
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization control of antisolvent-free perovskite films using alkali metal additives for improving efficiency and extending applicability of perovskite solar cells†
Min Jun
Choi‡
a,
Veera Murugan
Arivunithi‡
a,
So Jeong
Shin
a,
Gyeong G.
Jeon
a,
Hye W.
Chun
a,
Inho
Bae
a,
Dong Won
Kim
 *ab and
Jong H.
Kim
*a
*ab and
Jong H.
Kim
*a
aDepartment of Molecular Science and Technology, Ajou University, Suwon 16499, Republic of Korea. E-mail: blueocean07@naver.com; jonghkim@ajou.ac.kr
bAI-Superconvergence KIURI Translational Research Center, Ajou University, Suwon, 16499, Republic of Korea
First published on 11th June 2025
Abstract
The antisolvent-free fabrication of perovskite solar cells (PSCs) is a promising approach to secure their reproducibility and scalability. However, achieving high efficiency and uniform crystallization without antisolvent remains a critical challenge. In this study, we introduce alkali metal salts as additives to control the crystallization process and enhance the photovoltaic (PV) properties of antisolvent-free PSCs. The incorporation of KPF6 effectively modulates the perovskite growth kinetics, resulting in improved grain size, reduced defect density, and enhanced charge transport properties. As a result, the optimized PSCs exhibit a significant improvement in power conversion efficiency (PCE) compared to the reference devices without KPF6. Moreover, the addition of KPF6 enabled large-area and semi-transparent antisolvent-free perovskite layers with great uniformity. This work provides valuable insights into rational additive engineering for crystallization control to achieve high efficiency antisolvent-free PSCs, paving the way for the development of scalable PSCs and their broad applications.
1. Introduction
Organic–inorganic metal halide perovskites have garnered significant attention as a next-generation light absorber in photovoltaics (PVs) due to the exceptional optoelectronic properties of perovskites, including broadband light absorption, low exciton binding energy, long charge-carrier diffusion lengths, and a tunable bandgap. To date, the record power conversion efficiency (PCE) of perovskite solar cells (PSCs) has reached 27.0% with a remarkable improvement from 3.8% in 2009.1 This substantial enhancement in device performance has been driven by advancements in the engineering of perovskite composition,2 the development of efficient charge transport materials,3 and the process optimizations.4 Among them, one of the most successfully developed processes is the antisolvent-dropping method during deposition of perovskite solution, which enabled remarkable PCE enhancement over 26% through increasing crystallinity of the resulting perovskite films.5However, this process is very complex and highly susceptible to various experimental factors, resulting in poor reproducibility of PSC performances.6–8 These issues limit the large-area perovskite film preparation and scalable production of PSCs.9
As a result, alternative approaches have been suggested to manipulate crystal nucleation and grain growth without the use of antisolvents, including gas flow,10 vacuum flash processing,11 sequential deposition,12 and additive engineering.13 Of these methods, additives, including alkali metal (Li+, Na+, K+, Rb+, and Cs+) salts, have been widely employed in perovskite film preparation as they not only regulate perovskite crystallization but also provide additional benefits after crystal growth, such as the improvement of charge extraction properties and stability.14,15 In this regard, alkali metal salts are representative additives used for fabricating high-efficiency antisolvent-free PSCs.16 Despite such superiority of alkali metals, the influence of their cation sizes on crystallization of antisolvent-free perovskite films has not been studied, which affects the PV properties of PSCs most critically.
In this work, we introduced alkali metal hexafluorophosphates (LiPF6, NaPF6, and KPF6) as additives for perovskite precursors, and studied the effect of cation sizes on crystallization, morphology, and PV properties of antisolvent-free PSCs.
Through the morphological and absorption spectral characterizations, we found that the addition of KPF6 delayed perovskite crystallization and enabled uniform and pinhole-free film formation even without the antisolvent process. These improvements enabled KPF6-based antisolvent-free PSCs to achieve a higher PCE of 19.94%/36.10% compared to the non-additive devices (18.48%/29.65%), under standard 1 sun and indoor LED illumination conditions. Furthermore, incorporation of KPF6 allowed uniform antisolvent-free perovskite film formation suitable for both large-area and semi-transparent applications.
These results suggest that crystallization control based on rational selection of alkali metal additives offers a promising strategy for improving the efficiency of antisolvent-free PSCs and broadening their applications.
2. Results and discussion
2.1. Effect of alkali metal salt additives on crystallization and film morphology
We adopted LiPF6, NaPF6, and KPF6 additives as a source of alkali metals to examine the cation size effect on antisolvent-free perovskite film formation. We note that PF6−, commonly used in three additives, plays a role in suppressing halide vacancies within perovskite structures.17 Since the ionic radii of the used alkali metals (Li+ (76 pm), Na+ (102 pm), and K+ (138 pm)) are smaller than those of A-site cations in perovskites such as FA+ (279 pm), MA+ (270 pm), and Cs+ (167 pm), they are not expected to replace the A-site of the perovskite structure but rather to occupy interstitial positions within the perovskite lattice.18,19In antisolvent-free perovskite processing, controlling the crystallization rate during annealing is crucial for the preparation of morphologically uniform films.20 Thereby, first, we investigated the effect of alkali metal salt additives on the crystallization of antisolvent-free perovskite films. In this work, antisolvent-free perovskite films were prepared by spin-coating additive-added precursor solutions without the antisolvent-dropping process, while the reference films were prepared by the same recipe without additives. We denote the LiPF6, NaPF6, and KPF6 added perovskite films as LiPF6-PVSK, NaPF6-PVSK, and KPF6-PVSK, respectively. For this study, we performed in situ UV-Vis absorption measurements (Fig. 1) and monitored spectral changes of perovskite films with different additives during thermal annealing. As shown in Fig. 1a–d, all the films exhibited a rapid increase in absorption in the 500–700 nm range after 15 s of annealing and reached saturation after 40 s of annealing. This rapid change is associated with the nucleation and crystal growth processes of the perovskite, resulting from the increased concentration exceeding the saturation concentration. To investigate crystal growth kinetics, we evaluated the phase transition time (defined as the annealing time required for an increase of absorption from 10% to 90% of its maximum) at 700 nm, responsible for perovskite crystallization (Fig. 1e).21
 | ||
| Fig. 1 (a) In situ UV-Vis absorption spectra of the perovskite intermediate state for (a) reference, (b) LiPF6, (c) NaPF6, and (d) KPF6 during the annealing process. (e) Line profile of UV-Vis absorption intensity at a wavelength of 700 nm. The black dashed lines correspond to 10% and 90% of crystallization during the annealing process, respectively. | ||
The transition time of the reference film (without an additive) was 12.7 s, while LiPF6-PVSK exhibited the shortest time of 10.3 s. In contrast, as the ionic radius of added alkali metals (Na+ and K+) increased, the transition time increased to 11.0 s and 13.3 s, respectively. These results indicate that LiPF6 induces the fastest crystallization rate, whereas KPF6 leads to the slowest crystallization among the additives.
To support these observations, the Stokes–Einstein equation ( , where D is the diffusivity, kB is the Boltzmann constant, T is the absolute temperature, η is the dynamic viscosity of the solution, and r is the ionic radius) was used. This relation indicates that smaller ions diffuse more rapidly.22 Therefore, a smaller alkali metal ion (Li+) can diffuse faster into perovskite lattices compared to relatively large ions (Na+ and K+). Thus, it can be inferred that different cation sizes affected the crystallization speed and faster diffusion of Li+ into the perovskite lattice would accelerate crystal growth.23
, where D is the diffusivity, kB is the Boltzmann constant, T is the absolute temperature, η is the dynamic viscosity of the solution, and r is the ionic radius) was used. This relation indicates that smaller ions diffuse more rapidly.22 Therefore, a smaller alkali metal ion (Li+) can diffuse faster into perovskite lattices compared to relatively large ions (Na+ and K+). Thus, it can be inferred that different cation sizes affected the crystallization speed and faster diffusion of Li+ into the perovskite lattice would accelerate crystal growth.23
Prior to morphology analysis, we conducted concentration screening using KPF6, and found that a concentration of 1.5 mol% resulted in the most uniform morphology and superior optoelectronic properties (Fig. S1 and S2†). Based on these results, we selected the additive concentration at 1.5 mol% for all additive-based conditions.
Next, to explore the effect of crystallization rates on the morphology of resulting perovskite films, we carried out scanning electron microscopy (SEM) measurements for antisolvent-free perovskite films prepared with three additives.
As shown in Fig. 2a, the reference film with no additive exhibited non-uniform features with pinholes, which were consistently found in LiPF6-PVSK and NaPF6-PVSK. In contrast, the incorporation of KPF6 significantly enhanced film uniformity and reduced pinhole formation, implying that delayed crystallization allows homogeneous growth of perovskite grains. Grain size distributions (Fig. S3†) revealed that the addition of KPF6 improved the film uniformity and increased the crystalline domain size. These tendencies were also observed across the whole perovskite layer as shown in the cross-sectional SEM images (Fig. S4†).
 | ||
| Fig. 2 (a) SEM and (b) AFM morphology images of the perovskite films with different additives. (c) XRD patterns, (d) PL spectra, and (e) time-resolved PL spectra of the perovskite films with different additives. | ||
Additionally, atomic force microscopy (AFM) measurement was performed to analyze the surface roughness of each film (Fig. 2b). The high root-mean-square roughness of the reference (37.0 nm), LiPF6-PVSK (73.6 nm), and NaPF6-PVSK (51.4 nm) was effectively reduced to 28.0 nm by using KPF6. The pinhole-less feature with a smooth (reduced roughness) surface of KPF6-PVSK is a strong basis for enhancing PV performances due to reduced contact resistance at the interfaces of perovskite/charge transport layers.24
To probe the crystallinity of the antisolvent-free perovskite films depending on the used alkali metals, we conducted X-ray diffraction (XRD) analyses (Fig. 2c). After addition of the three additives, PbI2 diffraction peaks were reduced, compared to the reference film, due to the interaction of PF6− with Pb2+.16,25 In addition, the KPF6-added perovskite film exhibited the highest crystallinity with the highest intensity of the (100) diffraction peak due to the delayed crystallization effect.26
To gain further evidence for enhanced crystallinity of PF6-added perovskite films, we evaluated the crystal size (D) using Scherrer's equation D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where λ is the X-ray wavelength, β is the FWHM, and θ is the Bragg diffraction angle (Fig. S5†). The calculated crystal size of the KPF6 film was 78.17 nm, which is larger than those of LiPF6 (69.81 nm) and NaPF6 (68.70 nm).27 We note that the shift of the (310) plane peak to a lower angle after the addition of additives suggests perovskite lattice expansion due to interstitial occupancy of alkali metal cations.28
cosθ, where λ is the X-ray wavelength, β is the FWHM, and θ is the Bragg diffraction angle (Fig. S5†). The calculated crystal size of the KPF6 film was 78.17 nm, which is larger than those of LiPF6 (69.81 nm) and NaPF6 (68.70 nm).27 We note that the shift of the (310) plane peak to a lower angle after the addition of additives suggests perovskite lattice expansion due to interstitial occupancy of alkali metal cations.28
To further confirm the incorporation of additives into the perovskite lattice, we conducted X-ray photoelectron spectroscopy (XPS) measurements with the KPF6 additive (Fig. S6†). In the Pb 4f spectrum, KPF6-PVSK exhibited a noticeable shift in the Pb2+ 4f7/2 and 4f5/2 peaks to lower binding energies (from 138.52 to 138.37 eV and from 143.68 to 143.23 eV, respectively), suggesting chemical interaction between KPF6 and uncoordinated Pb2+. This interaction contributes to the stabilization of the perovskite structure and suppression of PbI2 formation.29 In addition, distinct F 1s and K 2p signals were observed only in KPF6-PVSK, confirming the intercalation of both PF6− and K+ ions into the perovskite lattice, consistent with the observed lattice expansion in XRD (Fig. 2c).
Subsequently, to study charge carrier recombination, we measured steady-state photoluminescence (PL) spectra for the films. Higher PL intensity of KPF6-PVSK compared to LiPF6-PVSK and NaPF6-PVSK indicates effectively suppressed non-radiative recombination by defects or pinholes (Fig. 2d).30
A longer average PL decay time (τavg = 1000.8 ns) of the KPF6-perovskite film determined from time-resolved PL (TRPL) measurements (Fig. 2e and Table S1†), further corroborates reduced defects by slow crystallization (c.f. τavg values for the reference, LiPF6-PVSK, and NaPF6-PVSK are 814.8, 433.9, and 648.8 ns, respectively).
Overall, the addition of KPF6 delayed the crystallization process, facilitating the formation of compact and pinhole-free perovskite films. The resulting improvement in morphology and crystallinity effectively suppressed non-radiative recombination, allowing high-efficiency antisolvent-free PSCs.
2.2. Photovoltaic properties of KPF6-based PSCs
We examined PV properties of antisolvent-free PSCs using a KPF6 additive based on the n–i–p device architecture of FTO/SnO2/perovskite/spiro-OMeTAD/Au (Fig. 3a). The PCE of reference PSCs (18.48%) without an additive was considerably increased to 19.94% after using KPF6 in the perovskite layer (Fig. 3b). As summarized in Fig. S7† and Table 1, overall PV performance parameters (open circuit voltage (VOC), short circuit density (JSC), and fill factor (FF)) were improved due to reduced defects by KPF6 addition. Moreover, the hysteresis index of KPF6-PSCs was effectively diminished to 0.45% (c.f. HI of reference PSCs: 7.84%) due to suppressed ion migration by K+.31
of KPF6-PSCs was effectively diminished to 0.45% (c.f. HI of reference PSCs: 7.84%) due to suppressed ion migration by K+.31
 | ||
| Fig. 3 (a) Schematic illustration of the n–i–p structure PSCs. (b) J–V curves under AM 1.5 G 1 sun illumination for PSCs with and without KPF6. (c) EQE spectra and (d) SPO measurements for PSCs with and without KPF6. J–V curves under LED illumination conditions with different intensities (e) without and (f) with KPF6. (g) Schematic illustration of the p–i–n structure PSCs. J–V curves of p–i–n based PSCs with KPF6 under (h) 1 sun and (i) different LED lux illumination. | ||
| Condition | Scan direction | V OC [V] | J SC [mA cm−2] | FF | PCE [%] |
|---|---|---|---|---|---|
| a The average values in the parentheses are the averaged ones for ten tested devices. | |||||
| Reference | FS | 1.11 | 22.25 | 0.69 | 17.04 |
| RS | 1.13 (1.10) | 22.41 (22.60) | 0.73 (0.72) | 18.49 (17.93) | |
| KPF6 | FS | 1.15 | 23.01 | 0.75 | 19.85 |
| RS | 1.15 (1.15) | 23.12 (22.90) | 0.75 (0.75) | 19.94 (19.52) | |
Fig. 3c presents the external quantum efficiency (EQE) of PSCs with and without KPF6. The incorporation of KPF6 enhanced the EQE level, resulting in an enhancement in JSC across the entire wavelength range. The calculated Jsc from the EQE spectra was consistent with the JSC obtained from the current density (J)–voltage (V) curves. The stable steady-state power output (SPO) and J at the maximum power point (MPP) confirmed the reliability of the PV properties (Fig. 3d). The KPF6-PSCs exhibited a stabilized PCE of 19.47% and a stabilized photocurrent density of 21.20 mA cm−2, both higher than those of the reference device (17.95% and 19.95 mA cm−2, respectively). Additionally, as shown in Fig. S9,† the performance difference became even more evident under the harsher MPP conditions. While the reference devices degraded relatively quickly, the KPF6-based PSCs retained their performance for a longer duration. Furthermore, the KPF6-treated devices retained their efficiency more effectively under ambient conditions.
The PV performance of KPF6-PSCs was further evaluated under low-intensity indoor LED illumination with various light intensities (400, 600, 800, and 1000 lux) (Fig. 3e, f and Table 2). The emission power and photon flux of the LED used for the measurements are presented in Fig. S8.† KPF6-PSCs achieved much more enhanced indoor PCEs (iPCEs) of 35.47, 35.69, 35.37, and 36.10% at 400, 600, 800, and 1000 lux, respectively, compared to those of reference devices (22.04, 25.78, 27.94, and 29.65%, respectively), in which the ratio of efficiency increase is more noticeable than that under standard 1 sun illumination conditions. In addition, the improvement of VOC and FF mainly contributed to the efficiency increase. These results indicate that KPF6 additives efficiently reduce defects in the perovskite layer through controlled crystallization, the effect of which is more significant under low-intensity light illumination conditions because of fewer excited carriers being more detrimentally affected by defects.32,33 While antisolvent-free methods have been reported to achieve high PCE,34–36 this work demonstrates additive-assisted PSCs achieving high efficiency under both 1 sun and indoor LED illumination, highlighting their broad applicability across lighting conditions.
| Condition | Light intensity [lux] | V OC [V] | J SC [μA cm−2] | FF | iPD [μW cm−2] | iPCE [%] |
|---|---|---|---|---|---|---|
| Reference | 400 | 0.90 | 52.09 | 0.59 | 27.66 | 22.04 |
| 600 | 0.93 | 82.29 | 0.63 | 48.21 | 25.78 | |
| 800 | 0.94 | 108.57 | 0.68 | 69.40 | 27.94 | |
| 1000 | 0.95 | 136.68 | 0.70 | 90.89 | 29.65 | |
| KPF6 | 400 | 0.97 | 60.38 | 0.76 | 44.51 | 35.47 |
| 600 | 0.98 | 88.46 | 0.77 | 66.75 | 35.69 | |
| 800 | 0.99 | 115.25 | 0.77 | 87.86 | 35.37 | |
| 1000 | 0.99 | 139.71 | 0.80 | 110.65 | 36.10 |
To examine the wide applicability of KPF6, we also assessed p–i–n PSCs consisting of FTO/MeO-2PACz/perovskite/C60/BCP/Ag structure (Fig. 3g). Under 1 sun illumination, the device exhibited a PCE of up to 20.12% (Fig. 3h). These PSCs also maintained excellent iPCEs of 25.07%, 25.16%, 26.09%, and 25.90% under 400, 600, 800, and 1000 lux LED illumination, respectively (Fig. 3i and Table S2†). These results highlight the general applicability of the KPF6 additive for achieving high PCE with versatile device structures and different illumination environments.
2.3. Characterization of charge recombination and transport
To scrutinize the origin of PV performance enhancement by KPF6 being involved in charge recombination, we assessed changes of trap density (Nt) in PSCs through space-charge-limited current (SCLC) measurements using an electron-only device consisting of FTO/SnO2/perovskite/C60/Ag (Fig. 4a). Following the relationship Nt = 2ε0εRVTFL/qL2,37 where ε0 is the vacuum permittivity, εR is the relative dielectric constant, VTFL is the trap-filled limit voltage, q is the electron charge, and L is the film thickness, respectively, we found that Nt of KPF6-PSC significantly decreased to 1.59 × 1015 cm−3, compared to the reference PSC (2.60 × 1015 cm−3). These results indicate that KPF6 effectively reduced trap density in the perovskite layer.38 In addition, the dependence of VOC on the light intensity (I) was measured to gain more information on the type of recombination process (Fig. 4b). Following the relationship of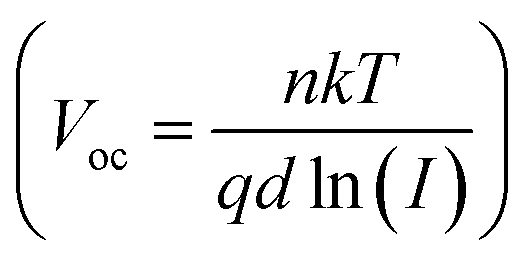 , the slope of the Vocversus dln(I) plot, for the KPF6-PSCs was determined to be 1.56kT/q which is closer to 1.0 compared to the value of the reference PSCs (1.62kT/q). This is indicative of effective suppression of trap-induced recombination by KPF6.39 Fitting the JSC ∝ Iα curves provided the exponent α of 0.92 and 0.90 for KPF6 and reference PSCs, suggesting a comparable degree of bimolecular recombination in the two devices (Fig. 4c).40
, the slope of the Vocversus dln(I) plot, for the KPF6-PSCs was determined to be 1.56kT/q which is closer to 1.0 compared to the value of the reference PSCs (1.62kT/q). This is indicative of effective suppression of trap-induced recombination by KPF6.39 Fitting the JSC ∝ Iα curves provided the exponent α of 0.92 and 0.90 for KPF6 and reference PSCs, suggesting a comparable degree of bimolecular recombination in the two devices (Fig. 4c).40
 | ||
| Fig. 4 (a) Space charge-limited current (SCLC) curves of the electron-only devices. Light intensity dependence of (b) VOC and (c) JSC for PSCs with and without KPF6. (d) Nyquist plots of the EIS measurement and (e) Mott–Schottky plots for PSCs. (f) Transient photovoltage (TPV) curves and (g) transient photocurrent (TPC) curves of PSCs with and without KPF6. Photo-CELIV transient curves of PSCs (h) without and (i) with KPF6. The inset shows the photogenerated extraction charge (Qext). | ||
Electrochemical impedance spectroscopy (EIS) analysis was employed to further investigate charge transport and recombination processes. The Nyquist plots in Fig. 4d show charge-transport resistance (RCT) and recombination resistance (RREC) at high frequency regions and low frequency regions, respectively, based on the equivalent circuit diagram in the inset. As summarized in Table S3,† the KPF6-PSCs exhibited a lower RCT and higher RREC compared to the reference PSCs, which suggests that KPF6 facilitated charge transport efficiency while alleviating recombination.41 Furthermore, the built-in potential (Vbi) of the KPF6-PSC based on the Mott–Schottky measurements was 0.96 V, higher than that of the reference PSC (0.92 V), as shown in Fig. 4e.42 In addition, carrier dynamics of the device were investigated through characterization of transient photovoltage (TPV) and transient photocurrent (TPC) dynamics for the two PSCs. After the incorporation of KPF6, the charge recombination lifetime (τr) of the PSC significantly increased from 15.7 to 49.0 μs (Fig. 4f), while charge transfer lifetimes (τt) decreased from 7.24 and 6.49 μs (Fig. 4g). To compare the charge transport capabilities of the two devices, we extracted charge mobility (u) and photogenerated extraction charge (Qext) through photo-CELIV measurements (Fig. 4h and i). The u value of KPF6-PSCs (8.44 × 10−3 cm2 V−1 s−1) was higher than that of the reference one (7.49 × 10−3 cm2 V−1 s−1), while a larger Qext value (5.00 × 1020 cm−3) was attained from KPF6-PSCs as well (c.f. Qext of the reference PSC: 4.16 × 1020 cm−3).43
All these characterization results support that the addition of KPF6 efficiently reduced defects in antisolvent-free perovskite films. This enables significant reduction of trap-induced recombination and enhancement of charge transport/extraction capability, resulting in highly improved efficiency under various lighting conditions.
2.4. Fabrication of large-area antisolvent-free perovskite films
One of the most critical applications of the antisolvent-free process is to prepare homogeneous perovskite films over large areas, which is hard to achieve through antisolvent-dropping due to the difficulty in uniform treatment.44 In this regard, using KPF6-added precursor solution can be a simple and effective approach to prepare uniform and pinhole-free large-area perovskite films without antisolvent.Thus, we fabricated 5 cm × 5 cm perovskite films using KPF6 as an additive without the antisolvent process. As shown in Fig. 5a, in contrast to the reference perovskite film without KPF6, the KPF6-perovskite film exhibited significantly improved uniformity, particularly near the film edges. For a more detailed investigation of film uniformity, we cut the large-area perovskite films into 9 pieces (Fig. S10†) and measured the thickness, UV-Vis spectra, steady-state PL, and SEM for different positions of each piece. As shown in Fig. 5b, the reference perovskite film exhibited a large variation in thickness across the entire films; however, the KPF6-added one displayed much more uniform thickness. In addition, the KPF6-perovskite film exhibited much more uniform UV-Vis absorbance values at 660 nm across different positions with low deviation, compared to the reference film (Fig. 5c and S11†). This trend was also observed in the distribution of PL spectra (Fig. 5d).
 | ||
| Fig. 5 (a) Optical image of the large-area perovskite films (yellow arrows: inhomogeneous region on the perovskite films). (b) Thickness uniformity of a 5 × 5 cm2 size perovskite film. (c) Absorbance uniformity test of the large-area perovskite film by UV-Vis absorption at 660 nm. (d) PL spectra of 9 pieces cut from a large-area perovskite sample with and without KPF6. | ||
Furthermore, SEM images for the 9 pieces of the reference and KPF6-perovskite films (Fig. S12 and S13†) show that the KPF6 addition enabled much more uniform morphology with reduced pinholes. These results demonstrate that the KPF6 additive ensures enhanced quality and uniformity of large-area antisolvent-free perovskite films by controlling crystallization rate, which is promising for the scalable fabrication of perovskite films.
2.5. PSCs with semi-transparent perovskite films
Finally, to investigate the feasibility of using KPF6 in fabricating semi-transparent perovskite films for potential application in semi-transparent PSCs (ST-PSCs), we fabricated perovskite films by controlling concentrations (Fig. 6a).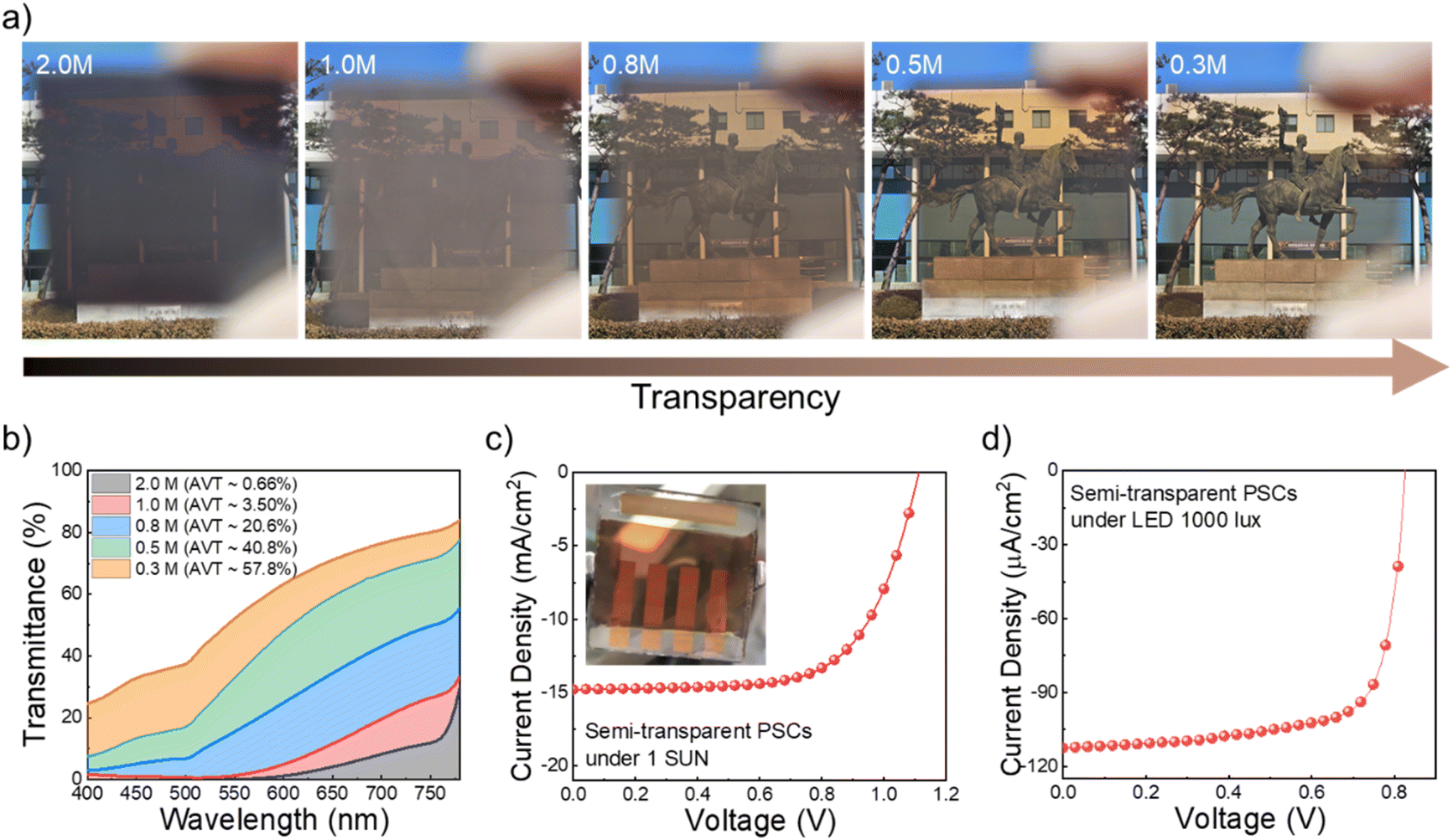 | ||
| Fig. 6 (a) Photograph images of antisolvent-free perovskite films. (b) Transmittance spectra of the perovskite films with different molar concentrations. J–V curve of PSCs with semi-transparent perovskite films under (c) 1 sun and (d) LED 1000 lux illumination. The inset shows a photograph of PSCs incorporating a transparent perovskite layer. | ||
First, we evaluated the average visible transmittance (AVT) of antisolvent-free perovskite films with different concentrations (Fig. 6b). For the 2.0 M perovskite film, the AVT was approximately 0.7%; however, the values gradually increased to 3.5%, 20.6%, 40.8% and 57.8% as the concentration decreased to 1.0, 0.8, 0.5 and 0.3 M, respectively. Considering that the requirement of minimum AVT for semi-transparent solar cells is 20–30%,45 we fabricated the devices using 0.8 M perovskite film (AVT: 20.6%) using an antisolvent-free process. As shown in Fig. 6c, the device exhibited a PCE of 10.7% with a VOC of 1.12 V, JSC of 14.78 mA cm−2, and FF of 0.65 under 1 sun conditions. In addition, a high iPCE of 22.27% was also achieved from the same device under 1000 lux LED illumination conditions (Fig. 6d).
3. Conclusions
In this work, we studied the effects of alkali metal hexafluorophosphate (LiPF6, NaPF6, and KPF6) additives on the crystallization, morphology, and photovoltaic properties of antisolvent-free perovskite solar cells (PSCs). The addition of KPF6 enabled significant improvements in film uniformity and crystallinity by delaying perovskite crystallization, leading to the formation of smooth, pinhole-free films. These morphological enhancements effectively suppressed trap-assisted recombination while improving charge extraction properties, resulting in a substantial efficiency increase under both standard 1 sun and indoor LED illumination conditions. Furthermore, the incorporation of KPF6 enabled the fabrication of large-area and semi-transparent perovskite films with excellent uniformity, highlighting its potential for scalable production. Our achievement suggests that rational selection of alkali metal additives, particularly KPF6, can be an effective strategy for optimizing PV performance of antisolvent-free PSCs and extending their applicability to various device architectures and illumination conditions.4. Experimental
4.1. Materials
Formamidinium iodide (FAI), formamidinium bromide (FABr), and phenylethylammonium iodide (PEAI) were purchased from Dyesol. All solvents, cesium iodide (CsI), lead chloride (PbCl2), bathocuproine (BCP), C60, alkali metal salts, and n-hexyl ammonium iodide (HAI) were purchased from Sigma-Aldrich. Spiro-OMeTAD was purchased from LumTech. Lead iodide (PbI2) and [2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid (MeO-2PACz) were purchased from TCI. Tin(IV) oxide colloid precursor (15% in H2O colloidal dispersion, SnO2) was purchased from Alfa Aesar.The 2 M FA0.83Cs0.17PbI3 perovskite precursor solutions were prepared by dissolving FAI 285.6 mg, PbI2 922 mg, CsI 88.4 mg, PbCl2 55.6 mg and the PF6 additive (1.5 mol%) in 1 ml of DMF: 192 μl of NMP. The spiro-OMeTAD solution was prepared by dissolving 90 mg spiro-OMeTAD in 1 ml chlorobenzene, followed by the addition of 23 μl Li-TFSI (from 520 mg per mL stock acetonitrile solution) and 39.5 μl 4-tert-butylpyridine.
4.2. Fabrication
:DIW ratio of 1:3) was spin-coated at 3000 rpm for 30 s twice on the FTO substrate. The films were annealed at 150 °C for 30 min. KCl doping solution (10 mg ml−1 in DIW) was spin-coated on top of the SnO2 layer. Then it was annealed at 100 °C for 10 min.
Perovskite layers were spin-coated on the KCl/SnO2/FTO substrate at 5000 rpm for 50 s. Then it was annealed at 70 °C for 2 min and further annealed at 150 °C for 10 min. The perovskite surface was passivated with a mixed salt of HAI and FABr (1:1) dissolved in IPA. The mixed salt solution was spin-coated at 3000 rpm for 30 s. After that, the films were annealed at 150 °C for 1 min. Spiro-OMeTAD solution was coated at 4000 rpm for 30 s. Finally 100 nm-thick gold electrodes were deposited using thermal evaporation at 5.0 × 10−6 Torr. The active area was 0.105 cm2.
The semi-transparent perovskite film-based solar cells were fabricated in an n–i–p architecture. Except for the perovskite concentration, all other processing steps were identical to the above n–i–p PSC fabrication.
4.3. Characterization
Perovskite morphology and cross-sectional images of the perovskite films were obtained through field-emission scanning electron microscopy (FE-SEM, Hitachi, S-4800). The morphology of the perovskite films was measured through AFM (Park Systems, XE-100). X-ray diffraction was employed to monitor the FACsPbI3 film formation using an X-ray diffractometer (Rigaku, SmartLab). The UV-Vis absorption and fluorescence spectra were recorded with a UV-visible/NIR spectrophotometer (JASCO, V-770) and a fluorescence spectrophotometer (Edinburgh, FS5), respectively, at room temperature. In steady-state PL measurement, the samples were excited by a pulsed LED with a wavelength of 460 nm. A picosecond pulsed laser diode (EPL-405) was used to measure time-resolved PL. The perovskite thickness was measured with a microfigure measuring instrument (Kosaka Laboratory Ltd, ET200A). The XPS spectra were measured using a K alpha X-ray photoelectron spectrometer (Thermo Fisher Scientific).The J–V curves of the devices were measured using a potentiostat (Compactstat, IVIUM) under AM 1.5 G (100 mW cm−2) illumination with a solar simulator (Oriel LCS-100, 94011A, Newport). Low-intensity light J–V measurements were performed using a source meter (Keithley 4200) under illumination intensities of 400, 600, 800, and 1000 lux with an illumination system (K3000, McScience). The lux levels of the LED lamp were measured using a luxmeter (GL Spectrolux meter, GL Optic).
A semiconductor parameter system (T4000, McScience) was used to measure TPV, TPC, and photo-CELIV curves. The lifetimes of the photocurrent and photovoltage were fitted using single exponential equations. Charge mobility (μ) was extracted from the photo-CELIV measurement using the following equation:46
 | (1) |
The extracted charge (Qext) was obtained from photo-CELIV measurements by integrating the transient photocurrent over time, reflecting the total amount of photogenerated carriers extracted under a linearly increasing voltage.47
The SCLC measurement was conducted using a source meter (Keithley 4200) with an electron-only device (FTO/SnO2/perovskite/C60/BCP/Ag). Electrochemical impedance spectroscopy and Mott–Schottky analysis were performed using a potentiostat (Compactstat, IVIUM). The built-in potential (Vbi) in the Mott–Schottky plot was estimated from the interception of the Mott–Schottky plot using the equation:48
 | (2) |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. J. Choi: experimental work, investigation, and writing – original draft. V. M. Arivunithi: experimental work, investigation, and writing – original draft. S. J. Shin, G. G. Jeon, and H. W. Chun: data analysis, characterization, and data curation. D. W. Kim: data analysis and writing – original draft. J. H. Kim: conceptualization, supervision, investigation, data analysis, and writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00436187) and Korea Electric Power Corporation (R22XO02-03).Notes and references
- NREL chart, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf.
- W. Li, M. U. Rothmann, Y. Zhu, W. Chen, C. Yang, Y. Yuan, Y. Y. Choo, X. Wen, Y.-B. Cheng, U. Bach and J. Etheridge, Nat. Energy, 2021, 6, 624–632 CrossRef CAS.
- P. Han and Y. Zhang, Adv. Mater., 2024, 36, 2405630 CrossRef CAS PubMed.
- T. Webb, S. J. Sweeney and W. Zhang, Adv. Funct. Mater., 2021, 31, 2103121 CrossRef CAS.
- J. Sun, F. Li, J. Yuan and W. Ma, Small Methods, 2021, 5, 2100046 CrossRef CAS PubMed.
- Y. Yang, Z. Huang, H. Gao, Z. Xu, W. Fang, Y. Chen, Y. Hu, Z. Yi, J. Huang and H. Zhu, RSC Adv., 2024, 14, 32370–32388 RSC.
- K.-M. Lee, C.-J. Lin, B.-Y. Liou, S.-M. Yu, C.-C. Hsu, V. Suryanarayanan and M.-C. Wu, Sol. Energy Mater. Sol. Cells, 2017, 172, 368–375 CrossRef CAS.
- A. D. Taylor, Q. Sun, K. P. Goetz, Q. An, T. Schramm, Y. Hofstetter, M. Litterst, F. Paulus and Y. Vaynzof, Nat. Commun., 2021, 12, 1878 CrossRef CAS PubMed.
- T. Bu, X. Liu, J. Li, W. Huang, Z. Wu, F. Huang, Y.-B. Cheng and J. Zhong, Sol. RRL, 2020, 4, 1900263 CrossRef CAS.
- K. Geistert, S. Ternes, D. B. Ritzer and U. W. Paetzold, ACS Appl. Mater. Interfaces, 2023, 15, 52519–52529 CAS.
- J. Gao, F. Fei, Y. Xu, S. Wang, Y. Li, K. Du, H. Sun, X. Dong, N. Yuan, L. Li and J. Ding, ACS Appl. Mater. Interfaces, 2024, 16, 38017–38027 CrossRef CAS PubMed.
- X. Ma, P. Chen, J. Luo, N. Yang, D. Luo, R. Su and K. Chen, ACS Appl. Energy Mater., 2024, 7, 4540–4548 CrossRef CAS.
- X. Gu, C. Shan, X. Xu, Q. Liu and A. K. K. Kyaw, Small, 2024, 20, 2307840 CrossRef CAS PubMed.
- M. Abdel-Shakour, T. H. Chowdhury, K. Matsuishi, M. A. Karim, Y. He, Y. Moritomo and A. Islam, ACS Appl. Energy Mater., 2021, 4, 12515–12524 CrossRef CAS.
- L. A. Castriotta, E. Calabrò, F. Di Giacomo, S. H. Reddy, D. Takhellambam, B. Paci, A. Generosi, L. Serenelli, F. Menchini, L. Martini, M. Tucci and A. D. Carlo, Nano Energy, 2023, 109, 108268 CrossRef CAS.
- T. Bu, J. Li, H. Li, C. Tian, J. Su, G. Tong, L. K. Ono, C. Wang, Z. Lin, N. Chai, X. Zhang, J. Chang, J. Lu, J. Zhong, W. Huang, Y. Qi, Y. Cheng and F. Huang, Science, 2021, 372, 1327–1332 CrossRef CAS PubMed.
- X. Guo, Z. Lin, W. Cao, Y. Xu, Q. Wang, B. Zhang, Y. Hao and J. Chang, J. Mater. Chem. C, 2023, 11, 9144–9152 RSC.
- D.-Y. Son, S.-G. Kim, J.-Y. Seo, S.-H. Lee, H. Shin, D. Lee and N.-G. Park, J. Am. Chem. Soc., 2018, 140, 1358–1364 CrossRef CAS PubMed.
- J. Cao, S. X. Tao, P. A. Bobbert, C. P. Wong and N. Zhao, Adv. Mater., 2018, 30, 1707350 CrossRef PubMed.
- T. Nie, Z. Fang, T. Yang, K. Zhao, J. Ding and S. Liu, Angew. Chem., Int. Ed., 2024, 63, e202400205 CrossRef CAS PubMed.
- H. Hu, Z. Ren, P. W. Fong, M. Qin, D. Liu, D. Lei, X. Lu and G. Li, Adv. Funct. Mater., 2019, 29, 1900092 CrossRef.
- Y. Zhou, M. Yang, O. S. Game, W. Wu, J. Kwun, M. A. Strauss, Y. Yan, J. Huang, K. Zhu and N. P. Padture, ACS Appl. Mater. Interfaces, 2016, 8, 2232–2237 CrossRef CAS PubMed.
- Z. Chen, Q. Dong, Y. Liu, C. Bao, Y. Fang, Y. Lin, S. Tang, Q. Wang, X. Xiao, Y. Bai, Y. Deng and J. Huang, Nat. Commun., 2017, 8, 1890 CrossRef PubMed.
- X. Xu, X. Ji, R. Chen, F. Ye, S. Liu, S. Zhang, W. Chen, Y. Wu and W. H. Zhu, Adv. Funct. Mater., 2022, 32, 2109968 CrossRef CAS.
- D. Zheng, F. Yi, Q. Zhang, Q. Guo, Q. Tang and J. Duan, Energy Technol., 2022, 10, 2200290 CrossRef CAS.
- C. Fei, N. Li, M. Wang, X. Wang, H. Gu, B. Chen, Z. Zhang, Z. Ni, H. Jiao, W. Xu, Z. Shi, Y. Yan and J. Huang, Science, 2023, 380, 823–829 CrossRef CAS PubMed.
- R. S. Bobba, N. Ghimire, M. B. Faheem, S. Mabrouk, A. Baniya, N. Narwal, Y. Zhang, P. I. Kaswekar, H. Li, M. B. Saud, M. Kumar and Q. Qiao, Sol. RRL, 2023, 7, 2300191 CrossRef CAS.
- W. J. Jang, E. H. Kim, J. H. Cho, D. Lee and S. Y. Kim, Adv. Sci., 2024, 11, 2406657 CrossRef CAS PubMed.
- F. Li, X. Deng, Z. Shi, S. Wu, Z. Zeng, D. Wang, Y. Li, F. Qi, Z. Zhang and Z. Yang, Nat. Photon., 2023, 17, 478–484 CrossRef CAS.
- L. Fu, H. Li, L. Wang, R. Yin, B. Li and L. Yin, Energy Environ. Sci., 2020, 13, 4017–4056 RSC.
- C. A. Aranda, A. O. Alvarez, V. S. Chivrony, C. Das, M. Rai and M. Saliba, Joule, 2024, 8, 241–254 CrossRef CAS.
- E. Han, M. Lyu, E. Choi, Y. Zhao, Y. Zhang, J. Lee, S. M. Lee, Y. Jiao, S. H. A. Ahmad, J. Seidel, J. S. Yun, J. Yun and L. Wang, Small, 2024, 20, 2305192 CrossRef CAS PubMed.
- C. Zhang, C. Liu, Y. Gao, S. Zhu, F. Chen, B. Huang, Y. Xie, Y. Liu, M. Ma, Z. Wang, S. Wu, R. E. I. Schropp and Y. Mai, Adv. Sci., 2022, 9, 2204138 CrossRef CAS PubMed.
- Z. Zhang, M. Li, R. Li, X. Zhuang, C. Wang, X. Shang, D. He, J. Chen and C. Chen, Adv. Mater., 2024, 36, 2313860 CrossRef CAS PubMed.
- Y. Yang, Y. Wang, Z. Qu, K. Zhang, T. Liang, S. Chen, W. Lv, F. Min, Y. Chen and Y. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202300971 CrossRef CAS PubMed.
- D.-K. Lee, K.-S. Lim, J.-W. Lee and N.-G. Park, J. Mater. Chem. A, 2021, 9, 3018–3028 RSC.
- J. Liang, Z. Chen, G. Yang, H. Wang, F. Ye, C. Tao and G. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 23152–23159 CrossRef CAS PubMed.
- M. Azam, Z. Ke, J. Luo, Z. Wan, A. Hassan and C. Jia, Chem. Eng. J., 2024, 483, 149424 CrossRef CAS.
- Y. Wang, J. Wu, C. Deng, F. Liu, M. Guo, J. Xu, L. Gao, M. Huang, Z. Lan and P. Gao, Adv. Funct. Mater., 2025, 2419868 CrossRef CAS.
- Y. Zhang, B. Yu, Y. Sun, J. Zhang, Z. Su and H. Yu, Angew. Chem., Int. Ed., 2024, 63, e202404385 CrossRef CAS PubMed.
- Y. Lei, H. Li, X. Liu, H. Wang, H. Dan, Y. Ni, W. Zou, Y. Tang, S. Liu and Y. Peng, J. Alloys Compd., 2023, 969, 172427 CrossRef CAS.
- Z. Zheng, F. Li, J. Gong, Y. Ma, J. Gu, X. Liu, S. Chen and M. Liu, Adv. Mater., 2022, 34, 2109879 CrossRef CAS PubMed.
- D. H. Kim, H. J. Lee, S. H. Lee, Y. J. Kang, S. N. Kwon, D. H. Kim and S. I. Na, Sol. RRL, 2024, 8, 2400067 CrossRef CAS.
- Y. Chen, L. Zhang, Y. Zhang, H. Gao and H. Yan, RSC Adv., 2018, 8, 10489–10508 RSC.
- T. M. Koh, H. Wang, Y. F. Ng, A. Bruno, S. Mhaisalkar and N. Mathews, Adv. Mater., 2022, 34, 2104661 CrossRef CAS PubMed.
- J. Peng, Y. Chen, K. Zheng, T. Pullerits and Z. Liang, Chem. Soc. Rev., 2017, 46, 5714–5729 RSC.
- D. H. Kim, H. J. Lee, S. H. Lee, Y. J. Kang, S. N. Kwon, D. H. Kim and S. I. Na, Sol. RRL, 2024, 8, 2400067 CrossRef CAS.
- Y. Bai, Z. Zhou, Q. Xue, C. Liu, N. Li, H. Tang, J. Zhang, X. Xia, J. Zhang and X. Lu, Adv. Mater., 2022, 34, 2110587 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00421g |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |