
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent cross-linking approaches for all-trans retinoic acid-loaded thermo-responsive hydrogels†
Xueli
Mei
a,
Robert C.
Stewart
b,
Xiao Zhen
Zhou
 bcd,
Kun Ping
Lu
be and
Elizabeth R.
Gillies
*af
bcd,
Kun Ping
Lu
be and
Elizabeth R.
Gillies
*af
aDepartment of Chemistry, The University of Western Ontario, 1151 Richmond St., London, Ontario N6A 5B7, Canada. E-mail: egillie@uwo.ca
bDepartments of Biochemistry and Oncology, The University of Western Ontario, 1151 Richmond St., London, Ontario N6G 2V4, Canada
cDepartment of Pathology and Laboratory Medicine, The University of Western Ontario, 1151 Richmond St., London, Ontario N6G 2V4, Canada
dLawson Health Research Institute, The University of Western Ontario, London, ON N6G 2V4, Canada
eRobarts Research Institute, The University of Western Ontario, 1151 Richmond St., London, Ontario N6G 2V4, Canada
fDepartment of Chemical and Biochemical Engineering, The University of Western Ontario, 1151 Richmond St., London, Ontario N6A 5B9, Canada
First published on 15th May 2025
Abstract
All-trans retinoic acid (ATRA) is a promising therapeutic for the treatment of a wide range of cancers. However, its short half-life, poor water-solubility, and low stability in vivo hinder its use. The development of injectable controlled release systems for ATRA delivery can potentially address these challenges. Building on a poly(caprolactone-co-lactide)–poly(ethylene glycol)–poly(caprolactone-co-lactide) (PCLA–PEG–PCLA) triblock copolymer system that undergoes thermo-responsive gelation at 37 °C, we explore and compare different approaches to stabilize the gels through covalent bonding. The attempted cross-linking of methacrylate end-capped PCLA–PEG–PCLA through thiol-Michael addition reactions using small molecule and 4-arm-PEG thiols led to precipitation rather than gelation. However, azide end-capped PCLA–PEG–PCLA was gelled using 5 kg mol−1 4-arm-PEG with terminal dibenzocyclooctyne (DIBAC) groups by strain-promoted azide–alkyne cycloaddition. This hydrogel was then compared with previously reported methacrylate end-capped PCLA–PEG–PCLA hydrogels cross-linked by free radical chemistry, as well as non-covalently cross-linked hydrogels. The azide–alkyne hydrogels exhibited properties intermediate between the free radical and non-covalently cross-linked gels. Incorporation of ATRA substantially disrupted the free radical cross-linking, but imparted only modest changes in the azide–alkyne gels. ATRA was released over about two weeks. The proliferation of MDA-MB-468 cells in the presence of ATRA-loaded and control azide–alkyne gels was investigated. The ATRA-loaded gel released active drug, while the unloaded gel did not affect proliferation.
Introduction
Cancer greatly affects the quality of life of patients and leads to millions of deaths each year worldwide.1 However, many current therapies suffer from harmful side effects as chemotherapeutics also kill healthy cells, motivating the ongoing search for more targeted and effective drugs.2,3 Peptidyl-prolyl isomerase (Pin1) is a promising therapeutic target for cancer, as its overexpression aids in the dysregulation of oncoproteins and tumour suppressors, thereby promoting cancer, cancer stem, and stromal cells.4,5 Zhou, Lu and coworkers recently discovered that all-trans retinoic acid (ATRA, Fig. 1a), one of the first targeted cancer therapies, inhibits and promotes the degradation of Pin1.4,6,7 In tamoxifen-resistant breast cancer cells, they found that ATRA treatment overcame drug resistance, suppressing cell viability and proliferation.8 However, ATRA has a half-life of only 45 minutes in humans, which is insufficient for the treatment of solid tumours.9 Furthermore, it is highly hydrophobic,10 and susceptible to photodegradation.11 Thus, ATRA could benefit from the protective and sustained release effects provided by a delivery vehicle. | ||
| Fig. 1 (a) Chemical structure of ATRA; (b) schematic representation of gelation of a thermo-responsive triblock copolymer; (c) thermal gelation accompanied by free radical covalent cross-linking from previous work;12 (d) approaches explored in this work using small molecule or multi-arm macromolecules as cross-linkers. | ||
Thus far, several delivery vehicles have been explored for ATRA. Tablets composed of cholesterol, cellulose, lactose, phosphates and stearates were used to release ATRA over 21 days, thereby reducing the growth of tamoxifen-resistant human breast cancer xenografts in mice.8 ATRA-loaded poly(L-lactic acid) microparticles had a stronger anti-tumor effect than the slow-release ATRA tablets in a xenograft mouse model of hepatocellular carcinoma.13 Pluronic F127, a triblock copolymer of poly(ethylene glycol) (PEG)–poly(propylene glycol)–PEG was conjugated to ATRA to form a micellar prodrug.14 When combined with cisplatin, pluronic-ATRA suppressed breast tumor growth in mice. Hydrogel ATRA formulations have also been explored. Hydrogels were formed through electrostatically-induced partial denaturation and self-assembly of albumin. ATRA was incorporated through its binding to albumin and after an initial burst release, exhibited a linear release over 10 days.15 The bioactivity of released ATRA was confirmed by its ability to inhibit human aortic smooth muscle cell migration in a scratch wound assay. In another example, hydrogel meshes were prepared by 3D printing of alginate-gelatin methacryloyl (GelMA) hydrogels containing ATRA-loaded polycaprolactone microparticles.16 ATRA has also been incorporated into zeolite nanoparticles, which were then incorporated into ε-poly(L-lysine) grafted GelMA hydrogels.17 These materials showed promise for localized injection to treat periodontitis.
Recently, Gillies and coworkers prepared an injectable hydrogel based on poly(caprolactone-co-lactide)–PEG–poly(caprolactone-co-lactide) (PCLA–PEG–PCLA) triblock copolymers with methacrylate end groups.12 The copolymer formulation was thermo-responsive, behaving as a liquid suspension of polymer assemblies at room temperature and below, while undergoing spontaneous gelation at physiological temperature (37 °C) (Fig. 1b).18–20 The addition of N,N,N′,N′-tetramethylethylenediamine (TEMED) and potassium persulfate (KPS) induced free radical polymerization of the methacrylate groups, leading to covalent cross-linking (Fig. 1c).12 The formulation was successfully injected into the joints of rats and horses.12,21 The covalent gelation process improved the stability and mechanical properties of the hydrogel, and enabled the sustained release of celecoxib in horse joints for at least 60 days.12 These properties are particularly important for intra-articular delivery, where the hydrogel is subjected to repeated mechanical loading and where the delivery system can only be administered every few months. The hydrogels were also well tolerated in vivo. However, the use of free radical initiators for gelation in vivo requires further study in terms of potential toxic side effects. Moreover, since ATRA has five conjugated double bonds and is known to react with free radicals,22 it may be incompatible the use of free radical chemistry for its direct incorporation into the hydrogel.
In this context, we were motivated to investigate alternative covalent cross-linking mechanisms to stabilize the hydrogels. Here we explore the use of two different covalent cross-linking reactions (Fig. 1d). One approach uses thiol-containing small molecules or multi-arm PEG-thiols to react with the terminal methacrylate groups on the triblock copolymer by thiol-Michael addition reactions.23 The other approach involves modification of the triblock copolymer with terminal azide groups and use of dibenzoazacyclooctyne (DIBAC) functionalized multi-arm PEG to cross-link the azides.24 Initial gelation tests were systematically performed to screen for the best method and cross-linker pair. Characterization of the resulting hydrogels by syneresis, degradation, and mechanical was performed. Then the encapsulation of ATRA was investigated, followed by studies of its effect on the properties of the hydrogels, evaluation of its in vitro release rates, and in vitro effects on cell proliferation.
Experimental
General materials
The syntheses of 4-arm-PEG-ester-SH and 4-arm-PEG-DIBAC are included in the ESI.† Uncapped PCLA–PEG–PCLA,25 acetate end-capped PCLA–PEG–PCLA (Ac–PCLA–PEG–PCLA–Ac),25,26 and methacrylate end-capped PCLA–PEG–PCLA (M–PCLA–PEG–PCLA–M)12 were synthesized as previously reported. 2-Azidoacetic acid27 and dibenzoazacyclooctyne-acid (DIBAC-CO2H)28 were synthesized as previously reported. p-Toluenesulfonic acid, concentrated hydrochloride acid, pentaerythritol tetrakis (3-mercaptopropionate) (PETMP) were purchased from Sigma (Oakville, Canada). NaCl, LiBr, MgSO4, diethyl ether, and toluene were purchased from Thermo Fisher Scientific (Mississauga, Canada). 3-Mercaptopropionic acid, cyclohexane, were purchased from Alfa Aesar (Massachusetts, US). Na2HPO3, KH2PO3, CH2Cl2, and N,N-dimethylformamide (DMF) were purchased from Caledon (Halton Hills, Canada). NEt3 was purchased from Supelco (Pennsylvania, US) and KCl was purchased from EMD Millipore (Oakville, Canada). N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) was purchased from Advanced ChemTech (Louisville, US). 4-Dimethylaminopyridine (DMAP) was purchased from AK Scientific. 4-arm-PEG2000 and 4-arm-PEG5000 were purchased from JenKem Technology USA Inc. (Plano, US). All chemicals were used as received unless otherwise noted. Phosphate-buffered saline (PBS) was prepared from deionized water with 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl. The pH was adjusted to 7.4 with concentrated NaOH. Under a nitrogen atmosphere, toluene was distilled over sodium, while NEt3 and CH2Cl2 were distilled over CaH2.General procedures
Column chromatography was performed using silica gel (0.040–0.063 mm particle size, 230–430 mesh). NMR spectroscopy was conducted on a Bruker AvIII HD 400 MHz Spectrometer (1H 400.09 MHz, 13C 100.5 MHz). The 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane and were calibrated against CHCl3 (7.26 ppm) and CDCl3 (77 ppm) respectively. Coupling constants (J) are expressed in Hertz (Hz). Fourier transform infrared (FTIR) spectra were recorded using a PerkinElmer Spectrum Two FTIR spectrometer with an attenuated total reflectance (ATR) attachment and a single reflection diamond. Size-exclusion chromatograms (SEC) were obtained in DMF containing 10 mM LiBr and 1% (v/v) NEt3. The instrument was composed of a Waters 515 HPLC pump, Waters In-Line Degasser AF, Waters Temperature Control Module II equipped with two PLgel mixed-D 5 μm (300 × 1.5 mm) columns attached to a corresponding PLgel guard column (Agilent Technologies), and a Wyatt Optilab Rex RI detector. Samples were dissolved in the mobile phase solution at a concentration of ∼5 mg mL−1 and filtered through a 0.2 μm polytetrafluoroethylene syringe filter prior to injection using a 50 μL loop. Samples were run at a flow rate of 1 mL min−1 for 30 min at 85 °C. Molar masses of the samples were calculated based on PEG standards.Synthesis of azide end-capped PCLA–PEG–PCLA (Az–PCLA–PEG–PCLA–Az)
To a flame-dried three neck 100 mL round bottom flask, 2.0 g (0.64 mmol, 1.0 equiv.) of PCLA–PEG–PCLA25 was added under N2 flow, followed by 10 mL of dry CH2Cl2. To this solution, 0.74 g of EDC·HCl (3.8 mmol, 6.0 equiv.) and 0.090 g of DMAP (0.77 mmol, 1.2 equiv.) were added, and the reaction mixture was stirred until fully dissolved. The flask was then placed in an ice bath. Next, 0.39 g (3.8 mmol, 6.0 equiv.) of 2-azidoacetic acid was carefully added dropwise into the cooled flask under N2. The reaction was protected from light and stirred under N2 overnight. The reaction mixture was then diluted with 50 mL of CH2Cl2 and washed with 0.1 M aqueous NaHCO3 (50 mL) three times, followed by 0.1 M aqueous citric acid (50 mL) three times. The organic layer was collected and dried over MgSO4. The functionalized polymer was obtained by precipitation in 50 mL of cold pentane. Yield = 1.3 g, 60%. 1H NMR (CDCl3, 400 MHz): δ 5.30–5.01 (m, 6.7 H), 4.39–4.27 (m, 2.5 H), 4.21 (t, J = 4.0 Hz, 3.4 H), 4.18–4.08 (m, 8.2 H), 4.04 (t, J = 8.0 Hz, 17.4 H), 4.00–3.83 (m, 5.5 H), 3.63 (s, 132 H), 2.45–2.36 (m, 7.3 H), 2.29 (t, J = 8.0 Hz, 18.1 H), 1.76–1.60 (m, 53.4 H), 1.60–1.47 (m, 18.1 H), 1.47–1.28 (m, 25.4 H). IR: 2940, 2965, 2110, 1730, 1580 cm−1. DMF SEC: Mn = 2302 g mol−1, Mw = 2951 g mol−1, Đ = 1.28.Hydrogel preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methacrylate ratios of 1:1, 2:1, and 4:1. The mixture was vortexed for 5 s and then placed in a 37 °C oven for 10 min. For the macromolecular cross-linkers, 4-arm-PEG2000-ester-SH (12.9 mg, 25.8 mg or 51.6 mg for 1, 2 and 4 equivalents relative to the methacrylate respectively) or 4-arm-PEG5000-ester-SH (29.2 mg, 58.4 mg or 117 mg for 1, 2 and 4 equivalents relative to the methacrylate respectively) was first dissolved with vortexing in 30 μL of deionized water and cooled to 4 °C. Then, 200 mg of the cold (∼4 °C) hydrogel formulation was added to the vial. It was then vortexed for 5 s and then placed in the 37 °C oven for 10 min.
:1 azide:alkyne ratio. 50 μL of pre-cooled MilliQ water was added to the vial followed by 10 s vortexing to dissolve the polymer. This solution was then placed in an ice bath for another 5 min. 100 mg of the cold (∼4 °C) hydrogel formulation was then added to the vial followed by 5 s vortexing and then placed in the 37 °C oven for 20 min.
:methacrylate ratios of 1:1, 2:1, and 4:1. The mixture was vortexed for 5 s and then placed in a 37 °C oven for 10 min. For the macromolecular cross-linkers, 4-arm-PEG2000-ester-SH (12.9 mg, 25.8 mg or 51.6 mg for 1, 2 and 4 equivalents relative to the methacrylate respectively) or 4-arm-PEG5000-ester-SH (29.2 mg, 58.4 mg or 117 mg for 1, 2 and 4 equivalents relative to the methacrylate respectively) was first dissolved with vortexing in 30 μL of deionized water and cooled to 4 °C. Then, 200 mg of the cold (∼4 °C) hydrogel formulation was added to the vial. It was then vortexed for 5 s and then placed in the 37 °C oven for 10 min.
:1 azide:alkyne ratio. 50 μL of pre-cooled MilliQ water was added to the vial followed by 10 s vortexing to dissolve the polymer. This solution was then placed in an ice bath for another 5 min. 100 mg of the cold (∼4 °C) hydrogel formulation was then added to the vial followed by 5 s vortexing and then placed in the 37 °C oven for 20 min.
Preparation of ATRA-loaded hydrogels
All hydrogels were prepared in a dark room and were protected from light using aluminum foil.Measurement of hydrogel syneresis
Hydrogels (0.10 g total formulation) were prepared in 3 mL vials as described above. The mass of each hydrogel was accurately measured. The vials were then placed in a 37 °C oven. At specified time points, the vials were removed, uncapped, and inverted for one min on a paper towel, allowing released water to flow from the vial. The vials were then re-weighed before being recapped and placed back in the oven. Measurements were performed in triplicate and the results are reported as the mean ± standard deviation.Measurement of hydrogel degradation
Hydrogels (0.10 g total formulation) were prepared in 3 mL vials as described above. The mass of each hydrogel was accurately measured. Then, 3.0 mL of PBS was added to each vial, and the vial was placed in the 37 °C oven. At specified time points, the vials were removed, uncapped, and inverted for 3 min, allowing the PBS to drain completely onto a paper towel, and then the vials were re-weighed. Next, 3.0 mL of fresh PBS was added to the vial, and it was placed back in the 37 °C oven until the next time point. The measurements were performed in triplicate and the results are reported as the mean ± standard deviation.Measurement of Young's moduli under unconfined compression
The covalently cross-linked hydrogels (0.50 g total formulation) were prepared in 5 mL vials as described above except that after 5 s of vortexing, the formulation was taken up in a pre-cooled 1 mL syringe. The syringe was then placed in a 37 °C oven for 20 min. Next, the cylindrical hydrogel samples were taken out of the syringes and cut into 100 mg cylindrical samples. The hydrogel without covalent cross-linking was prepared in a 5 mL glass vial without cutting as described above, and then maintained at 37 °C continually until measurement due to the reversible sol–gel transition. Right before the measurement, hydrogel was quickly taken out of the vial and put on the testing stub. Mechanical testing of hydrogel samples was performed using a CellScale Univert (Waterloo, ON, Canada), using a 0.5 N load cell. Samples were immersed in PBS at 37 °C and then loaded with uniaxial compression to a total strain of 30%, at a constant rate of 4% s−1. Secant moduli, calculated as the slope between 10 and 20% strain, were determined. All systems were measured in triplicate and the results are reported as the mean ± standard deviation.In vitro release of ATRA in PBS with 2% (w/w) Tween 80
ATRA-loaded hydrogels were prepared as described above in 5 mL glass vials. Free ATRA (10 mg) in a nylon mesh bag (37-micron pore size, Supreme Rosin, Canada) or ATRA-loaded hydrogels (100 mg) were immersed in 3.0 mL of release medium (pH 7.4 PBS containing 2% w/w Tween 80) in a vial. All vials were covered with aluminum foil to protect the samples from light and kept in a 37 °C oven. At specific time points, the release medium of each system was sampled and replaced with fresh release medium. The ATRA concentrations in the release media were determined based on the absorbance at 350 nm using a Varian Cary 300 Bio UV-visible spectrometer (Varian, USA) based on an extinction coefficient of 7.57 × 104 M−1 cm−1, while keeping the samples as protected as possible from light. The experiments were performed in triplicate and the results are reported as the mean ± standard deviation.In vitro release of ATRA in cell culture media
ATRA-loaded hydrogels were prepared as described above except that 50 mg was prepared and only 0.5 mg of ATRA (1% (w/w) relative to the whole gel mass) was incorporated. These release experiments were performed as described for the studies in PBS with 2% Tween 80, except that the release medium was 7.0 mL of Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum but without phenol red. Measurements were performed in triplicate and the results are reported as the mean ± standard deviation.In vitro proliferation assay of ATRA-loaded SPAAC hydrogel
The hydrogels were prepared as described above in sterile 5 mL glass vials except that 100 mg was prepared and only 1.0 mg of ATRA (1% (w/w) relative to the whole gel mass) was incorporated. 100 mg of non-drug-loaded (control) SPAAC hydrogel without ATRA was also prepared as the vehicle control group. All the hydrogels were prepared in a biosafety cabinet using sterile PBS, Az–PCLA–PEG–PCLA–Az formulation, vials and syringes. For ATRA treatment groups, the concentration was set to 10, 20, 30, 40 and 50 μM. Then, in a biosafety cabinet, ATRA loaded SPAAC gel was taken out of the vial, carefully weighed on an analytical balance and cut into single piece samples with the weight of 1.2 mg, 2.4 mg, 3.6 mg, 4.8 mg and 6.0 mg to match the previously mentioned concentration targeted for each group. For the vehicle control group, 6.0 mg of control SPAAC gel was carefully weighed, cut and added to match the mass of the hydrogel added in the highest concentration ATRA treatment group. MDA-MB-468 cells (shared by Dr Allison Alan's lab, Western University) were maintained in DMEM (Wisent, Saint-Jean-Baptiste, Canada) containing 10% FBS (Wisent, Saint-Jean-Baptiste, Canada) until 70–90% confluence. The cells were lifted using 1× TrypLE (12604013) (Invitrogen, Waltham, USA) and seeded at a density of 50000 cells per well in a 6 well plate. For ATRA treatment groups, either free ATRA (10, 20, 30, 40 and 50 μM) or 1% (w/w) ATRA-loaded SPAAC hydrogels (1.2 mg, 2.4 mg, 3.6 mg, 4.8 mg and 6.0 mg, respectively) were added to the wells (triplicate for each ATRA concentration). For the vehicle control group, 6.0 mg control SPAAC hydrogel was added to the cells (triplicate). After 5 days in a shaker incubator, the cells were fixed in formalin for 15 min and subsequently stained with Hoechst 33342 (Thermo Fisher Scientific, Mississauga, Canada) for 15 min. Cell counts were generated by imaging and analysis using a BioTek Cytation 1 cell imaging multi-mode reader (Agilent Technologies, Inc., Santa Clara, US) with Gen5 software.
Results and discussion
Polymer synthesis
PCLA–PEG–PCLA and its methacrylated derivative M–PCLA–PEG–PCLA–M (Scheme 1) were synthesized as previously reported.12,25,26 The batch of M–PCLA–PEG–PCLA–M used in the present work had an Mn of 3889 g mol−1, PCLA/PEG mass ratio of 0.98, and a CL/LA ratio of 1.63 as determined based on 1H NMR spectroscopy (Fig. S1, ESI†). SEC indicated an Mn of 2207 g mol−1 and a Đ of 1.39 relative to PEG standards (Fig. S2, ESI†). For the preparation of Ac–PCLA–PEG–PCLA–Ac, hydroxyl-terminated PCLA–PEG–PCLA was reacted with acetic anhydride in the presence of NEt3, as previously reported (Scheme 1).25 The resulting polymer had an Mn of 4104 g mol−1, PCLA/PEG mass ratio of 1.74, and a CL/LA ratio of 1.58 as determined based on 1H NMR spectroscopy (Fig. S3, ESI†). SEC indicated an Mn of 1957 g mol−1 and a Đ of 1.44 (Fig. S4, ESI†). For the preparation of Az–PCLA–PEG–PCLA–Az, the hydroxyl-terminated PCLA–PEG–PCLA was reacted with 2-azidoacetic acid using EDC·HCl and DMAP (Scheme 1) with 1H NMR spectroscopic analysis indicating an Mn of 3504 g mol−1, PCLA/PEG mass ratio of 1.34, and a CL/LA ratio of 1.32 (Fig. S5, ESI†). The product had an Mn of 2302 g mol−1 and a Đ of 1.28 as determined by SEC (Fig. S6, ESI†). Functionalization was confirmed by 1H NMR spectroscopy based on the appearance of new peaks from 4.0–3.8 ppm, corresponding to the methylene groups on the azidoacetate end groups. In addition, the FTIR spectrum showed a new sharp peak at 2110 cm−1, corresponding to the stretch of the new azide group (Fig. S7, ESI†). | ||
| Scheme 1 Synthesis of the Ac–PCLA–PEG–PCLA–A, M–PCLA–PEG–PCLA–M and Az–PCLA–PEG–PCLA–Az. | ||
Selection and synthesis of cross-linkers
Successful gelation requires three-dimensional network formation. It has been hypothesized, that the initial polymer “solution” is a suspension of copolymer micelles.20 For intra-micelle cross-linking, the cross-linker should be small enough and preferably hydrophobic so that it can penetrate the hydrophobic cores of the polymer assemblies to introduce covalent bonds. To realize inter-micelle covalent network formation in water, the cross-linker should be water soluble and contain at least two reactive functional groups. For the thiol-Michael cross-linking approach, PETMP (Fig. 2) was selected as a small molecule tetra-thiol because it has been commonly used in thiol–ene cross-linking reactions.29–31 It exhibits low water solubility (3.7 mg L−1 in water at 20 °C), but it was anticipated that it would be able to diffuse into the PCLA domains of the hydrogel to induce intra-micellar cross-linking (Fig. 1d). | ||
| Fig. 2 Chemical structures of PETMP, 4-arm-PEG-ester-SH, and 4-arm-PEG-DIBAC. | ||
PEG is a widely used cross-linker for hydrogel preparation.32–34 Therefore, 2000 g mol−1 and 5000 g mol−1 4-arm-PEGs were chosen as starting materials for inter-micellar cross-linking. Following a modification of a procedure previously reported by Truong and coworkers,35 the alcohol-terminated 4-arm-PEGs were functionalized with thiols by refluxing with excess 3-mercaptopropionic acid in in the presence of p-toluenesulfonic acid as a catalyst, to afford the 4-arm-PEG2000-ester-SH and 4-arm-PEG5000-ester-SH (Fig. 2 and Scheme S1, ESI†). The structures of the products were confirmed by 1H and 13C NMR spectroscopy, SEC, and FT-IR spectroscopy (Fig. S8–S15, ESI†). To synthesize the SPAAC cross-linker, 4-arm-PEG2000 and 4-arm-PEG-5000 were reacted with 6 equiv. of DIBAC-CO2H, using EDC·HCl as a coupling agent, and DMAP as a catalyst to give 4-arm-PEG2000-DIBAC and 4-arm-PEG5000-DIBAC (Fig. 2 and Scheme S2, ESI†). The products were characterized by 1H NMR spectroscopy, SEC, and FT-IR spectroscopy (Fig. S16–S21, ESI†).
Gelation
Qualitative gelation tests were initially performed. Successful gelation was defined by the well-established vial-tilt test.26 The formulation was incubated in a glass vial for 30 min at 37 °C, then the vial was inverted, and examined after 10 min. Gelation was indicated by the absence of flow. The observations from these experiments are described in Table 1. First, PETMP, 4-arm-PEG2000-ester-thiol, and 4-arm-PEG5000-ester-thiol were investigated as cross-linkers with M–PCLA–PEG–PCLA–M as it was anticipated that they may cross-link the methacrylate groups via thiol-Michael addition reactions. Unfortunately, all three of these cross-linkers led to precipitation rather than gelation (Fig. S22, ESI†). 4-arm-PEG5000-ester-thiol caused the least phase separation but qualitatively the product did not appear to be a gel. Thus, the thiol-Michael addition approach was deemed unsuccessful.| Cross-linker | Triblock copolymer | PCLA–PEG–PCLA end group:cross-linker functional group ratio |
||
|---|---|---|---|---|
| 1:1 |
1:2 |
1:4 |
||
| PETMP | M–PCLA–PEG–PCLA–M | White precipitate, no gel | White precipitate, no gel | White precipitate, no gel |
| 4-arm-PEG2000-ester-thiol | M–PCLA–PEG–PCLA–M | White precipitate, no gel | Translucent precipitate, no gel | Translucent precipitate, no gel |
| 4-arm-PEG5000-ester-thiol | M–PCLA–PEG–PCLA–M | Translucent precipitate, no gel | Translucent precipitate, no gel | Translucent precipitate, no gel |
| 4-arm-PEG2000-DIBAC | Az–PCLA–PEG–PCLA–Az | White precipitate, no gel | Viscous liquid, no gel | Viscous liquid, no gel |
| 4-arm-PEG5000-DIBAC | Az–PCLA–PEG–PCLA–Az | Gel | ND | ND |
For the SPAAC approach, 4-arm-PEG2000-DIBAC and 4-arm-PEG5000-DIBAC were investigated as cross-linkers with Az–PCLA–PEG–PCLA–Az with the goal to form covalent bonds between the azide end groups and the DIBAC group on the cross-linker. At all three of the azide–alkyne ratios, 4-arm-PEG2000-DIBAC led to either a precipitate or viscous liquid. However, for the 4-arm-PEG5000-DIBAC, with an azide:alkyne ratio of 1:1, a gel was successfully and reproducibly formed (Fig. S23, ESI†). Higher amounts of 4-arm-PEG5000-DIBAC were not added in this case because the mass ratio of PEG to triblock copolymer was already quite high and the solution was very viscous initially before cross-linking.
There are several possible scenarios that can explain the failure of many of the systems to gel. If the copolymers were self-assembled into micelles before addition of the cross-linkers, one possibility is that the length of cross-linker “arms” were insufficient to penetrate multiple micelles. The hydrophilic domain of each of the PCLA–PEG–PCLA copolymers is 1500 g mol−1 PEG, which corresponds to ∼34 ethylene oxide repeat units. In the flower-like micelle morphology, the folded PEG domain would be about 750 g mol−1, or 17 repeating units. Among our cross-linker candidates, the 4-arm-PEG2000 and 4-arm-PEG5000's arm lengths (PEG domains) were approximately 11 and 22 repeating units, respectively. Thus, only the 4-arm-PEG5000 would be expected to penetrate multiple micelles. Indeed, the 4-arm-PEG5000 performed better than the 4-arm-PEG2000. It is also possible that after one or two arms of the PEG react with the PCLA–PEG–PCLA copolymers, the formation of new amphiphilic macromolecules disrupts the assemblies, resulting in precipitation. This would be consistent with the formation of white precipitate upon mixing the copolymer formulation with the 4-arm-PEG-ester-SH cross-linker solutions. Finally, another issue may be that the cross-linker solutions were very viscous, which could result in a heterogenous distribution of molecules throughout the system in the initial time period after combining the solutions. The success of the SPAAC with the 4-arm-PEG5000-DIBAC cross-linker may relate to its higher reaction rate constant (∼0.31/M−1 s−1 in MeOH) compared to that of the thiol-Michael reaction (5.49 × 10−5 M−1 s−1 in 1,5-diazabicyclo[4.3.0]non-5-ene)36 or to the higher hydrophobicity of the DIBAC group compared to the thiol, which may favor their localization in the PCLA domains. In the case of the PETMP cross-linker, its poor water solubility may prevent it from diffusing rapidly into the PCLA domains and reacting.
Hydrogel properties
The effect of the cross-linking method on the hydrogel properties was investigated by comparing the hydrogels prepared from Ac–PCLA–PEG–PCLA–Ac by only thermo-responsive gelation without covalent cross-linking (non-covalent gel),25 thermo-responsive gelation with covalent free radical cross-linking of M–PCLA–PEG–PCLA–M (free radical gel),12 and thermo-responsive gelation with SPAAC covalent cross-linking of Az–PCLA–PEG–PCLA–Az (azide–alkyne gel). First, hydrogel syneresis was investigated. Syneresis is the spontaneous decrease in the gel volume, accompanied by the release of water that was initially entrapped by the network.37,38 The extent of syneresis affects the mechanical properties and long-term stability of the hydrogel.38 Moreover, changes in the gel structure caused by syneresis can affect the pathways of cargo diffusion and therefore their release rates. As shown in Fig. 3a, the water loss from the non-covalent gel was around 45% (w/w) after 24 h. In contrast, free radical gel lost only about 5% of its mass. These data are in reasonable agreement with those previously reported for similar hydrogels.12 The azide–alkyne gel lost about 15% of its mass, indicating that it was less stable than the free radical gel but more stable than the non-covalent gel.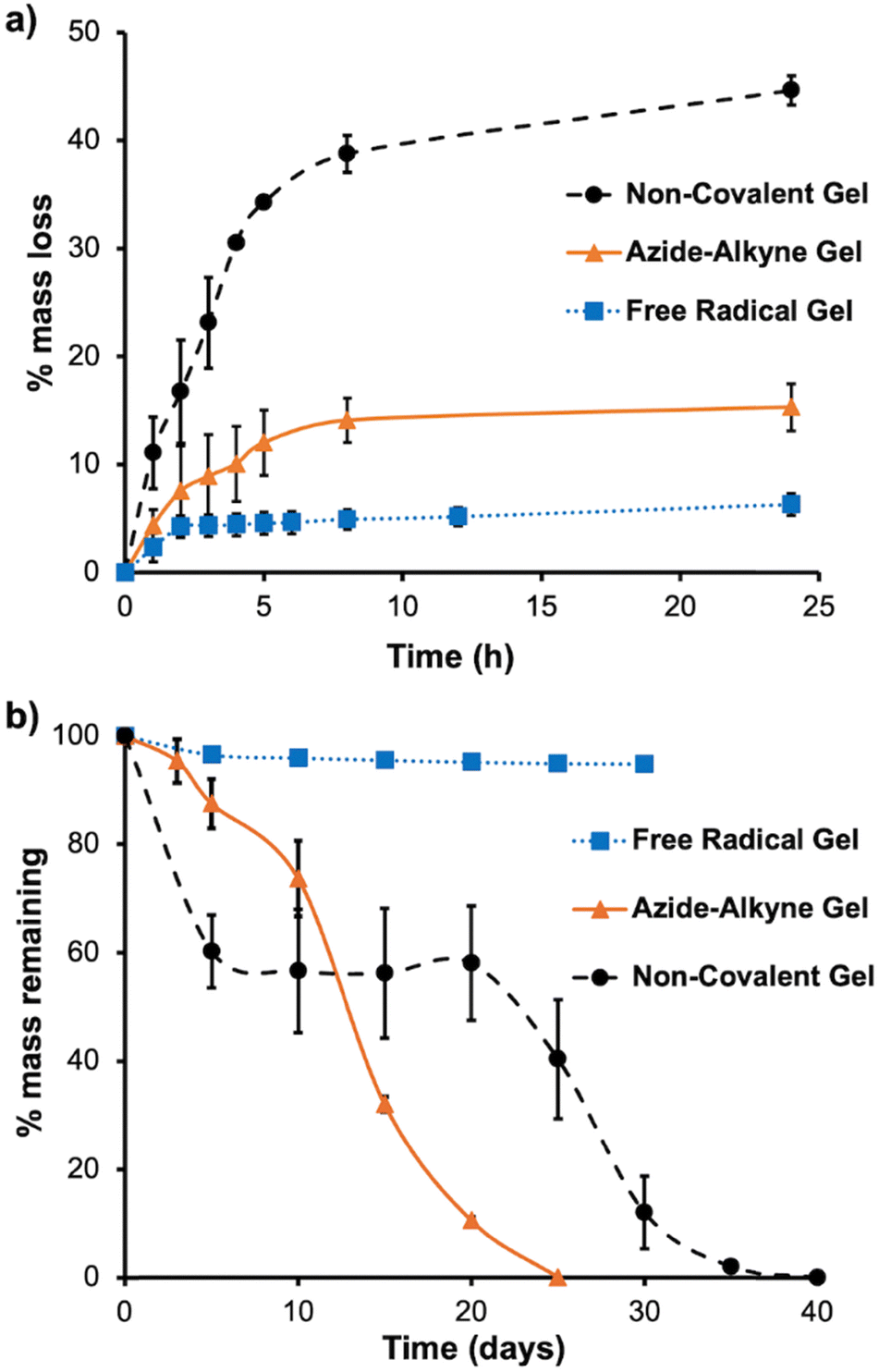 | ||
| Fig. 3 (a) Hydrogel syneresis, as measured by the mass of water lost from the gel upon incubation at 37 °C over the first 24 h; (b) hydrogel degradation upon immersion in PBS and incubation at 37 °C, as measured by a loss of hydrogel mass. The measurements were performed on triplicate samples of the gels and the error bars correspond to the standard deviations. | ||
Next, the hydrogel degradation in PBS at 37 °C was investigated. As shown in Fig. 3b, the free radical gel retained 96% of its mass over 30 days and maintained mechanical integrity during this time. The azide–alkyne gel lost about 50% of its mass in 12 days and was fully degraded by 25 days. It is possible that the onset of more rapid mass loss at about 10 days can be attributed to hydrolysis of the ester bonds between the DIBAC and PEG in the cross-linker. Up until 10 days, the azide–alkyne gel maintained its mechanical integrity, after which it began to erode, with the progressive loss of gel fragments from the surface of the material. The non-covalent gel showed mechanical integrity until around day 20, and then decreased integrity was observed with some gel fragmentation during the weighing measurements after this time point. It had completely degraded in 40 days. These results indicate that the newly developed azide–alkyne gel was more stable than the non-covalent gel over the first 10 days, but less stable with respect to degradation from about 15 days onward.
Finally, the Young's moduli of the hydrogels were determined under unconfined compression. As shown in Table 2, free radical gel had a significantly higher Young's modulus of 194 ± 1.5 kPa, compared to 17 ± 1.9 kPa for the azide–alkyne gel and 13.2 ± 0.5 kPa for the non-covalent gel. Thus, it is clear that the SPAAC approach didn’t lead to nearly as robust covalent cross-linking as the free radical cross-linking approach. For previous applications in intra-articular drug delivery,12 achieving a high modulus and robust mechanical properties was important due to the ongoing subjection of the hydrogel to mechanical forces in the joint. Indeed, the modulus of the free radical gel was just below that of normal articular cartilage (∼250–1600 kPa).39 On the other hand, for anti-cancer drug delivery a high modulus should not be required and may even be undesired for subcutaneous or intra-muscular injection due to a mismatch of the mechanical properties of the gel with those of surrounding soft tissues. Muscles have moduli around 1–30 kPa,40 which is similar to the moduli of the azide–alkyne gel and non-covalent gel. Thus, these hydrogels may be suitable for injectable anti-cancer drug delivery applications, with a dual barrel syringe approach likely being most promising to separate the azide and alkyne components before injection.
| Young's modulus (kPa) | Free radical gel | Azide–alkyne gel | Non-covalent gel |
|---|---|---|---|
| Unloaded hydrogel | 194 ± 1.5 | 17 ± 1.9 | 13.2 ± 0.5 |
| Hydrogel loaded with 10% (w/w) of ATRA | 18.8 ± 0.2 | ND | ND |
ATRA loading and its effects on hydrogel properties
To study the effect of ATRA loading on the properties of the hydrogels, 10% (w/w) of ATRA was loaded into the three different hydrogel systems. First, their degradation in PBS at 37 °C was investigated. As shown in Fig. 4, all systems degraded in about 20 days. This rate of degradation was similar to that observed for the azide–alkyne gel without ATRA, indicating the ATRA loading had only a modest effect on this hydrogel. However, for free radical gel and non-covalent gel, the degradation rates were faster than those observed in the corresponding unloaded hydrogels where the time for complete degradation was 40 days for non-covalent gel and the free radial gel retained 96% of its mass over 30 days (Fig. 3). These results indicated that incorporation of ATRA particularly lowered the stability of the free radical gel. Such a dramatic change in degradation rate was not previously observed upon incorporation of celecoxib into the free radical gel.12 Taking into consideration the cross-linking mechanism of the free radical gel, it is likely that ATRA served as radical trap, as retinoids are well-established to react with free radicals.22 For example, retinoids can react with radicals to form adducts or can form retinoid radical cations via single electron transfer. | ||
| Fig. 4 Degradation of the hydrogels loaded with 10% (w/w) ATRA as measured in PBS at 37 °C. The measurements were performed on triplicate samples of ATRA-loaded gels and the error bars correspond to the standard deviations. | ||
As shown in Table 2, upon ATRA loading, the Young's modulus of the free radical gel significantly decreased (∼10 fold) to 18.8 ± 0.2 kPa, a modulus similar to that of the azide–alkyne gel without ATRA loaded. In contrast, upon incorporation of celecoxib into the free radical gel in previous work, no such decrease in the compressive modulus was observed.12 This large reduction in modulus upon ATRA incorporation is further evidence that ATRA disrupted the free radical cross-linking of the methacrylate groups. The non-covalent gel and azide–alkyne gel loaded with 10% (w/w) ATRA were not handleable for compression testing, indicating that the incorporation of ATRA also disrupted the hydrogel's mechanical integrity to some extent. However, they did maintain their shape when immersed in PBS, indicating that they were still gels.
In vitro release of ATRA
First ATRA release from the hydrogels was studied in pH 7.4 PBS at 37 °C. To facilitate the dissolution of the hydrophobic drug, 2% (w/w) of Tween 80 was added to the sink solution.12,18 As shown in Fig. 5a, the non-covalent gel released ATRA most rapidly, with complete release in 14 days. Free radical gel and azide–alkyne gel had similar release profiles, with complete ATRA release at 16 days and 17 days, respectively. For all systems, after a small burst release of about 10% over the first two days, the ATRA release profile could be fit quite well to a zero-order release model up until twelve days (Fig. S24, ESI†). After twelve days, the release accelerated as substantial degradation of the gel was observed, resulting in loss of its integrity. This behavior indicates that release was likely not dominated by simple diffusion of the drug out of the hydrogel but rather a more complex mechanism involving entrapment of ATRA within hydrophobic PCLA domains in the hydrogel, combined with hydrogel erosion.41 Examining the UV-visible spectra of ATRA released from the azide–alkyne hydrogels compared to pure ATRA (Fig. S25, ESI†), the maximum absorption wavelength remained constant and only a very slight broadening of the absorption peak was observed, indicating that there wasn’t any substantial degradation of ATRA during its encapsulation and release. ATRA released from the free radical gel exhibited more peak broadening, which can potentially be attributed to the generation of ATRA byproducts during the free radical reaction. | ||
| Fig. 5 ATRA release from (a) the three different hydrogels in PBS with 2% (w/w) Tween 80 at 37 °C; (b) azide–alkyne gel in DMEM containing 10% fetal bovine serum at 37 °C. All measurements were performed on triplicate samples of ATRA-loaded gels, and the error bars correspond to the standard deviations. | ||
Notably, we previously observed a much slower release of celecoxib from free radical gel, with only about 20% of the drug released over 22 days.12 Again, these drug release data suggest that ATRA, but not celecoxib, disrupted the free radical cross-linking, resulting in properties similar to a more lightly cross-linked (i.e., azide–alkyne gel) or non-cross-linked gel (i.e., non-covalent gel). Although the ATRA release was much faster than that of celecoxib from this series of hydrogels, the release times are likely well suited to the delivery of anti-cancer drugs, where weekly injections are commonly used and it is beneficial to be able to adjust the dose on this basis to address patient side effects.42 However, of the three evaluated hydrogels, participation of ATRA in the free radical chemistry is highly undesirable, as it may modify the chemical structure of any released drug. Thus, the azide–alkyne gel or the non-covalent gel are more suitable for further study of ATRA delivery.
In preparation for the in vitro evaluation of the ATRA-loaded hydrogels, to understand the release rate of ATRA from the hydrogels under cell culture conditions, the ATRA release from the azide–alkyne gel was studied in DMEM containing 10% fetal bovine serum. Compared to the use of Tween, such conditions would better mimic the environment in the human body, where proteins can bind to drugs, thereby enhancing their solubility.43 The experiment focused on the azide–alkyne gel because the injection of the non-covalent hydrogel formulation in culture media at low hydrogel concentration did not allow for gelation and if the hydrogel was prepared in advance, it underwent a gel–sol transition during any manipulation at ambient temperature. On the other hand, the azide–alkyne gel could be prepared to form a stable, non-reversible gel, and then added to cells. The ATRA loading was lowered to 1% (w/w) to allow accurately measurable quantities of hydrogel, and thus accurate amounts of ATRA, to be added to the cells. Under these conditions, ∼78% of ATRA was released over 7 days (Fig. 5b). The gel maintained its mechanical integrity over this time period in the presence of serum, similar to its behavior in PBS. However, the release was notably faster in the presence of serum compared to PBS with Tween (∼25% over one week), indicating that serum components such as albumin and lipids may bind to ATRA and/or the hydrogel, thereby facilitating the drug's release. With these results, the ATRA loaded azide–alkyne gel was considered suitable for short term (4 days to 1 week) cell studies.8,13
In vitro evaluation of ATRA loaded azide–alkyne gel
To study the in vitro effect of the ATRA loaded azide–alkyne gel, the MDA-MB-468 cell line was selected because it is a previously investigated cancer cell line sensitive to ATRA treatment.44 Free ATRA was added to cells at concentrations ranging from 10–50 μM for comparison with the same concentrations of ATRA incorporated in the hydrogel. Control hydrogel (azide–alkyne gel with no ATRA loaded) with the same mass as that used to achieve the highest ATRA concentration (50 μM) for the ATRA loaded azide–alkyne gel was also used as the vehicle control. The cells were incubated for 5 days in the presence of the drug/hydrogel. As the concentration of free ATRA increased from 0–50 μM, the cell proliferation dropped to 53% of the control (cells exposed only to the cell culture media) (Fig. 6). For the ATRA-loaded azide–alkyne gel, as the ATRA concentration added to the cells increased to 50 μM, cellular proliferation decreased about 25% relative to vehicle control. The lower anti-proliferative effect of the ATRA encapsulated in the hydrogel can be attributed to the fact that over the 5-day incubation with cells, only about 50% of the encapsulated ATRA would have been released. Thus, the efficacy of ATRA-loaded hydrogels at 50 μM could be expected to be similar to that of 25 μM free ATRA, which is consistent with the experimental result. Incubation with the control hydrogel did not lead to any significant change (p = 0.5, two-sample T test) in the cell count compared to cells only exposed to culture media (>99% of the control), indicating that it did not have any significant negative effects on the cells. Overall, these results suggested that the azide–alkyne gel itself had no significant effect on cellular proliferation, and that the azide–alkyne gel can release active drug. | ||
| Fig. 6 In vitro proliferation assay of ATRA loaded azide–alkyne gel compared to free ATRA and a control gel without ATRA, after incubation for 5 days at varying ATRA concentrations or equivalent ATRA concentrations incorporated into the hydrogel. The quantity of control gel added to cells corresponds to the same amount of azide–alkyne gel required to deliver 50 μM of ATRA but without ATRA. The number of cells were quantified by Hoechst staining and image analysis, with cells only exposed to culture media defined as the 100% control. All measurements were performed in triplicate, and the error bars correspond to the standard deviations. | ||
Conclusions
We compared different cross-linking approaches for ATRA-loaded thermo-responsive hydrogels prepared from PCLA–PEG–PCLA triblock copolymers. In the absence of ATRA, free radical cross-linking led to hydrogels with the lowest degree of syneresis, slowest degradation, and highest Young's moduli, while non-covalently cross-linked hydrogels had the highest syneresis, most rapid degradation over the first 10 days, and the lowest Young's moduli. SPAAC cross-linking to afford azide–alkyne gels using azide terminal groups on the copolymers and a 4-arm-PEG-DIBAC cross-linker provided intermediate properties between those of the free radical gel and the non-covalent gel. Gelation using both small molecule and 4-arm-PEG tetra-thiols was unsuccessful. The incorporation of ATRA inhibited free radical cross-linking, making the properties of these hydrogels similar to the azide–alkyne gel and the non-covalent gel. All three hydrogels released ∼100% encapsulated ATRA over about 2 weeks in PBS containing 2% Tween at 37 °C, but the azide–alkyne gel was selected for further evaluation to avoid the use of free radical chemistry, while affording gels that could be handled for cell culture experiments. This hydrogel released 80% of loaded ATRA over 7 days in DMEM containing 10% FBS at 37 °C. Initial cell culture data showed that unloaded azide–alkyne gel was well tolerated by the MDA-MB-468 cells. The azide–alkyne gel loaded with 1% ATRA inhibited cell proliferation somewhat less compared to an equivalent dose of free ATRA, likely due to the release of ∼50% of ATRA over the 5-day cell culture experiment. Overall, this newly developed hydrogel should make a promising candidate for ATRA delivery via subcutaneous or intra-muscular injection via a dual barrel syringe, providing a slow-release depot of the drug. It can also potentially be used for other anti-cancer drug delivery applications, but future studies using animal tumor models will be needed to evaluate anti-cancer efficacy of our controlled release hydrogel system. In addition, while previous research has demonstrated that the SPAAC reaction can be applied in vivo for applications such as drug delivery and imaging,45 it will be important to fully evaluate the biological effects of the hydrogel and any residual functional groups in the context of the specific applications.Author contributions
XM: conceptualization, investigation, methodology, formal analysis, writing – original draft; RCS: investigation, methodology, formal analysis; XZZ: conceptualization, methodology, supervision, funding acquisition, writing – review & editing; KPL: conceptualization, methodology, supervision, funding acquisition, writing – review & editing; ERG: conceptualization, methodology, supervision, funding acquisition, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Canadian Institutes of Health Research (FRN 169677, 518390), the Natural Sciences and Engineering Research Council of Canada (RGPIN-2021-03950), Canada Research Chair program (CRC-2020-00101), Canada Foundation for Innovation (Grants 43822 and 43257), and Ontario Institute for Cancer Research (Grant 1240) for funding.References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef CAS PubMed.
- W. M. C. van den Boogaard, D. S. J. Komninos and W. P. Vermeij, Cancers, 2022, 14, 627 CrossRef CAS PubMed.
- X. Z. Zhou and K. P. Lu, Nat. Rev. Cancer, 2016, 16, 463–478 CrossRef CAS PubMed.
- K. P. Lu and X. Z. Zhou, Sci. Signaling, 2024, 17, eadi8743 CrossRef CAS PubMed.
- S. Wei, S. Kozono, L. Kats, M. Nechama, W. Li, J. Guarnerio, M. Luo, M. H. You, Y. Yao, A. Kondo, H. Hu, G. Bozkurt, N. J. Moerke, S. Cao, M. Reschke, C. H. Chen, E. M. Rego, F. Lo-Coco, L. C. Cantley, T. H. Lee, H. Wu, Y. Zhang, P. P. Pandolfi, X. Z. Zhou and K. P. Lu, Nat. Med., 2015, 21, 457–466 Search PubMed.
- S. Kozono, Y. M. Lin, H. S. Seo, B. Pinch, X. Lian, C. Qiu, M. K. Herbert, C. H. Chen, L. Tan, Z. J. Gao, W. Massefski, Z. M. Doctor, B. P. Jackson, Y. Chen, S. Dhe-Paganon, K. P. Lu and X. Z. Zhou, Nat. Commun., 2018, 9, 3069 CrossRef PubMed.
- S. Huang, Y. Chen, Z. M. Liang, N. N. Li, Y. Liu, Y. Zhu, D. Liao, X. Z. Zhou, K. P. Lu, Y. Yao and M. L. Luo, Front. Cell Dev. Biol., 2019, 7, 322 CrossRef PubMed.
- J. Jing, C. Nelson, J. Paik, Y. Shirasaka, J. K. Amory and N. Isoherranen, J. Pharmacol. Exp. Ther., 2017, 361, 246–258 CrossRef CAS PubMed.
- E. Z. Szuts and F. I. Harosi, Arch. Biochem. Biophys., 1991, 287, 297–304 CrossRef CAS PubMed.
- A. F. Ourique, A. Melero, C. de Bona da Silva, U. F. Schaefer, A. R. Pohlmann, S. S. Guterres, C. M. Lehr, K. H. Kostka and R. C. Beck, Eur. J. Pharm. Biopharm., 2011, 79, 95–101 Search PubMed.
- D. A. Prince, I. J. Villamagna, A. Borecki, F. Beier, J. R. de Bruyn, M. Hurtig and E. R. Gillies, ACS Appl. Bio Mater., 2019, 2, 3498–3507 CrossRef CAS PubMed.
- D. Yang, W. Luo, J. Wang, M. Zheng, X. H. Liao, N. Zhang, W. Lu, L. Wang, A. Z. Chen, W. G. Wu, H. Liu, S. B. Wang, X. Z. Zhou and K. P. Lu, J. Controlled Release, 2018, 269, 405–422 CrossRef CAS PubMed.
- Y. H. Zhu, N. Ye, X. F. Tang, M. I. Khan, H. L. Liu, N. Shi and L. F. Hang, Front. Pharmacol., 2019, 10, 447 CrossRef CAS PubMed.
- K. Baler, R. Michael, I. Szleifer and G. A. Ameer, Biomacromolecules, 2014, 15, 3625–3633 CrossRef CAS PubMed.
- B. Mirani, E. Pagan, S. Shojaei, J. Duchscherer, B. D. Toyota, S. Ghavami and M. Akbari, Eur. J. Pharmacol., 2019, 854, 201–212 CrossRef CAS PubMed.
- J. Xiang, X. Zhou, Z. Xia, Z. Zhang, K. Xu, S. Hu, Z. Zhang, J. Liu, W. Yang, L. Yu and J. Wang, Chem. Eng. J., 2024, 500, 156915 CrossRef CAS.
- A. Petit, M. Sandker, B. Müller, R. Meyboom, P. van Midwoud, P. Bruin, E. M. Redout, M. Versluijs-Helder, C. H. van der Lest, S. J. Buwalda, L. G. de Leede, T. Vermonden, R. J. Kok, H. Weinans and W. E. Hennink, Biomaterials, 2014, 35, 7919–7928 CrossRef CAS PubMed.
- A. Petit, E. M. Redout, C. H. van de Lest, J. C. de Grauw, B. Müller, R. Meyboom, P. van Midwoud, T. Vermonden, W. E. Hennink and P. R. van Weeren, Biomaterials, 2015, 53, 426–436 CrossRef CAS PubMed.
- R. Fan, Y. Cheng, R. Wang, T. Zhang, H. Zhang, J. Li, S. Song and A. Zheng, Polymers, 2022, 14, 2379 CrossRef CAS PubMed.
- E. Hadzic, X. Mei, H. Dupuis, G. Blackler, C. T. Appleton, E. Gillies and F. Beier, Osteoarthritis Cartilage, 2024, 32, S47–S48 CrossRef.
- R. Edge and T. G. Truscott, Antioxidants, 2018, 7, 5 CrossRef PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chem. – Eur. J., 2021, 27, 5057–5073 CrossRef CAS PubMed.
- D. A. Prince, I. J. Villamagna, C. C. Hopkins, J. R. de Bruyn and E. R. Gillies, Polym. Int., 2019, 68, 1074–1083 CrossRef CAS.
- A. Petit, B. Müller, R. Meijboom, P. Bruin, F. van de Manakker, M. Versluijs-Helder, L. G. J. de Leede, A. Doornbos, M. Landin, W. E. Hennink and T. Vermonden, Biomacromolecules, 2013, 14, 3172–3182 CrossRef CAS PubMed.
- N. Brabez, R. M. Lynch, L. Xu, R. J. Gillies, G. Chassaing, S. Lavielle and V. J. Hruby, J. Med. Chem., 2011, 54, 7375–7384 CrossRef CAS PubMed.
- S. A. McNelles, J. L. Pantaleo and A. Adronov, Org. Proc. Res. Dev., 2019, 23, 2740–2745 CrossRef CAS.
- S. Kasetaite, S. De la Flor, A. Serra and J. Ostrauskaite, Polymers, 2018, 10, 439 CrossRef PubMed.
- A. K. O'Brien, N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2007–2014 CrossRef.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- J. Shi, L. Yu and J. Ding, Acta Biomater., 2021, 128, 42–59 CrossRef CAS PubMed.
- S. Sun, Y. Cui, B. Yuan, M. Dou, G. Wang, H. Xu, J. Wang, W. Yin, D. Wu and C. Peng, Front. Bioeng. Biotechnol., 2023, 11, 1117647 CrossRef PubMed.
- Z. Wang, Q. Ye, S. Yu and B. Akhavan, Adv. Healthcare Mater., 2023, 12, 2370102 CrossRef.
- V. X. Truong, I. Donderwinkel and J. E. Frith, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1872–1876 CrossRef CAS.
- T. Luu, K. Gristwood, J. C. Knight and M. Jörg, Bioconjugate Chem., 2024, 35, 715–731 CrossRef CAS PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- S. Panja, B. Dietrich and D. J. Adams, Angew. Chem., 2022, 134, e202115021 CrossRef.
- N. Petitjean, P. Canadas, P. Royer, D. Noël and S. Le Floc'h, J. Biomed. Mater. Res., Part A, 2023, 111, 1067–1089 CrossRef CAS PubMed.
- G. Singh and A. Chanda, Biomed. Mater., 2021, 16, 062004 CrossRef CAS PubMed.
- M.-L. Laracuente, H. Y. Marina and K. J. McHugh, J. Controlled Release, 2020, 327, 834–856 CrossRef CAS PubMed.
- P. Smita, P. A. Narayan and P. Gaurav, Front. Oncol., 2022, 12, 1015200 CrossRef CAS PubMed.
- E. N. Hoogenboezem and C. L. Duvall, Adv. Drug Delivery Rev., 2018, 130, 73–89 Search PubMed.
- K. M. Coyle, S. Maxwell, M. L. Thomas and P. Marcato, Sci. Rep., 2017, 7, 16684 CrossRef PubMed.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00228a |
| This journal is © The Royal Society of Chemistry 2025 |