 1f and S6b†).25
C vibration and C–N tensile vibration of the pyrrole ring,19,20 respectively, while the weak characteristic peak at 1044 cm−1 corresponds to plane deformation vibration of the N–H.21 These characteristic peaks are also observed in the FTIR spectrum of Cu/PPy, suggesting that the hybridization does not change the molecular structure of PPy.22,23 Raman spectra display similar results, where peaks at 1365 cm−1 and 1580 cm−1 assigned to C–C and CC vibration are both observed before and after Cu loading (Fig. S6a†).24 For the PTh and Cu/PTh samples, FTIR peaks at 1651 cm−1 and 1456 cm−1 correspond to the CC stretching vibration of the thiophene ring,14 and that at 625 cm−1 corresponds to the C–S bending vibration.19 Raman spectra show the characteristic peaks of CC planar expansion and contraction vibration at 1511 cm−1 and two modes of C–C expansion and contraction vibration at 1175 cm−1 and 1216 cm−1, confirming that the original structure of PTh is also kept after Cu hybridization (Fig. 1f and S6b†).25
The element composition and valence state have been further evaluated using X-ray photoelectron spectroscopy (XPS) of Cu/PPy and Cu/PTh (Fig. 1g–i and S7†). According to survey scans, Cu, O, N and C elements are observed in Cu/PPy, while S is observed in Cu/PTh instead of N, suggesting successful hybridization between Cu and polymers. In detail, deconvolution of Cu 2p spectra shows Cu 2p3/2 peaks at 932.6 eV and 934.7 eV, Cu 2p1/2 peaks at 952.6 eV and 954.7 eV and their satellites, which can be ascribed to Cu0 and Cu2+, respectively (Fig. 1g).26 Since XPS peaks of Cu0 2p and Cu1+ 2p are hard to distinguish, Cu LMM spectra are also tested and deconvoluted, where both Cu/PPy and Cu/PTh display three peaks assigned to Cu elements with valence of 0, 1 and 2 (Fig. S7b†). However, XPS spectra of C, N and S show that none of these elements form chemical bonds with Cu elements.27,28 In detail, C is present as C–C, C–N, and CN for Cu/PPy at the binding energies of 284.8 eV, 286.4 eV and 287.6 eV, respectively and as C–C, C–S and COOH for Cu/PTh at the binding energies of 284.6 eV, 286 eV and 288 eV, respectively (Fig. 1h). N is present as C–N and NH– bonds at 399.7 eV and 397.8 eV (Fig. 1i), and S as S–O, C–S–C, and S–C bonds at the binding energies of 168.4 eV, 163.8 eV and 162.4 eV (Fig. 1i), originating from PPy or PTh itself, which is consistent with the PPy and PTh polymer structures in Fig. 4g. Therefore, presence of Cu1+ and Cu2+ might result from oxidation of the ambient environment,26 while only Cu metal phase could be observed in XRD because this surface oxidation layer is ultrathin and amorphous.
Electrochemical performance of Cu–polymer composites
The eCO2RR performance of the as-prepared Cu–polymer hybrids has been evaluated in a three-chamber flow-type cell using various electrochemical techniques,29 with 1 M KOH as the electrolyte, saturated Ag/AgCl/KCl as the reference electrode and nickel foam as the counter electrode. The loss of total FE is due to the inevitable incomplete collection of gas and liquid products over 800 mA cm−2. Firstly, linear scan voltammetry (LSV) shows that Cu/PPy exhibits a slightly higher transient-state current density than Cu/PTh (Fig. S8†). To further compare the performance, polarization curves are plotted using steady-state current densities obtained using chronoamperometry (v–t, Fig. 2a, b and S9†). It is found that Cu/PPy shows a similar trend to Cu/PTh, while the product distribution is quite different (Fig. 2c–f). The FEC2+ of the Cu/PPy catalyst is much higher than that of Cu/PTh and pure Cu particles without polymers in a wide working range (Fig. 2e and S16b†), and faradaic efficiency of hydrogen (FEH2, Fig. S10†) is much lower. In detail, Cu/PPy displays a broad working window for C2+ chemical production from 300 to 900 mA cm−2 with a FEC2+ above 60.0%, and an optimal value of 80.0% is achieved at 700 mA cm−2. | ||
| Fig. 2 (a) Chronoamperometry testing in a current-density range from 50 to 1000 mA cm−2; (b) polarization curves plotted using steady-state current densities and corresponding potentials; (c and d) Faraday efficiency of different eCO2RR products; (e) Faraday efficiency of C2+; (f) partial current density for C2H5OH, C2H4 and C2+ products; (g) electrochemical impedance spectroscopy of Cu/PPy and Cu/PTh; (h) durability measurements for Cu/PPy in 1 M KOH at 200 mA cm−2; (i) comparison of the eCO2RR activity (details are given in Table S3†). | ||
In addition, faradaic efficiencies of monocarbon products (FEC1, for example, CO and CH4, Fig. S11†) of Cu/PPy and Cu/PTh samples are similar, while those of ethanol and ethylene products (FEethanol and FEC2H4) vary a lot, where Cu/PPy displays much higher EtOH and C2H4 selectivity (Fig. S12 and S13†). This suggests that the local environment modulated by polymers around Cu active centers affects the performance of eCO2RR → C2+ products and the side-reaction HER significantly.30 It is demonstrated that the difference in eCO2RR performance is due to the effect of the surface microenvironment brought by different polymers, rather than the size and structure of Cu catalysts. In addition, the Cu ratio of both Cu/PPy and Cu/PTh has been varied in eCO2RR (Fig. S14 and S15†). With increasing the theoretical Cu ratio from 20 to 30, 40, 50 and 60 wt% in the Cu/PPy composite (Table S1†), the FEC2H4 firstly increases and then decreases, achieving the highest value of 40.9% at 40 wt% Cu. The Cu/PTh composite shows the same trend with the highest FEC2H4 value of 38.2% at 40 wt% Cu loading. To quantify the actual Cu loading of Cu/PPy and Cu/PTh accurately, an inductively coupled plasma optical emission spectrometer (ICP-OES) is used. As a result, the Cu percentage of Cu40/PPy is 48.76 wt%, close to that of Cu/PTh (51.47 wt%, Table S2†). This suggests that the eCO2RR difference originates from the different proton-promoting effect of PPy and PTh in composites, not from the different Cu loading.
In this case, why could the Cu ratio still affect the eCO2RR performance? This is related to the balance of the number of protons and catalytic sites that consume protons. It is known that pure polymers show negligible eCO2RR activity;31,32 therefore, the Cu20/PPy and Cu30/PPy samples show high FEH2 possibly due to the good proton transport ability of PPy and insufficient active sites for eCO2RR (Fig. S14b†). Since Cu is the catalytic center, the FEC2+ of the Cu60 and Cu80 composites is lower than that of the Cu40 one, indicating that it requires an optimal ratio between active centers and local proton conductors to achieve the best eCO2RR → C2+ performance, especially at the industrial level. By comparing the FEC2H4, Cu40/PPy shows the maximum value of 40.9% at a current density of 600 mA cm−2 (Fig. S14d†). According to the polarization curve (Fig. 2f), the partial current densities of C2H4, C2H5OH and all C2+ products reach the diffusion-control area, which suggests that the eCO2RR activity is limited by mass diffusion of CO2 reactants, protons, etc., rendering the local mass-transport-promoter highly significant in the large current-density range.
Electrochemical impedance spectra (EIS) of as-prepared catalyst constructed GDEs have also been recorded (Fig. 2g), where Cu/PPy shows a similar system resistance (Rs) to but smaller charge-transfer resistance (Rct) than Cu/PTh, indicating that the charge transport is greatly promoted during eCO2RR with the modification of conductive polymer PPy, as compared with PTh even with the same reaction sites. The durability of the eCO2RR promoted by Cu/PPy is also tested, which remains over 50% FEC2+ for stable 50 hours in alkaline media at an industrial-level current density of 200 mA cm−2 (Fig. 2h). Further comparing the above eCO2RR → C2+ performance with reported values, the Cu/PPy stands among the best (Fig. 2i and Table S3†). Additionally, the electrochemical surface area (ECSA) of the as-prepared electrodes has been measured, where the double-layer capacitance of Cu/PPy (0.27 mF cm−2) is about five times that of Cu/PTh (0.05 mF cm−2, Fig. S17 and S18†), beneficial for the good dispersion of Cu catalysts.
Addition of polymer components to the CL of GDEs inevitably affects the local microenvironment of the reactive interface. Firstly, a seating droplet method is used to measure the wetting ability of Cu/PPy and Cu/PTh constructed GDEs (Fig. S19†). The Cu/PPy GDE shows a contact angle of 162.8°, which decreases slightly to 162.3° after prolonged wetting, and that of the Cu/PTh sample displays a similar change from 161.4° to 160.3°. The little difference in hydrophobicity suggests that structural modulation of the triple-phase interface is not responsible for the large difference in the eCO2RR activity between PPy and PTh composites. Besides, the stable hydrophobicity of the Cu/Polymer GDE preventing water access macroscopically and the local proton-transport promoter enriching protons microscopically, together with the preferred CO2 adsorption over hydrogen, result in promoted eCO2RR while suppressing the HER simultaneously.
The eCO2RR mechanism using Cu/polymer electrocatalysts
As discussed previously, there are no significant interactions between Cu particles and polymers (no chemical bonding, only van der Waals forces) in the composites; therefore, the pure-Cu structure is used for DFT calculations. The thermodynamics of eCO2RR catalyzed by Cu/PPy in electrolytes is also investigated by DFT calculations. As shown in Fig. 3a, eCO2RR is endothermic in a pH 0 medium, with energy-demanding steps such as *CO2 protonation to *COOH, dimerization of two *CO, and hydrogenation to *COCOH with free energy changes (ΔG) of 2.61 eV, 1.77 eV, and 1.51 eV, respectively. After this, continuous hydrogenation steps from *COCOH to the final products C2H4 and CH3CH2OH are exothermic. Importantly, the ΔG of *CH2CHO to *C2H4 is −1.29 eV, more negative than the −0.54 eV value for *CH3CHO and CH3CH2OH, suggesting that the eCO2RR promoted by Cu/PPy more likely proceeds via the C2H4 pathway. Among all steps, the potential-determining step (PDS) is the first *CO2 protonation with the highest uphill energy barrier of 2.61 eV. The eCO2RR pathway in a pH 14 medium was calculated using a pH-correction method using eqn (1) to compare with the current performance in actual experiments.| G = E + ZTE − TS + GpH | (1) |
GpH = kBT × pH × ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 10 | (2) |
 | ||
| Fig. 3 (a and b) Free energy diagrams of the eCO2RR promoted by Cu/PPy at pH 0 and 14 electrolytes; (c and d) Pourbaix diagrams for the C2H4 and C2H5OH products; ΔG variation for (e) C2H4 and (f) C2H5OH under different potentials and pH values. | ||
As shown in Fig. 3b, for the eCO2RR in a pH 14 electrolyte, the three steps (*CO2 protonation to *COOH, dimerization of two *CO, and hydrogenation to *COCOH) are also endothermic, with ΔG of 1.78, 0.94 and 0.69 eV, respectively. The energy barrier of its PDS is much lower than that of eCO2RR at pH 0 (1.78 eV vs. 2.61 eV), while the C2H4 and CH3CH2OH selectivity remains the same with similar exothermic energy. The energy barrier for H3O+via the hopping mechanism (Fig. S22†) at the N site of PPy (+0.31 eV) is lower than that of the S site of PTh (+0.53 eV), suggesting that the proton transfer on Cu/PPy via the Grotthuss mechanism is more facile than on Cu/PTh.
Furthermore, the eCO2RR selectivity to different C2+ products has been explored by using a Pourbaix diagram, which shows the ΔG values of the PDS changing with pH (Fig. 3c). The ΔG of the C2H4 pathway is consistently lower than that of the CH3CH2OH pathway, indicating the higher C2H4 selectivity of eCO2RR promoted by Cu/PPy at all pH values. A potential correction is also applied (Fig. 3d), where the ΔG of the PDS for the eCO2RR to C2H4 and CH3CH2OH both decrease as the potential decreases, with that of C2H4 lower than that of the CH3CH2OH pathway (0.75 eV), still suggesting favorable C2H4 selectivity. In addition, heat maps are also plotted to compare the ΔG of PDS of eCO2RR to different C2+ products, C2H4 (Fig. 3e) and CH3CH2OH (Fig. 3f). Specifically, the eCO2RR → C2+ pathway is preferred in media with higher pH values under more negative potentials,33 which is consistent with the experimental results that the Cu/PPy composite is advantageous for C2+ chemical production, especially C2H4.
The transport mechanism of protons and electrons
A homemade four-electrode device has been constructed to measure the proton and electron conductivity of Cu/PPy and Cu/PTh samples (Fig. 4a–d and S23, S24†), by decoupling the proton and electron transport behavior through EIS tests at different temperatures. Specifically, using the above homemade device, intrinsic proton and electron conductivity can be obtained by fitting the high and low-frequency impedance of EIS plots, respectively. As expected, the electronic conductivity increases with temperature, with the electronic conductivity of the Cu/PPy sample higher than that of Cu/PTh (Fig. 4a).34 Further, the electron transport mechanism has been investigated by fitting the Arrhenius eqn (3).| σ = σ0exp(−Ea/(kBT)) | (3) |
 | ||
| Fig. 4 (a) Electronic conductivity and (b) Arrhenius plot; (c) proton conductivity and (d) Arrhenius plot; (e) RDF for N from PPy and O from H2O or S from PTh and O from H2O molecules; (f) RDF for N and H from H2O or S and H from H2O; (g) details of the simulation box; (h and i) hydrogen bond statistics. | ||
Therefore, the calculated activation energy of electron transport for the PPy composite is 0.27 eV, slightly higher than the 0.26 eV of the PTh composite (Fig. 4b). These low activation energies (less than 0.4 eV) suggest that the electrons in Cu/polymer composites transport via a hopping mechanism.35,36 The temperature-dependence is also observed in the proton conductivity of the prepared hybrids (Fig. 4c), and the proton conductivity of Cu/PPy increases from 9 mS cm−1 at 40 °C to 24 mS cm−1 at 80 °C, higher than that of the PTh sample. Similarly, linearly fitting the Arrhenius eqn (4), the activation energy of proton transport for the PPy and PTh composites is obtained to be 0.26 eV and 0.34 eV, respectively.
| σT = Aexp(−Ea/(kBT)) | (4) |
As shown, the values are also less than 0.4 eV, indicating that intra-electrode protons mainly transfer by jumping through the Grotthuss mechanism.37 As compared to Cu/PTh, the higher proton conductivity and lower activation energy of proton transport for Cu/PPy are possibly due to the higher electronegativity of the “N” from PPy than the “S” from PTh, which could act as the proton hopping site via “HN1⋯H2O N2H” hydrogen bonding formation and “HN1 OH2⋯N2H” breakage. This gives Cu/PPy a unique proton and electron dual-conductive property, greatly promoting the eCO2RR → C2+ pathway, especially under industrial-level current densities.
To further verify the advantageous proton transport of the PPy composite, theoretical MD simulation is also conducted (Fig. 4e–i). In the simulation, the chemical structures of PPy, PTh, water molecules, protons and hydrated protons (H3O+, H5O+, etc.) as well as their combined models, including 3 polymer chains with 500 water molecules and 100 hydrated protons (Fig. 4g). Firstly, mean-square displacement (MSD) is used to evaluate the transport kinetics of water and hydrated hydrogen ions during eCO2RR. As shown in Fig. S25 and S26,† the diffusion coefficient of water molecules in PPy and PTh composites is obtained by calculating the MSD of water, which is 0.5947 Å2 ps−1 and 0.6037 Å2 ps−1, respectively. Similarly, the diffusion coefficient of hydrated protons is 0.353 Å2 ps−1 and 0.3607 Å2 ps−1 for Cu/PPy and Cu/PTh, respectively. As compared to Cu/PTh, despite the lower diffusion efficiency of water and protons in the Cu/PPy system, more facile proton transport is achieved because its main transport mechanism is the Grotthuss mechanism, namely, hopping mechanism rather than the vehicle one, according to the low activation energy (0.26 eV and 0.34 eV, less than 0.4 eV). To analyze the reason of facile proton transport, radial distribution function (RDF) of related groups and molecules is also investigated, as shown in Fig. 4e–f. The RDF g(A–B)(r) refers to the probability of finding atom B at a distance r from atom A in the MD trajectory. The number of oxygen atoms in the water molecules near the –NH– group of PPy is larger than that near the S of PTh (Fig. 4e). In eCO2RR, water is the main source of protons under high current densities, and this local hydrophilic property of the –NH– group is more favorable to form hydrogen bonds with water molecules in the microenvironment to transfer protons. This speculation is further supported by Fig. 4f, where hydrogen atoms in water molecules are also more distributed in the vicinity of –NH– groups. Based on hydrogen bonding statistics on the MD trajectory, the average number of hydrogen bonds counted for the PPy simulation box is 737, higher than the 696 for the PTh one. Therefore, it can be concluded that PPy modulates the local proton transport in the electrochemical microenvironment, through the –NH– hopping site via a Grotthuss mechanism.13
In situ proton characterization using SECM
In addition to the above ex situ proton transport study of Cu/polymer composites, SECM is performed, which is a technique that qualitatively and quantitatively offers precise local information of an electrochemical reaction on the electrode surface during operation.38–41 To confirm the enhanced proton transport on Cu/PPy, an in situ SECM analysis has been performed during eCO2RR promoted by Cu/polymer catalysts in a CO2-saturated 1 M KOH solution. In this work, the surface-generation tip-collection mode of SECM is firstly employed to identify eCO2RR products and monitor local pH changes (Fig. 5a). Here, a Pt ultramicroelectrode (Pt-UME) with a 13.3 μm radius (Fig. S27†) is put in the diffusion layer above the substrate electrode. A negative potential of −1.6 V vs. RHE is applied to the sample for 50 seconds, followed by recording a cyclic voltammogram (CV) of the Pt-UME. And the sensitivity of platinum voltammetry to trace amounts of chemisorbed CO and its effectiveness as a catalyst for hydrogen oxidation reaction (HOR), facilitate the detection of both CO and H2 using the Pt-UME.42 The results presented in Fig. 5b illustrate the CV of the Pt-UME after applying −1.6 V to the substrate electrode, with oxidation peaks denoting the characteristic response of CO (anodic sweep, 0.9 V to 1.3 V) and H2 (cathodic sweep, 1.1 V to 0.3 V), respectively. Significantly, the Pt-UME showed a more pronounced oxidation peak of H2 for Cu/PTh compared to Cu/PPy, indicating a higher HER side reaction for Cu/PTh (Fig. 5c). When the eCO2RR does not occur, no corresponding oxidation peak is detected at the platinum electrode (Fig. S28†), which further suggests that the oxidation peaks in Fig. 5b and c are from the oxidation of eCO2RR products. This observation is consistent with the previous eCO2RR performance of these catalysts on GDEs, as depicted in Fig. 2c and d. | ||
| Fig. 5 (a) Schematic of the generation-collection mode of SECM and potentials applied during testing; (b and c) CVs of the Pt-UME recorded during (b) Cu/PPy and (c) Cu/PTh promoted eCO2RR (tip-substrate distance d = 20 μm, scan rate = 0.05 V s−1); (d) schematic of local pH detection at different tip-substrate distances and applied potentials during the eCO2RR process; HER LSV current ratio of the Pt-UME positioned at different tip-substrate distances (2, 20, 50 and 100 μm) with (e) no potentials and (f) −1.6 V vs. RHE applied to the Cu/PPy and Cu/PTh electrodes, with a scan rate of 0.05 V s−1. The electrolyte is CO2-saturated 1 M KOH. | ||
Subsequently, localized fluctuation of H+ concentration during eCO2RR within the catalyst's diffusion zone at varying tip-substrate distances is investigated using SECM by assessing the HER response of the Pt-UME tip electrode, as shown in Fig. 5d. The onset potential (Eonset) of the HER is correlated with the solution pH (Fig. S29†);43,44 therefore, that of the Pt-UME is selected to indicate the concentration changes of local protons. In the absence of potential applied to the catalyst electrode, the LSV of the Pt-UME largely overlaps, indicating a consistent microenvironment pH without eCO2RR occurring on Cu/PPy and Cu/PTh (Fig. 5e). In contrast, upon initiation of eCO2RR by applying −1.6 V to the substrate electrode, distinct LSV behaviors of the Pt UME are observed for Cu/PPy and Cu/PTh samples. The shift in Eonset of the Pt-UME HER curve indicates a change in the microenvironment pH on the catalyst surface (Fig. 5f). A noticeable pH decrease is observed with a decreasing tip-substrate distance in both Cu/PPy and Cu/PTh systems, suggesting a higher concentration of H+ in closer proximity to the catalyst electrode, owing to the enrichment of H+ around both Cu/PPy and Cu/PTh catalysts. Compared to Cu/PTh, the shift of Eonset to a negative potential in the Cu/PPy system indicates a higher pH above the sample. A plausible explanation is that the high proton-transport efficiency of Cu/PPy allows H+ to be transferred from the electrolyte bulk to the diffusion layer and then hopping sites around the active sites to facilitate eCO2RR, leading to a H+ concentration decrease in the area above the electrode surface. Conversely, the slightly poor proton transport efficiency of Cu/PTh makes it harder for protons to take part in eCO2RR, resulting in enriched H+ above the electrode surface and thus a pH decrease. In conclusion, the in situ SECM examination of the local pH highlights the significant impact of the superior proton transport promotion by Cu/PPy according to the local reaction microenvironment change, elucidating the outstanding eCO2RR performance of Cu/PPy.
Conclusions
In summary, a local proton-transport-promotion strategy was developed by hybridizing Cu catalyst sites with proton and electron dual-conductive polymers, to overcome the mass-diffusion limitation for industrial-level eCO2RR. As compared with Cu/PTh, Cu/PPy exhibited an outstanding eCO2RR performance with a high FEC2+ of 80.0% under a large current density 700 mA cm−2 and a broad working window for C2+ chemical production, which could be ascribed to the efficient proton supply locally via the N hopping site from PPy. The proton transport was found to occur via the Grotthuss mechanism with a small activation energy (0.26 eV), which means that the mass diffusion is no longer the limiting factor because anhydrous and hydrous protons transfer via hydrogen bond formation and breakage during eCO2RR, such as “HN1⋯H2O N2H to HN1 OH2⋯N2H”. Significantly, the ex situ experimental and theoretical results were further confirmed using in situ SECM testing, where the proton concentration in the diffusion layer of the Cu/PPy electrode was much lower than that of Cu/PTh due to its fast proton transport and consumption at the local active sites. Interestingly, the PDS was identified (the first protonation step of adsorbed *CO2 to *COOH), and thus, a selectivity Pourbaix diagram was proposed using the ΔG of the PDS as a measure, which might also be applicable to other related electrocatalytic reactions. The multi-scale study of proton transport behavior in the eCO2RR system using DFT and MD, together with ex situ four-probe testing and in situ SECM, could not only address the pH effect on the reaction thermodynamics and kinetics, but also precisely clarify the proton transport mechanism of industrial-level eCO2RR, greatly promoting its commercial success.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Haiyi Guo and Qi Huang contribute equally to this manuscript. Haiyi Guo is responsible for manuscript writing, electrochemical testing, physical characterization, and molecular simulations. Qi Huang, Di Li and Sheng Chen are responsible for DFT simulation. Shiyu Dai and Wei Ma are responsible for SECM in situ detection of local protons transport. Kang Yang is responsible for the physical characterization. Qiang Li and Jingjing Duan are responsible for conceptualization, manuscript writing, and theoretical analysis of this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (NSFC 52376193, 52488201, and 92163124), International Young Talent Program of China (QN2022182001L), National Key R&D Program of China (2021YFF0500700), Jiangsu Natural Science Foundation (No. BK20190460), Fundamental Research Funds for the Central Universities (30921013103), and Jiangsu Innovative/Entrepreneurial Talent Program.References
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- S. R. Hui and P. D. Luna, Matter, 2021, 4, 1555–1557 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Curr. Opin. Electrochem., 2017, 1, 104–109 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray and F. G. L. Parlane, Nat. Rev. Mater., 2021, 7, 55–64 CrossRef.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- A. J. Welch, A. Q. Fenwick, A. Böhme and H.-Y. Chen, J. Phys. Chem. C, 2021, 125, 20896–20904 CrossRef CAS.
- M. K. Aslam, H. Wang, S. Chen, Q. Li and J. Duan, Mater. Today Energy, 2023, 31, 101196 CrossRef CAS.
- Q. Chen, X. Wang, Y. Zhou, Y. Tan, H. Li, J. Fu and M. Liu, Adv. Mater., 2024, 36, 2303902 CrossRef CAS PubMed.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- K. W. Kimura, R. Casebolt, J. Cimada DaSilva, E. Kauffman, J. Kim, T. A. Dunbar, C. J. Pollock, J. Suntivich and T. Hanrath, ACS Catal., 2020, 10, 8632–8639 CrossRef CAS.
- K. Yang, M. Li, T. Gao, G. Xu, D. Li, Y. Zheng, Q. Li and J. Duan, Nat. Commun., 2024, 15, 7060 CrossRef CAS.
- D. Çirmi, R. S. Karatekin, R. Aydın and F. Köleli, Synth. Met., 2022, 289, 117102 CrossRef.
- J. Cheng, L. Chen, X. Xie, K. Feng, H. Sun, Y. Qin, W. Hua, Z. Zheng, Y. He, W. Pan, W. Yang, F. Lyu, J. Zhong, Z. Deng, Y. Jiao and Y. Peng, Angew. Chem., Int. Ed., 2023, 62, e202312113 CrossRef CAS.
- F. Li, S.-F. Zhao, L. Chen, A. Khan, D. R. MacFarlane and J. Zhang, Energy Environ. Sci., 2016, 9, 216–223 RSC.
- Y. Liang, J. Zhao, Y. Yang, S. F. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang, D. Appadoo, L. Zhang, Z. Geng, F. Li and J. Zeng, Nat. Commun., 2023, 14, 474 CrossRef CAS.
- L. Shang, X. Lv and H. Shen, J. Colloid Interface Sci., 2019, 552, 426–431 CrossRef CAS PubMed.
- R. Yuksel, E. Alpugan and H. E. Unalana, Org. Electron., 2018, 52, 272–280 CrossRef CAS.
- S. T. Navale, G. D. Khuspe and M. A. Chougule, J. Phys. Chem. Solids, 2014, 75, 236–243 CrossRef CAS.
- Y. Ji, C. Yang and L. p. Qian, J. Colloid Interface Sci., 2021, 600, 847–853 CrossRef CAS PubMed.
- Z. Li, K. Hu and M. Yang, Nano Energy, 2019, 58, 852–861 CrossRef CAS.
- N. Wang, H. Dai and D. Wang, Mater. Sci. Eng., C, 2017, 76, 139–143 CrossRef CAS PubMed.
- H. Kashani, L. Chen and Y. Ito, Nano Energy, 2016, 19, 391–400 CrossRef CAS.
- P. Pascariu, D. Vernardou and M. P. Suchea, Mater. Des., 2019, 182, 108027 CrossRef CAS.
- J. Lv, M. Jouny, W. Luc and W. Zhu, Adv. Mater., 2018, 30, 1803111 CrossRef.
- I. M. Minisy, N. Gavrilov, U. Acharya and Z. Morávková, J. Colloid Interface Sci., 2019, 551, 184–194 CrossRef CAS PubMed.
- T. Yuan, J. Ruan, W. Zhang and Z. Tan, ACS Appl. Mater. Interfaces, 2016, 8, 35114–35122 CrossRef CAS PubMed.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS.
- S. Jia, Q. Zhu, M. Chu, S. Han, R. Feng, J. Zhai, W. Xia, M. He, H. Wu and B. Han, Angew. Chem., 2021, 60, 10977–10982 CrossRef CAS PubMed.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- M. Ma, L. Xiong, Y. Dong, Q. Bai, W. Hua, Z. Zheng, F. Lyu, Y. Lian, Z. Wei, H. Yuan, Z. Jiao, J. Cheng, D. Song, M. Wang, Z. Xing, J. Zhong, S. Han, Z. Deng and Y. Peng, Adv. Funct. Mater., 2024, 2315667 CrossRef CAS.
- K. Liu, Z. Xu, J. Mei, J. Han and F. Zheng, J. Mater. Chem. C, 2023, 11, 4966–4992 RSC.
- X. Wu, J. J. Hong, W. Shin, L. Ma and T. Liu, Nat. Energy, 2019, 4, 123–130 CrossRef CAS.
- H. Sun, H. Wang, Y. M. Fu and X. Meng, Dalton Trans., 2022, 51, 4798–4805 RSC.
- Z. Lu, C. Yang, L. He, J. Hong, C. Huang, T. Wu, X. Wang and Z. Wu, J. Am. Chem. Soc., 2022, 144, 9624–9633 CrossRef CAS PubMed.
- W. Ma, K. Hu, Q. Chen, M. Zhou, M. V. Mirkin and A. J. Bard, Nano Lett., 2017, 17, 4354–4358 CrossRef CAS PubMed.
- J. Hui, S. Pakhira, R. Bhargava, Z. J. Barton, X. Zhou, A. J. Chinderle, J. L. Mendoza-Cortes and J. Rodríguez-López, ACS Nano, 2018, 12, 2980–2990 CrossRef CAS PubMed.
- T. Kai, M. Zhou, Z. Duan, G. A. Henkelman and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 18552–18557 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- G. García and M. T. M. Koper, ChemPhysChem, 2011, 12, 2064–2072 CrossRef PubMed.
- S. H. Salleh, S. Thomas, J. A. Yuwono, K. Venkatesan and N. Birbilis, Electrochim. Acta, 2015, 161, 144–152 CrossRef CAS.
- W. Hua, T. Liu, Z. Zheng, H. Yuan, L. Xiao, K. Feng, J. Hui, Z. Deng, M. Ma, J. Cheng, D. Song, F. Lyu, J. Zhong and Y. Peng, Angew. Chem., Int. Ed., 2024, 63, e202315922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04672b |
| This journal is © The Royal Society of Chemistry 2025 |