
Enhancing photoactivity of defective g-C3N4via self-polarization effect of tourmaline for CO2 reduction†
Jiangpeng
Wang
,
Chao
Huang
,
Deng
Liu
,
HuiHui
Peng
,
Qiong
Luo
,
Dimin
Yang
,
Xuelian
Yu
 * and
Yingmo
Hu
*
* and
Yingmo
Hu
*
Engineering Research Center of Ministry of Education for Geological Carbon Storage and Low Carbon Utilization of Resources, Beijing Key Laboratory of Materials Utilization of Nonmetallic Minerals and Solid Wastes, National Laboratory of Mineral Materials, School of Materials Science and Technology, China University of Geosciences, Beijing, 100083, China. E-mail: xlyu@cugb.edu.cn; huyingmo@cugb.edu.cn
First published on 18th November 2024
Abstract
Graphite carbon nitride (g-C3N4) has been extensively studied as a non-metallic catalyst for photocatalytic reduction of CO2. However, its efficiency and selectivity in CO2 reduction still require further enhancement. In this study, we have incorporated the silicate mineral tourmaline, known for its spontaneous polarization properties, into g-C3N4 with nitrogen defects. The novel composite catalyst, named TM/CN(NH), was synthesized by a two-step method of high-temperature calcination. The optimal composite ratio of the sample (25TM/CN(NH)) can achieve a CO yield rate of 118.17 μmol g−1 h−1, which is 6.4 times that of the bulk g-C3N4(CN) and 2.9 times that of g-C3N4 containing N defects (CN(NH)). Our findings indicate that the self-polarization effect of tourmaline and the introduction of nitrogen vacancies can remarkably upgrade the photocatalytic efficiency of g-C3N4. On one hand, the nitrogen vacancies can broaden the light absorption range of g-C3N4, optimize its band gap structure, and improve its efficiency in utilizing light energy. On the other hand, the electric field generated by the self-polarization effect of tourmaline can enhance the migration of electrons in the lattice of g-C3N4, promote the migration and separation of electrons and holes, and thus increase the reduction efficiency of CO2 by g-C3N4. This research innovatively integrates cost-effective mineral materials into g-C3N4, significantly elevating the photocatalytic capabilities of g-C3N4. Furthermore, it paves the way for the rational design of abundant and inexpensive catalysts, aiming to achieve efficient photocatalytic carbon dioxide reduction.
Introduction
Photocatalytic CO2 reduction technology is considered a visionary approach to solving the global environmental and energy crises, because of its capability to convert CO2 into valuable chemicals and fuels.1–4 However, the actual utilization of this technology is still restricted owing to the poor efficiency of the developed photocatalysts in dissociating chemically stabilized CO2 molecules with high C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding energy.5–7 Various metal oxides,8 sulfides,9 layered double hydroxides,10 and metal nitrides,11etc. have been explored as photocatalytic materials for CO2 photoconversion. Among them, g-C3N4 has evolved into a focus of research due to its unique electronic structure, excellent chemical stability, and activity in reducing CO2 under visible light.12–14 Nevertheless, due to its poor specific surface area and high photogenerated carrier recombination rate, the efficiency of conventional bulk g-C3N4 synthesized by pyrolysis of nitrogen-containing precursors is moderate.15,16 Various methods such as functional group modification,17 elemental doping18 and defect engineering have been developed to enhance the photocatalytic activity of g-C3N4.19 Among them, introducing vacancy defects can remarkably optimize the band gap of g-C3N4 to achieve visible light absorption, thereby enhancing the performance of g-C3N4 for CO2 photoreduction by diminishing the photo-generated carrier recombination.20,21 Wang et al.22 established that nitrogen vacancies stabilize the intermediate *COOH during the photocatalytic reduction of CO2 to CO, facilitating the conversion of *COOH to *CO and ultimately boosting CO production. Tu et al.23 discovered, through experiments and theoretical calculations, that increased nitrogen vacancy density creates an intermediate band gap beneath g-C3N4 conduction band. This narrowing of the band gap enhances visible light absorption and diminishes photo-generated carrier recombination. Nevertheless, the potential of g-C3N4 in CO2 photoreduction is still to be developed.
O bonding energy.5–7 Various metal oxides,8 sulfides,9 layered double hydroxides,10 and metal nitrides,11etc. have been explored as photocatalytic materials for CO2 photoconversion. Among them, g-C3N4 has evolved into a focus of research due to its unique electronic structure, excellent chemical stability, and activity in reducing CO2 under visible light.12–14 Nevertheless, due to its poor specific surface area and high photogenerated carrier recombination rate, the efficiency of conventional bulk g-C3N4 synthesized by pyrolysis of nitrogen-containing precursors is moderate.15,16 Various methods such as functional group modification,17 elemental doping18 and defect engineering have been developed to enhance the photocatalytic activity of g-C3N4.19 Among them, introducing vacancy defects can remarkably optimize the band gap of g-C3N4 to achieve visible light absorption, thereby enhancing the performance of g-C3N4 for CO2 photoreduction by diminishing the photo-generated carrier recombination.20,21 Wang et al.22 established that nitrogen vacancies stabilize the intermediate *COOH during the photocatalytic reduction of CO2 to CO, facilitating the conversion of *COOH to *CO and ultimately boosting CO production. Tu et al.23 discovered, through experiments and theoretical calculations, that increased nitrogen vacancy density creates an intermediate band gap beneath g-C3N4 conduction band. This narrowing of the band gap enhances visible light absorption and diminishes photo-generated carrier recombination. Nevertheless, the potential of g-C3N4 in CO2 photoreduction is still to be developed.
The most vital issue in photocatalysis is the spatially effective separation of photo-generated carriers for catalyzing redox reactions. Introducing an electric field into the catalytic reaction system has become an effective tactic to foster the migration and separation of photogenerated electron–hole pairs. The electric field can be used as a driving force to push these charges to move in different directions, ultimately improving the photocatalytic performance.24,25 At present, charge separation strategies for external field enhancement are mainly assisted by applied electric fields, piezoelectric or ferroelectric materials.26–28 Tourmaline is a ring silicate mineral composed of boron and various other elements such as Na, Mg, Fe, Li, and Al. Depending on the diversity in composition, tourmaline exhibits unique properties for a wide range of applications.29,30 The most critical characteristic of tourmaline is the non-overlapping centers of positive and negative charges within its crystal cell, creating a perpetual spontaneous polarization electric field along the c-axis.31–33 At present, numerous studies have been conducted on the composite of tourmaline with typical photocatalysts (TiO2, ZnO, WO3, BiOBr, etc.), which are mainly applied in the photocatalytic degradation of pollutants, and there is a need to further broaden the scope of its application in photocatalysis. The polarized electric field of tourmaline can provide a sufficiently large driving force for the migration and separation of photogenerated electron–hole pairs during photocatalysis. Therefore, coupling tourmaline with g-C3N4 can provide a strong driving force for carrier separation during the photocatalytic reaction and accelerate the separation efficiency of photogenerated charge carriers, which leads to increased photocatalytic CO2 reduction performance.
Herein, the natural mineral tourmaline was used to build a heterostructure with g-C3N4 to investigate the contribution of self-polarization to the photoactivity of a semiconductor material. We found that nitrogen vacancy defects effectively increase the light absorption range, and the spontaneous polarization electric field of tourmaline further accelerates the separation and migration of these photogenerated carriers, thus enhancing the utilization efficiency of photogenerated electrons. The 25TM/CN(NH) heterostructure exhibited a significantly enhanced capability for CO2 photoreduction, achieving a high CO yield rate of 118.17 μmol g−1 h−1, which was 6.4 times higher than that of pristine CN. The essential roles of the self-polarization field and nitrogen vacancies in performance improvement were fundamentally investigated. Our work can provide a new avenue toward developing high-efficiency and low-cost catalysts for the photocatalytic reduction of CO2.
Experimental
Synthesis of g-C3N4
The bulk g-C3N4 was synthesized from melamine using a conventional thermal polymerization approach by reacting at 550 °C for 4 h. Upon completion of the calcination process, collect the sample, grind it into a powder, labeling it as CN.Synthesis of TM/g-C3N4 sample
0.5 g of CN was added to 50 mL of deionized water, followed by a certain amount of tourmaline (TM) powder. The mixture was sonicated for one hour to ensure that the g-C3N4 and tourmaline were completely mixed. The solution was then stirred to dryness in a water bath at 80 °C and the sample was collected to obtain the xTM/CN sample. The dried sample was calcined in an Ar/H2 environment at 550 °C for 4 hours to obtain the final composite sample. It was designated as xTM/CN(NH), where “x” denotes the mass percentage of tourmaline in the TM/g-C3N4(NH) composite. The steps for the CN(NH) samples were identical to the preparation of TM/CN(NH) except that no tourmaline was added.Characterization of catalyst
The crystal structures of the photocatalysts were analyzed using an X-ray diffractometer (XRD) (D8 Advance X-ray Diffractometer). Both Scanning Electron Microscopy (SEM, model JSM-7600F from JEOL) and Transmission electron microscopy (TEM, model JEOL JEM-2100) was used to characterize the microscopic surface morphology of the catalyst. The molecular structure of the material was characterized using the Fourier transform infrared spectroscopy (FTIR) (Bruker Tenson27 spectrometer). To determine the photocatalyst photoresponse range, absorption wavelength, and bandgap, we employed a UV-visible diffuse reflectance spectrometer (Hitachi U4150 from Japan) with a scanning range of 200–800 nm. The photoluminescence spectrometer (PL F-7000) was used to test the photoluminescence intensity of the sample under a 380 nm excitation wavelength. The specific surface area of the sample was obtained through N2 adsorption and desorption experiments conducted using the Micromeritics ASAP 2460 instrument under liquid nitrogen conditions. Thermo Scientific K-Alpha X-ray Photoelectron Spectrometer was utilized to collect X-ray photoelectron spectra (XPS), analyzing the elemental composition and chemical states within the sample. In situ Diffuse Reflectance Fourier Transform Infrared Spectroscopy (in situ DRIFTS) measurements were performed using a JASCO FTIR 6300 spectrometer in a custom chamber to determine the intermediates in the photocatalytic reduction of CO2.Photocatalytic reduction of CO2
The photocatalytic reduction activity of the catalyst for CO2 was assessed in a customized 200 mL quartz reactor. A total of 20 mg of the sample was dispersed in 30 mL of aqueous acetonitrile. 15 mg of 2,2′-bipyridine and 1 μmol of CoCl2 were added as a co-catalyst, and 6 mL of triethanolamine (TEOA) was added as a hole sacrificial. Apply ultrasonic treatment for 30 minutes to ensure thorough mixing. Then, introduce high-purity CO2 (99.999%) for another 30 minutes to allow for catalyst saturation. Initiate the photocatalytic reaction with a 300 W xenon lamp, stabilizing the reaction temperature at 6 °C using a recirculating chilled water system. These samples undergo rigorous analysis via a GC-7920 gas chromatograph, featuring FID and TCD detectors, to ascertain the products of photocatalytic CO2 reduction.Electrochemical testing
During electrochemical testing, we used the CHI650E workstation to acquire the transient photocurrent spectrogram (I–T), electrochemical impedance spectroscopy (EIS), and Mott–Schottky (M–S) curves for the catalyst. We employed a three-electrode setup with a 0.5 M Na2SO4 electrolyte (pH 6.8), the platinum counter electrode, Ag/AgCl reference electrode, and a working electrode crafted via drop-casting. To prepare the working electrode, we mixed 10 mg of catalyst in 2 mL of an aqueous ethanol solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 water to ethanol ratio) with 20 μL of naphthol solution, sonicated for 30 minutes, drop-casted onto a 1 × 1 cm2 FTO glass, and dried. We measured the transient photocurrent using chronoamperometry under a 300 W xenon lamp, EIS from 1 MHz to 100 MHz, and M–S curves at 500 Hz, 1000 Hz, and 2000 Hz.
:3 water to ethanol ratio) with 20 μL of naphthol solution, sonicated for 30 minutes, drop-casted onto a 1 × 1 cm2 FTO glass, and dried. We measured the transient photocurrent using chronoamperometry under a 300 W xenon lamp, EIS from 1 MHz to 100 MHz, and M–S curves at 500 Hz, 1000 Hz, and 2000 Hz.
Results and discussion
The preparation of xTM/CN (NH) heterostructured catalyst is illustrated in Scheme 1. Specifically, bulk g-C3N4 was synthesized via the high-temperature pyrolysis of melamine in a muffle furnace. After dispersing as-synthesized carbon nitride and mineral tourmaline powders in water by ultrasonication, the composite sample was fabricated using a water bath evaporation method. The collected sample was expressed as xTM/CN, where x means the weight percent of tourmaline to CN. Following calcination process under an Ar/H2 atmosphere was performed to synthesize TM/CN(NH) heterostructure with nitrogen vacancies. | ||
| Scheme 1 Schematic diagram of the synthesis process of xTM/CN (NH) composite catalyst. | ||
Characterization of the synthesized catalysts
XRD analysis was employed to investigate the phase composition of catalysts. As shown in Fig. S1,† g-C3N4 displays two distinct characteristic peaks. Specifically, the diffraction peak at 13.1° corresponds to the (100) crystal plane of CN, representing the triazine ring structure, while the peak at 27.8° was attributed to its (002) crystal plane, signifying the interlayer stacking of aromatic rings.34,35 In the case of CN(NH) with nitrogen defects, the diffraction peak of its (100) crystal plane becomes weaker and broader compared to the pristine CN. This observation aligns with a decreased stacking order within the in-plane structure of g-C3N4, suggesting the formation of additional nitrogen defects during the calcination process in an Ar/H2 atmosphere.36Fig. 1a illustrates the XRD patterns of tourmaline and its composite. The diffraction pattern of tourmaline closely matches the standard card for Schorl (PDF # 70-1877), and the absence of noticeable impurity peaks indicates high purity and good crystallinity of the tourmaline material. In the composite sample 25TM/CN(NH), the retention of characteristic peaks for both g-C3N4 and tourmaline confirms the successful synthesis of the composite. The formation of defects in CN(NH) can be further confirmed by EPR analysis. As depicted in Fig. 1b, g-C3N4 demonstrates unpaired electrons stemming from sp2 carbon atoms on the aromatic ring.37 Following heat treatment in an Ar/H2 environment, the signal intensity of CN(NH) is notably stronger than that of CN, suggesting the creation of numerous nitrogen vacancies within CN. Similarly, the elevated signal intensity in 25TM/CN(NH) compared to CN indicates abundant nitrogen vacancies in the composite sample as well. | ||
| Fig. 1 (a) XRD patterns of CN, CN(NH), 25TM/CN(NH) and TM; (b) EPR spectra of CN, CN(NH) and 25TM/CN(NH); (c) FTIR spectroscopy of CN, CN(NH), 25TM/CN(NH) and TM; (d) N2 adsorption and desorption of CN, CN(NH), TM and 25TM/CN(NH) isotherms and the table for corresponding data (inset). | ||
FTIR was employed to investigate the molecular structure changes in the catalyst. It is obvious from the Fig. 1c that tourmaline exhibits distinct peaks. Specifically, the stretching vibration peak at 470 cm−1 corresponds to the Si–O bond. The peaks at 709 cm−1 and 779 cm−1 represent the stretching vibration of Fe–O or Al–O bonds within the tourmaline structure.38 Moreover, the peaks at 971 and 1027 cm−1 are ascribed to the stretching vibration of O–Si–O and Si–O–Al in tourmaline, respectively. Additionally, the peaks at 1332 cm−1 and 1243 cm−1 correspond to the asymmetric stretching vibration of BO3,39 while the peak at 3567 cm−1 signifies the surface hydroxyl group of tourmaline. The presence of these peaks reflects the characteristic structure of tourmaline. Examining the infrared absorption spectra of CN and CN(NH), a typical vibrational mode of triazine units was observed at 810 cm−1. The characteristic peak ranging from 1270 to 1670 cm−1 aligns with the stretching vibration of C–N heterocyclic compounds, whereas the stretching vibration peak between 2900–3400 cm−1 corresponds to the stretching vibration of N–H and OH.40 In the composite sample 25TM/CN(NH), the typical characteristic peak closely matches that of CN(NH), accompanied by a broadening of the Si–O bond peak at 470 cm−1 and the disappearance of the peak at 3567 cm−1. This suggests a possible binding between tourmaline and CN(NH) through surface hydroxyl groups. Generally, a larger specific surface area of a catalyst facilitates the exposure of adsorption and active sites for catalytic reactions. As depicted in Fig. 1d, the N2 adsorption isotherm of the prepared sample exhibits Type IV with an H3 hysteresis loop, indicating the stacking of catalyst flake structures.41 According to the table in Fig. 1d, the specific surface area (SBET) of 25TM/CN(NH) (26.75 cm2 g−1) is higher compared to the CN(NH) (15.29 cm2 g−1) and TM (4.82 cm2 g−1). This enlarged specific surface area will be advantageous for the adsorption of CO2 by 25TM/CN(NH) during photocatalytic reduction of CO2.
 | ||
| Fig. 2 TEM images of (a) CN(NH), (b) TM and (c) 25TM/CN(NH); (d) HRTEM images of 25TM/CN(NH); (e–l) EDX elements mapping of 25TM/CN(NH). | ||
To further evaluate the morphology and microstructure, the morphology of TM, CN, CN(NH), and 25TM/CN(NH) was analyzed by SEM and TEM. In Fig. S2,† the morphology of the tourmaline is clearly block structure. In the 25TM/CN(NH) composite (Fig. S3†), tourmaline can be clearly seen attached to g-C3N4, indicating the successful preparation of the composite catalyst. As can be seen from Fig. 2a, CN(NH) exhibits a transparent lamellar structure, which is different from the morphology of the bulk CN (Fig. S4†), confirming the exfoliation of the bulk CN. The pristine tourmaline is an irregular particle with a size of about 1 μm (Fig. 2b). For 25TM/CN(NH) (Fig. 2c), it can be clearly seen that tourmaline is successfully loaded on the lamellar CN(NH). Based on the HRTEM image (Fig. 2d), a region with distinct lattice stripes (0.808 nm) can be observed, corresponding to the (110) crystalline surface of tourmaline. The interface in the region of disordered atomic arrangement corresponds to the structure of low crystalline CN(NH). This indicates the formation of a tight interfacial contact between tourmaline and CN(NH), which should be favorable for the formation of the spontaneous surface electric field. EDX elemental analysis reveals that 25TM/CN(NH) is composed of C, N, O, B, Si, Al, and Fe elements and they are homogeneously distributed in the composite (Fig. 2e–l).
XPS was used to reveal the elemental composition and chemical state of the catalyst surface. Based on Fig. 3a, both TM and 25TM/CN(NH) encompass Si, B, C, O, and Fe elements, confirming the successful combination between tourmaline and CN(NH) via interfacial interactions. Fig. 3b presents the Si 2p spectra of TM and 25TM/CN(NH). The featuring peaks at 102.22 eV and 103.34 eV can be ascribed to Si4+ and Si–OH in SiO2, respectively.42 The peak component of Si–OH in 25TM/CN(NH) is lower than that of TM, suggesting the possible binding between CN(NH) and the Si–OH groups of tourmaline. For the O 1s spectra of TM and 25TM/CN(NH), the peaks at 532.81 eV, 531.65 eV, and 530.21 eV correspond to hydroxyl oxygen, bridge oxygen and lattice oxygen (Fig. S5†), respectively.43 For the 25TM/CN(NH) sample, these peaks shifted towards higher binding energies, indicating a possible influence of the tourmaline polarized electric field on the electron transfer at the catalyst surface. Further examination of the C 1s spectra for CN(NH) and 25TM/CN(NH) revealed peaks at 284.80 eV and 288.34 eV, attributing to the C–C bond and N–CN bond (Fig. 3c), respectively. A novel peak at 286.11 eV in the 25TM/CN(NH) spectrum suggests the presence of C–OH, potentially associated with the binding of CN(NH) with hydroxyl groups on the tourmaline surface. As displayed in Fig. 3d, in the N 1s spectra of CN and CN(NH), the peaks at 398.60 eV, 399.67 eV, and 401.07 eV correspond to CN–C, N–(C)3 and NHx, respectively.44 Notably, the CN–C/N–(C)3 ratio decreased from 2.74 in CN to 1.88 in CN(NH), indicating the release of nitrogen-containing substances and the formation of nitrogen defects under the Ar/H2 atmosphere,45 aligning with the EPR results. For the 25TM/CN(NH) sample, the N 1s peak shifted towards a higher binding energy compared to CN(NH), indicating the influenced interfacial interactions between self-polarization tourmaline and nitrogen-deficient CN(NH).
 | ||
| Fig. 3 (a) XPS full spectra of CN, CN(NH), 25TM/CN(NH) and TM; (b) XPS Si 2p spectra of TM and 25TM/CN(NH); (c) XPS C 1s spectra of CN(NH) and 25TM/CN(NH); (d) XPS N 1s spectra of CN, CN(NH) and 25TM/CN(NH). | ||
Superior photocatalytic CO2 reduction performance of 25TM/CN(NH)
The performance of heterostructured catalysts for CO2 reduction was then assessed. Fig. 4a illustrates that pristine tourmaline has no photocatalytic activity, the catalytic activity of bulk CN is very low, and the activity of nitrogen-deficient CN(NH) is still moderate. Interestingly, 25TM/CN(NH) exhibited excellent photocatalytic performance, achieving a CO release rate of 118.17 μmol g−1 h−1, which is 6.4 and 2.9 times higher than that of CN and CN(NH), respectively. The nitrogen vacancy performed an essential role in enhancing the activity since the CO yield of the 25TM/CN sample was only 23.9 μmol g−1 h−1. Profited from the accelerated separation and migration of photogenerated carriers by polarized electric field, the selectivity (Fig. 4b) of CO product reaches 95.56%, much higher than that of bulk CN (48.27%). Thus, the introduction of a polarized electric field can effectively suppress the competing reaction of H2 evolution. No CO was detected without light and catalyst, highlighting that both light and catalyst are essential for the photocatalytic process. When replacing CO2 with Ar gas, the absence of CO product confirms that CO2 is the sole carbon source for the photocatalytic reaction (Fig. 4c). There is an optimal ratio of tourmaline is 25% (Fig. S6†), since no further improvement is achieved by increasing the content of tourmaline. Moreover, 25TM/CN(NH) heterostructure exhibits superior performance compared to published g-C3N4-based composite catalysts for photocatalytic CO2 reduction (Table S1†). We finally conducted stability tests on the 25TM/CN(NH) catalyst for photocatalytic CO2 reduction (Fig. 4d and S7†). During the four reaction cycles, only a slight decrease in the production amount of CO is observed for 25TM/CN(NH), which might be attributed to the depletion of triethanolamine (TEOA) in the reaction system. Upon adding 2 mL of TEOA after the fourth cycle test, CO production notably rebounded during the fifth cycle test. Overall, throughout the 15 h reaction process, CO production steadily increased, ultimately attaining a level of 1578 μmol g−1. The catalysts collected after the cyclic testing were analyzed by XRD (Fig. S8†) and FTIR (Fig. S9†). The results revealed no substantial alterations in 25TM/CN(NH) after the reaction, indicating the long-term stability of the catalysts. These findings offer a viable approach for the efficient utilization of natural minerals in CO2 reduction, holding promising potential for future practical applications. | ||
| Fig. 4 (a) CO production data of different products over time under full spectrum irradiation; (b) CO2 photoreduction activity and CO selectivity of CN, CN(NH), and 25TM/CN(NH); (c) CO2 reduction performance of 25TM/CN(NH) under various conditions; (d) cycle stability test of 25TM/CN(NH). | ||
Efficient charge separation and transfer driven by the 25TM/CN(NH)
The separation, migration, and recombination of charges during the photocatalytic process are crucial determinants of photocatalyst performance. To demonstrate the existence of a polarized electric field in the composite sample 25TM/CN(NH), the surface potential of the 25TM/CN(NH) sample was tested using Kelvin probe force microscopy (KPFM). The morphology of the 25TM/CN(NH) sample can be observed with a total height of about 680 nm, which is shown in Fig. 5a. More significantly, the presence of a significant potential difference in the 25TM/CN(NH) sample (Fig. 5b) and its corresponding surface potential difference of about 70 mV (inset of Fig. 5b) proves the existence of a polarized electric field.46 The ferroelectric hysteresis loops are closely related to spontaneous polarization.47 To deeply investigate the effect of the polarized electric field of tourmaline in the composites on the photogenerated electron–hole separation and migration during CN(NH) photocatalysis, we carefully analyzed the ferroelectric hysteresis loops of the pristine TM and 25TM/CN(NH) composites. As shown in Fig. 5c, both the pristine TM and 25TM/CN(NH) samples show obvious nonlinear hysteresis loops, in which the remanent polarization (0.018 μC cm−2) in the 25TM/CN(NH) sample is close to half of that in the pristine tourmaline (0.038 μC cm−2), a feature that confirms the existence of spontaneous polarization in tourmaline,48 and further suggests that the presence of a polarized electric field can influence the directional motion of photogenerated electrons and holes. | ||
| Fig. 5 The pictures of (a) KPFM height and (b) surface potential of 25TM/CN(NH) (inset is corresponding surface potential profiles); (c) ferroelectric hysteresis loops of TM and 25TM/CN(NH); (d) transient photocurrent response, (e) electrochemical impedance spectra, (f) PL spectra of CN, CN(NH), and 25TM/CN(NH). | ||
We then employed transient photocurrent measurements (I–T), electrochemical impedance spectroscopy (EIS), Linear Sweep Voltammetry (LSV) curves, and photoluminescence spectroscopy (PL) to investigate the effect of the polarized electric field of tourmaline on the separation and migration of photogenerated carriers. Fig. 5d presents the transient photocurrent response spectra of CN, CN(NH), and 25TM/CN(NH). The photocurrent density remains consistent and stable across five switching cycles for all samples. Notably, 25TM/CN(NH) exhibits the highest photocurrent intensity, followed by CN(NH), suggesting superior photogenerated electron–hole separation efficiency for 25TM/CN(NH). Fig. 5e shows the electrochemical impedance spectra (EIS) of the prepared samples and equivalent circuits consisting of resistance through solution resistance (R1), constant phase element (CPE), and charge transfer resistance through the electrode/electrolyte interface (R2). The composite catalysts with the smallest radius of the semicircular arcs for the 25TM/CN(NH) composites indicate the lowest charge transfer resistance, which accelerates the kinetics of the photocatalytic surface reactions. Furthermore, Fig. 5f showcases the PL spectra, measured at an excitation wavelength of 380 nm. CN exhibits the highest PL intensity, whereas the PL intensity of 25TM/CN(NH) in the composite material is notably lower. This finding underscores the composite catalyst proficiency in preventing the recombination of photogenerated electrons and holes, thereby enhancing the photocatalytic performance. Additionally, Fig. S10† shows the LSV curves of the samples, with the LSV curve for 25TM/CN(NH) showing a higher current density and adjusted onset potential. This disparity widens as the applied voltage increases. The lower overpotential signifies effective interface electron diffusion in 25TM/CN(NH) due to a reduced reliance on external driving forces. Based on the above analysis, it was demonstrated that the existence of a polarized electric field 25TM/CN(NH) could enhance the separation and migration of photogenerated carriers in the 25TM/CN(NH) catalysts, ultimately facilitating the effective photoreduction of CO2.
Mechanism analysis for CO2 photoreduction of 25TM/CN(NH)
The optical properties and energy band structures of pristine CN, CN(NH), and 25TM/CN(NH) were investigated by UV-visible diffuse reflectance spectroscopy. As illustrated in Fig. 6a, CN(NH) displays a notable redshift compared to CN. This shift may be attributed to nitrogen vacancies formed during the Ar/H2 atmosphere treatment of the CN. The introduction of tourmaline further extends its absorption edge to 540 nm, indicating that tourmaline enhances the light absorption capacity of CN(NH). The bandgap widths for pristine CN, CN(NH), and 25TM/CN(NH) are calculated to be 2.48 eV, 2.35 eV, and 1.92 eV, respectively (Fig. S11†). To determine the band structures of samples, from the XPS valence band spectra in Fig. 6b the valence band potentials (VB) of CN, CN(NH), and 25TM/CN(NH) were 1.86 eV, 1.60 eV, and 0.98 eV, respectively. Based on the value of bandgap and VB potential of each sample,49 the conduction bands (CB) of CN, CN(NH), and 25TM/CN(NH) can be calculated by ECB = EVB − Eg and the corresponding results were −0.62 eV, −0.71 eV, and −0.98 eV respectively. The energy band structure diagram in Fig. 6c illustrates that nitrogen vacancies in CN(NH) cause a redistribution of charges, making its conduction band more negative. The introduction of tourmaline further shifts the conduction band of 25TM/CN(NH) to a more negative potential, significantly altering both the valence and conduction band positions. It is hypothesized that the polarization electric of tourmaline induces directional charge movement in CN(NH), ultimately modifying its energy band structure. To more deeply understand the reaction process of photocatalytic reduction of CO2, in situ DRIFTS spectroscopy was employed to analyze the reaction intermediates produced during the catalytic process of 25TM/CN(NH). First, the 25TM/CN(NH) sample was passed through high-purity CO2 gas in darkness for 30 min to achieve the equilibrium of CO2 adsorption, and then a xenon lamp was turned on and illuminated for 60 min to detect the reaction intermediates of photocatalytic reduction of CO2.50 As illustrated in Fig. 6d, the characteristic peaks at 1510 cm−1 and 1714 cm−1 represent the monodentate carbonate (m-CO32−) adsorbed on the catalyst surface during the whole reaction process. The peaks at 1435 cm−1 and 1464 cm−1 correspond to the *HCO3 group, and the peaks at 1341 cm−1 and 1373 cm−1 correspond to the bidentate carbonate (b-CO32−). It is noteworthy that during the photocatalytic reduction of CO2, distinct characteristic peaks appeared at 1552 cm−1 and 1681 cm−1, which corresponded to the intermediate *COOH, and their peak intensities increased markedly with the extension of the illumination time, which is in agreement with the previous reports.51–53 These results suggest that CO2 is adsorbed on 25TM/CN(NH) in the form of m-CO32− species, and *COOH is a key intermediate in the photoreduction of CO2 to produce CO.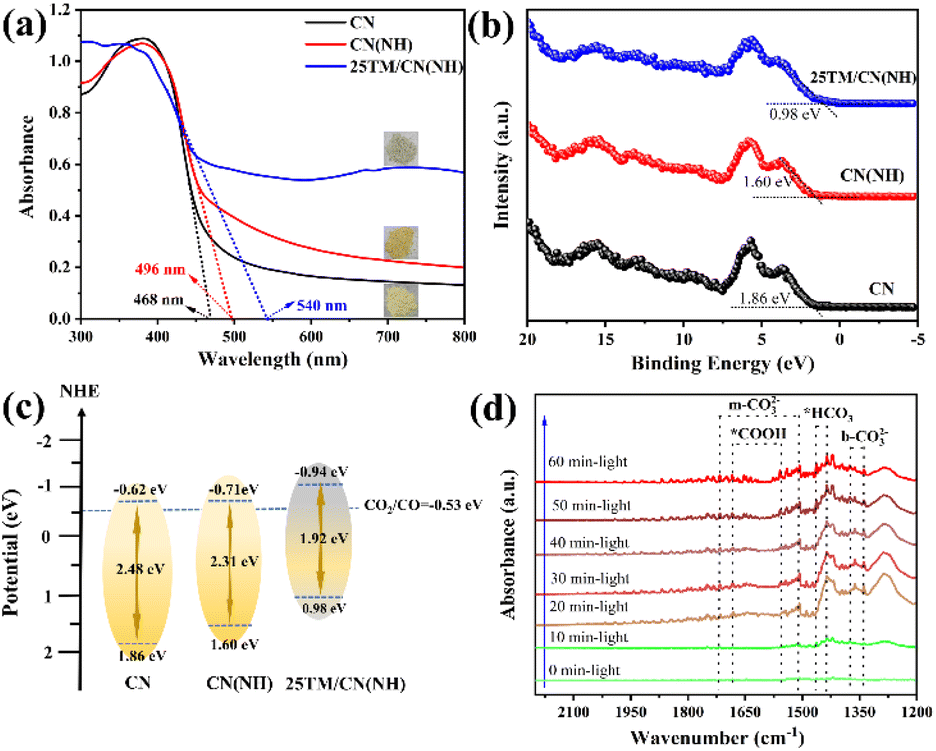 | ||
| Fig. 6 (a) UV visible diffuse reflectance spectra; (b) VB-XPS spectra, and (c) band structure diagrams of CN, CN(NH) and 25TM/CN(NH); (d) in situ DRIFTS measurements of 25TM/CN(NH). | ||
Based on the preceding analysis, a plausible mechanism for augmenting the photocatalytic reduction of CO2via g-C3N4 is proposed, leveraging the synergy between nitrogen vacancies and the polarization electric field generated by tourmaline. As shown in Scheme 2, the composite catalyst-enhanced photocatalytic reduction of CO2 can be ascribed to two key factors. Firstly, the nitrogen vacancies in CN(NH) expand its light absorption range and optimize its band structure. Secondly, the introduction of tourmaline not only facilitates the migration and separation of photo-generated electrons and holes at the interface, ensuring a higher number of electrons reaching the catalyst surface for CO2 reduction but also favorably influences the interface charge distribution of g-C3N4. This, in turn, optimizes its band energy structure and boosts both light absorption and the capacity for photo-generated electron reduction.
 | ||
| Scheme 2 Schematic diagram of CO2 reduction reaction mechanism of 25TM/CN(NH) photocatalyst. | ||
Conclusion
In summary, tourmaline with a self-polarized electric field was coupled with CN(NH) to construct the 25TM/CN(NH) heterostructures for photocatalytic reduction of CO2. Owing to the enhanced separation and migration of electron–hole pairs during the photocatalytic process by the polarized electric field of tourmaline, the 25TM/CN(NH) catalyst exhibited impressive activity and selectivity in the photocatalytic reduction of CO2, with a CO generation rate as high as 118.17 μmol g−1 h−1 and a selectivity up to 95.56%. Mechanistic analysis showed that introducing nitrogen vacancies enhanced the light absorption range and optimized its energy band structure, while the self-polarized electric field introduced by tourmaline could boost the separation and migration of photogenerated charge carriers, resulting in highly efficient photocatalytic CO2 reduction performance. This investigation emphasizes the significance of the self-polarized electric field in enhancing the photocatalytic activity of CO2 reduction catalysts.Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 42377227, 52272094) and Beijing Natural Science Foundation (No. 2232061).References
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- Y. Shi, Z. Zhao, D. Yang, J. Tan, X. Xin, Y. Liu and Z. Jiang, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- J. Xiong, M. Zhang, M. Lu, K. Zhao, C. Han, G. Cheng and Z. Wen, Chin. Chem. Lett., 2022, 33, 1313–1316 CrossRef CAS.
- X. Tong, W. Zhang, G. Cheng and J. Xiong, Mol. Catal., 2024, 555, 113877 CrossRef CAS.
- G. Yang, P. Qiu, J. Xiong, X. Zhu and G. Cheng, Chin. Chem. Lett., 2022, 33, 3709–3712 CrossRef CAS.
- T. Zhang, T. Li, M. Gao, W. Lu, Z. Chen, W. L. Ong, A. S. W. Wong, L. Yang, S. Kawi and G. W. Ho, Adv. Energy Mater., 2024, 14, 2400388 CrossRef CAS.
- G. Yang, J. Xiong, M. Lu, W. Wang, W. Li, Z. Wen, S. Li, W. Li, R. Chen and G. Cheng, J. Colloid Interface Sci., 2022, 624, 348–361 CrossRef CAS.
- J. Chen, Y. Ren, Y. Fu, Y. Si, J. Huang, J. Zhou, M. Liu, L. Duan and N. Li, ACS Nano, 2024, 18, 13035–13048 CrossRef CAS PubMed.
- X. Liang, X. Wang, X. Zhang, S. Lin, M. Ji, Q. Liu and M. Wang, ACS Catal., 2024, 14, 4648–4655 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef CAS.
- L. Yang, Y. Peng, X. Luo, Y. Dan, J. Ye, Y. Zhou and Z. Zou, Chem. Soc. Rev., 2021, 50, 2147–2172 RSC.
- Y. Lv, D. Ma, K. Song, S. Mao, Z. Liu, D. He, X. Zhao, T. Yao and J.-W. Shi, J. Mater. Chem. A, 2023, 11, 800–808 RSC.
- D. Ma, J. Chen, Z. Zhang, J. Li and J.-W. Shi, J. Mater. Chem. A, 2024, 12, 13647–13671 RSC.
- Z. Li, J. Ao, Z. Wang, Z. Huang, Z. Xu, X. Wu, Z. Cheng and K. Lv, Sep. Purif. Technol., 2024, 338, 126577 CrossRef CAS.
- D. Ma, Z. Zhang, Y. Zou, J. Chen and J.-W. Shi, Coord. Chem. Rev., 2024, 500, 215489 CrossRef CAS.
- C. Yang, Y. Hou, G. Luo, J. Yu and S. Cao, Nanoscale, 2022, 14, 11972–11978 RSC.
- Z. Jiang, M. Guo, Z. Yang, R. Fang, Z. Wang and J. Ran, J. Mater. Chem. A, 2024, 12, 11591–11601 RSC.
- Z. Yang, M. Yuan, Z. Cheng, B. Liu, Z. Ma, J. Ma, J. Zhang, X. Ma, P. a. Ma and J. Lin, Angew. Chem., Int. Ed., 2024, 63, e202401758 CrossRef CAS PubMed.
- M. Chen, M. Sun, X. Cao, H. Wang, L. Xia, W. Jiang, M. Huang, L. He, X. Zhao and Y. Zhou, Coord. Chem. Rev., 2024, 510, 215849 CrossRef CAS.
- Y. Li, Z. He, L. Liu, Y. Jiang, W. J. Ong, Y. Duan, W. Ho and F. Dong, Nano Energy, 2023, 105, 108032 CrossRef CAS.
- Y. Wang, C. Ban, Y. Feng, J. Ma, J. Ding, X. Wang, L. Ruan, Y. Duan, M. G. Brik, L. Gan and X. Zhou, Nano Energy, 2024, 124, 109494 CrossRef CAS.
- W. Tu, Y. Xu, J. Wang, B. Zhang, T. Zhou, S. Yin, S. Wu, C. Li, Y. Huang, Y. Zhou, Z. Zou, J. Robertson, M. Kraft and R. Xu, ACS Sustain. Chem. Eng., 2017, 5, 7260–7268 CrossRef CAS.
- Z. Dong, H. Feng, L. Li, Y. Hu, T. Yang and S. Xue, J. Alloys Compd., 2023, 968, 172226 CrossRef CAS.
- C. Yang, Y. Chen, T. Chen, S. Rajendran, Z. Zeng, J. Qin and X. Zhang, Fuel, 2022, 314, 122758 CrossRef CAS.
- J. Joy, T. Stuyver and S. Shaik, J. Am. Chem. Soc., 2020, 142, 3836–3850 CrossRef CAS PubMed.
- X. Zhou, B. Shen, A. Lyubartsev, J. Zhai and N. Hedin, Nano Energy, 2022, 96, 107141 CrossRef CAS.
- S. Wang, Q. Li, S. Sun, K. Ge, Y. Zhao, K. Yang, Z. Zhang, J. Cao, J. Lu, Y. Yang, Y. Zhang, M. Pan, Z. Lin and L. Zhu, J. Mater. Chem. A, 2022, 10, 5350–5360 RSC.
- Y. Liang, X. Tang, Q. Zhu, J. Han and C. Wang, Chemosphere, 2021, 281, 130780 CrossRef CAS.
- L. Yu, C. Wang, F. Chen, J. Zhang, Y. Ruan and J. Xu, J. Mol. Catal. A: Chem., 2016, 411, 1–8 CrossRef CAS.
- C. Zhao, L. Liao, Z. Xia and X. Sun, Vib. Spectrosc., 2012, 62, 28–34 CrossRef CAS.
- B. Niu, N. Wang, Y. Chen, M. Yu, Z. Hou, Z. Li and Y. Zheng, Sep. Purif. Technol., 2021, 257, 117893 CrossRef CAS.
- L. Sun, Y. Guo, Y. Liu, R. Ni, G. Chen, X. Wei, Z. An and Z. Jiao, Sens. Actuators, B, 2022, 371, 132533 CrossRef CAS.
- T. Yang, X. Mao, Y. Zhang, X. Wu, L. Wang, M. Chu, C.-W. Pao, S. Yang, Y. Xu and X. Huang, Nat. Commun., 2021, 12, 6022 CrossRef CAS.
- Y. Xia, H. Yang, W. Ho, B. Zhu and J. Yu, Appl. Catal., B, 2024, 344, 123604 CrossRef CAS.
- X. An, Q. Tang, H. Lan, H. Liu, X. Yu, J. Qu, H. Lin and J. Ye, Angew. Chem., Int. Ed., 2022, 61, e202212706 CrossRef CAS.
- Z. Chen, Y. Gao, F. Chen and H. Shi, Chem. Eng. J., 2021, 413, 127474 CrossRef CAS.
- Y. Chen, S. Wang, Y. Li, Y. Liu, Y. Chen, Y. Wu, J. Zhang, H. Li, Z. Peng, R. Xu and Z. Zeng, J. Colloid Interface Sci., 2020, 575, 367–376 CrossRef CAS.
- M. Hao, H. Li, L. Cui, W. Liu, B. Fang, J. Liang, X. Xie, D. Wang and F. Wang, Environ. Chem. Lett., 2021, 19, 3573–3582 CrossRef CAS.
- J. Park, H. Liu, G. Piao, U. Kang, H. W. Jeong, C. Janáky and H. Park, Chem. Eng. J., 2022, 437, 135388 CrossRef CAS.
- X. Wang, S. Hong, H. Lian, X. Zhan, M. Cheng, Z. Huang, M. Manzo, L. Cai, A. Nadda, Q. V. Le and C. Xia, J. Hazard. Mater., 2021, 420, 126565 CrossRef CAS PubMed.
- S. P. Chenakin, G. Melaet, R. Szukiewicz and N. Kruse, J. Catal., 2014, 312, 1–11 CrossRef CAS.
- D. Wang, H. Xu, J. Ma, X. Lu, J. Qi and S. Song, Chem. Eng. J., 2018, 354, 113–125 CrossRef CAS.
- G. Alnaggar, A. Hezam, Q. A. Drmosh and S. Ananda, Chemosphere, 2021, 272, 129807 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- Y. Jiang, H. Y. Chen, J. Y. Li, J. F. Liao, H. H. Zhang, X. D. Wang and D. B. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- J. An, C. Xu, L. Li and B. Tang, Nano Res., 2024, 17, 3571–3585 CrossRef CAS.
- G. Zhou, K. Chen, X. Gai, H. Liu, C. Zhao, K. Shen and Z. Fan, Ferroelectrics, 2017, 514, 89–96 CrossRef CAS.
- W. Liu, Y. Dong, J. Liu, L. Zhang, Y. Lu and H. Lin, Chem. Eng. J., 2023, 451, 138666 CrossRef CAS.
- H. Jiang, W. Wang, L. Sun, T. Kong, Z. Lu, H. Tang, L. Wang and Q. Liu, J. Catal., 2022, 416, 1–10 CrossRef CAS.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- Y. Shang, M. Zheng, H. Liu, X. Jin, C. Yan, L. Song, Z. Qi, F. Jing, P. Song, X. Zhou, G. Chen and C. Lv, ACS Catal., 2023, 13, 14530–14539 CrossRef CAS.
- Y. Wang, Y. Zhang, Y. Wang, C. Zeng, M. Sun, D. Yang, K. Cao, H. Pan, Y. Wu, H. Liu and R. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 40629–40637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of CN and CN(NH); SEM images of TM and 25TM/CN(NH); TEM images of CN; XPS spectra of TM and 25TM/CN(NH): O 1s; time-dependent CO yields produced in the presence of different photocatalysts; the plot of CO and H2 yields of 25TM/CN(NH) over a continuous 15 h period; XRD patterns of 25TM/CN(NH) before and after the cyclic experiment; FTIR spectra of 25TM/CN(NH) before and after the cyclic experiment; polarization curve of CN, CN (NH), and 25TM/CN (NH); Tauc spectra of CN, CN(NH), and 25TM/CN(NH); photocatalytic CO2 reduction properties of previously reported g-C3N4 based photocatalysts. See DOI: https://doi.org/10.1039/d4ta06709f |
| This journal is © The Royal Society of Chemistry 2025 |