
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 hydrogenation on Ni(111): microkinetic modelling vs. kinetic Monte Carlo simulations – choosing the right approach for unravelling reaction kinetics†
Alejandro
Gracia
 ,
Pablo
Lozano-Reis
,
Fermín
Huarte-Larrañaga
,
Pablo
Gamallo
* and
Ramón
Sayós
,
Pablo
Lozano-Reis
,
Fermín
Huarte-Larrañaga
,
Pablo
Gamallo
* and
Ramón
Sayós
Departament de Ciència de Materials i Química Física, Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, C. Martí i Franquès 1, 08028 Barcelona, Spain. E-mail: gamallo@ub.edu
First published on 30th May 2025
Abstract
While density functional theory (DFT) provides a very helpful tool for the microscopic domain, kinetic methods that study the evolution of the system with time are needed for the complete study of catalytic systems normally including complex reaction networks. Herein, we study the complex CO2 hydrogenation reaction over Ni(111) under different catalytic conditions by means of two kinetic methods, namely kinetic Monte Carlo (kMC) and Microkinetic Modelling (MkM) simulations. Predictions on relevant macroscopic magnitudes such as turnover frequencies and coverages have been obtained from analysing the two kinetic approaches. Moreover, the reaction mechanisms have been analysed scrutinizing where the differences between the two methods come from. The simulations suggest that, for systems with low coverage of adsorbed species, the MkM approach is a very suitable option. It provides results very similar to those of the more complex kMC simulations but with significantly lower computational cost. Nevertheless, for a deeper understanding of the system's behaviour, spatially resolved kMC simulations are a better choice. Precisely, the use of kMC simulations is of paramount importance when dealing with systems with high coverages in which adsorbate–adsorbate interactions and the topological arrangement of the adsorbates are more influential. MkM simulations that include a lateral potential yield results that align more closely with those of kMC. However, the local considerations in kMC lead to differences in macroscopic properties and reaction mechanisms. These findings are likely applicable to other catalytic systems and aim to guide theoreticians in selecting the most suitable kinetic approach.
Sustainability spotlightThis study contributes to the understanding of CO2 conversion into light fuels from a microscopic point of view. Microkinetic modelling and kinetic Monte Carlo simulations have been performed under different conditions to better understand the catalytic hydrogenation of CO2 over Ni(111) trying to unravel the differences observed in both methods. A deep understanding of how CO2 catalytic conversion proceeds microscopically is essential to propose new catalysts that could improve the process. Moreover, by converting CO2 we can repurpose this greenhouse gas into a resource, effectively closing the loop in the carbon cycle, achieving a circular economy, and thus enhancing sustainability. This work is aligned with the United Nations Sustainable Development Goals (SDGs) 7 (Affordable and Clean Energy) and 13 (Climate Action). |
1. Introduction
Heterogeneous catalysis plays a fundamental role in the chemical industry, being pivotal in the design and production of many important materials and chemicals. There are numerous industrial heterogeneously catalysed reactions1–3 that are crucial for the production of a vast number of chemicals used in daily life. However, the ever-growing demand for energy and manufactured goods, along with the issue of climate change, makes it paramount to explore novel catalysts with high conversion, activity, and selectivity, while also ensuring low environmental impact and economic cost. Therefore, a deep understanding of the principles of heterogeneous catalysis is essential for the rational design of novel catalysts to optimize industrial processes and develop new and innovative technologies. To this end, theoretical chemistry has proven to be very helpful in studying the behaviour of heterogeneous catalytic systems. Focusing on the quantum mechanical regime, ab initio quantum methods—most notably DFT calculations—have become essential tools for understanding reaction mechanisms and the catalytic properties of materials at the atomistic level. By calculating the electronic structure and energies of a system, DFT can be used to define the energetics of the various elementary processes involved in catalytic reactions, which helps identify the rate-limiting steps, the role of each intermediate, and the most active sites on the catalytic surface. Nevertheless, the purely atomistic information that DFT provides is often not enough to fully understand the time evolution of the reaction under real working conditions. Therefore, DFT calculations must be coupled with kinetic modelling techniques4–8—either via Microkinetic Modelling (MkM) or kinetic Monte Carlo (kMC) simulations—to unveil the dominant reaction mechanisms.It is well known that the main source of energy for humanity continues to be carbon-rich fuels, such as oil and gas.9 The combustion of these energy sources results in increased carbon dioxide emissions10 to the atmosphere, which is a widely recognized climate issue that covers from the greenhouse effect to the ocean's acidification,11 among others. Therefore, CO2 conversion is one of the most relevant modern scientific challenges, essential for achieving a circular economy, and hence, enhancing sustainability. In a traditional linear economy, CO2 is typically considered a waste product, contributing significantly to global warming and environmental degradation. However, by converting CO2, we can repurpose this greenhouse gas into a valuable resource, effectively closing the loop in the carbon cycle. In this context, CO2 valorisation has attracted increasing attention as an efficient and economical source to obtain C1 carbon species. Among the different strategies for CO2 recycling, power-to-gas (PtG) technology12 has garnered significant interest, as it offers a promising route for sequestering CO2 from the atmosphere and transforming it into a usable fuel. The PtG process is based on the use of surplus renewable energy to split water and produce H2, which is subsequently used for CO2 hydrogenation. Depending on the choice of metal and support, it is possible to tune the selectivity of CO2 hydrogenation toward the desired product. For instance, methanol can be produced using Au,13,14 Pd15 and Cu-based16,17 catalysts; CO can be obtained through Pt,18 Fe,19 Rh20 or Ni-based21 catalysts; and methane is mostly formed via complete CO2 hydrogenation using Ru,22,23 Ni,24–27 Pt,18 and Pd-based28 catalysts. Although the current environmental urgency has renewed industrial interest in CO2 methanation, this reaction was originally discovered by the French scientist Paul Sabatier in the early twentieth century.
Regarding the CO2 methanation reaction, Ni-based catalysts are the most widely used, as they offer a good balance between catalytic activity and economic cost compared to other noble metals.29,30 Therefore, a deep understanding of the mechanism that governs the reaction over the Ni(111) facet, the most stable one, is required for the proper design of new Ni-based catalysts. In this sense, several theoretical studies have addressed the CO2 hydrogenation reaction over the Ni(111) surface31–35 but only a few have performed kinetic simulations, which are necessary to unveil the reaction mechanism and the time evolution of the system under real working conditions. For instance, Vogt et al.36 performed a Microkinetic Modelling study of the CO2 hydrogenation reaction over a variety of Ni surfaces, including the Ni(111) facet, and found some extent of CH4 formation. In contrast, Lozano-Reis et al.37 also studied the CO2 hydrogenation reaction over Ni(111) by means of kinetic Monte Carlo simulations and concluded that this surface was unable to produce CH4 by itself and instead only CO was observed. However, in the former study,36 only methane was allowed to desorb, but no other possible intermediate products such as CO, formaldehyde or methanol, which were allowed to desorb in the latter study.37 Therefore, it remains unclear whether the differences observed between the two studies stem from the use of different mechanism-related assumptions or from the different kinetic methods employed.
In order to unravel the source of the disparities in the abovementioned studies, the CO2 hydrogenation reaction over Ni(111) has been simulated by means of Microkinetic Modelling and kinetic Monte Carlo simulations using two different approaches: (i) all possible intermediate products are allowed to desorb and (ii) some intermediate products are not allowed to desorb. The first approach mimics the conditions used in the work of Lozano-Reis et al.,37 while the latter replicates the conditions used by Vogt et al.36 Moreover, in the simulations of Vogt et al.,36 very large surface coverages were obtained. Under these conditions, the local lateral interactions and the mean-field approach, considered in the kMC and MkM simulations, respectively, could be key factors affecting performance. The impact of these assumptions highlights the need for careful analysis under such conditions to better understand the differences between the two kinetic approaches. Therefore, a detailed analysis has been performed to assess how coverages, product formation, and the main reaction mechanism vary between the two approaches. Finally, the advantages and disadvantages of both methods have been outlined. It is worth noting that restricting the desorption of possible intermediate products is something that should be avoided in reliable kinetic simulations. However, such restrictions can significantly alter surface coverages, thereby providing different surface environments, which is of interest when analysing both methods at different situations. Although other studies have previously compared MkM and kMC models,38,39 we believe that the extensive reaction network employed in our work (41 non-diffusive reversible steps) contrasts with these other studies. Furthermore, the inclusion of lateral interactions in our kMC model, along with the diverse testing conditions, allows for a more comprehensive comparison aimed at uncovering the sources of disparity between the two methods.
2. Methods and computational details
2.1. Kinetic simulations and the reaction model
The main goal of kinetic simulations is to study the evolution of a system under real working conditions. Particularly, in the context of heterogeneous catalysis, the main interests are to see how coverages and productions evolve with time, as well as the mechanisms that govern the overall reaction. In that sense, some basic ingredients are needed to perform kinetic simulations. First of all, one has to define the relevant species of the specific catalytic process, including all reactants, products and intermediate species. Then, the different elementary processes that can take place are depicted, and for each process a rate of transition or transition probability is associated, which can be derived from Transition State Theory (TST) and DFT data. Note that, depending on the type of process, the transition probability has a specific form, as TST implies.40 Finally, one has to set up the temperature and pressure of interest and portray the catalytic surface on which the overall reaction happens. These are the basic ingredients that kinetic methods, such as Microkinetic Modelling or kinetic Monte Carlo, require. However, the technical implementation and the approaches on which they rely are different, and the details of each of these methods are explained below.To study the CO2 hydrogenation reaction over Ni(111) either via MkM or kMC simulations, the same reaction model has been used. This model covers different pathways of the CO2 hydrogenation reaction and considers the possible formation and desorption of various products such as carbon monoxide, methane, methanol and formaldehyde, as shown in Fig. 1. This involves considering 19 intermediate species, 7 gas-phase species, and a total of 86 elementary processes (including adsorption, desorption, bond breaking/forming, and diffusion processes), which have been energetically defined in terms of their initial and final states and their corresponding transition states. For the elementary processes that involve two co-adsorbed species, either as reactants or products, the interaction (i.e., repulsion or attraction) between these two co-adsorbed species has been considered in the energetic characterization of the co-adsorbed state. Note that some simulations carried out throughout the present study sometimes involve small modifications to the reaction network to evaluate how these assumptions affect the outcome of the two kinetic methods. Thus, the reaction network has been examined as follows: (i) taking into consideration the desorption of possible products (CO, CH2O, and CH3OH) and (ii) restricting some of these desorption processes. The DFT data are directly taken from our previous work,37 where details on the software used and further specifications can be found. Also, readers interested in thermal contributions to entropy and the expressions for Gibbs free energy used are referred to the literature.37,40
 | ||
| Fig. 1 Considered reaction network for the CO2 hydrogenation reaction including formation of different products. Red, dark blue and green arrows stand for the dissociative, the HCOO-mediated and COOH-mediated pathways, respectively. Black lines are for elementary steps that connect different pathways. Dark yellow and dark green stand for reactants and products, respectively, of the CO2 hydrogenation reaction. Reversible steps are represented by double ended arrows. Reprinted (adapted) with permission from ref. 37. Copyright 2020 American Chemical Society. | ||
All the simulations performed consider an initial mixture of H2 and CO2 with a partial molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which corresponds to the stoichiometric ratio for complete CO2 hydrogenation to methane. Simulations have been carried out at total pressures of 1 and 10 bar, and temperatures of 573.15 and 673.15 K. Readsorption of products is not allowed, in order to mimic a constant-flow reactor, although we did not model the reactor explicitly. From the results of both the MkM and kMC simulations, the coverages of all the species have been obtained, along with the rate of transition or event frequency for the different processes, defined as the number of times a process is executed per second and per site (TOF refers to the event frequency of the elementary step that leads to product formation). We then define the net event frequency of a process as the difference between its event frequency in the forward direction and its event frequency in the reverse direction. The analysis of the net event frequencies is relevant to identify the most plausible mechanisms under different conditions and methods. Note that in the case of MkM simulations, the event frequencies span values between 106 and 10−30 event per s per site due to the nature of the method. Therefore, for a better comparison between the two methods, we have considered that the relevant elementary steps used to derive the reaction mechanisms for the MkM simulations are those events that have a net event frequency larger than that of the process with the lowest net event frequency in the kMC simulations multiplied by a factor of 10−2. For instance, in the case where the smallest net even frequency in kMC simulations is 10−4 event per s per site, the considered events in the MkM simulations are those with a net event frequency larger than 10−6 event per s per site.
:1, which corresponds to the stoichiometric ratio for complete CO2 hydrogenation to methane. Simulations have been carried out at total pressures of 1 and 10 bar, and temperatures of 573.15 and 673.15 K. Readsorption of products is not allowed, in order to mimic a constant-flow reactor, although we did not model the reactor explicitly. From the results of both the MkM and kMC simulations, the coverages of all the species have been obtained, along with the rate of transition or event frequency for the different processes, defined as the number of times a process is executed per second and per site (TOF refers to the event frequency of the elementary step that leads to product formation). We then define the net event frequency of a process as the difference between its event frequency in the forward direction and its event frequency in the reverse direction. The analysis of the net event frequencies is relevant to identify the most plausible mechanisms under different conditions and methods. Note that in the case of MkM simulations, the event frequencies span values between 106 and 10−30 event per s per site due to the nature of the method. Therefore, for a better comparison between the two methods, we have considered that the relevant elementary steps used to derive the reaction mechanisms for the MkM simulations are those events that have a net event frequency larger than that of the process with the lowest net event frequency in the kMC simulations multiplied by a factor of 10−2. For instance, in the case where the smallest net even frequency in kMC simulations is 10−4 event per s per site, the considered events in the MkM simulations are those with a net event frequency larger than 10−6 event per s per site.
2.2. Microkinetic modelling simulations
In the MkM simulations, the time evolution of the system is expressed as the rate of change of each adsorbate coverage as a function of the instantaneous coverage of all the species in the model, represented as a system of ordinary non-linear differential equations: | (1) |
2.3. Kinetic Monte Carlo simulations
In the present study, the graph-theoretical kMC approach40,44 combined with cluster expansion Hamiltonians45,46 has been used as implemented in the Zacros software46,47 (version 3.01). In order to represent the Ni(111) surface, a two-dimensional hexagonal periodic grid of size (8 × 8) has been used, comprising a total of 64 points that mirror the hexagonal symmetry of the Ni(111) surface. Note that in this model, each lattice point corresponds to a coarse-grained Ni(111) site that can interact with its six nearest neighbours. Convergence with respect to lattice size has been already tested in our previous work.37 Denticity of the adsorbed species (i.e., the number of atoms bound to the surface) is established according to DFT calculations. First nearest-neighbour pairwise adsorbate–adsorbate interactions (interaction energies, IE) between all possible reactant/product pairs and among the most relevant surface species have been included in the cluster expansion. Although interaction energies (IE) are essentially lateral interactions between adsorbates, the term ‘IE’ is used to distinguish them from the lateral potential employed in MkM simulations. This expansion comprises a total of 19 one-body terms and 51 two-body terms, that are summarized in Tables S2 and S3, in Sections S2 and S3, respectively, in the ESI.† In a few specific cases throughout this study, adsorbate–adsorbate interactions have been intentionally omitted from the kMC model. These instances are clearly indicated in the text and correspond to simulations in which all the values in Table S3† have been set to zero. Further information on how formation energies and interaction energies (IE), corresponding to pairwise adsorbate–adsorbate lateral interactions, were calculated can be found in Sections S2 and S3 in the ESI,† respectively. Moreover, for very fast, quasi-equilibrated processes, transition probabilities (i.e., reaction rates, rj) have been scaled using a scaling factor α < 1 to avoid exceedingly large simulation times. This pragmatic approach ensures that the final outcome remains unaffected and has been successfully applied in previous studies.37,44,47–50 Coverage-dependent energy barriers have been treated by means of the Brønsted–Evans–Polanyi relations, as implemented in the Zacros software (see Section S4 of the ESI†). Under each working condition, three independent simulations were performed, differing only in the sequence of random numbers used. This approach allows for improved sampling of the configurational space. In general, all simulations were run until at least 1000 product molecules were generated under steady-state conditions. The choice of 1000 molecules is somewhat arbitrary but has been selected to ensure a sufficiently large sample size for statistically meaningful results. However, in certain cases – explicitly noted throughout the manuscript – the number of produced molecules was significantly lower (i.e., ∼10 molecules), and obtaining a statistically robust number would require prohibitively long simulations. In such instances, the corresponding results should not be interpreted from a statistical standpoint, but rather as qualitative insights.3. Results and discussion
3.1. MkM vs. kMC simulations in a low coverage environment
In general, very similar results have been obtained for both MkM and kMC simulations when all possible products can desorb, a situation which leads to a low coverage environment. As schematically shown in Fig. 2 at 673.15 K and 10 bar, only CO(g) formation has been observed. Note that similar results have been obtained under other working conditions (Table 1). As shown in Fig. 2 both methods agree well about the predicted mechanism showing that almost all the CO (i.e., >99.95%) is formed following the dissociative pathway (i.e., direct CO2 dissociation) while the rest is formed through the COOH-mediated pathway. Note however, that the COOH-mediated pathway plays almost an insignificant role in CO production. In the case of MkM simulations, the degree of rate control (DRC)51 analysis was performed, pointing out that the CO2 dissociation reaction is the rate-determining step by a vast margin, something already expected due to the importance of this route in the final CO production, in agreement with previous kMC results.37 | ||
| Fig. 2 Schematic representation of the most plausible mechanism for (a) CO2 hydrogenation and (b) H2(g) transformation into H2O(g) over Ni(111) at T = 673.15 K and P = 10 bar with a CO2:H2 molar ratio of 1:4 for kMC simulations with (blue) and without (green) IE and MkM (red) simulatons without lateral potential. Coloured arrows refer to produced events while grey arrows are for events that do not occur (continuous) or occur at the same rate in both forward and backward directions (dashed). The arrow's thickness symbolizes the net event frequency of each elementary process. For details on the calculations for the arrow's thickness, the reader is referred to Section S5.† | ||
:H2 molar ratio is 1:4. Values in parentheses correspond to the kMC results, while the values outside the parentheses stand for the MkM simulations. Note that the kMC values refer to the mean value obtained from three different kMC runs
| MkM (kMC) | ||||
|---|---|---|---|---|
| T = 573.15 K | T = 573.15 K | T = 673.15 K | T = 673.15 K | |
| P = 1 bar | P = 10 bar | P = 1 bar | P = 10 bar | |
| TOFCO (molecules per s per site) | 0.014 (0.028 ± 0.003) | 0.119 (0.147 ± 0.004) | 0.151 (0.485 ± 0.002) | 1.410 (2.92 ± 0.01) |
| θ H (%) | 2.954 (12.79 ± 0.04) | 8.772 (17.38 ± 0.02) | 1.602 (7.78 ± 0.03) | 4.895 (12.79 ± 0.04) |
| θ CO (%) | 0.007 (0.02 ± 0.01) | 0.061 (0.049 ± 0.004) | 0 (0.006 ± 0.002) | 0.008 (0.011 ± 0.002) |
| θ O (%) | 0.019 (0.086 ± 0.007) | 0.054 (0.101 ± 0.004) | 0.013 (0.511 ± 0.007) | 0.034 (0.46 ± 0.02) |
| θ CO2 (%) | 0 (0.76 ± 0.02) | 0.002 (0.786 ± 0.006) | 0 (0.708 ± 0.005) | 0.002 (0.749 ± 0.006) |
| θ * (%) | 97.020 (86.1 ± 0.1) | 91.111 (81.3 ± 0.1) | 98.384 (90.4 ± 0.1) | 95.062 (85.6 ± 0.1) |
Let us analyse why only CO is formed among all the other possible products. When CO is formed it can desorb, dissociate or hydrogenate either to form HCO or COH, with energy barriers of 1.56, 2.98, 1.42 and 2.28 eV, respectively, as shown in Table S4, Section S6 in the ESI.† Regarding the possible processes, both COH formation and CO dissociation exhibit high energy barriers, making them very improbable processes. In the case of HCO formation and CO desorption, the former has a slightly lower energy barrier, but it is a bimolecular reaction less likely to occur than the unimolecular CO desorption. Moreover, the HCO formation reaction is a very endothermic reaction with a low reverse energy barrier of 0.21 eV, which suggests that whenever HCO would be formed it will rapidly dissociate to again form CO and H. Indeed, in both kMC and MkM simulations HCO formation is observed, but this reaction always goes backward to CO (see Fig. S5a in the ESI†), which eventually desorbs, thus hindering evolution to other possible products.
Focusing on quantitative aspects, it can be seen in Table 1 that for both methods the calculated TOFs and coverages follow similar trends when varying the working conditions while the absolute magnitudes slightly differ. MkM and kMC both predict TOFs in the same order of magnitude while kMC estimates a slightly larger CO production. To further investigate the difference in TOFCO between the MkM and kMC simulations when CO desorption is allowed, we have calculated the probability that adsorbed CO2 undergoes any process different to desorption, as detailed in Table S5 in the ESI† (i.e., 0.098 and 0.20‰ for MkM and kMC, respectively). Interestingly, the probability for kMC simulations is twice the MkM one (Table S5†). Thus, when a CO2 molecule adsorbs on the Ni(111) surface, the probability of adsorption and not immediate desorption is doubled for the kMC approach, with the concomitant increase of the probability of CO2 dissociation leading to this difference in TOFCO.
Regarding coverages, very similar values have been obtained although again kMC predicts slightly higher coverages with this difference more pronounced in H coverage. The lower coverage obtained from MkM simulations may be attributed to the MFA as, in this approach, species are more available and reactants do not have to be strictly in close proximity to react, making them more reactive. For instance, for H, the MFA makes the direct associative desorption H + H → H2(g) much more probable with the concomitant decrease in H coverage. Following this line of thought, in kMC simulations two neighbouring H adatoms are needed for the reaction to occur, a less likely situation as hydrogen can easily diffuse over the surface (see Fig. S5a in the ESI†), thus increasing H coverage. This larger H coverage for kMC simulations also explains the disparity in the water formation mechanism between both methods (Fig. 2). In kMC simulations, 100% of the produced water comes from OH hydrogenation, whereas in MkM, the majority of the water is produced through the recombination of two OH adsorbates, although OH hydrogenation also contributes (see Fig S6a in the ESI†).
Another difference between MkM and kMC simulations is the inclusion of interaction energies, which are considered only in kMC simulations. Their influence can be important as they can create special spatial arrangements of species that cannot be captured with mean-field approaches with the concomitant effect on the final coverage. Moreover, interaction energies can modify the energy barrier of a given elementary process, which eventually may produce a change in the mechanism and consequently, in the product formation. Therefore, another source of variety between kMC and MkM methods, apart from the MFA, can be attributed to the absence of interaction energies in the MkM simulations with respect to kMC. To take a first glance at the influence of lateral interactions we included a CO lateral potential (ElatCO) in our microkinetic model which affected both CO and CO2 desorption rates, following the model by Zijlstra et al.,43 as briefly explored in Section 2.2. This procedure allows us to evaluate how the lateral potential included in MkM simulations mimics the the interaction energies included in kMC simulations. MkM simulation results including this lateral potential are reported in Table S6, Section S9, of the ESI† showing that the values are very similar (i.e., differences of 10% maximum under all conditions) to those presented in Table 1, where lateral potential was not included in the microkinetic model. This fact proves that in the case of simulations with low surface coverage, the inclusion of a lateral potential in MkM does not affect the microkinetic model. The lateral potential included in the MkM simulations derive, in fact, in lowering the TOF values obtained for MkM simulations without lateral potential, moving away from kMC results. This is due to the modification produced by the lateral potential in the CO2 adsorption and desorption rate constants (see eqn (S6) and (S7) in Section S1 of the ESI†), which translates into a reduction of the CO2 coverage and, subsequently, into a decrease in the CO coverage. The CO coverage directly affects the TOFCO because the CO desorption rate is proportional to the CO coverage in MkM simulations.
For a deeper insight into this low coverage situation, we performed two additional types of simulations. First, the CO2 dissociation energy barrier was manually changed in the MkM simulations, as this reaction is the rate-determining step. The particular choice of the energy barrier value for each new MkM simulation is set according to the calculated mean value of the CO2 dissociation energy barrier obtained from kMC simulations (see Fig. S2†). Note that the lateral potential (ElatCO) affecting the CO and CO2 desorption rate was not included in these MkM simulations. And second, these adsorbate–adsorbate interaction energies have been completely removed in kMC simulations. As these new simulations do not include the effect of interaction energies, it is predicted that their results regarding macroscopic magnitudes such as TOFCO and coverages are similar. Indeed, Table 2 shows that the coverage predictions for these new simulations are almost identical to the previous MkM results (1st column of Table 2). The kMC simulations without interaction energies produce H coverages very similar to those from MkM simulations, strongly suggesting that H coverage differences between MkM and kMC arise from the inclusion of interaction energies in the latter. A repulsive interaction of 0.27 eV between two neighbour H adsorbates (see Table S3†) makes the situation very unlikely. Therefore, H tends to diffuse away reducing the probability of H2(g) formation and increasing H coverage. This lower H coverage in kMC simulations without interaction energies also explains the difference in the water formation mechanism compared to previous kMC simulations (Fig. 2). Water formation through a combination of 2 OH adsorbates is now minimally present in kMC simulations, contrasting with kMC simulations with interaction energies, where it was non-existent.
:H2 molar ratio of 1:4 for the MkM and kMC simulations with and without the consideration of local potential and interaction energies, respectively. The kMC simulations without including interaction energies correspond to simulations where all IE (see Table S3, Section S3 in the ESI) have been set to 0 eV. Note that the lateral potential has been incorporated in the MkM simulations by artificially changing the CO2 dissociation energy barrier using the value shown in Fig. S2d, Section S4 in the ESI. The kMC values refer to the mean value obtained from three different kMC runs
| T = 673.15 K | ||||
|---|---|---|---|---|
| P = 10 bar | MkM | MkM with
|
kMC | kMC with IE |
| a Note that the implementation of lateral potential in MkM in this section refers only to the change in the CO2 dissociation barrier according to Fig. S2, Section S4 in the ESI, and without the lateral potential (ElatCO) described in Section S1 in the ESI. | ||||
| TOFCO (molecules per s per site) | 1.41 | 0.79 | 8.33 ± 0.04 | 2.92 ± 0.01 |
| θ H (%) | 4.89 | 4.84 | 4.87 ± 0.02 | 12.79 ± 0.04 |
| θ O (%) | 0.03 | 0.02 | 0.37 ± 0.02 | 0.46 ± 0.02 |
| θ CO2 (%) | 0 | 0 | 0.73 ± 0.01 | 0.75 ± 0.01 |
| θ * (%) | 95.06 | 95.13 | 93.8 ± 0.1 | 85.6 ± 0.1 |

Regarding TOFs, kMC simulations without IE present the largest CO production out of all simulations presented in Table 2. This is due to the fact that for these kMC simulations the CO2 dissociation energy barrier is set to its zero-coverage value, which is always lower than the one kMC originally computed considering interaction energies (Fig. S2 in the ESI†). As CO2 dissociation is the rate-determining step, changes in the energy barrier of that process produce an overall change in the final TOFs, so, as expected, the larger the energy barrier the smaller the TOF. Analogously, MkM simulations where the CO2 dissociation energy barrier was manually set to the mean value of the CO2 dissociation energy barrier obtained from kMC simulations present the lowest TOFCO as shown in Table 2.
3.2. MkM vs. kMC simulations in a medium to large coverage environment
As shown above, both MkM and kMC simulations produce very similar results for the CO2 hydrogenation reaction over Ni(111) as the overall coverage is low and the effect of lateral interactions is less likely to be important. However, for the situation in which only methane can desorb, previous results36 have shown very high coverages, a situation in which the effect of lateral interactions could be more relevant. In this section, the desorption of some products is restricted to evaluate how this assumption could affect the final results for both methods, following literature suggestions where CO(g) is not a product for the CO2 hydrogenation reaction over Ni(111).24–27,36,52 Therefore, four different MkM simulations have been performed in which the desorption of the following species was inhibited: (I) CO, (II) CO and CH2O, (III) CO and CH3OH and (IV) CO, CH2O and CH3OH. In these MkM simulations, both the (I) and (III) conditions as well as the (II) and (IV) conditions produce similar results, showing that CH3OH is by far the least formed molecule with an almost negligible formation while the CH2O formation is preferred rather than the CH4 one when both gaseous products are possible. As this information suggests, extra kMC simulations were performed using the setups (I) and (II). Note that all these different simulations were performed under four different sets of working conditions, such as pressure and temperature.Table 3 summarizes the CH2O(g) and CH4(g) TOFs along with the coverage of some relevant species under the conditions when CO cannot desorb for both the MkM and kMC simulations at a temperature of 673.15 K and a total pressure of 10 bar. Note that the discussion is focused on this working condition as it is where TOFs exhibit the highest values. A reader who may be interested in gaseous production and coverages under other working conditions is referred to Table S7, Section S10 in the ESI.† On comparing the results obtained from both kinetic methods, opposite trends and larger differences are observed with respect to the results displayed in the previous section. First and foremost, methane is produced under both conditions in the kMC simulations while it is not observed in the MkM simulations as shown in Table 3. In addition, kMC simulations produce TOFs four orders of magnitude larger than the ones obtained from MkM simulations while the overall kMC predicted coverage is now lower. These observations contrast with those reported in the previous section as the TOF values for both methods are around the same order of magnitude although kMC presented higher coverages. However, both methods predict very low product formation under all the conditions studied. It is important to note that the low gaseous outcome is strongly connected to a high CO coverage and a low H coverage over the Ni(111) surface, a situation that is even more evident in the case of MkM simulations. In fact, MkM simulations produce an almost 100% overall coverage, hindering gaseous production. The high CO coverage obstructs H2 activated adsorption, thus reducing the H present on the surface with the concomitant inhibition of the hydrogenation reactions to CH2O and CH4 (see the difference in event frequencies for H2 activated adsorption between Figs. S5 and S6 in the ESI for both methods†). Interestingly, the higher production obtained for kMC simulations is also associated with the coverage, as the CO coverage is lower while H coverage is higher (i.e., ∼30% and ∼4%, respectively) in comparison to the MkM simulations, and thus it is easier to hydrogenate CO. Water formation, which is stoichiometrically equivalent to CO production when the RWGS mechanism governs the reaction as reported in the previous section, is also drastically decreased under the new simulation conditions. Gaseous water is now produced at the same rate as CH2O, as shown in Fig. S6† for both methods, strongly suggesting that the mechanism followed corresponds to the following stoichiometry: CO2 + 2H2 → CH2O + H2O. A net hydrogen adsorption rate twice as large as the CO2 one further confirms the proposed stoichiometry.
:H2 molar ratio of 1:4. The condition label states which adsorbates cannot desorb from the Ni(111) surface: (I) CO, (II) CO and CH2O. The kMC values refer to the average value obtained from three different kMC runsa
| T = 673.15 K | Condition (I) | Condition (II) | ||
|---|---|---|---|---|
| P = 10 bar | MkM | kMC | MkM | kMC |
| a The desorption of CH2O is inhibited in these simulations. | ||||
| TOF CH2O (molecules per s per site) | 6.57 × 10−7 | (5.7 ± 0.9) × 10−3 | * | * |
| TOF CH4 (molecules per s per site) | 0 | (4 ± 1) × 10−5 | 0 | (6 ± 1) × 10−5 |
| θ CO (%) | 99.55 | 31.2 ± 0.3 | 99.91 | 44.4 ± 0.5 |
| θ H (%) | 0.02 | 3.53 ± 0.08 | 4.49 × 10−3 | 1.8 ± 0.1 |
| θ * (%) | 0.44 | 65.2 ± 0.5 | 0.08 | 53.2 ± 0.4 |
When CO adsorbed species are not able to desorb, both MkM and kMC point out the relevance of the HCO intermediate in the final formation of CH2O and methane production (Fig. 3a). The processes in which HCO participates are the dissociations into CO + H or CH + O and the hydrogenations to CH2O or HCOH with energy barriers of 0.21, 1.10, 0.71 and 0.93 eV, respectively. Clearly, the most plausible process is the dissociation into CO + H followed by CH2O formation. As shown in the previous section, the very low energy barrier for the former HCO dissociation is a hurdle for the reaction to result in other products besides CO. However, in these simulations, CO cannot desorb favouring the evolution through the HCO intermediate. A careful analysis of the simulations shows that most of the time CO adsorbed species hydrogenate to HCO, although they rapidly dissociate again into CO while eventually, some HCO is hydrogenated to CH2O which further desorbs to CH2O(g), as shown in Fig. S5 and S6 in the ESI.† HCO dissociation to CH + O is even more rare, and is 106 times slower than the CH2O formation reaction, which explains the lower CH4(g) formation reported in Table 3. It is important to consider that while these events are occurring, CO2 adsorbs and dissociates to CO + O leading to an increase in the CO coverage. As CO cannot desorb and the evolution of HCO to either CH2O or CH is quite unfavoured, the surface gets covered with CO with the associated decrease in the TOFs. Even if the CO coverage is around 30% for these simulations, the CO aggregation tendency in the Ni(111) surface starts to be notable (Fig. S7†). Interestingly, in Fig. 3a for kMC simulations there is an alternative path that involves the initial CO hydrogenation to COH instead of HCO (Fig. S6a†), which contributes to the final CH4 production and, to a lower extent, to CH2O formation through CH2OH. In this sense, after COH is formed it hydrogenates to HCOH, which can either hydrogenate to CH2OH with a barrier of 0.63 eV or surpass a 0.72 eV energy barrier to dissociate to CH, that further hydrogenates until CH4(g) formation. As both processes have similar energy barriers, the rates of both elementary steps are very similar. The event frequencies for all these processes can be found in Fig. S6 of Section S7 of the ESI.†
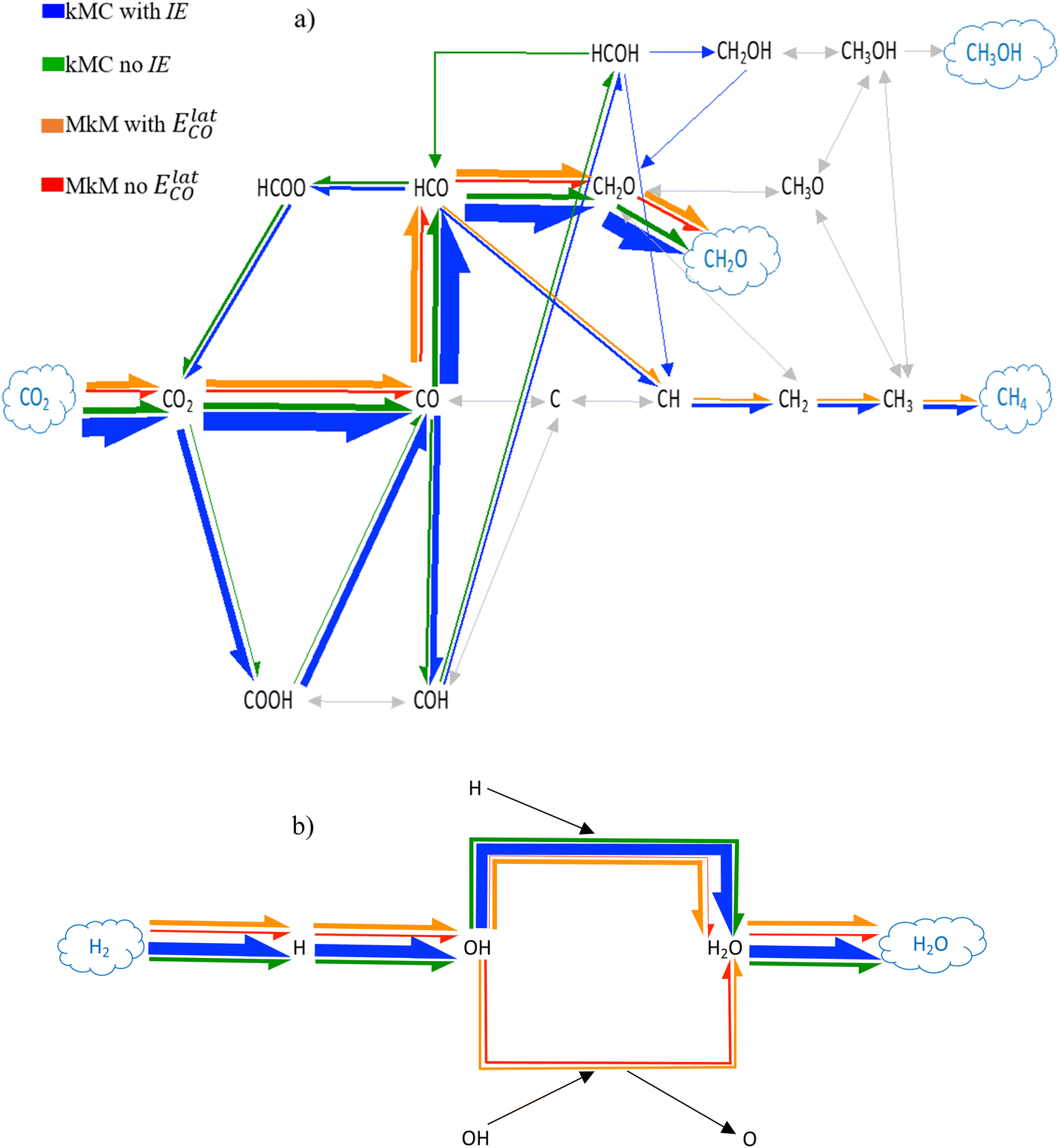 | ||
| Fig. 3 Schematic representation of the most plausible mechanism for (a) CO2 hydrogenation and (b) H2(g) transformation into H2O(g) over Ni(111) at T = 673.15 K and P = 10 bar with a CO2:H2 molar ratio of 1:4 under condition (I), in which CO is not allowed to desorb. Coloured arrows refer to produced events while grey arrows are for events that do not occur. The arrow's thickness symbolizes the net event frequency of each elementary process. MkM and kMC results in red and blue, respectively. MkM and kMC results without lateral interactions in red and green, respectively. MkM with coverage-dependent lateral potential (ElatCO) and kMC results with IE in orange and blue, respectively. For details on the calculations for the arrow's thickness the reader is referred to Section S5.† | ||
To assess the effect of adsorbate–adsorbate interactions in both kinetic methods we carried out two additional sets of simulations. We first ran simulations including the lateral potential (ElatCO) mentioned above in the microkinetic model, modifying the CO2 adsorption and desorption rates as a function of surface coverage. Details on the expressions of these laterally affected rates can be seen in Section S1 of the ESI.† Then, some kMC simulations with no interaction energies have been also performed. The results of these simulations at 673.15 K and 10 bar can be seen in Table 4. The TOFs and coverages shown in Table 4 show the importance of lateral interactions in the correct description of the system. The lateral potential allows MkM simulations to achieve physically meaningful CO coverage in contrast with the 99.55% value presented in previous MkM simulations. This is translated into an increase in CH2O(g) production of almost 2 orders of magnitude, as the lowering in CO coverage allows for the dissociative adsorption of H2(g) with the concomitant increase in H adsorbates, enabling further CO hydrogenation steps. This increase in H coverage also allows CO2 hydrogenation into methane. In addition, a larger number of H adsorbates in MkM simulations changes the mechanism by which water is formed (Fig. 3), pointing at OH hydrogenation and coming closer to kMC predictions. Even if the mean-field consideration of lateral potential improves the microkinetic model, the TOFs are still 2 orders of magnitude away from kMC values. The lower CO coverage and the higher H coverage kMC simulations present are thought to be one of the sources for disparity, as together, these enhance CO hydrogenation into more hydrogenated products (i.e., CH2O(g) and CH4(g)). Overall, the mean-field consideration of lateral potential improves the original microkinetic model, but topologic consideration of these interactions is needed to correctly describe systems with medium to high surface coverage.
:H2 molar ratio of 1:4 under condition (I) in which CO cannot desorb. The kMC values refer to the mean value obtained from three different kMC runs
| T = 673.15 K | Without lateral interactions | With lateral interactions | ||
|---|---|---|---|---|
| P = 10 bar condition (I) | MkM | kMC | MkM with ElatCO | kMC with IE |
| TOF CH2O (molecules per s per site) | 6.57 × 10−7 | (1.5 ± 0.6) × 10−5 | 3.69 × 10−5 | (4.4 ± 0.9) × 10−3 |
| TOF CH4 (molecules per s per site) | 0 | 0 | 5.44 × 10−7 | (4 ± 1) × 10−5 |
| θ CO (%) | 99.55 | 91.5 ± 0.2 | 52.77 | 31.2 ± 0.3 |
| θ H (%) | 0.02 | 2.34 ± 0.07 | 2.32 | 3.53 ± 0.08 |
| θ HCO (%) | 0 | 0.5 ± 0.1 | 0 | 0 |
| θ * (%) | 0.44 | 5.7 ± 0.2 | 44.91 | 65.2 ± 0.5 |
We also carried out kMC simulations without IE to get the true weight of the local lateral interactions in the system description. For these, the CO coverage increases significantly and gaseous production lowers notably following MkM suggestions. The notable increase in the CO coverage between both kMC simulations hinders all surface processes lowering their rates. The reason behind IE's influence on the final CO coverage is that when these interactions are included, a repulsive CO–CO interaction of 0.14 eV (see Table S3, Section S3 in the ESI†) between neighbouring CO causes them to diffuse in such a way that the overall energy of the system is minimized. Following this line of thought, the repulsive interaction eases CO desorption with the concomitant decrease in the CO coverage. However, when the interaction is neglected, the CO species adopt positions in which CO adsorbed species form CO islands (Fig. S7c†) that can also explain why kMC simulations without IE lead to a notable decrease in CH2O production, as only CO molecules from the borders of these islands are actually reactive. The CO surface control also affects the mechanism, inhibiting many processes, which demand the presence of other adsorbates such as H, an issue clearly illustrated in Fig. 3. The main difference in the kMC mechanisms when IE are allowed or not (see blue and green paths in Fig. 3, respectively) is that methane is no longer formed under the same conditions without interaction energies and only formaldehyde is formed.
Careful analysis of the energy barriers computed by kMC with IE shows that they influence the system in a way which favours gaseous production. First, histograms shown in Fig. S3,† show that interaction energies affect a path that leads both to methane and formaldehyde: CO hydrogenation to COH. The mean value of the forward energy barrier for this elementary step lowers by 0.15 eV from 2.28 to 2.13 eV while the mean value of the backwards energy barrier increases by 0.13 eV from 1.21 to 1.34 eV as shown in Fig. S3a and b.† The combination of these changes in the energy barriers enhances COH production, which appears to be a crucial intermediate towards methane production through HCOH. This effect of the interaction energies in the energy barriers also explains why MkM simulations with the lateral potential (ElatCO) are not able to identify the CO → COH → HCOH → CH → CH2 → CH3 → CH4 path as opposed to kMC. Our mean-field consideration of lateral interactions, which affects only desorption processes, cannot account for these changes in energy barriers. This translates into the omission of a path that produces methane at the same rate as HCO scission to CH in the kMC simulations. Table S5 in the ESI† shows that it is much more probable for a COH adsorbate not to immediately undergo dehydrogenation back to CO + H in kMC simulations instead of in MkM. This continues to prove that, even if they enhance the description of the system compared to conventional MkM simulations, mean-field lateral interactions are not able to fully report catalytic behaviour of medium to high coverage systems. In contrast, the local consideration of lateral interactions that kMC simulations provide allows for a better setup that allows us to better describe this kind of system.
Another difference in the mechanism derived from Fig. 3 is that the HCOH dissociation towards HCO is only observed in kMC simulations without IE. This process blocks further HCOH transformation either into CH2O(g) or CH4(g). This blocking of HCOH transformation only occurs in kMC simulations without IE because more HCOH is being formed from HCO when adsorbates interact with each other due to the lowering of the HCO hydrogenation barrier from 0.93 eV to 0.81 eV (see Fig. S2c, Section S4 in the ESI†). In addition, interaction energies slightly increase the energy barrier of HCOH dissociation to HCO from 0.53 to 0.56 eV as shown in Fig. S2d.† This small increase, added to the fact that more HCOH is being produced from HCO when interaction energies are considered, is suggested to be enough to allow other processes starting from HCOH to occur, instead of HCOH dissociating towards HCO and truncating other processes (i.e., HCOH dissociation to CH and its hydrogenation towards CH2OH), which eventually lead to methane and formaldehyde.
Let us now analyse condition (II) in which CO and CH2O desorptions are restricted. Fig. 4 shows the more probable processes when CO and CH2O are not allowed to desorb in MkM and kMC simulations. The reader may notice that no red arrows corresponding to MkM simulations are present in the diagram and this is because rates for MkM simulations were so low that the discerning method used for constructing the diagrams neglected them (see Section 2.1). Therefore, for this setup the MkM simulations suggest that Ni(111) is inactive for CO2 hydrogenation to methane according to the outstandingly high CO coverage observed (99.91% at 673.15 K and 10 bar). For kMC simulations, the methane production exhibits a slightly higher rate than that in the situation where only CO could not leave the nickel surface. CH4 was a secondary product for the situations where CO could not desorb while CH2O was the main product by far. As CH2O cannot desorb now, the events that lead to further hydrogenation into methane are more frequent. Again, the main processes involving adsorbed CO are the hydrogenation to CH2O through HCO. Nevertheless, CH2O quickly dehydrogenates back to HCO due to its small barrier (0.45 eV) and the fact that CH2O desorption is blocked in these simulations. Thus, even though the sequence CO → HCO → CH2O and their reverse reactions are by far the most executed processes, the net balance is 0 and no methane is produced from this path. The main part of methane is produced by the sequence CO → COH → HCOH → CH → CH2 → CH3 → CH4, although some methane is also formed through HCO formation and further dissociation to CH, which is finally, hydrogenated to methane.
 | ||
| Fig. 4 Schematic representation of the most plausible mechanism for (a) CO2 hydrogenation and (b) H2(g) transformation into H2O(g) over Ni(111) at T = 673.15 K and P = 10 bar with a CO2:H2 molar ratio of 1:4 under condition (II), in which CO and CH2O are not allowed to desorb. Coloured arrows refer to produced events while grey arrows are for events that do not occur. The arrow’s thickness symbolizes the net event frequency of each elementary process. MkM with coverage-dependent lateral potential (ElatCO) and kMC results with interaction energies (IE) in orange and blue, respectively. For details on the calculations for the arrow's thickness the reader is referred to Section S5.† | ||
Looking at the mean energy barrier values of some important steps (see histograms in Fig. S4†) is useful for understanding the source of these mechanistic differences. For instance, due to the interaction energies the mean energy barrier of COH formation is reduced from 2.28 eV, at the zero-coverage limit, to 2.04 eV (see Fig. S4a†). On the other hand, backward reactions are more hindered when interaction energies act, as the 1.43 eV value contrasts with the 1.21 eV value at the zero-coverage limit (see Fig. S4b†). Consequently, it is easier to achieve COH when interaction energies are active to then hydrogenate through COH + H → HCOH, whose barrier also decreases from 0.82 eV to 0.74 eV due to interaction energies (see Fig. S4c†). Interestingly, the effect of the interaction energies as a response of the new coverage favours the execution of these important elementary steps. This translates into an increase in COH production and further hydrogenation to HCOH to finally produce methane, which becomes the most favoured path (see Fig. 4). This fact contrasts with the results shown in Fig. 3a in which both paths contribute similarly to the final methane production. Additionally, as the path towards formaldehyde from HCOH is now impeded as CH2O cannot desorb, HCOH tends to dissociate towards CH with the associated increase in methane formation. Thus, easing the CO → COH and COH → HCOH paths is the reason for this mechanistic divergence between MkM and kMC for the situation in which neither CO nor CH2O can desorb. Note that no statistical outcome should be extracted from these simulations where CO and CH2O cannot desorb as the methane production is minimal (<10 molecules). As in previous sections, we applied a coverage-dependent lateral potential to MkM simulations (Section 3.1) to assess the importance of lateral interactions. As the orange path in Fig. 4 shows, the incorporation of mean-field lateral interactions allows MkM to capture methane formation following kMC and literature36 suggestions. We reach a TOFCH4 100 times smaller than the literature value36 at 673.15 K, and the H coverage seems to be the difference. Our simulations present 2% of H coverage against the 20% presented in ref. 36. Interestingly, as for the situations where only CO is not able to desorb, this coverage-dependent lateral potential (ElatCO) is not able to account for methane formation through COH and HCOH. Now this contrast between the system's behaviour when lateral interactions are considered either locally or by means of a mean-field approximation is more crucial. The reasoning behind this is that the path of MkM simulations with the coverage-dependent lateral potential omission is the majoritarian path towards methane. The way the local consideration of lateral interactions unravels their effect on energy barriers as explained above allows for a better description of the system. Finally, new kMC simulations without interaction energies were run for the situation in which neither CO nor CH2O can desorb in order to quantify the effect of adsorbate–adsorbate lateral interactions. Again, the CO coverage significantly increases hindering methane formation in agreement with MkM simulations.
Finally, regarding the computational cost of both approaches it is worth mentioning the huge difference between them. MkM simulations are by far less demanding, spending only a few seconds to find the steady-state solution. Even when calculating the DRC of several reactions, the computational time spent by MkM is at most around one minute for such a complex reaction network. In contrast, kMC simulations could run several days to reach the steady-state and obtain statistically meaningful results. Nevertheless, even if they are clearly more computationally demanding, they provide more detailed information about the catalytic processes occurring under real working conditions than MkM simulations.
4. Conclusions
The complex CO2 hydrogenation reaction over the Ni(111) surface has been thoroughly studied by means of two different kinetic approaches, kinetic Monte Carlo and Microkinetic Modelling simulations, in order to unravel the differences between them and identify the situations in which each approach is more suitable. With this objective in mind, different simulations have been performed in which all the possible products of the CO2 hydrogenation reaction can desorb, while in some simulations the desorption process of certain products has been blocked. Although neglecting the possible desorption of intermediate products should be avoided when performing kinetic simulations, this provides situations with very different coverages, allowing for a better understanding of the differences between the two kinetic approaches.For the simulations with low coverage, corresponding to the situation in which all products can desorb, both kinetic methods produce similar results in TOF, coverage, and mechanism under different working conditions, with CO being the major product. The lower coverages predicted by MkM simulations are associated with the use of the mean-field approximation which makes species generally more reactive, resulting in a concomitant decrease in their coverage. Moreover, as CO is mainly produced from CO2 dissociation, a lower CO2 coverage translates into a lower CO production, which explains the lower TOF observed for the MkM simulations compared to the kMC ones. In addition, a careful analysis of the effect of adsorbate–adsorbate interactions reveals their impact on the final coverages and product distributions and highlights the importance of lateral interactions in better defining the catalytic properties, even if their effect seems to be minimized under low-coverage conditions. The relevance of these lateral interactions is further studied through simulations in which the desorption of different products is restricted, leading to higher surface coverages where adsorbate–adsorbate interactions are more significant. In these high-coverage simulations, a coverage-dependent lateral potential applied in MkM simulations allows for a better description of the system than conventional MkM simulations. Nevertheless, a local treatment of interaction energies, as provided by kMC, is required to fully account for their influence on the system's behaviour.
The present results suggest that MkM simulations provide results comparable to those from more intricate kMC simulations for systems with low coverages, where the effect of lateral interactions is less likely to be significant. Therefore, given the substantial time savings offered by MkM simulations compared to kMC and their relatively accurate representation of system behaviour, MkM appears to be a viable option for an initial assessment of a system's performance. However, for systems with high coverages, kMC simulations incorporating lateral interactions seem to be the more appropriate choice for an accurate representation of system behaviour. Finally, these findings are likely to be relevant in other types of catalysts and reactions and could guide theoreticians in choosing the most appropriate kinetic method.
Data availability
The data supporting this article have been included as part of the ESI.† These data include information about the lateral potential applied to the microkinetic model, formation energy calculations and the set of reference species used, the cluster expansion Hamiltonians, the calculation process and values for lateral interactions, reaction energies and reaction energy barriers for all elementary processes, the Brønsted–Evans–Polanyi relationship, histograms regarding relevant energy barriers and TOFs and coverages for the most relevant species under different temperature and pressure conditions. The input and output files for both MkM and kMC simulations under all the working conditions sampled are available on GitHub (https://github.com/alejandrograciag/CO2_hydro_Ni111).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study has been supported by the Spanish Ministry of Science and Innovation (MICIN) through the project PID2022-138180OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by the European Union through ERDF A way of making Europe. Additional support has been provided by the CEX2021-001202-M María de Maeztu Unit of Excellence through the A. G. PhD grant and by the Generalitat de Catalunya (project 2021SGR00079 and P. G. Serra Hunter Associate Professorship). Computational resources provided by Red Española de Supercomputación (RES) and by Consorci de Serveis Universitaris de Catalunya (CSUC) are gratefully acknowledged, the latter with financial support from Universitat de Barcelona.References
- Haber-Bosch and Other Industrial Processes, in Catalysts for Nitrogen Fixation, ed. G. J. Leigh, Springer, Netherlands, Dordrecht, 2004, pp. 33–54 Search PubMed.
- W. Moeller and K. Winkler, J. Air Pollut. Control Assoc., 1968, 18, 324–325 CrossRef CAS.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev. - Sci. Eng., 1999, 41, 255–318 CrossRef CAS.
- P. Lozano-Reis, H. Prats, R. Sayós and F. Illas, J. Catal., 2023, 425, 203–211 CrossRef CAS.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, Nat. Catal., 2019, 2, 659–670 CrossRef CAS.
- M. Pineda and M. Stamatakis, J. Chem. Phys., 2022, 156, 120902 CrossRef CAS PubMed.
- L. Grajciar, C. J. Heard, A. A. Bondarenko, M. V. Polynski, J. Meeprasert, A. E. Pidko and P. Nachtigall, Chem. Soc. Rev., 2018, 47, 8307–8348 RSC.
- A. H. Motagamwala and J. A. Dumesic, Chem. Rev., 2021, 121, 1049–1076 CrossRef CAS PubMed.
- H. Ritchie, M. Roser and P. Rosado, Energy mix, Published online at https://OurWorldInData.org, 2020.
- National Oceanic and Atmospheric Administration, Global Monitoring Laboratory.
- I. Nagelkerken and S. D. Connell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13272–13277 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC.
- Y. Hartadi, D. Widmann and R. J. Behm, ChemSusChem, 2015, 8, 456–465 CrossRef CAS PubMed.
- H. Sakurai and M. Haruta, Catal. Today, 1996, 29, 361–365 CrossRef CAS.
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef CAS.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, J. Catal., 2016, 343, 115–126 CrossRef CAS.
- A. G. Kharaji, A. Shariati and M. A. Takassi, Chin. J. Chem. Eng., 2013, 21, 1007–1014 CrossRef CAS.
- H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, Appl. Catal., A, 2001, 205, 285–294 CrossRef CAS.
- L. Yang, L. Pastor-Pérez, S. Gu, A. Sepúlveda-Escribano and T. R. Reina, Appl. Catal., B, 2018, 232, 464–471 CrossRef CAS.
- V. Jiménez, P. Sánchez, P. Panagiotopoulou, J. L. Valverde and A. Romero, Appl. Catal., A, 2010, 390, 35–44 CrossRef.
- S. Eckle, H. G. Anfang and R. J. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CrossRef CAS.
- S. W. Chang, M. S. Kuo, M. T. Tsay and M. C. Hsieh, Appl. Catal., A, 2003, 247, 309–320 CrossRef.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- J. N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- W. Wei and G. Jinlong, Front. Chem. Sci. Eng., 2011, 5, 2–10 CrossRef.
- C. Zhi, Q. W. Wang, B. Wang, D. Li and R. Zhang, RSC Adv., 2015, 5, 66742–66756 RSC.
- M. Zhou and B. Liu, ChemCatChem, 2015, 7, 3928–3935 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- D. W. Blaylock, T. Ogura, W. H. Green and G. J. O. Beran, J. Phys. Chem. C, 2009, 113, 4898–4908 CrossRef CAS.
- R. C. Catapan, A. M. M. Oliveira, Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2012, 116, 20281–20291 CrossRef CAS.
- C. Vogt, M. Monai, E. B. Sterk, J. Palle, A. E. M. Melcherts, B. Zijlstra, E. Groeneveld, P. H. Berben, J. M. Boereboom, E. J. M. Hensen, F. Meirer, I. A. W. Filot and B. M. Weckhuysen, Nat. Commun., 2019, 10, 5330 CrossRef PubMed.
- P. Lozano-Reis, H. Prats, P. Gamallo, F. Illas and R. Sayós, ACS Catal., 2020, 10, 8077–8089 CrossRef CAS.
- M. Andersen, C. P. Plaisance and K. Reuter, J. Chem. Phys., 2017, 147, 152705 CrossRef PubMed.
- M. J. Hoffmann and T. Bligaard, J. Chem. Theory Comput., 2018, 14, 1583–1593 CrossRef CAS PubMed.
- H. Prats, F. Illas and R. Sayós, Int. J. Quantum Chem., 2018, 118, e25518 CrossRef.
- N. Mehra and W. F. Schneider, Catal. Sci. Technol., 2024, 14, 4869–4881 RSC.
- I. A. W. Filot, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2014, 53, 12746–12750 CrossRef CAS PubMed.
- B. Zijlstra, R. J. P. Broos, W. Chen, G. L. Bezemer, I. A. W. Filot and E. J. M. Hensen, ACS Catal., 2020, 10, 9376–9400 CrossRef CAS.
- M. Stamatakis and D. G. Vlachos, J. Chem. Phys., 2011, 134, 214115 CrossRef PubMed.
- M. Stamatakis and D. G. Vlachos, ACS Catal., 2012, 2, 2648–2663 CrossRef CAS.
- J. Nielsen, M. d'Avezac, J. Hetherington and M. Stamatakis, J. Chem. Phys., 2013, 139, 224706 CrossRef PubMed.
- H. Prats, S. Posada-Pérez, J. A. Rodriguez, R. Sayós and F. Illas, ACS Catal., 2019, 9, 9117–9126 CrossRef CAS.
- H. Prats, L. Álvarez, F. Illas and R. Sayós, J. Catal., 2016, 333, 217–226 CrossRef CAS.
- S. Piccinin and M. Stamatakis, ACS Catal., 2014, 4, 2143–2152 Search PubMed.
- P. Lozano-Reis, P. Gamallo, R. Sayós and F. Illas, ACS Catal., 2024, 14, 2284–2299 CrossRef CAS PubMed.
- C. T. Campbell, ACS Catal., 2017, 7, 2770–2779 CrossRef CAS.
- E. B. Sterk, A. Nieuwelink, M. Monai, J. N. Louwen, E. T. C. Vogt, I. A. W. Filot and B. M. Weckhuysen, JACS Au, 2022, 12, 2714–2730 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00240k |
| This journal is © The Royal Society of Chemistry 2025 |