
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting bifunctional oxygen electrocatalysis by integrating Fe–Nx moieties and FeNi nanoparticles for highly efficient and long-life rechargeable zinc–air batteries†
Zubair
Ahmed
a,
Jekaterina
Kozlova
b,
Kaupo
Kukli
b,
Arvo
Kikas
b,
Vambola
Kisand
 b,
Alexey
Treshchalov
b,
Maike
Käärik
a,
Jaan
Leis
a,
Jaan
Aruväli
c and
Kaido
Tammeveski
*a
b,
Alexey
Treshchalov
b,
Maike
Käärik
a,
Jaan
Leis
a,
Jaan
Aruväli
c and
Kaido
Tammeveski
*a
aInstitute of Chemistry, University of Tartu, Ravila 14a, Tartu 50411, Estonia. E-mail: kaido.tammeveski@ut.ee
bInstitute of Physics, University of Tartu, W. Ostwald Str. 1, Tartu 50411, Estonia
cInstitute of Ecology and Earth Sciences, University of Tartu, Vanemuise 46, Tartu 51014, Estonia
First published on 25th February 2025
Abstract
Realizing high-performance and long-life rechargeable zinc–air batteries (ZABs) requires developing efficient and robust bifunctional electrocatalysts for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), as the low efficiency and short lifetime of bifunctional oxygen electrocatalysts greatly limit the practical application of rechargeable ZABs. Herein, based upon multi-component-dependent electrocatalytic activity and selectivity, we propose to synthesize a promising bifunctional oxygen electrocatalyst enriched with highly ORR-active atomically dispersed Fe–Nx sites and exceptionally efficient FeNi nanoparticles for the OER. Owing to this integration, the developed catalyst Ni3@Fe-N-GNS exhibits a small potential gap for catalyzing both the ORR and OER, accordingly making it an ideal electrocatalyst for rechargeable Zn–air batteries. Impressively, when used as an air electrode, the corresponding ZAB exhibits a high peak power density of 171 mW cm−2, a small charge–discharge voltage gap of 0.71 V at 5 mA cm−2, and excellent charge–discharge cycling stability without much deviation after 180 h of a continuous run. The present work proposes a new avenue for the rational design of bifunctional electrocatalysts to make advances in electrochemical energy technologies.
1. Introduction
In search of green electrochemical energy systems, rechargeable Zn–air batteries (ZABs) have attracted wide scientific attention as energy storage devices because of their high energy density, good overall performance, and eco-friendliness.1–4 They have been demonstrated to be a potential source for handy and high energy storage devices, where the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are the most consequential electrochemical reactions. In this regard, efficient bifunctional oxygen electrocatalysts are critical for the air electrode in ZABs.5–8 They are desirable so that reactions are activated independently and maintain conditions close to the equilibrium of the O2/OH− couple in opposite directions. Numerous research groups are dedicated to exploring high-performance and cost-efficient bifunctional ORR/OER electrocatalysts to boost metal–air battery performance and advance their practical application.9–11At present, Ir/Ru-based oxides are the most efficient catalysts for the OER, while Pt gives the best electrocatalytic activity for the ORR. Unfortunately, none of them have the necessary bifunctional activity to catalyze both the ORR and OER at the same time, which makes them unsuitable catalysts for rechargeable ZABs. Along with this, the high cost, scarcity, and unstable active sites restrict their further commercial application on large scale.12–16 Therefore, exploration of alternative low-cost catalysts with earth-abundant elements and high electrocatalytic activity that can be upgraded in the single cathode is of scientific interest and economic value. Emerging as a class of alternative non-precious metal electrocatalysts, transition metals and their compounds such as alloys,17–19 oxides,20–22 hydroxides,23,24etc. are considered as a group of eco-friendly and low-cost OER and ORR electrocatalysts, some of which have demonstrated high electrocatalytic performances. Bimetallic catalysts, in particular FeNi-based materials, exert a unique synergistic effect, which generates abundant active sites for OH− adsorption during the OER process.25–28 However, poor ORR performance and low stability are apparent problems with these FeNi catalysts for practical application in rechargeable ZABs.29 Among the most widely used approaches, incorporating FeNi nanostructures on heteroatom-doped carbon nanomaterials or creating heterostructures are the most widely used methods to improve their ORR performance, suggesting that these nanostructures could serve as catalysts in multiple systems.6,30,31 For instance, Lai et al.32 reported that S modulation of Ni3FeN nanoparticles increases the number of ORR-active sites which in turn enhances their bifunctional OER/ORR performance. Liu et al.33 reported NiFe nanoparticles encapsulated within oxygen-doped carbon quantum dots as a bifunctional OER/ORR electrocatalyst. Li et al.34 demonstrated alloyed FeNi nanoparticles encapsulated within N-doped layered carbon nanosheets for achieving high efficiency in ZABs. Morales et al.35 reported an MnOx incorporating strategy for the activation of the Fe–Ni catalyst toward the ORR and thus, fabricating them as bifunctional oxygen electrodes. Similarly, Lan et al.36 explored the synergetic effect of NiFe-MOF/NiFe2O4 heterostructures for ORR and OER bifunctional electrocatalysis. On the other hand, atomically dispersed Fe coordinated to nitrogen in the carbon framework (Fe–N–C) structure further optimizes the O2-adsorption and reduction on the active sites.37,38 These intrinsic features of such electrocatalysts have compelled researchers to explore various strategies to construct hybrid catalysts consisting of atomically dispersed sites in order to overcome the large potential barriers between oxygen evolution and reduction reactions. Xu et al.39 recently developed a hybrid CoyNix@Fe–N–C material by connecting the OER-active CoyNix alloy to the ORR-active Fe–Nx sites. It was found that the electron transfer pathway between the heterogeneous CoNi alloy and Fe–Nx moieties contributes to superior ORR/OER bifunctional performance.
Inspired by these findings, we demonstrate a systematic strategy based on multi-component-dependent activity and selectivity, and their optimized structures, which outperform single-component systems in bifunctional OER/ORR activity with high stability, meeting the requirements for practical application. Herein we integrate iron phthalocyanine-derived atomically dispersed Fe–Nx moieties and FeNi nanoparticles into a Ni3@Fe-N-GNS hybrid material. The prepared Ni3@Fe-N-GNS material is demonstrated to be a highly active and stable bifunctional electrocatalyst for the ORR and OER. Notably, a rechargeable zinc–air battery consisting of a Ni3@Fe-N-GNS air electrode is fabricated and exhibits a high peak power density of 171 mW cm−2, an ultrahigh specific capacity of 894 mA h g−1 (at 20 mA cm−2), and excellent cycling stability for 180 h, making it superior to commercial Pt/Ru-containing catalysts. The study provides a new pathway to design bifunctional electrocatalysts for electrochemical applications and related energy devices.
2. Experimental section
2.1. Material synthesis
2.2. Material characterization
The X-ray diffraction (XRD) analysis was done using a Bruker D8 Advance diffractometer with Ni-filtered Cu Kα radiation (λ = 1.54 Å). Micro-Raman spectra were recorded in back-scattering geometry (Renishaw spectrometer) in conjunction with a confocal microscope (Leica Microsystems CMS GmbH, 50× objective) and an argon ion laser operated at 514.5 nm. The N2 adsorption–desorption isotherms of the samples were obtained using a Quantachrome NOVAtouch LX2 instrument after overnight drying at 200 °C prior to analysis. The specific surface area was estimated according to the Brunauer–Emmett–Teller (BET) theory in the P/P0 range of 0.02–0.2 and the pore size distribution (PSD) was calculated by the quenched solid density functional theory (QSDFT) model for slit-type pores. Scanning electron microscopy (SEM) was performed with a Helios NanoLab 600 (FEI) to study surface morphology. The detailed examination of morphological features was done with scanning transmission electron microscopy (STEM) using a Titan Themis 200 (FEI). Both bright field (BF) and high-angle annular dark field (HAADF) images were acquired on the sample at an accelerating voltage of 200 kV. The elemental mapping in STEM was acquired using a four-quadrant Super-X energy dispersive X-ray (EDX) system (FEI/Bruker). Elemental composition of the samples was measured using an INCA Energy 350 EDX spectrometer (Oxford Instruments) in a Helios NanoLab 600 SEM. To investigate the elemental states and surface chemical composition, the X-ray photoelectron spectroscopy (XPS) analysis was done employing an electron energy analyzer (SCIENTA SES-100) and Mg Kα radiation (1253.6 eV) from a non-monochromatic twin anode X-ray tube, Thermo XR3E2. The binding energy calibration was done with respect to the C 1s peak and fitted with Lorentzian–Gaussian peak functions and a mixture of linear and Shirley backgrounds using the Casa XPS software.2.3. Electrochemical measurements
All the electrochemical tests for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) were performed under ambient conditions in O2-saturated 0.1 M KOH solution using a Metrohm Autolab potentiostat/galvanostat, PGSTAT30. The experiments were conducted in a three-electrode setup with the rotating disc electrode (RDE) and rotating ring-disc electrode (RRDE) as working electrodes of 0.196 and 0.164 cm2 geometric area, respectively. For RDE tests, a CTV101 (Radiometer) was used as a speed control unit and an AFMSRX rotator together with an MSRX speed controller (Pine Research) was employed for RRDE measurements. The saturated calomel electrode (SCE) connected via a salt bridge was used as the reference electrode and a graphitic rod served as the counter electrode. The potentials were converted to the reversible hydrogen electrode (RHE) scale using the equation: ERHE = ESCE + 0.241 V + 0.059 V × pH.To prepare the working electrode, 2 mg of catalyst material was dispersed in 495 μL of isopropanol and 5 μL of Nafion ionomer solution (5 wt% Sigma-Aldrich). The catalyst inks were sonicated until a homogeneous dispersion was obtained. 10 μL of the above ink was then drop-cast onto the cleaned glassy carbon (GC) surface and dried in an oven at 60 °C, to attain a catalyst loading of 0.2 mg cm−2. Pt/C (20 wt% Sigma-Aldrich) and RuO2 (99%, Alfa Aesar) ink was prepared by a similar procedure with a catalyst loading of 0.1 mg cm−2. The ORR polarization curves were recorded in the potential range from 1.1 to 0 V vs. RHE at a scan rate (ν) of 10 mV s−1. The RDE polarization curves were recorded at various rotation speeds (ω) from 600 to 3000 rpm. The Koutecky–Levich (K–L) analysis of the RDE polarization data for O2 reduction at various potentials is provided in the ESI.† The OER polarization curves were recorded in the potential range from 1.2 to 1.8 V vs. RHE at 1600 rpm with a scan rate of 10 mV s−1. Before each electrochemical test, the electrodes were activated by fast scanning in the same potential window for 20 cycles, and iR-correction was applied to the polarization curves with Nova 2.1 software. The peroxide percentage yield (% HO2−) and the electron transfer number (n) in the ORR process were calculated using eqn (1) and (2), respectively.40
 | (1) |
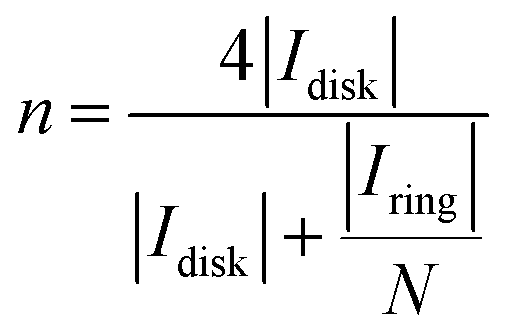 | (2) |
2.4. Zn–air battery (ZAB) assembly
In-house built liquid-state ZABs identical to the setup used in the previous investigation42 were assembled using a zinc plate (99.9%, Auto-plaza) of 0.2 mm thickness, a solution of 6 M KOH with 0.2 M zinc acetate (98%, Fisher Scientific) as the electrolyte, and a catalyst-coated gas diffusion layer (GDL, Sigracet BB39) of 1.8 cm × 1.8 cm as the air electrode with Ni mesh as the current collector. For air electrode preparation, 7 mg of catalyst was dispersed in 800 μL (200 μL water and 600 μL ethanol) of solution with an additional 20 μL of 5% Nafion solution as a binder and sonicated until a uniform dispersion was achieved. The ink was then pipetted onto the GDL to obtain 2 mg cm−2 catalyst loading and dried at 60 °C. As a standard material, the PtRu/C (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:25; Alfa Aesar) catalyst electrode was prepared using the same procedure but with a catalyst loading of 1 mg cm−2. The catalyst area exposed to the electrolyte solution at the air-electrode is 0.79 cm2. Specific capacity of the ZAB was measured at 20 mA cm−2 current density during total discharge over time to the weight loss of the Zn anode. The assembled rechargeable ZAB's stability was assessed using a galvanostatic discharge–charge cycle at 5 mA cm−2 (10 min per cycle). The calculation for specific capacity and round-trip efficiency is provided in the ESI. The EIS measurements for the ZAB were conducted at a discharging potential of 1.25 V, with a frequency range from 100 kHz to 0.1 kHz and an amplitude of 0.01 V.
:25:25; Alfa Aesar) catalyst electrode was prepared using the same procedure but with a catalyst loading of 1 mg cm−2. The catalyst area exposed to the electrolyte solution at the air-electrode is 0.79 cm2. Specific capacity of the ZAB was measured at 20 mA cm−2 current density during total discharge over time to the weight loss of the Zn anode. The assembled rechargeable ZAB's stability was assessed using a galvanostatic discharge–charge cycle at 5 mA cm−2 (10 min per cycle). The calculation for specific capacity and round-trip efficiency is provided in the ESI. The EIS measurements for the ZAB were conducted at a discharging potential of 1.25 V, with a frequency range from 100 kHz to 0.1 kHz and an amplitude of 0.01 V.
3. Results and discussion
3.1. Physical characterization
Scheme 1 illustrates the preparation process of the Ni3@Fe-N-GNS catalyst materials. First, graphene nanostructures (GNS) were prepared using commercial graphene nanoplatelets and carbon nanotubes (CNTs), as discussed in Subsection 2.1.1. Such hetero-structured carbon support materials are crucial to intrinsic electrical conductivity and active site distribution due to their high surface area and stability, which in turn enhances electrocatalytic performance.43–45 The iron phthalocyanine (containing Fe–N4 moiety) as an Fe source and nickel nitrate as a Ni source were added in a methanol solution containing well-dispersed GNS. The suspension was then sonicated and dried, and the obtained powder was pyrolyzed in an inert atmosphere, leading to the formation of FeNi nanoparticles with abundant Fe–Nx moieties. | ||
| Scheme 1 A schematic representation of the Ni3@Fe-N-GNS catalyst preparation process. | ||
To characterize the crystallographic structure of all the catalysts, XRD studies were performed (Fig. 1). The XRD peak at 26.5° in all the catalysts corresponds to the (002) planes of graphitic carbon.46 The XRD pattern of Fe-N-GNS shows peaks at 44.7° and 65.0° that can be indexed to the reflection from the (110) and (200) planes indicating the existence of metallic Fe (PDF 04-007-9753). The Fe-N-GNS also displays peaks that can be attributed to Fe3C (JCPDS no. 65-2412) and Fe3O4 (JCPDS no. 01-090-3358), suggesting that the Fe-N-GNS material consists of Fe/Fe3C species with a small amount of Fe3O4. The XRD pattern of Ni1@Fe-N-GNS, Ni2@Fe-N-GNS, Ni3@Fe-N-GNS, and Ni4@Fe-N-GNS displays peaks at 43.49°, 50.67°, and 74.54° that are assigned to the (111), (200), and (220) planes, respectively, comparable to Fe0.5Ni0.5 alloy nanoparticles (PDF#06-6296).29 In addition, Ni1@Fe-N-GNS also shows the presence of Fe–Fe3C phases, which diminished from Ni1@Fe-N-GNS to Ni4@Fe-N-GNS due to the higher amount of Fe0.5Ni0.5 formed. Furthermore, the catalyst composition was analyzed by Rietveld refinement using Topas 6 software, which shows that Ni3@Fe-N-GNS consists of the highest amount of FeNi phase among all the samples (Table S1†). To explore the defects, Raman spectroscopy measurements were carried out (Fig. S1†). It can be seen that all catalysts possess the D and G bands located at 1365 and 1590 cm−1, according to the disordered and graphitic structure, respectively.47 Generally, in the CNT-containing composites, the G band intensity appears to be higher than that of other carbon-based materials, which is due to large compressive stress affecting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond present in highly defective carbon nanotube walls.48 Moreover, the ratio of D and G peak intensity (ID/IG) increases in the order of Fe-N-GNS < Ni1@Fe-N-GNS < Ni2@Fe-N-GNS < Ni3@Fe-N-GNS < Ni4@Fe-N-GNS, which is attributed to heavily loaded Ni/Fe moieties, while the ID/IG ratio of Ni3@Fe-N-GNS is close to 0.5, suggesting that the CNT structure is not disrupted during catalyst synthesis.
C bond present in highly defective carbon nanotube walls.48 Moreover, the ratio of D and G peak intensity (ID/IG) increases in the order of Fe-N-GNS < Ni1@Fe-N-GNS < Ni2@Fe-N-GNS < Ni3@Fe-N-GNS < Ni4@Fe-N-GNS, which is attributed to heavily loaded Ni/Fe moieties, while the ID/IG ratio of Ni3@Fe-N-GNS is close to 0.5, suggesting that the CNT structure is not disrupted during catalyst synthesis.
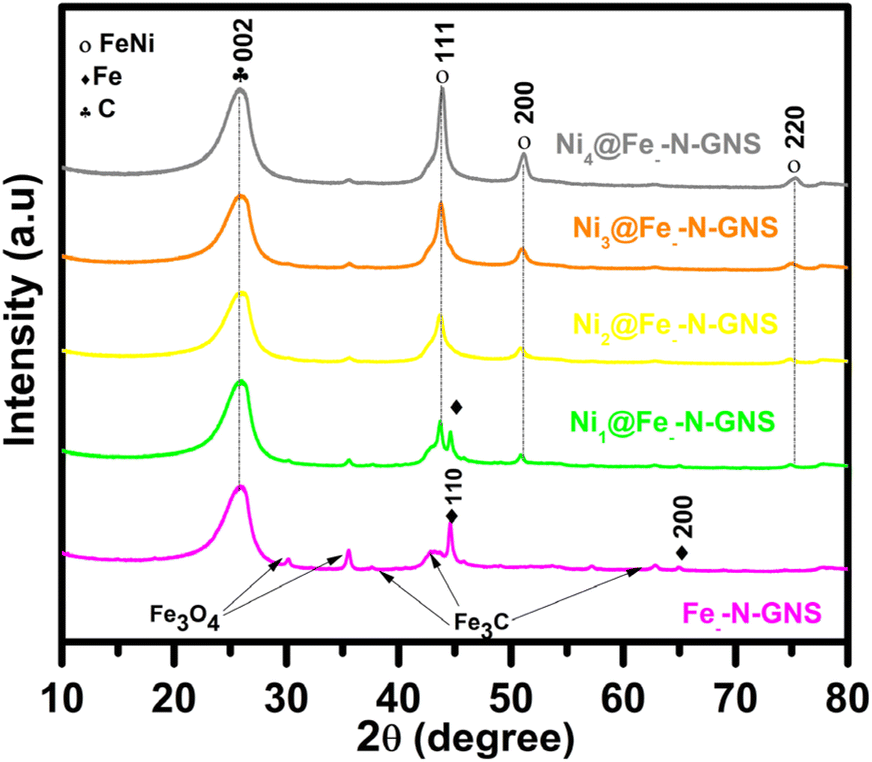 | ||
| Fig. 1 XRD patterns of Ni1@Fe-N-GNS, Ni2@Fe-N-GNS, Ni3@Fe-N-GNS, Ni4@Fe-N-GNS, and Fe-N-GNS (synthesized without the nickel precursor). | ||
To further investigate the surface chemical composition and the valence state of transition metals in the prepared catalysts, X-ray photoelectron spectroscopy (XPS) analysis was performed. Fig. 2a shows the survey spectrum of Ni3@Fe-N-GNS depicting the coexistence of Ni, Fe, C, N and O elements. In general, the N atoms in a two-dimensional carbon framework initiate electron flow to carbon's π-orbital, causing high charge density in the neighboring carbon atom. Meanwhile, the second or third neighboring C atoms possess a high spin density, and this combination favors the ORR kinetics.49,50 The high-resolution N 1s spectrum (Fig. 2b) was deconvoluted into five characteristic peaks, which are imine (∼397.8 eV), pyridinic-N (∼398.6 eV), M–Nx (∼399.7 eV), pyrrolic-N (∼400.6 eV), and graphitic-N (∼402.0 eV). A comparison of N 1s XPS spectra among all the catalysts (see Fig. 2b and S2†) suggests that Ni3@Fe-N-GNS is rich in M–Nx centers, which plays a key role in enhancing the ORR electrocatalytic activity of the catalyst.51–54 The high-resolution Fe 2p spectrum is shown in Fig. 2c, where the peak located at 706.9 eV is allocated to Fe0, while the peak at 710.9 eV is assigned to Fe–N species in Ni3@Fe-N-GNS, and the shoulder peaks at 713.8 eV indicate the presence of Fe3+ species.55,56 The appearance of Fe3+ is most likely due to surface oxidation by exposure to the air, thus illustrating that Fe in Ni3@Fe-N-GNS maintains its original iron phthalocyanine-derived Fe–Nx structure. For Ni 2p (Fig. 2d) the XPS spectrum of Ni2+ was identified by the Ni 2p3/2 (855.7 eV) and Ni 2p1/2 (873.1 eV) peaks with two satellites at 861.3 and 879.9 eV,57 while the peak at 852.8 eV indicates the presence of a small amount of metallic Ni.58 Notably, the binding energy of Fe 2p peaks displays a positive shift after introduction of Ni as compared to the undoped Fe-N-GNS sample due to augmented electronic interactions involving Fe and Ni (Fig. 2c and S3†). The binding energy shift is Ni/Fe ratio dependent, which facilitates the non-uniform charge distribution, leading to faster OER kinetics.59 In addition, the Ni 2p XPS spectra of the other prepared catalysts are shown in Fig. S4,† and surface elemental content and concentrations of various moieties [are presented in Table S2†].
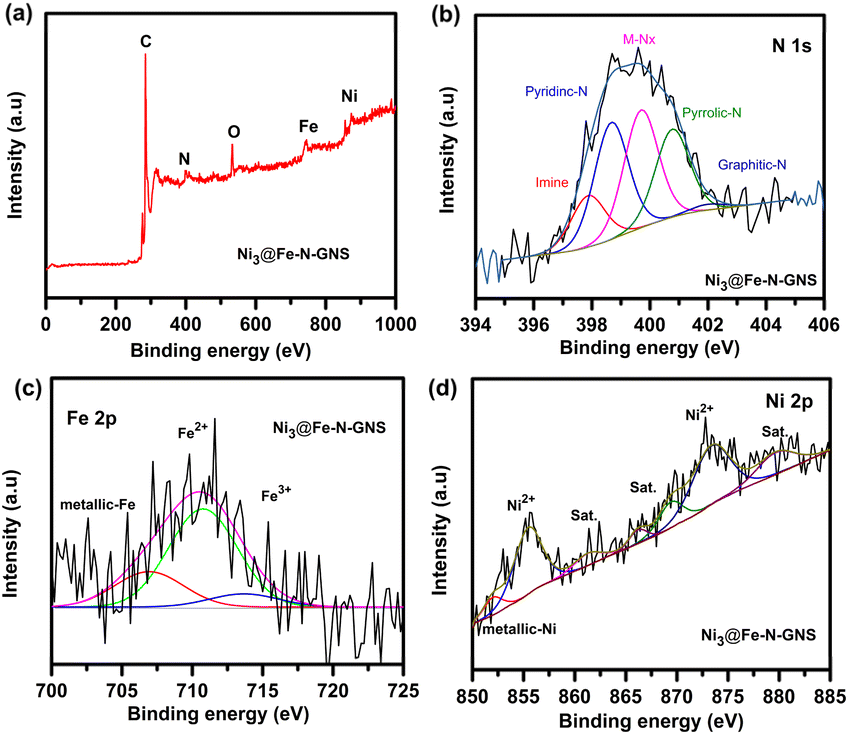 | ||
| Fig. 2 XPS spectra of Ni3@Fe-N-GNS: (a) survey spectrum, core-level XPS spectra in the (b) N 1s, (c) Fe 2p, and (d) Ni 2p regions. | ||
The morphology of the prepared Fe-N-GNS, Ni3@Fe-N-GNS, and Ni4@Fe-N-GNS catalysts was first investigated via scanning electron microscopy (SEM). All the prepared materials possess a porous graphene sheet wrapped with CNTs creating a CNT/graphene heterostructure-based network that holds the FeNi nanoparticles. Such structures are favorable for the enhanced mass-transfer properties during electrocatalysis.60 The morphology of Fe-N-GNS (Fig. 3a and b) appears denser compared to the other two samples. Following the introduction of nickel, the catalysts exhibited a rougher surface, which partially triggered the formation of additional CNTs in the Ni3@Fe-N-GNS (Fig. 3c and d) and Ni4@Fe-N-GNS (Fig. 3e and f) samples during the pyrolysis process. Additionally, SEM-EDX was conducted to analyze the bulk elemental composition of the prepared catalysts and the analysis results are presented in Table S3.† The Fe and Ni content in Ni3@Fe-N-GNS was approximately 4.68 and 4.86 wt%, respectively, which is close to the Fe and Ni content that was added during its synthesis step. The N2 adsorption–desorption isotherms were used to access the specific surface area and pore size distribution of the material as the porous nature of the catalyst favors mass transport and enhances electrocatalytic performance.61,62
 | ||
| Fig. 3 SEM images of (a and b) Fe-N-GNS, (c and d) Ni3@Fe-N-GNS, and (e and f) Ni4@Fe-N-GNS samples with different magnifications. | ||
The Fe-N-GNS, Ni3@Fe-N-GNS, and Ni4@Fe-N-GNS materials display a high specific surface area of 394, 352, and 300 m2 g−1, respectively (Fig. 4a). The reduced surface area of Ni3@Fe-N-GNS and Ni4@Fe-N-GNS compared to Fe-N-GNS may be due to a collapsed carbon skeleton caused by optimizing the Fe/Ni ratio through increased Ni loading. Furthermore, the pore size distribution curve (Fig. 4b) confirms the mesoporous structure of the prepared catalysts, with pore sizes ranging from 2 to 10 nm. The overall textural properties of the catalysts are provided in Table S4.† The bright field (BF)-STEM image confirms the uniform confinement of FeNi nanoparticles within the graphene nanostructures in Ni3@Fe-N-GNS as shown in Fig. S5.† The high-angle annular dark-field (HAADF)-STEM image (Fig. 5a) displays a lattice fringe of 0.207 nm, corresponding to the (111) plane in the cubic FeNi phase in Ni3@Fe-N-GNS. Fig. 5b shows abundant isolated bright spots, indicating atomically dispersed metal sites anchored on the graphene nanostructures. Furthermore, the elemental mapping (Fig. 5c–f) reveals the uniform distribution of Ni, Fe, and N, suggesting good dispersion of all the elements. Compared to Ni3@Fe-N-GNS, the STEM image of Fe-N-GNS (Fig. 5g) shows the presence of a single Fe nanoparticle with a less wide lattice fringe of 0.201 nm, which is well-consistent with the d-spacing of the (110) planes of Fe. Fig. 5h shows evenly scattered bright spots on graphene nanostructures, demonstrating that the isolated metal atoms are atomically dispersed Fe–Nx sites anchored on the carbon substrate. Moreover, the elemental mapping images of Fe-N-GNS confirm the presence of the constituent elements and are shown in Fig. 5i–l.
 | ||
| Fig. 4 (a) N2 adsorption/desorption isotherms of Fe-N-GNS, Ni3@Fe-N-GNS, and Ni4@Fe-N-GNS, (b) pore size distribution of the samples. | ||
 | ||
| Fig. 5 (a) HAADF-STEM image of a single FeNi nanoparticle, (b) HAADF-STEM image showing atomically dispersed metals, (c–f) elemental mapping of Ni, Fe, N and composite maps in the HAADF-STEM image of Ni3@Fe-N-GNS, (g) STEM-HAADF image of a single Fe nanoparticle, (h) STEM image showing atomically dispersed Fe, (i–l) HAADF-STEM image and elemental maps of Fe, N, and composite maps in Fe-N-GNS. | ||
3.2. Electrocatalytic performance for the ORR and OER
The electrocatalytic ORR activity of the prepared catalysts was measured in O2-saturated 0.1 M KOH electrolyte in a standard three-electrode system. Cyclic voltammetry (CV) curves (Fig. S6†) show a well-defined cathodic peak in O2-saturated electrolyte and this peak disappeared when the CV curve was recorded in Ar-saturated electrolyte, indicating the ORR catalytic ability of Ni3@Fe-N-GNS. The RDE polarization curves (Fig. 6a) show that Ni3@Fe-N-GNS possesses the best ORR activity among the examined catalysts. Specifically, Ni3@Fe-N-GNS demonstrates a high ORR onset potential (Eonset) of 0.96 V, a half-wave potential (E1/2) of 0.834 V, and a limiting current density (JL) of 5.91 mA cm−2 at 1600 rpm, similar to the Pt/C benchmark catalyst (E1/2 = 0.838 V and JL = 5.99 mA cm−2). The Tafel slope value derived from the corresponding LSV curves (Fig. 6b) is −54 mV dec−1 for Ni3@Fe-N-GNS, which is lower than those of Fe-N-GNS (−68 mV dec−1) and Pt/C (−80 mV dec−1), as well as other examined catalysts, including Ni1@Fe-N-GNS (−63 mV dec−1), Ni2@Fe-N-GNS (−66 mV dec−1) and Ni4@Fe-N-GNS (−72 mV dec−1), as displayed in Fig. S7,† indicating that Ni3@Fe-N-GNS has a better ORR kinetics. Additionally, electrochemical impedance spectroscopy measurements were conducted in order to understand the superior performance of Ni3@Fe-N-GNS. The reduced charge transfer resistance of Ni3@Fe-N-GNS compared to Fe-N-GNS indicates its enhanced ORR kinetics (Fig. 6c). To investigate the ORR pathway, the RDE polarization curves were recorded at various rotation speeds (Fig. 6d and S8†). The obtained Koutecky–Levich (K–L) plots derived from the corresponding RDE polarization data are shown in Fig. 6e and S9.† The linearity of the K–L plots of Ni3@Fe-N-GNS (Fig. 6e) indicates the first-order kinetics toward dissolved O2 concentration and almost identical electron transfer number (n) during the ORR process. The n values obtained from the slope of the K–L plots at potentials of 0.3–0.7 V (vs. RHE) are in the range of 3.8–4.2, indicating a pre-dominant four-electron pathway involving the complete electroreduction of oxygen. The rotating ring-disk electrode (RRDE) investigations further confirm the high-efficiency 4e− reduction on Ni3@Fe-N-GNS by the n value of 3.7–3.8 and a low HO2− yield in the potential range of 0.2–0.7 V vs. RHE as compared to Fe-N-GNS (Fig. 6f). The ring current values of various catalysts are shown in Fig. S10.† To understand the nature of active sites of the ORR process, the ORR polarization curves were recorded in 0.1 M KOH electrolyte containing 10 mM NaCN. Cyanide ions are known to coordinate with M–Nx sites, which serve as active sites for O2 electroreduction. They exhibit a high affinity for these sites, forming a stable adduct that significantly reduces the catalyst's ORR activity.63 The similar and considerable decrease in ORR activity observed in both Ni3@Fe-N-GNS and Fe-N-GNS suggests that the abundant Fe–Nx centers derived from iron phthalocyanine precursors are primarily responsible for the enhanced ORR activity. The remaining ORR electrocatalytic activity can be attributed to the presence of metal nanoparticles and the nitrogen doping within the carbon matrix (Fig. S11†).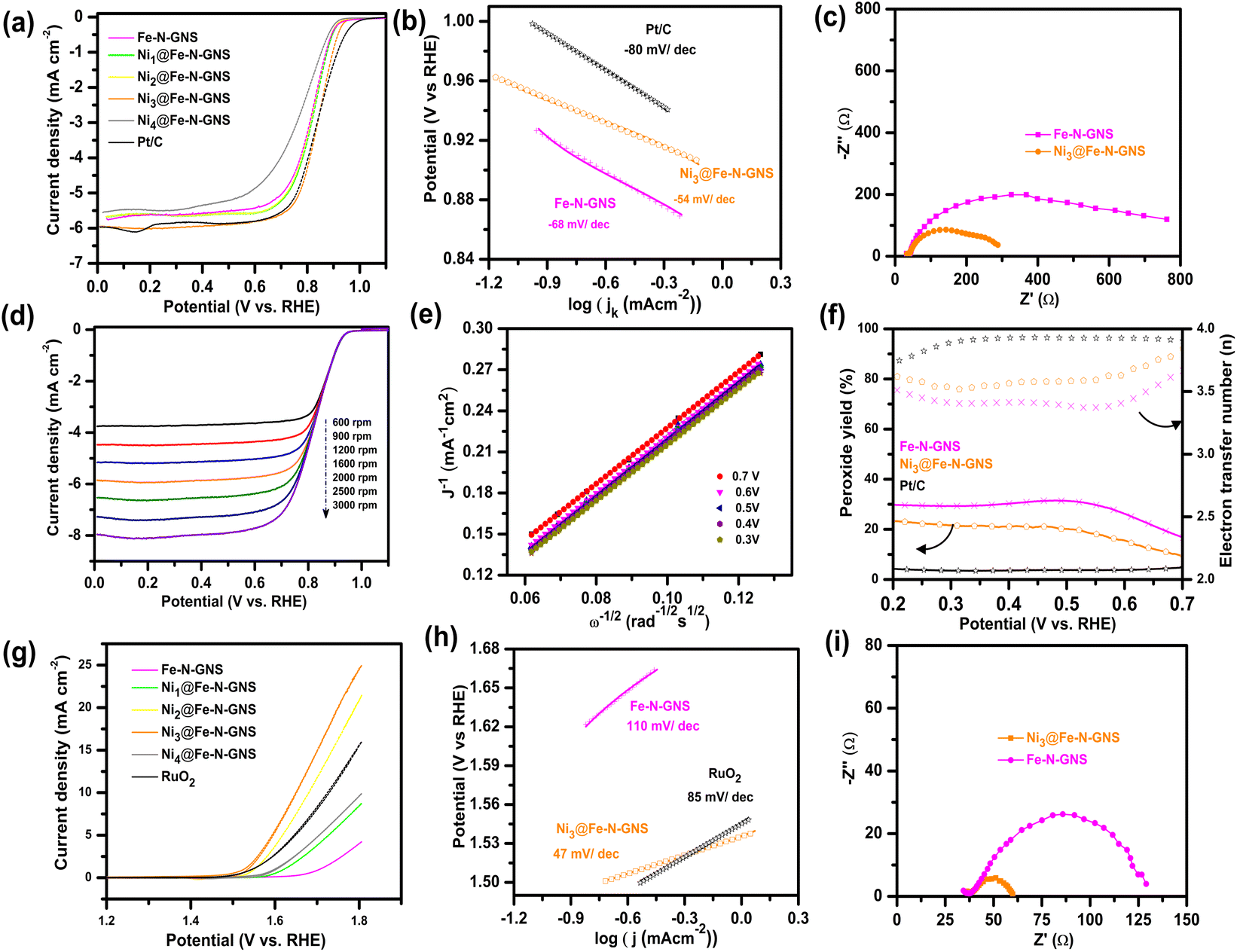 | ||
| Fig. 6 (a) ORR polarization curves for various catalysts in O2-saturated 0.1 M KOH solution at 1600 rpm, (b) Tafel plots for the ORR, (c) Nyquist plot of Ni3@Fe-N-GNS and Fe-N-GNS recorded at 0.8 V vs. RHE. (d) LSV curves for the ORR on Ni3@Fe-N-GNS at different rotation rates, (e) corresponding K–L plots for the ORR, (f) electron transfer number (n) and peroxide percentage yield as a function of potential, (g) LSV curves for the OER in 0.1 M KOH, (h) Tafel plots for the OER, (i) Nyquist plot of Ni3@Fe-N-GNS and Fe-N-GNS recorded at 1.65 V vs. RHE. | ||
The long-term durability of the catalysts was evaluated and after 10 h of chronoamperometric testing at 0.6 V vs. RHE, the Ni3@Fe-N-GNS catalyst maintains similar stability to the commercial Pt/C (Fig. S12†). The electrochemical stability test was followed by the STEM analysis of Ni3@Fe-N-GNS to investigate the morphological changes occurring in the material. Fig. S13† shows that the Ni3@Fe-N-GNS catalyst retains its initial architecture in the post-mortem analysis with no appreciable changes in the elemental distribution of the catalyst owing to corrosion resistance from the graphitic layers.
The reverse electrochemical conversion of OH− to O2 occurred through the OER and thus became significantly important for rechargeable Zn–air batteries. Fig. 6g demonstrates the OER polarization curves of Ni3@Fe-N-GNS along with other prepared materials. Ni3@Fe-N-GNS exhibits an OER potential (Ej10 = 10 mA cm−2) of 1.65 V, which is much better than that of all other investigated catalysts and commercial RuO2 (Ej10 = 1.726 V). The Tafel slope of Ni3@Fe-N-GNS is 47 mV dec−1, which is lower than that of RuO2 (85 mV dec−1) and other prepared catalysts (Fig. 6h and S14†), indicating the fast OER kinetics of the Ni3@Fe-N-GNS. Ni3@Fe-N-GNS also possesses a smaller charge transfer resistance as compared to Fe-N-GNS as displayed in the Nyquist plot (Fig. 6i). This indicates the fast OER kinetics of Ni3@Fe-N-GNS because of the integration of the Ni–Fe responsible for their superior OER performance. Fe–Nx are known to exhibit low OER activity, and the high OER activity of Ni3@Fe-N-GNS can be attributed to a higher amount of NiFe that was estimated from XRD studies. As evaluated from the CV curves of all the catalysts in the non-faradaic region (Fig. S15 and S16a†), the calculated double layer capacitance (Cdl) for Ni3@Fe-N-GNS is 3.7 mF cm−2, much higher than those of other investigated catalysts (Fig. S16b†). This shows the large electrochemically active surface area of the Ni3@Fe-N-GNS catalyst, which is beneficial for improved electrocatalytic performance.64,65 It is also important to note that the GC electrode is not electrochemically inert under OER conditions due to its rapid oxidation in alkaline media, which causes drastic loss of current density over the course of extended chronoamperometric stability tests,66 making it unsuitable for OER electrocatalyst stability analysis. Meanwhile, the Eonset, E1/2, jL, and Ej10 values of all the catalysts, along with Pt/C and RuO2, are listed in Table S5.† The OER/ORR bifunctional activity is largely evaluated based on the potential gap (ΔE = Ej10 − E1/2) as the bifunctional activity descriptor; the smaller the ΔE value, the higher the bifunctional activity. As shown in Fig. S17† the Ni3@Fe-N-GNS affords a ΔE of 0.81 V, which is better than that of Pt/C/RuO2 catalysts (ΔE = 0.89 V) and is highly competitive with most reported bifunctional OER/ORR electrocatalysts (Table S6†). Therefore, the interaction between Fe–Nx sites and FeNi enhances the involvement of active sites for both the OER and ORR, consequently making it a suitable bifunctional catalyst compared to others.
3.3. Zn–air battery performance
Owing to the decent bifunctional ORR/OER electrocatalytic activity of Ni3@Fe-N-GNS, a homemade rechargeable liquid ZAB was assembled with a Ni3@Fe-N-GNS catalyst coated on the GDL as the air electrode (Fig. 7a). To compare the ZAB performance of the prepared catalysts, the Fe-N-GNS and PtRu/C air electrodes were also assembled under the same conditions. The Ni3@Fe-N-GNS based ZAB shows an open-circuit voltage (OCV) of 1.47 V, which is nearly equivalent to that of the PtRu/C based Zn–air battery (1.48 V). The rechargeable ZAB assembled with a Ni3@Fe-N-GNS air electrode displayed a power density of 171 mW cm−2, which is higher than that of the battery based on Fe-N-GNS (152.7 mW cm−2) and PtRu/C (133.8 mW cm−2) air electrodes (Fig. 7b). Fig. 7c shows the galvanostatically discharged curves of Ni3@Fe-N-GNS, Fe-N-GNS, and PtRu/C based ZABs at 20 mA cm−2. Based upon the mass of Zn consumed, the ZAB with the Ni3@Fe-N-GNS air electrode shows a calculated specific capacity of 894 mA h g−1, which is much higher than that of the ZAB with PtRu/C (844 mA h g−1) and Fe-N-GNS (762 mA h g−1), indicating its promising application in rechargeable Zn–air batteries. The Ni3@Fe-N-GNS also displays highly competitive ZAB performance with other recently reported transition metal-based (especially Ni and Fe-containing) catalysts (see Table S7†). Apart from the battery output performance, durability is another important index for rechargeable ZABs; therefore, ZAB stability was measured by means of constant current charge–discharge cycling at 5 mA cm−2. As shown in Fig. 7d, the Fe-N-GNS equipped battery demonstrated a more stable performance for both discharge and charge processes and lasted for ca. 88 h, much longer than the PtRu/C, which lasted for ca. 28 h. The PtRu/C degrades much faster due to much higher current density in Zn–air batteries leading to severe carbon corrosion and oxidation or dissolution of Pt/Ru nanoparticles.67 In contrast, the ZAB with the Ni3@Fe-N-GNS air electrode shows remarkable cycling performance and exhibits superior long-life cycling stability of 180 h (Fig. 7e). In detail, for the initial cycle (Fig. 7f), the charge–discharge overpotential (ξ) of Ni3@Fe-N-GNS is 0.71 V, which is much lower than that of Fe-N-GNS (ξ = 0.84 V) and PtRu/C (ξ = 0.80 V), with round-trip efficiency (ε) of 62.9%, which is much better than that of Fe-N-GNS (58.8%) and PtRu/C (60.3%). Moreover, after only 20 h, the discharge voltage of PtRu/C drops to below 1.12 V, implying a significant loss in electrocatalytic activity (Fig. 7g). A significant loss of activity was also observed for Fe-N-GNS with considerably high charging and discharging voltage, which led to a high charge–discharge overpotential (ξ = 1.22 V) and reduced round-trip efficiency to 45.5%, thus degrading its performance (Fig. 7h). On the other hand, Ni3@Fe-N-GNS maintains a stable electrocatalytic behavior in the ZAB and shows only a slight change in voltage efficiency during discharge and charge cycling for over 150 h, with an increased voltage gap of ξ = 0.74 V and a slightly lower round-trip efficiency of ε = 60.6% (Fig. 7i). The excellent cycling stability of Ni3@Fe-N-GNS within the rechargeable ZAB operating environments suggests that the dissolved zinc ions will not precipitate onto the catalyst surface, thereby maintaining its activity. The changes in the Ni3@Fe-N-GNS-based ZAB were investigated through EIS measurements both before and after the stability test, following the replacement of the electrolyte and zinc anode. A small increase in the charge transfer resistance was observed after the ZAB stability measurement, which could be due to carbon corrosion from the catalyst or the used GDL surface (Fig. S18†). This performance can be significantly improved by modifying battery parameters, such as cell components and operating conditions, for the future design of rechargeable ZAB technology.68–70 Therefore, the Ni3@Fe-N-GNS catalyst not only possesses high intrinsic electrocatalytic activity and stability but also endows Zn–air batteries with excellent cycle life and high round-trip efficiency. | ||
| Fig. 7 (a) Photograph of the homemade liquid-state ZAB setup illustrating open-circuit voltage on the display screen with a Ni3@Fe-N-GNS air electrode. (b) Discharge polarization curves and power density curves of ZABs assembled with Ni3@Fe-N-GNS, Fe-N-GNS, and PtRu/C air electrodes. (c) Zn mass-normalized specific capacity at 20 mA cm−2. Galvanostatic discharge–charge cycling of ZAB with (d) Fe-N-GNS and PtRu/C electrodes and (e) Ni3@Fe-N-GNS air electrode at 5 mA cm−2. (f–i) Magnified plots of galvanostatic charge and discharge curves at different stages of ZABs with Ni3@Fe-N-GNS, Fe-N-GNS, and PtRu/C. | ||
4. Conclusions
In summary, we propose integrating iron phthalocyanine-derived Fe-Nx moieties and FeNi nanoparticles embedded within graphene nanostructures into a hybrid material that serves as a robust bifunctional oxygen electrocatalyst. Benefiting from this unique integration of atomically dispersed Fe–Nx active sites and FeNi nanoparticles, which is confirmed by STEM measurements, the Ni3@Fe-N-GNS with decent bifunctional electrocatalytic activity (ΔE = 0.81 V) outperforms commercial Pt/C + RuO2 (ΔE = 0.89 V). Moreover, the Ni3@Fe-N-GNS-based ZAB presents a high peak power density of 171 mW cm−2, surpassing that of the PtRu/C-based ZAB (133.8 mW cm−2). More importantly, the Ni3@Fe-N-GNS-based ZAB exhibits excellent long-term cycling performance, lasting 180 h in a single run, while delivering a low charge–discharge voltage gap (0.71 V @ 5 mA cm−2) and high reversibility (initial round-trip efficiency of 62.9%), uncovering a new method for exploring non-precious metal-based bifunctional oxygen electrocatalysts for Zn–air battery devices.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Zubair Ahmed: writing – review & editing, writing – original draft, methodology, investigation, data curation, and conceptualization. Arvo Kikas: investigation. Jaan Leis: methodology, funding acquisition, writing – review & editing. Vambola Kisand: funding acquisition. Jaan Aruväli: investigation. Alexey Treshchalov: investigation, methodology. Maike Käärik: investigation. Kaido Tammeveski: writing – review & editing, resources, methodology, funding acquisition. Jekaterina Kozlova: investigation, writing – review & editing, formal analysis. Kaupo Kukli: methodology, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Estonian Research Council (grants PRG2569, PRG2594, PRG1509, TEM-TA31, and SJD67). This work was also supported by the Estonian Ministry of Education and Research (TK210, Centre of Excellence in Sustainable Green Hydrogen and Energy Technologies).References
- N. Shang, K. Wang, M. Wei, Y. Zuo, P. Zhang, H. Wang, Z. Chen and P. Pei, J. Mater. Chem. A, 2022, 10, 16369–16389 RSC.
- L. Tang, H. Peng, J. Kang, H. Chen, M. Zhang, Y. Liu, D. H. Kim, Y. Liu and Z. Lin, Chem. Soc. Rev., 2024, 53, 4877–4925 RSC.
- Y. Wang, J. Liu, J. Liu, Z. Wang, B. Zhuang, N. Xu, X. Cui and J. Qiao, Sustainable Energy Fuels, 2024, 8, 3610–3616 RSC.
- X. Hao, Z. Jiang, B. Zhang, X. Tian, C. Song, L. Wang, T. Maiyalagan, X. Hao and Z.-J. Jiang, Adv. Sci., 2021, 8, 2004572 CrossRef CAS PubMed.
- X. Liu, G. Zhang, L. Wang and H. Fu, Small, 2021, 17, 2006766 CrossRef CAS PubMed.
- D. Qiu, H. Wang, T. Ma, J. Huang, Z. Meng, D. Fan, C. R. Bowen, H. Lu, Y. Liu and S. Chandrasekaran, ACS Nano, 2024, 18, 21651–21684 CrossRef CAS PubMed.
- S. Haller, C. de Chaby Ribeiro, F. Reinauer, S. Yadav, L. Ni, R. W. Stark, J. J. Schneider and U. I. Kramm, ACS Appl. Energy Mater., 2024, 7, 4698–4709 CrossRef CAS.
- Y. Kumar, M. Mooste and K. Tammeveski, Curr. Opin. Electrochem., 2023, 38, 101229 CrossRef CAS.
- Z. Huang, M. T. Nguyen, W. J. Sim, M. Takahashi, S. Kheawhom and T. Yonezawa, Sustainable Energy Fuels, 2022, 6, 3931–3943 RSC.
- D. Frattini, E. García Gaitán, A. Bustinza Murguialday, M. Armand and N. Ortiz-Vitoriano, Energy Environ. Sci., 2022, 15, 5039–5058 RSC.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- K. Sheng, Q. Yi, A. L. Chen, Y. Wang, Y. Yan, H. Nie and X. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 45394–45405 CrossRef CAS PubMed.
- H.-F. Wang, C. Tang and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1803329 CrossRef.
- L. Zan, H. M. A. Amin, E. Mostafa, A. A. Abd-El-Latif, S. Iqbal and H. Baltruschat, ACS Appl. Mater. Interfaces, 2022, 14, 55458–55470 CrossRef CAS PubMed.
- Z. Ahmed and V. Bagchi, New J. Chem., 2021, 45, 22012–22033 RSC.
- W. Orellana, C. Zuñiga, A. Gatica, M.-S. Ureta-Zanartu, J. H. Zagal and F. Tasca, ACS Catal., 2022, 12, 12786–12799 CrossRef CAS.
- D. Xie, D. Yu, Y. Hao, S. Han, G. Li, X. Wu, F. Hu, L. Li, H.-Y. Chen, Y.-F. Liao and S. Peng, Small, 2021, 17, 2007239 CrossRef CAS PubMed.
- G. Li, Y. Tang, T. Fu, Y. Xiang, Z. Xiong, Y. Si, C. Guo and Z. Jiang, Chem. Eng. J., 2022, 429, 132174 CrossRef CAS.
- H. Hu, Y. Xie, F. M. D. Kazim, K. Qu, M. Li, Z. Xu and Z. Yang, Sustainable Energy Fuels, 2020, 4, 5188–5194 RSC.
- M. Mechili, C. Vaitsis, N. Argirusis, P. K. Pandis, G. Sourkouni and C. Argirusis, Renewable Sustainable Energy Rev., 2022, 156, 111970 CrossRef CAS.
- C. Goswami, K. K. Hazarika, Y. Yamada and P. Bharali, Energy Fuels, 2021, 35, 13370–13381 CrossRef CAS.
- S. Li, X. Zhou, G. Fang, G. Xie, X. Liu, X. Lin and H.-J. Qiu, ACS Appl. Energy Mater., 2020, 3, 7710–7718 CrossRef CAS.
- S. A. Chala, M.-C. Tsai, W.-N. Su, K. B. Ibrahim, B. Thirumalraj, T.-S. Chan, J.-F. Lee, H. Dai and B.-J. Hwang, ACS Nano, 2020, 14, 1770–1782 CrossRef CAS PubMed.
- F. Chang, H. Du, P. Su, Y. Sun, R. Ye, Q. Tian, G. Zhang, H. Li and J. Liu, Small Struct., 2024, 5, 2300111 CrossRef CAS.
- C. Gan, Q. Jiang, X. Wu and J. Tang, Mater. Today Chem., 2023, 27, 101330 CrossRef CAS.
- Y. Chen, Q. Li, Y. Lin, J. Liu, J. Pan, J. Hu and X. Xu, Nat. Commun., 2024, 15, 7278 CrossRef CAS PubMed.
- M. A. Kazakova, D. M. Morales, C. Andronescu, K. Elumeeva, A. G. Selyutin, A. V. Ishchenko, G. V. Golubtsov, S. Dieckhöfer, W. Schuhmann and J. Masa, Catal. Today, 2020, 357, 259–268 CrossRef CAS.
- D. Chen, J. Zhu, X. Mu, R. Cheng, W. Li, S. Liu, Z. Pu, C. Lin and S. Mu, Appl. Catal., B, 2020, 268, 118729 CrossRef CAS.
- Z. Wang, J. Ang, J. Liu, X. Y. D. Ma, J. Kong, Y. Zhang, T. Yan and X. Lu, Appl. Catal., B, 2020, 263, 118344 CrossRef CAS.
- M. Jiang, Z. Tan and M. Cao, Int. J. Hydrogen Energy, 2021, 46, 15507–15516 CrossRef CAS.
- J.-T. Ren, L. Chen, Y.-S. Wang, W.-W. Tian, L.-J. Gao and Z.-Y. Yuan, ACS Sustain. Chem. Eng., 2020, 8, 223–237 CrossRef CAS.
- C. Lai, M. Gong, Y. Zhou, J. Fang, L. Huang, Z. Deng, X. Liu, T. Zhao, R. Lin, K. Wang, K. Jiang, H. Xin and D. Wang, Appl. Catal., B, 2020, 274, 119086 CrossRef CAS.
- M. Liu, Z. Liu, W. Chen, Z. Liu, Z. Li, X. Pi, Q. Du, X. Lai, Y. Xia and Y. Li, Inorg. Chem., 2023, 62, 11199–11206 CrossRef CAS PubMed.
- X. Li, Y. Liu, H. Chen, M. Yang, D. Yang, H. Li and Z. Lin, Nano Lett., 2021, 21, 3098–3105 CrossRef CAS PubMed.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- T. Lan, H. Du, Y. Li, K. Qu, J. Zhao, X. Zhang, Y. Dong, Y. Zhang, X. Zhang and D. Zhang, J. Alloys Compd., 2023, 943, 169144 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- S. Ding, J. A. Barr, Q. Shi, Y. Zeng, P. Tieu, Z. Lyu, L. Fang, T. Li, X. Pan, S. P. Beckman, D. Du, H. Lin, J.-C. Li, G. Wu and Y. Lin, ACS Nano, 2022, 16, 15165–15174 CrossRef CAS PubMed.
- Z. Xu, S. Wang, W. Tu, L. Shen, L. Wu, S. Xu, H. Zhang, H. Pan and X.-Y. Yang, Small, 2024, 20, 2401730 CrossRef CAS PubMed.
- Z. Ahmed, S. Akula, J. Kozlova, H.-M. Piirsoo, K. Kukli, A. Kikas, V. Kisand, M. Käärik, J. Leis, A. Treshchalov, J. Aruväli and K. Tammeveski, Int. J. Hydrogen Energy, 2024, 62, 849–858 CrossRef CAS.
- Z. Ahmed, Krishankant, R. Rai, R. Kumar, T. Maruyama, C. Bera and V. Bagchi, ACS Appl. Mater. Interfaces, 2021, 13, 55281–55291 CrossRef CAS PubMed.
- M. Mooste, Z. Ahmed, P. Kapitulskis, R. Ivanov, A. Treshchalov, H.-M. Piirsoo, A. Kikas, V. Kisand, K. Kukli, I. Hussainova and K. Tammeveski, Appl. Surf. Sci., 2024, 660, 160024 CrossRef CAS.
- Y. Kumar, E. Kibena-Põldsepp, M. Mooste, J. Kozlova, A. Kikas, J. Aruväli, M. Käärik, V. Kisand, J. Leis, A. Tamm, S. Holdcroft, J. H. Zagal and K. Tammeveski, ChemElectroChem, 2022, 9, e202200717 CrossRef CAS.
- J. Zhao, Q. Li, Q. Zhang and R. Liu, Chem. Eng. J., 2022, 431, 133730 CrossRef CAS.
- Y. Kumar, E. Kibena-Põldsepp, J. Kozlova, A. Kikas, M. Käärik, J. Aruväli, V. Kisand, J. Leis, A. Tamm and K. Tammeveski, ChemElectroChem, 2021, 8, 2662–2670 CrossRef CAS.
- S. Gupta, S. Zhao, O. Ogoke, Y. Lin, H. Xu and G. Wu, ChemSusChem, 2017, 10, 774–785 CrossRef CAS PubMed.
- V. Sivamaran, V. Balasubramanian, M. Gopalakrishnan, V. Viswabaskaran and A. G. Rao, Chem. Phys. Impact, 2022, 4, 100072 CrossRef.
- R. Kumar, M. Mooste, Z. Ahmed, S. Akula, I. Zekker, M. Marandi, M. Käärik, J. Leis, A. Kikas, A. Treshchalov, M. Otsus, J. Aruväli, V. Kisand, A. Tamm and K. Tammeveski, Ind. Chem. Mater., 2023, 1, 526–541 RSC.
- C.-X. Zhao, J.-N. Liu, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- S. K. Singh, K. Takeyasu and J. Nakamura, Adv. Mater., 2019, 31, 1804297 CrossRef PubMed.
- D. Wang, X. Pan, P. Yang, R. Li, H. Xu, Y. Li, F. Meng, J. Zhang and M. An, ChemSusChem, 2021, 14, 33–55 CrossRef CAS PubMed.
- K. Wang, J. Liu, Z. Tang, L. Li, Z. Wang, M. Zubair, F. Ciucci, L. Thomsen, J. Wright and N. M. Bedford, J. Mater. Chem. A, 2021, 9, 13044–13055 RSC.
- Y. Lei, G. Li, J. Yang, F. Zhang, Y. Shen, X. Zhang and X. Wang, Energy Fuels, 2023, 37, 11260–11269 CrossRef CAS.
- M. Muhyuddin, E. Berretti, S. A. Mirshokraee, J. Orsilli, R. Lorenzi, L. Capozzoli, F. D'Acapito, E. Murphy, S. Guo, P. Atanassov, A. Lavacchi and C. Santoro, Appl. Catal., B, 2024, 343, 123515 CrossRef CAS.
- X. Zhang, J. Wang, C. Dai, X. Jin, Y. Zhao and L. Qu, J. Power Sources, 2022, 538, 231563 CrossRef CAS.
- T. Li, Y. Hu, K. Liu, J. Yin, Y. Li, G. Fu, Y. Zhang and Y. Tang, Chem. Eng. J., 2022, 427, 131992 CrossRef CAS.
- Z. Li, X. Wu, X. Jiang, B. Shen, Z. Teng, D. Sun, G. Fu and Y. Tang, Adv. Powder Mater., 2022, 1, 100020 CrossRef.
- X. Liu, W. Liu, M. Ko, M. Park, M. G. Kim, P. Oh, S. Chae, S. Park, A. Casimir, G. Wu and J. Cho, Adv. Funct. Mater., 2015, 25, 5799–5808 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- M. Etesami, M. T. Nguyen, T. Yonezawa, A. Tuantranont, A. Somwangthanaroj and S. Kheawhom, Chem. Eng. J., 2022, 446, 137190 CrossRef CAS.
- X. He, Y. Zhang, J. Wang, J. Li, L. Yu, F. Zhou, J. Li, X. Shen, X. Wang, S. Wang and H. Jin, ACS Sustain. Chem. Eng., 2022, 10, 9105–9112 CrossRef CAS.
- M. Gopalakrishnan, W. Kao-ian, M. Rittiruam, S. Praserthdam, P. Praserthdam, W. Limphirat, M. T. Nguyen, T. Yonezawa and S. Kheawhom, ACS Appl. Mater. Interfaces, 2024, 16, 11537–11551 CrossRef CAS PubMed.
- G. Bae, H. Kim, H. Choi, P. Jeong, D. H. Kim, H. C. Kwon, K.-S. Lee, M. Choi, H.-S. Oh, F. Jaouen and C. H. Choi, JACS Au, 2021, 1, 586–597 CrossRef CAS PubMed.
- M. Alam, K. Ping, M. Danilson, V. Mikli, M. Käärik, J. Leis, J. Aruväli, P. Paiste, M. Rähn, V. Sammelselg, K. Tammeveski, S. Haller, U. I. Kramm, P. Starkov and N. Kongi, ACS Appl. Energy Mater., 2024, 7, 4076–4087 CrossRef CAS PubMed.
- K. Chen, S. Kim, R. Rajendiran, K. Prabakar, G. Li, Z. Shi, C. Jeong, J. Kang and O. L. Li, J. Colloid Interface Sci., 2021, 582, 977–990 CrossRef CAS PubMed.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32–40 CrossRef CAS.
- A. Pandikassala, M. Kurian, P. K. Gangadharan, A. Torris and S. Kurungot, Adv. Sustainable Syst., 2024, 8, 2400012 CrossRef CAS.
- J. Chang, G. Wang and Y. Yang, Small Sci., 2021, 1, 2100044 CrossRef CAS.
- K. Muuli, X. Lyu, M. Mooste, M. Käärik, B. Zulevi, J. Leis, H. Yu, D. A. Cullen, A. Serov and K. Tammeveski, Electrochim. Acta, 2023, 446, 142126 CrossRef CAS.
- S. Ding, L. He, L. Fang, Y. Zhu, T. Li, Z. Lyu, D. Du, Y. Lin and J.-C. Li, Adv. Energy Mater., 2022, 12, 2202984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00072f |
| This journal is © The Royal Society of Chemistry 2025 |