
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModification of a bioabsorbable carbon electrode on silk-fibroin carriers: setting the composition and adjustment of the working potential†
Kevin Alexander
Janus
 ab,
Madita
Zach
a,
Stefan
Achtsnicht
a,
Aleksander
Drinic
c,
Alexander
Kopp
c,
Michael
Keusgen
b and
Michael Josef
Schöning
*ad
ab,
Madita
Zach
a,
Stefan
Achtsnicht
a,
Aleksander
Drinic
c,
Alexander
Kopp
c,
Michael
Keusgen
b and
Michael Josef
Schöning
*ad
aInstitute of Nano- and Biotechnologies (INB), FH Aachen, Campus Jülich, 52428 Jülich, Germany. E-mail: schoening@fh-aachen.de
bInstitute for Pharmaceutical Chemistry, Philipps University of Marburg, 35032 Marburg, Germany
cFibrothelium GmbH, 52068 Aachen, Germany
dInstitute of Biological Information Processing (IBI-3), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
First published on 6th March 2025
Abstract
In this work, different surface treatment and modification procedures (KCl, Na2CO3, H2O2, O2 plasma, multi-walled carbon nanotubes (MWCNTs)) are applied to a screen-printed carbon-based electrode on bioabsorbable silk-fibroin, aiming to reduce the applied working potential in operation. The screen-printed carbon electrode houses the enzyme glucose oxidase for glucose monitoring, and is encapsulated by the biocompatible material Ecoflex. The working electrode is characterized amperometrically at different working potentials (0.6 to 1.2 V vs. the Ag/AgCl reference electrode) at physiological glucose concentrations ranging from 0.5 to 10 mM. The surface morphology of the electrode is analyzed utilizing scanning electron microscopy and contact angle measurements. Addition of 2 wt% MWCNTs to the carbon screen-printing paste allowed the reduction of the applied working potential from 1.2 to 0.8 V, resulting in a mean glucose sensitivity of 2.5 ± 0.6 μA cm−2 mM−1. Moreover, the bioabsorbability (i.e., the degradation behavior) of the different surface-treated carbon electrodes on silk-fibroin is studied over several months using the enzyme protease XIV from Streptomyces griseus.
Introduction
Personalized medicine can enable the prevention, more precise-targeted investigation and treatment of diseases, tailored to the individual.1 Here, the demand of “real-time insights” into the human body can be met, e.g., by implantable biosensors that collect data and are surgically removed at the end of their lifetime.2,3 Bioabsorbable, implantable biosensors are the next evolutionary step of implantable biosensors, however, in this case, the selected materials need to be both biocompatible and bioabsorbable so that the biosensor is absorbed by the human body when not needed anymore, eliminating the need for surgical removal.4 Depending on the utilized bioreceptor (e.g., immobilized enzyme) layer, such a biosensor might be tailored for different tasks, such as monitoring the healing process after a operation, like a skin-flap transplantation2 or heart surgery,5 or can be used, for example, to track the glucose, lactate or pyruvate levels in the brain energy metabolism.6 In addition to biocompatibility and bioabsorbability properties, the biosensor must also prevent inflammation, fibrous encapsulation or biofouling, and all of this without negatively affecting its biosensor characteristics.7 Furthermore, such implantable bioabsorbable biosensor should be flexible to adapt to its specific environment, like the brain or heart.Materials that combine these properties are, e.g., carbon and silk-fibroin. The latter is obtained from silk of the domesticated silkworm, Bombyx mori.8 The biopolymer silk-fibroin contains three components: the heavy chain (391 kDa), the light chain (25 kDa) and the glycoprotein P25 (30 kDa). The heavy chain, mainly responsible for the enhanced stability, crystallinity and mechanical properties due to the secondary structure (hydrophobic β-sheets with anti-parallel assembly), is linked via disulfide bonds to the light chain, which is more hydrophilic and elastic, leading to the flexibility of the resulting material. Both chains are stabilized by the glycoprotein P25, which prevents aggregation and enhances the solubility of silk-fibroin in water.9 These three proteins arrange themselves primarily into β-sheets and α-helices as secondary structures. The β-sheets act as physical cross-linkers between the protein-chains to ensure mechanical stability. The α-helices (and also random coil structures) contribute to the flexibility and elasticity of the material. The tertiary structure of silk-fibroin is described by β-crystallites embedded in a β-sheet- and α-helices-containing matrix. The ratio of the crystalline to the amorphous regions (β-sheet ratio) in this semi-crystalline biopolymer determines the properties of the material, where, for example, the mechanical strength and the water insolubility increase with increasing β-sheet ratio.8 Here, a solvent, such as water, can only slowly diffuse into the dense crystalline regions. For the same reason, the enzymatic degradation of silk-fibroin takes longer for a silk-fibroin membrane with a high β-sheet ratio compared to a silk-fibroin membrane with a low β-sheet ratio, as an enzyme, such as protease XIV, can only digest the silk-fibroin from the surface. The β-sheet ratio in silk-fibroin can be adjusted in various ways, like water annealing, alcohol treatment, or applying temperature and pressure to the material.10,11
These customizable characteristics combined with its inherent biocompatibility and bioabsorbability and a wide variety of processable morphologies such as sponges,12 membranes,13–15 scaffolds,16 hydrogels,17 powders,18 glues,19 or fibres,20 make silk-fibroin a preferred material for the development of bioabsorbable, implantable biosensors.
Such a bioabsorbable, implantable biosensor that is designed for later use in a patient should be absorbed by the human body through, for instance, enzymatic digestion, preventing an additional extraction surgery. The bioabsorbable material silk-fibroin can be digested by various enzymes, such as protease XIV, proteinase K, α-chymotrypsin or matrix metalloproteinase. The available type of enzyme as well as its accessibility to the cleavage sites, the silk-fibroin morphology, the cross-linking state and the crystallinity strongly influence the bioabsorbability.21–23 As a well-described model enzyme, protease XIV from Streptomyces griseus, belongs to the serine proteases and cleaves amide bonds adjacent to aliphatic, aromatic or hydrophobic residues.15,21,24 The enzyme digests dissolved silk-fibroin and solid silk-fibroin membranes equally with an affinity towards the amorphous polymer structures, breaking them down to peptides and amino acids.9,21
Therefore, silk-fibroin demonstrates a suitable substrate material for the development of implantable, bioabsorbable biosensors. As demonstrated in previous publications, silk-fibroin can be utilized as carrier material by housing screen-printed carbon electrodes, recently giving promising results towards the development of bioabsorbable, implantable amperometric glucose biosensors.13,25 However, in addition to the material aspects, an obstacle to realistic use in the human body as implantable biosensor is the high potential of the working electrode.13 This can result in several drawbacks, for example, creating a current due to oxygen reduction reactions,26 influencing the immobilized enzyme,27 damaging the working electrode over time,25 or triggering the oxidation/reduction of interfering electroactive species, like ascorbic acid28 and urea.29 In literature, different methods have been discussed to reduce the applied working potential of amperometric biosensors: one possibility is the activation of the carbon electrode surface using chemicals such as potassium chloride (KCl),30 disodium carbonate (Na2CO3)31 or hydrogen peroxide (H2O2).32 These electrochemical surface treatments aim to increase the surface functionalities and roughness (by removing the polymeric binder) or remove surface contaminations from the screen-printed carbon electrode.31 A similar effect can be achieved by exposing the electrode surface to an oxygen plasma (O2 plasma).33 In addition, non-toxic and bioabsorbable multi-walled carbon nanotubes (MWCNTs) represent a common way to increase the conductivity of the carbon working electrode and thus, to improve the overall sensor performance.34,35
The present study aims to reduce the applied working potential of our recently developed screen-printed carbon electrode on a flexible silk-fibroin substrate for amperometric glucose determination.13 For this, a series of surface treatment and electrode modification procedures (KCl, Na2CO3, H2O2, O2 plasma, MWCNTs) was applied. Homogeneity and surface morphology of the electrodes have been characterized via scanning electron microscopy (SEM) and contact angles measurements, while (chrono-)amperometry enabled to demonstrate the biosensor's functionality towards glucose sensing. Furthermore, the bioabsorbability (i.e., the degradation behavior) for each carbon electrode type was investigated using the enzyme protease XIV from Streptomyces griseus.
Experimental section
Materials
Bovine serum albumin (96%), glycerol (99.5%), disodium carbonate (analytical grade), glucose monohydrate (99%), glucose oxidase (GOx) from Aspergillus niger, disodium hydrogen phosphate dihydrate (analytical grade), sodium dihydrogen phosphate monohydrate (analytical grade), diiodomethane (99%), potassium chloride (99%), multi-walled carbon nanotubes (article number 901019), protease XIV from Streptomyces griseus (P5147-1G, 3.5 U mL−1) and phosphate buffered saline solution (PPB006-20PK) were purchased from Sigma-Aldrich (St. Louis, USA). Diiodomethane (99%, stabilized with copper) was ordered from abcr GmbH (Karlsruhe, Germany). Ecoflex 00-30 was obtained from KauPo Plankenhorn e.K. (Spaichingen, Germany). The two components of Ecoflex were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and degassed in vacuum. Hydrogen peroxide (35%) was acquired from MC Industrial Chemicals (Philadelphia, USA). Glutaraldehyde (25%) was purchased from Acros Organics (Geel, Belgium). To process silk-fibroin to an adequate carrier material, silk-fibroin aqueous solution was obtained using PureSilk® Technology (Fibrothelium GmbH, Aachen, Germany), enabling medical-grade quality production of silk on an industrial scale for a broad range of concentrations. Briefly, silk-fibroin was separated from silk-sericin by degumming Bombyx mori silk in a hot alkali solution before dissolving it in a proprietary non-toxic solvent system based on Ajisawa's reagent. The conductive carbon paste Custom Ink/CMC 12072021 was purchased from Cambridge Graphene Ltd. (Gloucestershire, England).
:1 ratio and degassed in vacuum. Hydrogen peroxide (35%) was acquired from MC Industrial Chemicals (Philadelphia, USA). Glutaraldehyde (25%) was purchased from Acros Organics (Geel, Belgium). To process silk-fibroin to an adequate carrier material, silk-fibroin aqueous solution was obtained using PureSilk® Technology (Fibrothelium GmbH, Aachen, Germany), enabling medical-grade quality production of silk on an industrial scale for a broad range of concentrations. Briefly, silk-fibroin was separated from silk-sericin by degumming Bombyx mori silk in a hot alkali solution before dissolving it in a proprietary non-toxic solvent system based on Ajisawa's reagent. The conductive carbon paste Custom Ink/CMC 12072021 was purchased from Cambridge Graphene Ltd. (Gloucestershire, England).
Fabrication of the bioabsorbable screen-printed carbon working electrode on silk-fibroin substrate
The screen-printed carbon working electrode was fabricated on the flexible silk-fibroin substrate by using a semiautomatic screen-printing machine from Hary Manufacturing Inc (Lebanon, USA) with a 40 μm thick stencil plate, as described in our previous publications: in short, the carbon paste was directly screen-printed onto the silk-fibroin substrate and cured at 160 °C for 1 h.13,25 Afterwards, the electrodes were separated and fixed onto a printed circuit board using Ecoflex as adhesive. Ecoflex is a biocompatible and bioabsorbable synthetic polymer with mechanical properties similar to human skin after curing at room temperature. The electrical contact was made by a silver conductive paste and silver foil. The whole electrode and the electrical contact were encapsulated with a precast Ecoflex foil with a 5 mm opening (for the glucose oxidase cocktail), fixed again by Ecoflex adhesive, see Fig. 1a).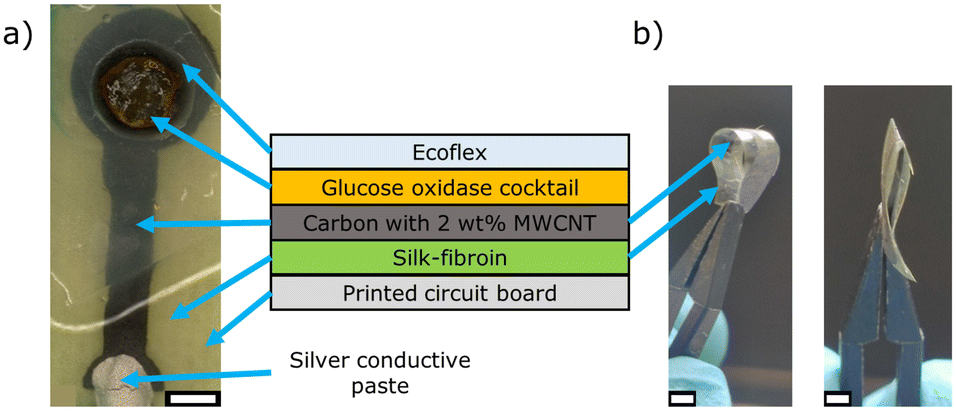 | ||
| Fig. 1 a) Microscopic image of the screen-printed carbon working electrode on silk-fibroin with immobilized glucose oxidase membrane, and cross-sectional schematic of the layer sequence of the working electrode (middle). b) Photos of a screen-printed (and cured) carbon electrode on silk-fibroin in bended (left image) and folded (right image) condition, respectively. The white scale bar corresponds to 2 mm. The glucose oxidase cocktail contains glucose oxidase, bovine serum albumin and glutaraldehyde/glycerol, MWCNT: multi-walled carbon nanotubes. | ||
Different working electrode treatments and/or modifications were applied, as described in section “Modification of the carbon working electrode”. Afterwards, 20 μL of the glucose oxidase cocktail were drop-coated in the 5 mm opening of the Ecoflex encapsulation layer. The enzyme cocktail consists of glucose oxidase, bovine serum albumin (as polymeric matrix) and glutaraldehyde/glycerol solution (as cross-linker) in a 1–2–2 ratio, see also study by Molinnus et al.25 The glutaraldehyde/glycerol stock solution was prepared by mixing 2.0 mL glycerol with 2.5 mL glutaraldehyde and 20.5 mL ultrapure water in a glass bottle.
The fabricated screen-printed carbon working electrode on silk-fibroin with immobilized glucose oxidase is schematically shown in Fig. 1. As demonstrated in Fig. 1b), the screen-printed carbon electrode on silk-fibroin remains highly flexible after the curing step at 160 °C for 1 h. The carbon electrode can be bent and even folded without losing its functionality.
Modification of the carbon working electrode
The surface of the carbon working electrode was treated with acid, basic and neutral reagents. This is intended to study their influence on reducing the applied working potential.Potassium chloride (KCl) treatment: the carbon electrode was electrochemically activated by applying 1.4 V vs. the Ag/AgCl reference electrode for 240 s in a 100 mM KCl solution.30
Disodium carbonate (Na2CO3) treatment: the carbon electrode was to be exposed for 360 s in saturated Na2CO3 solution, while applying 1.2 V vs. the Ag/AgCl reference electrode.31
Hydrogen peroxide (H2O2) treatment: the carbon electrode was immersed in 10 mM H2O2 in phosphate buffer solution, pH 7, and 25 consecutive voltammetric cycles at 0.1 V s−1 between −0.7 and +1.0 V vs. the Ag/AgCl reference electrode were performed.32
Oxygen plasma (O2 plasma) treatment: the carbon electrode was treated with O2 plasma at 100 W for 60 s (Diener Electronic GmbH + CO. Kg, Ebhausen, Germany).33,34,36
Multi-walled carbon nanotubes (MWCNT) treatment: different amounts of MWCNTs were directly added to the screen-printing paste and homogeneously dispersed with a vortex mixer. Concentrations were varied between 2 wt% and 4 wt%. A maximum amount of 2 wt% MWCNTs was found as the limit to fabricate homogeneous thick-film layers.
Electrochemical biosensor characterization
The sensor set-up was electrochemically characterized by (chrono-)amperometry in a three-electrode arrangement: the Ag/AgCl reference electrode (Deutsche Metrohm GmbH & Co. KG, Filderstadt, Germany), the platinum wire as the counter electrode (MaTeck, Jülich, Germany) and the functionalized screen-printed carbon working electrode were connected to a PalmSens3 potentiostat (Palm Instruments BV, GA Houten, The Netherlands) and operated by the software PSTrace. The applied working potential to the carbon working electrode varied between 0.6 and 1.2 V. The measurements were performed in phosphate buffer solution, pH 7.4, at 21 °C with glucose concentrations varying between 0.5 and 10 mM, using a 250 mM stock solution. Each concentration was measured for 10 minutes. The glucose sensitivity is determined by a linear fit in the linear concentration range of the biosensor between 0 and 4 mM glucose. For more details regarding the measurement principle, see previous works.13,25Degradation studies of the carbon working electrode on silk-fibroin
The bioabsorption (i.e., the degradation) of screen-printed carbon electrodes on silk-fibroin without Ecoflex encapsulation was studied in three different experiments: in the first experiment, the weight loss of differently treated samples was examined over a period of 17 days; in the second experiment, the samples were incubated until they were fully decomposed by the enzyme protease XIV. All samples were incubated at 37 °C in phosphate buffer saline solution (PBS), pH 7.4. The enzyme concentration was 3.5 U mL−1. The first and the second experiments were conducted with five different sample groups: 1) “reference silk-fibroin” and 2) “silk-fibroin”, both utilizing untreated silk-fibroin membranes; 3) “silk-fibroin 160 °C”, a silk-fibroin membrane cured at 160 °C for 1 h; 4) “silk-fibroin + C”, a silk-fibroin membrane composed with a screen-printed carbon electrode on top; and 5) “silk-fibroin + MWCNT”, a silk-fibroin membrane with a screen-printed carbon electrode modified with 2 wt% MWCNTs. The sample groups 4–5 were also cured at 160 °C for 1 h, however, after the screen-printing of the carbon electrode on top of the silk-fibroin membrane. Each sample group contained n = 3 samples. The sample groups were selected based on the different steps of electrode fabrication and are summarized in Table 1.| Number | Sample group name | Incubation solution |
|---|---|---|
| 1 | Reference silk-fibroin | PBS |
| 2 | Silk-fibroin | PBS + protease XIV |
| 3 | Silk-fibroin 160 °C | PBS + protease XIV |
| 4 | Silk-fibroin + C | PBS + protease XIV |
| 5 | Silk-fibroin + MWCNT | PBS + protease XIV |
The experimental procedure is schematically described in Fig. 2. First, the samples were weighed and placed in a polystyrene Petri dish (Fig. 2I and II). Then, the samples were immersed in 5 mL PBS containing 3.5 U mL−1 protease XIV except for the reference sample, followed by incubation in a laboratory oven at constant temperature of 37 °C during the whole experiment (Fig. 2III and IV).
 | ||
| Fig. 2 Schematic depiction of the bioabsorbability (degradation) test sequence for silk-fibroin containing samples (different groups, see Table 1) by protease XIV. The sample is weighed (I), placed in a Petri dish (II) immersed in PBS solution containing protease XIV (III), and incubated at constant 37 °C for up to 17 days (IV). At each data point (day 1, 3, 10, 17), the sample is removed from the laboratory oven, filtrated and washed with distilled water (V), and dried at 105 °C for 1 h (VI). Afterwards, the sample is weighed again and the weight loss is determined (VII). | ||
In the first bioabsorption experiment, named as “weight loss study over time”, the samples were removed, rinsed and filtered after a certain time (day 1, day 3, day 10 and day 17, Fig. 2V). The filter cake on the filtration paper was dried at 105 °C for 1 h and weighed (Fig. 2VI and VII). The samples were discarded afterwards. The second bioabsorption experiment “fully sample digestion” was also performed with three samples per group. The enzyme was replaced every two weeks from the sample surface and microscopic pictures were taken in order to examine how much of the particular sample was already left. Afterwards, the same sample was incubated again in the laboratory oven for further observation.
The third bioabsorption experiment studied the influence of the continuous enzymatically induced bioabsorption on the electrochemical sensor signal. Here, bare screen-printed carbon electrodes on the silk-fibroin substrate (fixed by Ecoflex to a printed circuit board) were used without an additional Ecoflex encapsulation layer and immobilized glucose oxidase. The electrodes were analyzed amperometrically on five consecutive days, using hydrogen peroxide concentrations from 0 to 4 mM (in phosphate buffer solution, pH 7.4, 21 °C) with an applied potential of 0.8 V vs. the Ag/AgCl reference electrode. Before and after each electrochemical characterization, microscopic images were taken. Afterwards, the electrodes were placed in phosphate buffer saline solution containing protease XIV (3.5 U mL−1) for 19 h and stored in a laboratory oven at 37 °C. The carbon electrodes on silk-fibroin were mounted in both cases in a 3D-printed holder to ensure that always the same area (round shaped “head” of the electrode, see Fig. 1) is submerged in the solution.
Results & discussion
Physical characterization of modified/surface-treated carbon electrodes on silk-fibroin
The surface of the modified/surface-treated carbon electrodes was analyzed by scanning electron microscopy (JEOL JSM-7800F, Freising, Germany) with an acceleration of 5 kV. The SEM images in Fig. 3 address a characteristic top view of the middle of the particular carbon electrode surface. No visual differences between an untreated (carbon electrode without any further modification, Fig. 3a)) and the KCl-, Na2CO3-, H2O2-, and the O2 plasma-treated samples (Fig. 3b)–e)) can be recognized. Regardless of the applied surface treatment method, the carbon electrode displayed a homogeneous, uniform 3D-structure with overlapping carbon flakes. In contrast, the sample with 2 wt% MWCNT (see Fig. 3f)), which was mixed into the carbon screen-printing paste, showed the carbon nanotubes overlapping the carbon flakes. In addition, a reduction of the electrical resistance of the carbon electrode by half to around 500 Ω was observed. The electrical resistance was determined by measuring the distance between two points on the electrode: the first point was located at the center of the electrode head (round area in Fig. 1a)), and the second point was the center of the contact pad at the electrode's end. The distance between the two points was approximately 2 cm. | ||
| Fig. 3 Top-view SEM images of screen-printed carbon electrodes a) without surface treatment and modified by b) KCl, c) Na2CO3, d) H2O2, or e) O2 plasma exposure and f) by adding of 2 wt% multi-walled carbon nanotubes (MWCNT). The black scale bars correspond to 1 μm. | ||
The surface free energy, calculated from the contact angle of water and diiodomethane, using the method of Owens, Wendt, Rabel and Kaelble (OWRK-model)37 and the water contact angle, as well as an exemplary mean water contact angle measurement (corresponding image of an untreated and a MWCNT-modified sample) are depicted in Fig. 4. For this experiment, the carbon paste was screen-printed on a microscopic glass slide to create a nearly homogeneous flat surface, and is treated as described before.
 | ||
| Fig. 4 a) Surface free energy (left side) obtained by contact angle measurements using water and diiodomethane of the screen-printing carbon paste applied to a microscopic glass slide and modified with either multi-walled carbon nanotubes (MWCNT, red), treated with H2O2 (green), KCl (blue), Na2CO3 (cyan) or O2 plasma (pink). Untreated (black) refers to a sample without further modification after the curing step. The corresponding mean water contact angle is shown on the right side. b) Mean water contact angle of an exemplary untreated sample (left) and a MWCNT sample (right) with the corresponding image of the water droplet on the carbon surface. | ||
The measurement of the water contact angle on the surface-treated electrode (or the determination of the resulting free surface energy) should provide preliminary insights into the efficacy of the various surface treatments to electrochemically activate the carbon electrode's surface. The activation of the carbon surface entails the introduction of polar groups at the electrode surface or the etching of the surface to enhance the roughness and surface porosity by removing binder or impurities. Such surface modification would result in a uniform potential formation across the entire electrode surface in contact with the electrolyte, while simultaneously enhancing the wettability of the surface when in contact to the electrolyte. Consequently, even small pores in the carbon surface (e.g., resulting from the overlapping of the carbon flakes, see Fig. 3) are wetted by the electrolyte, thereby increasing the available surface area for the electrochemical reaction. If there is a change in the contact angle (or corresponding surface free energy) after the modification procedure, it can be assumed that the surface treatment has altered the electrode surface, which should influence the electrochemical behavior in the later-on glucose measurements, see section “Electrochemical characterization of modified/surface-treated carbon working electrodes on silk-fibroin”.
The solvent-based surface treatments only slightly shift the surface free energy, compared to the untreated sample, see Fig. 4a), indicating a minor influence of these modification steps to the carbon surface. The O2 plasma treatment and the addition of MWCNTs strongly increased the surface free energy from 57 ± 5 mN m−1 to 75 ± 2 mN m−1 and 74 ± 3 mN m−1, respectively.
The mean water contact angle (n = 3) of the MWCNT sample decreased to 24 ± 3° (see Fig. 4b)) and at the same time the wettability of the carbon surface increased. This behavior might be explained due to the oxidation of the MWCNTs during the curing step, as reported by Kakade et al.38
Electrochemical characterization of modified/surface-treated carbon working electrodes on silk-fibroin
In previous publications of our group, 1.2 V vs. the Ag/AgCl reference electrode was applied to the bioabsorbable screen-printed carbon working electrode to determine the biosensor performance.13,25 The presented experiments were conducted to further investigate the extent to which a modification/surface treatment of the carbon working electrode has an influence on the setting: the behavior of the glucose biosensor was studied at different applied potentials (0.6 to 1.2 V vs. the Ag/AgCl reference electrode) for glucose concentrations between 0.5 to 10 mM. Six carbon-based working electrodes on silk-fibroin were fabricated for each studied working potential, i.e., 0.6, 0.8, 1.0 and 1.2 V. Treatment of the sensor before glucose oxidase immobilization included exposure to KCl, Na2CO3, H2O2, O2 plasma and modification with MWCNTs. Fig. 5a) shows as a selection the dynamic sensor response of the amperometric glucose biosensor with the MWCNT-modified carbon working electrode in the concentration range from 0.5 to 10 mM glucose. In addition, Fig. 5b) exemplary proves the good reproducibility, when comparing n = 5 biosensors achieved from the same fabrication batch. Note that a summary of all studied modification variants of the prepared carbon working electrode with regard to the resulting sensor characteristics is overviewed in Table 2. As can be seen in Fig. 5a), the sensor current raises with increasing glucose concentration, independent of the applied working electrode potential. At the same time, the signal strength decreases, when the working potential is set to lower values (starting from 1.2 V down to 0.6 V). Table 2 presents the calculated mean glucose sensitivity values with standard deviation (normalized per area) for 0.6, 0.8, 1.0, 1.2 V in the linear glucose range (linear fit from 0 to 4 mM). | ||
| Fig. 5 a) Dynamic response of carbon working electrodes with 2 wt% MWCNTs with immobilized glucose oxidase to different glucose concentrations (0.5–10 mM) at different applied potentials 0.6 V (black), 0.8 V (red), 1.0 V (blue) and 1.2 V (green) vs. the Ag/AgCl reference electrode. b) Mean calibration plot with n = 5 sensors at an applied potential of 0.8 V vs. the Ag/AgCl reference electrode. | ||
| Applied working potential to the working electrode vs. Ag/AgCl | Mean sensitivity and standard deviation (μA cm−2 mM−1) | Limit of detection (mM) |
|---|---|---|
| 0.6 V | 0.3 ± 0.3 | 2.5 |
| 0.8 V | 2.5 ± 0.6 | 0.7 |
| 1.0 V | 5.2 ± 0.5 | 0.4 |
| 1.2 V | 8 ± 2 | 0.7 |
Even though, the achieved sensitivity values are comparable to literature data,25 the linear concentration range was clearly expanded from up to 2 mM to 4 mM. The limit of detection (LoD) of the glucose biosensor was determined using a linear calibration function and the method as described in literature.39 As one example, Fig. 5b) shows the good reproducibility for n = 5 sensors (mean calibration plot with standard deviations), studied in the glucose range between 0.5 and 10 mM at a working potential of 0.8 V. The results are summarized in Table 2.
Fig. 6 compares the mean glucose sensitivities of the different surface-treated modified carbon working electrodes at the different applied working potentials, as for Fig. 5 and Table 2. Only the MWCNT sample group displayed a clear trend, where the mean sensitivity decreased with lowering the applied working potential. Despite the fact that for the other samples groups at lower working electrode potentials of 0.6 and 0.8 V higher mean glucose sensitivities were obtained (e.g., for KCl, Na2CO3) as for the MWCNT-type sensors, either strong variations (i.e., high standard deviations) were found or insufficient reproducibility in manufacturing existed. For some of the experiments, only one of the sensors was running at the end (e.g., KCl – 0.8 V, Na2CO3 – 0.8 V). One possible explanation might be the swelling behavior when in contact with the treatment solution. With reference to previous studies, the silk-fibroin membrane can absorb up to 300% of its own weight and changes its dimensions before depositing the carbon paste via thick-film technique.13
 | ||
| Fig. 6 Mean glucose sensitivity of carbon working electrodes modified by either KCl, Na2CO3, O2 plasma, H2O2 or multi-walled carbon nanotubes with different applied working potentials 0.6 V (black), 0.8 V (red), 1.0 V (green) and 1.2 V (blue) vs. the Ag/AgCl reference electrode. | ||
If all the parameters used and examined are taken into account, then the modification of the carbon working electrode with MWCNTs represents a good compromise, where the applied working potential vs. the Ag/AgCl reference electrode can be reduced to 0.8 V or even 0.6 V, respectively.
Bioabsorbability studies of screen-printed carbon electrode on silk-fibroin
One of the main advantages using silk-fibroin in biosensing applications is the inherent biocompatibility and bioabsorbability. The biodegradation behavior of silk-fibroin depends on factors such as the accessibility of the cleavage sites, its morphology, the cross-linking state, the crystallinity and the in situ enzymes involved in the biodegradation process.21–23During the biosensor fabrication, the silk-fibroin crystallinity can be affected by the water contact (solvent of the screen-printing paste) or heat exposure (curing of the screen-printing paste) leading to an increased β-sheet content. Therefore, each step of the electrode fabrication (without applying the Ecoflex encapsulation layer) was studied with regard to the bioabsorbability through the enzyme protease XIV from Streptomyces griseus. The results of weight loss over 17 days are displayed in Fig. 7a). Here, the mean weight loss of all samples incubated in the presence of protease XIV was significantly higher than the mean weight loss of the reference silk-fibroin sample, incubated in an enzyme-free buffer solution indicating the digestion of silk-fibroin by protease XIV. The about 10% weight loss of the reference sample mainly results from non-cross-linked, soluble silk-fibroin components (i.e., short polymer chains with 20–70 kDa), which was also reported in literature.23
 | ||
| Fig. 7 a) Mean weight loss (n = 3) of different treated silk fibroin-based samples representing the carbon electrode fabrication process over 17 days in the presence of protease XIV from Streptomyces griseus. b) Exemplary microscopic images of the sample “silk-fibroin + MWCNT” during its bioabsorbation over 63 days. The black scale bars correspond to 2 mm. “Reference silk-fibroin” = silk-fibroin membrane in protease-free buffer solution; “silk-fibroin” = silk-fibroin membrane; “silk-fibroin 160 °C/1 h” = silk-fibroin membrane cured at 160 °C for 1 h; “silk-fibroin + C” = silk-fibroin membrane with screen-printed carbon electrode (cured at 160 °C for 1 h); “silk-fibroin + MWCNT” = silk-fibroin membrane with screen-printed carbon electrode containing 2 wt% MWCNT (cured at 160 °C for 1 h). Except for the reference silk-fibroin sample, all samples were incubated in the presence of protease XIV (3.5 U mL−1) in phosphate buffer saline solution at 37 °C, pH 7.4. | ||
The silk-fibroin sample without further treatment (“silk-fibroin”) had the highest enzymatically induced weight loss of around 80% after 17 days. This is in accordance with results from Li et al., where only 30% of the original weight of an untreated silk-fibroin membrane was still present after 15 days of enzymatic incubation.23 In contrast, the heat-treated sample (“silk-fibroin 160 °C/1 h”) in this experiment lost 30% after 17 days. This difference to the untreated sample (“silk-fibroin”) indicates that the crystallinity ratio changed during the curing step in the electrode fabrication process.40 Screen-printing carbon electrodes on top of the silk-fibroin substrate also influence the degradation behavior. The “silk-fibroin + C” sample had an overall mean weight loss that is slightly lower compared to that of silk-fibroin without carbon paste. In case of the MWCNT-containing silk-fibroin carbon sample, the mean weight loss was about 50%.
Supporting microscopic observations revealed that the carbon electrode disappeared over time, see exemplary images in Fig. 7b) for day 1 and day 12. Nevertheless, none of these samples was completely digested by protease XIV within the first 17 days. Therefore, in a second experiment, again three samples of each sample group were incubated until their full digestion (enzyme solution was exchanged every two weeks). A sample of “silk-fibroin + MWCNT” was almost completely degraded by day 63 (see Fig. 7b)). The structural integrity, however, was already destroyed after just two weeks. Here, the electrode's “head” was separated from the electrode “tail”.
With regard to a future application as implantable bioabsorbable biosensor, it is not only important to know the degradation behavior of the sensor, but also to determine the influence of the enzymatic degradation on the electrochemical sensor signal. As an additional experiment, carbon electrodes on silk-fibroin were alternately recorded electrochemically and exposed to a degradation-forcing enzyme solution (containing protease XIV) for 19 hours and inspected via microscopy. The course of experiments for five successive days is summarized in Fig. 8.
 | ||
| Fig. 8 a) Sensitivity of screen-printed carbon electrodes on silk-fibroin towards H2O2 concentrations (0–4 mM) over five consecutive days. Between the electrochemical measurements, the electrodes were exposed to a protease XIV-containing solution, forcing the degradation of the silk-fibroin and the carbon electrode. b) Microscopic images of sensor 1 (left) and sensor 2 (right) after the electrochemical measurement on day 4 and day 3, respectively. The red circles highlight changes in the carbon electrodes, the red arrow depicts damage in the silk-fibroin substrate. c) Exemplary dynamic response of sensor 1 to different hydrogen peroxide concentrations on day 1, day 2 and day 4 with continuous decrease of overall H2O2 sensitivity. | ||
Both the current density and the sensitivity towards H2O2 decreased within the studied period. Sensor 1 operated during the entire duration of the experiment, while sensor 2 became inoperable on day 3. The H2O2 sensitivity of sensor 1 decreased from 17 μA cm−2 mM−1 to 5 μA cm−2 mM−1 (see Fig. 8a)); comparable data resulted from sensor 2. The microscopic images revealed two different types of electrode damage (Fig. 8b)): i.) cracks appeared in the silk-fibroin (see red arrow), but could not be identified as responsible for electrode damage, and ii.) cracks on the carbon electrode itself (see red circles); for instance, sensor 2 showed a continuous crack through its conducting line on day 3. Furthermore, it was noticed that small parts of the carbon electrode detached during the rinsing procedure with water, indicating that the adhesion to the silk-fibroin was influenced, too.
In all cases examined, the structural integrity of the silk-fibroin substrate was still intact within this 5-day period, even after sensor failure. This is consistent with the findings from Fig. 7, where the loss of structural integrity of the silk-fibroin was not observed until day 12. In addition, it has to be taken into account, that in vivo degradation of silk-fibroin is reported to be slower than for in vitro studies.41
In summary, the fully bioabsorbability of the proposed carbon electrodes modified with 2 wt% MWCNT on silk-fibroin for amperometric glucose measurements could be demonstrated. Such an electrode was completely digested under laboratory in vitro conditions within two months by protease XIV. In comparison, experiments showed that enzymatic degradation in a rat model, depending on the silk-fibroin fabrication conditions, can take up to 6 months.41
Conclusions
The biosensor performance (i.e., signal strength, sensitivity, cross-sensitivity, life-time and limit of detection) is influenced by the applied working potential in case of amperometric sensors. For this reason, the previously developed screen-printed carbon electrodes on flexible, bioabsorbable silk-fibroin substrates were modified/surface-treated using O2 plasma, MWCNTs, KCl, Na2CO3 or H2O2 aiming to reduce the applied working potential. Of the five treatments examined, only KCl, Na2CO3 and MWCNT exhibited sufficient biosensor signals to be measured adequately at lower potentials. At the same time, the solvent-based techniques resulted in a damage of the carbon electrode, which drastically reduced the reproducibility in manufacturing of the biosensors. The addition of 2 wt% MWCNT to the carbon paste resulted in a more viscose but still processable paste for screen-printing. Those biosensors enabled to reduce the working potential down to 0.8 or 0.6 V vs. the Ag/AgCl reference electrode. For example, in the physiological glucose concentration range (0.5 to 10 mM) at an applied working potential of 0.8 V a mean glucose sensitivity of 2.5 ± 0.6 μA cm−2 mM−1 with a limit of detection of 0.7 mM was achieved.Furthermore, the bioabsorbability of the surface-treated carbon electrodes on silk-fibroin (without Ecoflex encapsulation) was demonstrated. Therefore, their weight loss over time was studied. Protease XIV from Streptomyces griseus (3.5 U mL−1) induced a distinct weight loss for all types of sensors within the first three weeks. There was a distinct and complete digestion of the MWCNT-modified carbon electrode on silk-fibroin substrate within about two months. These enzymatically induced bioabsorption experiments were supported by electrochemical studies with screen-printed MWCNT-modified carbon electrodes on silk-fibroin for H2O2 detection.
This study demonstrates that a working electrode based on a silk-fibroin substrate and a MWCNT-modified carbon paste (using thick-film technology) is entirely biodegradable. Furthermore, the applied working electrode's potential for a future implantable sensor application has been successfully reduced to 0.8 V vs. the Ag/AgCl reference electrode. In order to obtain a fully implantable and bioabsorbable biosensor, however, the next steps will address the biocompatibility and bioabsorption of the enzymatic membrane. Currently, the glucose oxidase-based membrane cocktail for immobilization still contains components, such as glutaraldehyde as cross-linking agent, which are harmful to health. Here, the use of silk-fibroin as immobilization matrix might be advantageous. In addition, the transition from the rigid printed circuit board carrier to a flexible sensor system is envisaged.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Kevin Alexander Janus: conceptualization, investigation, validation, writing – original draft, writing – review & editing. Madita Zacha: investigation, validation. Stefan Achtsnicht: conceptualization, validation, writing – original draft. Aleksander Drinic: resources. Alexander Kopp: resources, writing – review & editing. Michael Keusgen: supervision, validation, writing – review & editing. Michael Josef Schöning: conceptualization, funding acquisition, resources, supervision, validation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank D. Rolka for assistance with the SEM images.Notes and references
- W. Evans, E. M. Meslin, J. Kai and N. Qureshi, J. Pers. Med., 2024, 14, 418 CrossRef PubMed.
- D. Özsoylu, K. A. Janus, S. Achtsnicht, T. Wagner, M. Keusgen and M. J. Schöning, Sens. Actuators Rep., 2023, 6, 100163 CrossRef.
- D. Yogev, T. Goldberg, A. Arami, S. Tejman-Yarden, T. E. Winkler and B. M. Maoz, APL Bioeng., 2023, 7, 31506 Search PubMed.
- F. Alam, M. A. Ahmed, A. H. Jalal, I. Siddiquee, R. Z. Adury, G. M. M. Hossain and N. Pala, Micromachines, 2024, 15, 475 CrossRef.
- R. Omar, W. Saliba, M. Khatib, Y. Zheng, C. Pieters, H. Oved, E. Silberman, O. Zohar, Z. Hu, V. Kloper, Y. Y. Brozar, T. Dvir, A. Grinberg Dana, Y. Wang and H. Haick, ACS Sens., 2024, 9, 126 Search PubMed.
- M. Gray, J. Meehan, C. Ward, S. P. Langdon, I. H. Kunkler, A. Murray and D. Argyle, Vet. J., 2018, 239, 21 CrossRef CAS PubMed.
- F. Ghorbanizamani, H. Moulahoum, E. G. Celik and S. Timur, Appl. Sci., 2023, 13, 4630 CrossRef CAS.
- L. Bitar, B. Isella, F. Bertella, C. Bettker Vasconcelos, J. Harings, A. Kopp, Y. van der Meer, T. J. Vaughan and L. Bortesi, Int. J. Biol. Macromol., 2024, 264, 130374 CrossRef CAS PubMed.
- S. Inoue, K. Tanaka, F. Arisaka, S. Kimura, K. Ohtomo and S. Mizuno, J. Biol. Chem., 2000, 275, 40517 CrossRef CAS PubMed.
- H.-J. Jin, J. Park, V. Karageorgiou, U.-J. Kim, R. Valluzzi, P. Cebe and D. L. Kaplan, Adv. Funct. Mater., 2005, 15, 1241 Search PubMed.
- Z. Wang, B. A. Serban and M. A. Serban, ACS Biomater. Sci. Eng., 2020, 6, 7004 CrossRef CAS PubMed.
- M. Sun, Q. Li, H. Yu, J. Cheng, N. Wu, W. Shi, F. Zhao, Z. Shao, Q. Meng, H. Chen, X. Hu and Y. Ao, Bioact. Mater., 2022, 8, 505 Search PubMed.
- K. A. Janus, S. Achtsnicht, L. Tempel, A. Drinic, A. Kopp, M. Keusgen and M. J. Schöning, Phys. Status Solidi A, 2023, 220, 2300081 Search PubMed.
- A. Kopp, L. Schunck, M. Gosau, R. Smeets, S. Burg, S. Fuest, N. Kröger, M. Zinser, S. Krohn, M. Behbahani, M. Köpf, L. Lauts and R. Rutkowski, Int. J. Mol. Sci., 2020, 21, 6704 Search PubMed.
- D. Molinnus, A. Drinic, H. Iken, N. Kröger, M. Zinser, R. Smeets, M. Köpf, A. Kopp and M. J. Schöning, Biosens. Bioelectron., 2021, 183, 113204 CrossRef CAS PubMed.
- A. Kopp, R. Smeets, M. Gosau, R. E. Friedrich, S. Fuest, M. Behbahani, M. Barbeck, R. Rutkowski, S. Burg, L. Kluwe and A. Henningsen, In Vivo, 2019, 33, 757 CrossRef CAS PubMed.
- H. Zheng and B. Zuo, J. Mater. Chem. B, 2021, 9, 1238 RSC.
- Y. Zhuang, Q. Zhang, J. Feng, N. Wang, W. Xu and H. Yang, Proc. Inst. Mech. Eng., Part H, 2017, 231, 337 CrossRef PubMed.
- S. H. Kim, Y. J. Lee, J. R. Chao, D. Y. Kim, M. T. Sultan, H. J. Lee, J. M. Lee, J. S. Lee, O. J. Lee, H. Hong, H. Lee, O. Ajiteru, Y. J. Suh, H. S. Choi, Y.-J. Cho and C. H. Park, NPG Asia Mater., 2020, 12, 46 Search PubMed.
- A. Kopp, R. Smeets, M. Gosau, N. Kröger, S. Fuest, M. Köpf, M. Kruse, J. Krieger, R. Rutkowski, A. Henningsen and S. Burg, Bioact. Mater., 2020, 5, 241 Search PubMed.
- J. Brown, C.-L. Lu, J. Coburn and D. L. Kaplan, Acta Biomater., 2015, 11, 212 CrossRef CAS PubMed.
- Y. Cao and B. Wang, Int. J. Mol. Sci., 2009, 10, 1514 CrossRef CAS PubMed.
- M. Li, M. Ogiso and N. Minoura, Biomaterials, 2003, 24, 357 CrossRef CAS.
- R. L. Horan, K. Antle, A. L. Collette, Y. Wang, J. Huang, J. E. Moreau, V. Volloch, D. L. Kaplan and G. H. Altman, Biomaterials, 2005, 26, 3385 CrossRef CAS PubMed.
- D. Molinnus, K. A. Janus, A. C. Fang, A. Drinic, S. Achtsnicht, M. Köpf, M. Keusgen and M. J. Schöning, Phys. Status Solidi A, 2022, 219, 2200100 CrossRef CAS.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 Search PubMed.
- M. J. Danilich, D. Gervasio and R. E. Marchant, Ann. Biomed. Eng., 1993, 21, 655 CrossRef CAS PubMed.
- Y.-J. Tu, D. Njus and H. B. Schlegel, Org. Biomol. Chem., 2017, 15, 4417 Search PubMed.
- M. M. Bani Amer, J. Med. Eng. Technol., 2002, 26, 208 CrossRef PubMed.
- J. Pilas, T. Selmer, M. Keusgen and M. J. Schöning, Anal. Chem., 2019, 91, 15293 CrossRef CAS PubMed.
- G. Cui, J. H. Yoo, J. S. Lee, J. Yoo, J. H. Uhm, G. S. Cha and H. Nam, Analyst, 2001, 126, 1399 Search PubMed.
- M. I. González-Sánchez, B. Gómez-Monedero, J. Agrisuelas, J. Iniesta and E. Valero, Electrochem. Commun., 2018, 91, 36 CrossRef.
- C. Colin, P. Levallois, U. Botsos-Margerit, F. Clément, D. Zigah and S. Arbault, Bioelectrochemistry, 2023, 154, 108551 CrossRef CAS PubMed.
- M. Şen, M. Oğuz and İ. Avcı, Talanta, 2024, 268, 125341 CrossRef PubMed.
- A. S. Ibrahim, D. A. M. Farage and G. A. M. Ali, Handbook of Biodegradable Materials, Springer, New York City, 2023 Search PubMed.
- M. I. González-Sánchez, B. Gómez-Monedero, J. Agrisuelas, J. Iniesta and E. Valero, J. Electroanal. Chem., 2019, 839, 75 CrossRef.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741 CrossRef CAS.
- B. A. Kakade and V. K. Pillai, J. Phys. Chem. C, 2008, 112, 3183 CrossRef CAS.
- C. A. Holstein, M. Griffin, J. Hong and P. D. Sampson, Anal. Chem., 2015, 87, 9795 CrossRef CAS PubMed.
- C. Geão, A. R. Costa-Pinto, C. Cunha-Reis, V. P. Ribeiro, S. Vieira, J. M. Oliveira, R. L. Reis and A. L. Oliveira, J. Mater. Sci.: Mater. Med., 2019, 30, 27 CrossRef PubMed.
- Y. Wang, D. D. Rudym, A. Walsh, L. Abrahamsen, H.-J. Kim, H. S. Kim, C. Kirker-Head and D. L. Kaplan, Biomaterials, 2008, 29, 3415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00371c |
| This journal is © The Royal Society of Chemistry 2025 |