
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enrichment of prostate-specific antigen using magnetic-silica antibody nanobioconjugates and fluorescence detection†
Tumelo
Msutu
a and
Philani
Mashazi
 *ab
*ab
aDepartment of Chemistry, Rhodes University, P.O. Box 94, Makhanda, 6140, South Africa. E-mail: p.mashazi@ru.ac.za
bInstitute for Nanotechnology Innovation, Rhodes University, P.O. Box 94, Makhanda, 6140, South Africa
First published on 6th November 2024
Abstract
Herein, we report the development of an immunosensor for the detection of prostate-specific antigen (PSA). The immunosensor platform was based on the immunometric sandwich protocol, using magnetic-silica nanoparticles for capture and pre-concentration of PSA. The preparation and application of the magnetic-silica nanobioconjugates and the use of phenylboronic acid for the immobilization of the capture antibody are the innovative steps of this report. The fluorescent sensing nanobioprobes contained 6% fluorescein in the fluorescein-doped silica nanoparticles. Silica nanoparticles can easily undergo alkaline dissolution for an enhanced fluorescence signal and thus ultrasensitive detection of PSA. The specificity of the immunosensor was achieved by the use of the anti-PSA monoclonal capture antibody (Ab1) bioconjugated in an oriented manner onto phenylboronic acid functionalized magnetic-silica nanoparticles. Non-specific binding sites were blocked with glucose to yield Fe3O4@SiO2-PBA-Ab1/glucose. Ab1 capture magnetic nanoparticles allowed for ease of separation using a magnet. For sensing, the polyclonal anti-PSA antibody (Ab2) was bioconjugated onto fluorescein-doped silica nanoparticles to form FITC@SiO2-PBA-Ab2/glucose. PSA was selectively isolated, enriched and purified from the serum samples using a magnetic nanobioconjugate (Fe3O4@SiO2-PBA-Ab1/glucose). A sandwich immunoreaction was achieved with FITC@SiO2-PBA-Ab2/glucose binding to the captured PSA. The alkali hydrolysis resulted in the disintegration of the nanoparticle thus releasing FITC molecules for fluorescence detection. This resulted in signal amplification. The analytical performance of the proposed immunosensor showed an excellent linear relationship between the fluorescence signal intensity and the concentration of PSA ranging from 2.0 pg mL−1 to 100 ng mL−1. The very low limit of detection (LOD) was 0.81 pg mL−1 and the limit of quantification (LOQ) was 2.46 pg mL−1. The immunosensor also exhibited good specificity and selectivity to PSA with 98.0–102.7% recovery rates. The detection was accomplished in newborn calf serum samples representing real samples.
1. Introduction
Prostate-specific antigen (PSA) is an androgen protease produced by both normal and cancerous cells.1,2 It is used clinically as an effective biomarker for prostate cancer screening and monitoring.3–5 Serum PSA levels below 4.0 ng mL−1 are considered normal for patients. However, PSA concentrations in the serum between 4.0 ng mL−1 and 10 ng mL−1 are considered to be a high risk for developing prostate cancer.6,7 For these patients an invasive confirmatory test involving a tissue biopsy is used for confirmation. Patients with PSA levels greater than 10 ng mL−1 are at advanced developmental stages of prostate cancer. Therefore, precise quantification of PSA is important for the early diagnosis and monitoring the development of prostate cancer. Other methods for prostate cancer diagnosis are highly invasive and these include tissue biopsy, which may miss cancerous tissue, and digital rectal examination, which require skilled personnel for diagnosis. Biosensors are sensing systems capable of detecting ultra-low concentrations of PSA in bodily fluids. They improve the early detection of prostate cancer. Zhao et al.8 designed a fluorescent immunosensor based on quantum dots as the detection probe. Quantum dots (QDs) contain heavy toxic metals like cadmium (Cd) in their preparation thus limiting their use due to the environmental effect9 of heavy metals as pollutants. Herein, we report the use of fluorescein-5-isothiocyanate (FITC) as an organic fluorophore doped into a silica nanoparticle matrix for stability and used as a sensing nanobioprobe. The magnetic-silica nanoparticles with antibody bioconjugates were also prepared and used as capture nanobioprobes. The non-toxic nature of magnetic-silica nanoparticles has been investigated for various medical applications.10–12 Their advantageous use in biodetection is due to their facile synthesis, small size, stability, good dispersity and high magnetic moment.11–13 The magnetic-silica nanoparticles provide an easy way to separate samples from solutions using a magnet.14–16 Magnetic-silica capture nanobioprobes contain iron oxide nanoparticles at the core and a silica shell. Iron oxide nanoparticles (Fe3O4NPs) alone are used in biomedical applications for magnetic resonance imaging (MRI), biomolecular separation, and for targeted drug delivery.10–18 This is due to their superparamagnetic properties, controllable nanoparticle size, and high specific surface area.10–19 Bare Fe3O4NPs are also prone to agglomeration, air oxidation, and biodegradation.20–22 To overcome such limitations, surface functionalization has been shown to be successful19–22 and silica (SiO2) is a widely used coating shell.11,12,15,20–25 SiO2 provides unique properties such as good stability, hydrophilicity, biocompatibility, and ease of surface modification, and helps prevent unwanted interactions with the magnetic core.19–26 Iron oxide–silica nanoparticles have been functionalized by direct covalent immobilization with phenylboronic acid for oriented antibody immobilization.25 The boronic acid functional groups form strong covalent boronate ester bonds with the N-glycans on the Fc region of the antibody27,28 exposing antigen binding sites.The use of phenylboronic acid for antibody immobilization is not widely reported even though it offers oriented immobilization. This is because antibodies with N-glycans are not widely produced. In this work, we exploit N-glycosylated antibodies for oriented immobilization. Both anti-PSA monoclonal (Ab1) and polyclonal (Ab2) antibodies were respectively immobilized onto magnetic nanoparticles for capture and enrichment of PSA and onto FITC-doped silica nanoparticles for fluorescence sensitive detection. A sandwich-type fluorescence immunoassay was developed and assessed for selective detection of PSA. The novelty of this work is in the preparation of the magnetic-silica nanobioconjugates and the use of phenylboronic acid for the immobilization of the capture antibody. Furthermore, the detection of PSA using sandwich immunoassay with fluorescent dye-doped silica nanoparticles and magnetic-silica nanoparticles (magnetic separation) for analyte capture and enrichment was demonstrated. The use of the magnetic-silica–Ab1 and FITC-doped silica–Ab2 nanoparticles is to our knowledge reported here for the first time.
2. Experimental
2.1 Materials and reagents
Iron(III) chloride (FeCl3·2H2O), sodium oleate, oleic acid (90%), 1-octadecene (90%), tetraethyl orthosilicate (TEOS, 99.9%), sodium chloride (NaCl), mouse anti-human IgG antibody (IgG), and 4-(1,1,3,3-tetramethylbutyl)-phenyl polyethylene glycol (Triton X-100) were purchased from Sigma-Aldrich (USA). Absolute ethanol (98.6%, w/v), sodium dihydrogen phosphate (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), and anhydrous D-glucose were purchased from SAARChem (South Africa). Cyclohexane (99.5%) and toluene were purchased from Merck (Germany). Hexan-1-ol was purchased from B&M Scientific (South Africa). Ammonium hydroxide (NH4OH, 25% wt) was purchased from Minema Chemicals (South Africa). The triethoxysilanepropyl-3-amido phenylboronic acid (TES-PBA) precursor was prepared and characterized as previously reported.29 Prostate-specific antigen (PSA), mouse anti-human prostate-specific antibody (Ab1, 7820-0370) and sheep anti-human prostate-specific antibody (Ab2, 7820-0154) were purchased from Bio-Rad Laboratories (USA).A coupling buffer, phosphate buffered saline (PBS, pH 7.4, 10 mM), was prepared using 0.63 g disodium hydrogen phosphate (Na2HPO4) and 60 mg sodium dihydrogen phosphate (NaH2PO4) in 400 mL of distilled water. A washing buffer (PBST, pH 7.4, 10 mM) was prepared using 1.2 g disodium hydrogen phosphate (Na2HPO4), 0.12 g sodium dihydrogen orthophosphate (NaH2PO4), and 100 μL of Tween 20 in 100 mL of ultra-pure Millipore water. All reagents and solvents in this study were of analytical grade and used as received from the supplier. Ultra-pure water with a resistivity of 18 MΩ cm (at 25 °C) was obtained from a Milli-Q water purification system (Millipore Corp. Bedford, MA, USA) and was used throughout the experiments. The synthesis of 6% FITC@SiO2NPs and bioconjugation of Ab2 as sensing nanobioprobes were performed following our previous work using FITC@SiO2-PBA-Ab2/glucose.29 The functionalization with the boronic group and antibody bioconjugation are reported in the ESI.†
2.2 Apparatus and instrumentation
Infra-red spectra were collected on a Perkin-Elmer Universal ART sampling accessory spectrum 100 FT-IR spectrometer. Zeta-potential measurements were carried out on a Malvern Zetasizer Nano series, Nano ZS90 series S equipped with a 633 nm He/Ne laser. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (SEM-EDS) and elemental mapping were carried out using an INCA penta FET coupled to the VAGA TESCAM using 20 kV accelerating voltage. The X-ray powder diffraction (XRD) patterns were recorded on a Bruker D8 Discover equipped with a Lynx Eye detector, using Cu-Kα radiation (1.5405 Å, nickel filter). The data were collected in the range from 2θ = 10° to 90°, scanning at 1° min−1 with a filter time-constant of 2.5 seconds per step and a slit width of 6.0 mm. Samples were placed on a silicon wafer slide. The XRD data were treated using the freely available Eva (evaluation curve fitting) software. Baseline correction was performed on each diffraction pattern by subtracting a spline fitted to the curve background. UV-visible (UV-vis) spectroscopy analysis was performed on a SHIMADZU UV-2550 spectrometer. The fluorescence spectra were recorded using a SpectraMax multi-mode spectrofluorometer and Synergy MX microplate reader.2.3 Preparation of magnetic-silica–Ab1 nanoparticles, Scheme 1
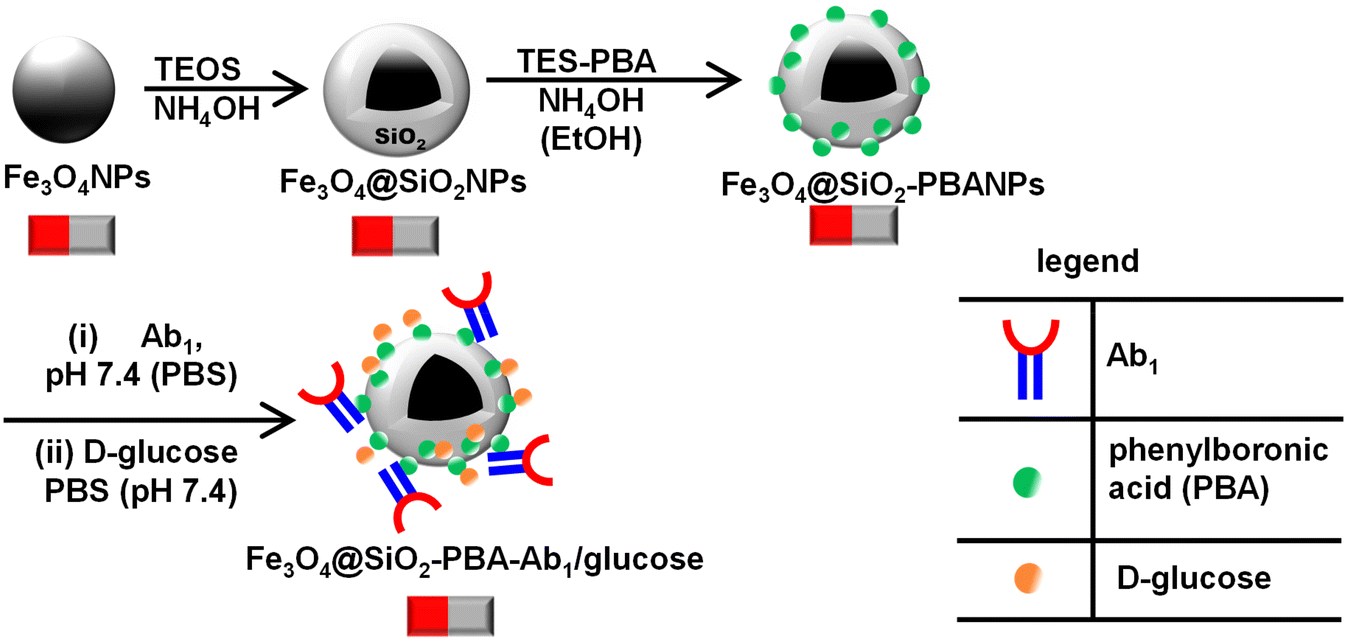 | ||
| Scheme 1 Step-by-step preparation of the Fe3O4@SiO2-PBA-Ab1/glucose capture nanobioconjugates. | ||
FT-IR (ATR, τmax/cm−1): 2918 (C–H), 2849 (C–H), 1545 (C–C), 1409 (C–H), 562 (Fe–O).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 volume) solution and collected by magnetic separation. The magnetic-silica nanoparticles were dried in the oven overnight at 50 °C and collected as a brown powder.
:3 volume) solution and collected by magnetic separation. The magnetic-silica nanoparticles were dried in the oven overnight at 50 °C and collected as a brown powder.
FT-IR (ATR, τmax/cm−1): 3285 (O–H), 1046 (Si–O–Si), 940 (Si–O), 797 (Si–OH), 562 (Fe–O).
:1) dry EtOH and toluene were added. The mixture reacted for 24 h at room temperature and resulted in the phenylboronic functionalized magnetic-silica nanoparticles (Fe3O4@SiO2-PBANPs). A magnet was used to separate the product after several washing steps with a 1:1 mixture of water and ethanol. The product was dried at 50 °C for 48 h.
FT-IR [(ATR), τmax/cm−1]: 3872 (N–H), 1649 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1541 (N–H), 1386 (B–O), 1056 (Si–O–Si), 949 (Si–O), 785 (Si–OH), 567 (Fe–O).
O), 1541 (N–H), 1386 (B–O), 1056 (Si–O–Si), 949 (Si–O), 785 (Si–OH), 567 (Fe–O).
FT-IR [(ATR), τmax/cm−1]: 3355 (N–H), 1639 (CO, amide I), 1546 (CO, amide II), 1390 (B–O), 1064 (Si–O–Si), 985 (Si–O), 795 (Si–OH), 567 (Fe–O).
The Bradford assay was used for the quantification of the antibody (Ab1) bioconjugated onto Fe3O4@SiO2-PBANPs to yield Fe3O4@SiO2-PBA-Ab1. The experimental conditions are outlined in the ESI.†
2.4 Immunoassay procedure for detection of PSA
The capture nanobioconjugates, Fe3O4@SiO2-PBA-Ab1/glucose (100 μL, 100 μg mL−1), were added into 50 μL of spiked serum in pH 7.4 PBS with different concentrations of PSA. The mixture was gently shaken for 50 min at room temperature. This resulted in the capture of PSA, Fe3O4@SiO2-PBA-Ab1<PSA, which was magnetically separated. The captured PSA, Fe3O4@SiO2-PBA-Ab1<PSA was washed with PBS (pH 7.4) three times. The sensing nanobioprobes, FITC@SiO2-PBA-Ab2/glucose (100 μL, 200 μg mL−1), were added. The mixture was incubated in PBS (pH 7.4) with gentle shaking for 50 min at room temperature. Upon magnetic separation, the sandwich immunoassay (Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC) was obtained. The isolated immunoassay was washed three times with cold PBS buffer (pH 7.4) and a magnet was used for separation. The sandwiched PSA will result in the capture of the fluorescent nanoparticles and they were magnetically separated. The isolated Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC immunocomplex was dispersed in 50 μL of PBS buffer (pH 7.4,10 mM). NaOH (20 μL, 10 mM) was added and incubated at 30 °C. After 20 min, the fluorescence was measured at 480 nm excitation wavelength. The total assay time was 120 min and the fluorescence signal was recorded for each concentration of PSA.3. Results and discussion
3.1 Synthesis and characterization of Fe3O4@SiO2-PBA-Ab1/glucose
Magnetic-silica nanobioconjugates were prepared and accomplished following several steps. Initially, the synthesis of the stable magnetite (Fe3O4NPs) nanoparticles was done by (a) the thermal decomposition of the Fe(oleate)3 precursor to form a wüstite (Fe–O) nanomaterial and (b) the oxidation of Fe–O to form the Fe3O4 phase.14,30,31 A quaternary water-in-oil (W/O) reverse microemulsion method afforded a coat of Fe3O4NPs with a silica shell to yield Fe3O4@SiO2NPs. Oleic acid-capped magnetic nanoparticles were coated with the silica shell via ligand exchange of the oleic ligands and hydrolyzed tetraethyl orthosilicate (TEOS). TEOS undergoes polycondensation, forming Fe–O–Si and Si–O–Si bonds resulting in a silica shell.19,32 The Fe3O4@SiO2NPs were surface functionalized with triethoxysilanepropyl-3-amido phenylboronic acid (TES-PBA) to form Fe3O4@SiO2-PBANPs. The Fe3O4@SiO2-PBANPs were bioconjugated with Ab1 and non-specific binding sites were blocked with D-glucose to yield Fe3O4@SiO2-PBA-Ab1/glucose nanobioconjugates. The step-by-step preparation and characterization of Fe3O4@SiO2-PBA-Ab1/glucose were carried out.Fig. 1 shows the XRD diffractograms of (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, and (iii) Fe3O4@SiO2-PBANPs. The diffractogram in Fig. 1(i) showed the formation of the Fe3O4NPs. The diffraction patterns are observed at 2θ values of 30.6°, 35.8°, 43.8°, 57.7°, and 63.2° which are indicative of a crystalline cubic spinel structure of the magnetite (Fe3O4).19,33,34 The diffraction patterns show the characteristic of magnetite which were indexed to the following Miller Indices (220), (311), (400), (420), and (440).25,35 The diffraction patterns matched well with the Fe3O4NPs for the JCPDS-International Centre (JCPDS file No. 19-0629). Similarly, the same peaks are observed for the functionalized magnetic-silica nanoparticles, Fe3O4@SiO2NPs and Fe3O4@SiO2-PBANPs, with a new peak observed at 2θ = 18.1° from the amorphous SiO2 as shown in Fig. 1(ii) and (iii). This indicated that Fe3O4NPs were stable after the formation of the silica shell and post-surface functionalization with TES-PBA. The crystalline phase of the Fe3O4NPs after silica shell coating and surface functionalization remained intact. The average crystal size of the Fe3O4NP core, obtained by calculation using the Debye–Scherrer equation was calculated to be 8.7 nm for Fe3O4NPs, 15.9 nm for Fe3O4@SiO2NPs, and 16.5 nm for Fe3O4@SiO2-PBANPs. The increase in particles size was due to the silica shell coating the Fe3O4NP core. The functionalization with TES-PBA did not result in the increase in the particle size and this is due to the thin layer of TES-PBA forming Fe3O4@SiO2-PBANPs. The nanoparticles retained their magnetic properties.
 | ||
| Fig. 1 XRD diffractograms of (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, and (iii) Fe3O4@SiO2-PBANPs. The insert exhibits the photographs of Fe3O4@SiO2NPs and Fe3O4@SiO2-PBANPs in the presence of the magnetic field. | ||
Fig. 2 shows the TEM micrographs and size distribution histograms of (a) Fe3O4NPs, (b) Fe3O4@SiO2NPs, and (c) Fe3O4@SiO2-PBANPs. The TEM micrographs of the nanoparticles were spherical and exhibited monodispersity. For Fe3O4NPs, the average diameter sizes were 10 ± 2 nm, clumping together as shown in Fig. 2(a). The clumping together or self-association of magnetic nanoparticles was due to the magnetic properties of the iron oxide. A 5 nm increase in the particle size diameter to 15 ± 3 nm for Fe3O4@SiO2NPs is shown in Fig. 2(b). A 5 nm size increase was attributed to the silica shell formation. The magnetic properties remained even after coating with a silica shell. The reverse water-in-oil (W/O) microemulsion method induced the surfactant Triton-X100 to surround droplets that contained the Fe3O4NPs and subsequent coating of silica via hydrolysis of the TEOS precursor.34 A 6 nm average particle size increase was observed after functionalization with phenylboronic acid to form Fe3O4@SiO2-PBANPs. The average particle size distribution of Fe3O4@SiO2-PBANPs was 21 ± 3 nm as shown in Fig. 2(c). The increase in the particle size diameter was due to the introduction of TES-PBA onto the silica surface. Furthermore, the particle size distribution of Fe3O4@SiO2-PBA-Ab1 as compared to the Fe3O4@SiO2-PBANPs showed no size variations. The antibody being a protein molecule does not appear in the TEM measurement. The successive increase in particle size diameter at each stage of functionalization successfully confirms the coating with the silica shell, TES-PBA and Ab1.
 | ||
| Fig. 2 TEM images and corresponding size distribution histograms of (a) Fe3O4NPs, (b) Fe3O4@SiO2NPs, and (c) Fe3O4@SiO2-PBANPs. | ||
Further characterization of the as-prepared magnetic nanoparticles was performed by EDS-SEM, FT-IR analyses, and zeta-potential measurements. Fig. 3 shows the EDS spectra with corresponding SEM images of (a) Fe3O4NPs, (b) Fe3O4@SiO2NPs, and (c) Fe3O4@SiO2-PBANPs. Fe3O4NPs in Fig. 3(a) showed the presence of Fe K (33.9%), O K (27.8%), and C K (38.3%) confirming the formation of iron oxide nanoparticles. The carbon (C K) was from the oleic acid (C–H) chains from the iron(II) oleate precursor. After modifying with the silica shell, the EDX spectrum in Fig. 3(b) showed the emergence of the Si K. The elemental composition showed Fe K (29.5%), Si K (20.1%) and O K (50.4%). The increase in O K from 27.8% to 50.4% is due to the silica (SiO2) shell, and the Fe K decrease from 33.9% to 29.5% further confirms the formation of the silica (SiO2) shell. The presence of Si K and increased O peaks confirms the successful silica coating of magnetic nanoparticles. The C K disappeared after the formation of the silica shell. After surface functionalization with TES-PBA to yield Fe3O4@SiO2-PBANPs, the appearance of the boron (B K) peak (6.6%) was observed in Fig. 3(c). Further, the presence of the N K peak (3.8%) is due to the amide bond from TES-PBA and the C K peak (5.0%) from TES-PBA. The corresponding scanning electron microscopy (SEM) images and the EDX spectra showed the smooth particles of Fe3O4NPs. These became rough and porous after silica shell formation and functionalization with phenylboronic acid.
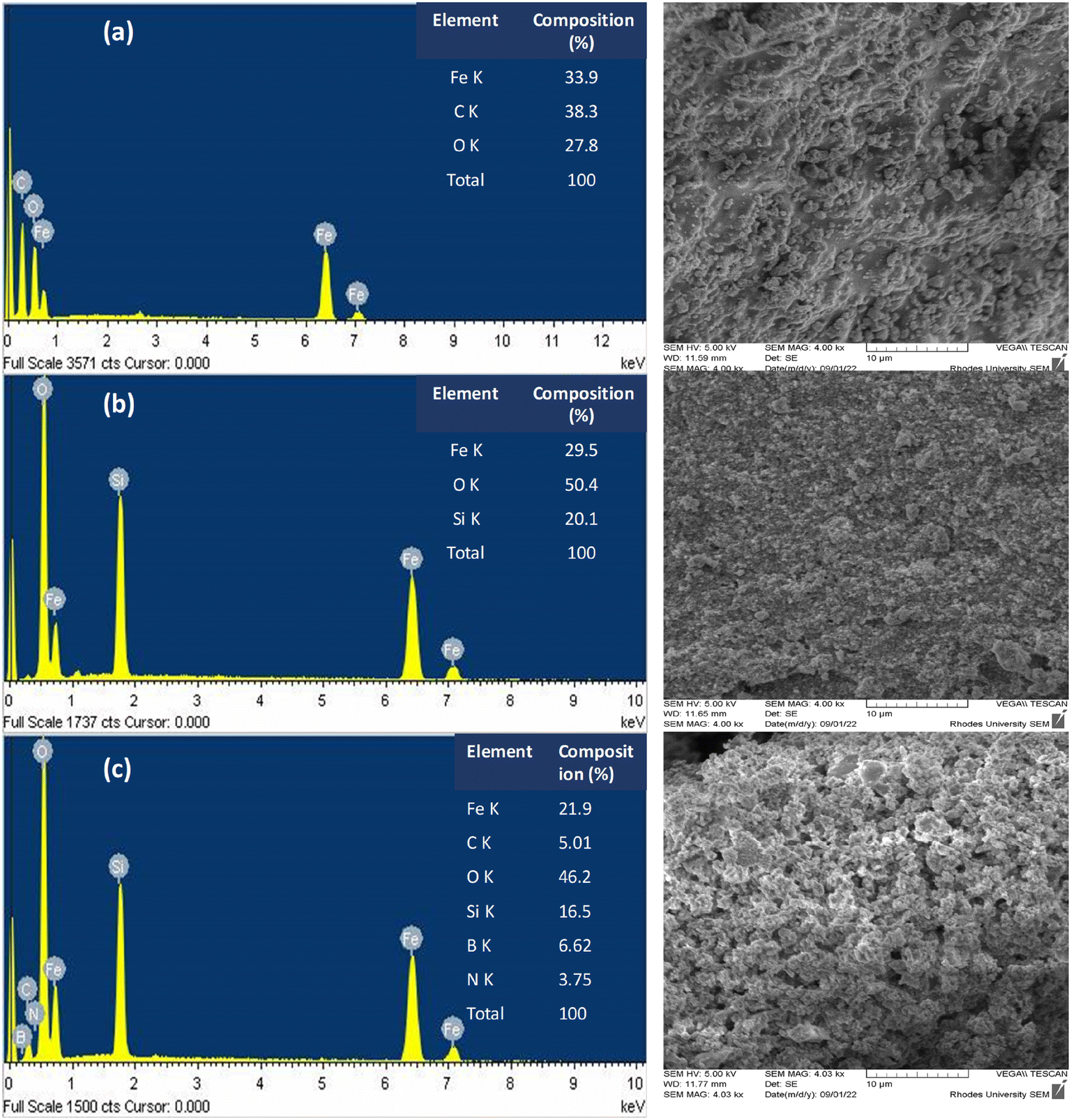 | ||
| Fig. 3 Energy dispersive X-ray (EDX) spectra with their corresponding SEM images for (a) Fe3O4NPs, (b) Fe3O4@SiO2NPs, and (c) Fe3O4@SiO2-PBANPs. | ||
Fig. 4 shows the FT-IR spectra of (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, (iii) Fe3O4@SiO2-PBANPs, and (iv) Fe3O4@SiO2-PBA-Ab1. The FT-IR spectrum of Fe3O4NPs in Fig. 4(i) shows the aliphatic C–H asymmetric and symmetric stretching bands at 2918 cm−1 and 2849 cm−1. The peaks at 1545 cm−1 and 1409 cm−1 are attributed to asymmetric and symmetric (–COO−) stretching vibrations, due to the carboxylates with Fe ions of Fe3O4NPs. Oleic acid (OA) is a stabilizer of Fe3O4NPs and is chemically bound to the magnetite (Fe3O4NPs) via oxygen atoms of the carboxylic acid functional group. An intense (Fe–O) peak is also observed at 562 cm−1 confirming the successful synthesis and coating of Fe3O4NPs with oleic acid. Then, it was followed by the coating with SiO2 shell to form Fe3O4@SiO2NPs. This is to enable the stability of the nanoparticles. After the formation of the silica shell, the peaks at 1710 cm−1, 1545 cm−1, and 1409 cm−1 disappeared as shown in Fig. 4(ii). This was due to the ligand exchange between TEOS and oleic acid ligands to yield Fe3O4@SiO2 core shell nanoparticles. Additionally, a broad (O–H) peak is observed at 3285 cm−1. Other peaks at 1046 cm−1, 940 cm−1, and 797 cm−1 appeared due to the asymmetric stretching vibration bands of siloxane (Si–O–Si), (Si–O), and silanol (Si–OH) groups, respectively. A decrease in the (Fe–O) peak is observed at 562 cm−1. This confirmed the successful synthesis of the Fe3O4@SiO2NPs. Surface functionalization of the magnetic-silica nanoparticles was achieved using TES-PBA through a covalent attachment forming siloxane bond. Fig. 4(iii) shows the Fe3O4@SiO2-PBANP spectrum which exhibits an N–H stretching peak at 3872 cm−1 corresponding to the N–H bending at 1541 cm−1 of the amide bond of TES-PBA. The (CO) absorption peak at 1649 cm −1 and the emergence of the absorption peak of the (B–O) vibrational stretching at 1386 cm−1 are observed. These indicate the successful surface modification of Fe3O4@SiO2NPs with TES-PBA onto the silica layer. The Fe3O4@SiO2-PBANPs were later reacted with anti-PSA-mAb (Ab1) to form Fe3O4@SiO2-PBA-Ab1. The immobilization of the antibody was achieved through cyclic boronate ester bonds between the cis-diol of the N-glycan moieties on the Fc region of the antibody and the (–OH) groups on the phenylboronic acid. The boronic acid bioconjugation method helps target the heavy chain antibody and this maintains the orientation of the antibody for enhanced antigen binding. Fig. 4(iv) shows the FT-IR spectrum of Fe3O4@SiO2-PBA-Ab1 and the appearance of intense stretching vibrational peaks of the (CO, amide I) and (CO, amide II) cyclic ester bonds of the anti-PSA antibody at 1639 cm−1 and 1546 cm−1 is observed. The absorption peak at 3355 cm−1 is assigned to the presence of the NH2 from the antibody. The shift of the B–O peak from 1386 cm−1 to 1390 cm−1 of the boronic acid confirmed the successful bioconjugation of the antibody to the phenylboronic acid. This confirms the successful preparation of Fe3O4@SiO2-PBA-Ab1 capture nanobioconjugates.
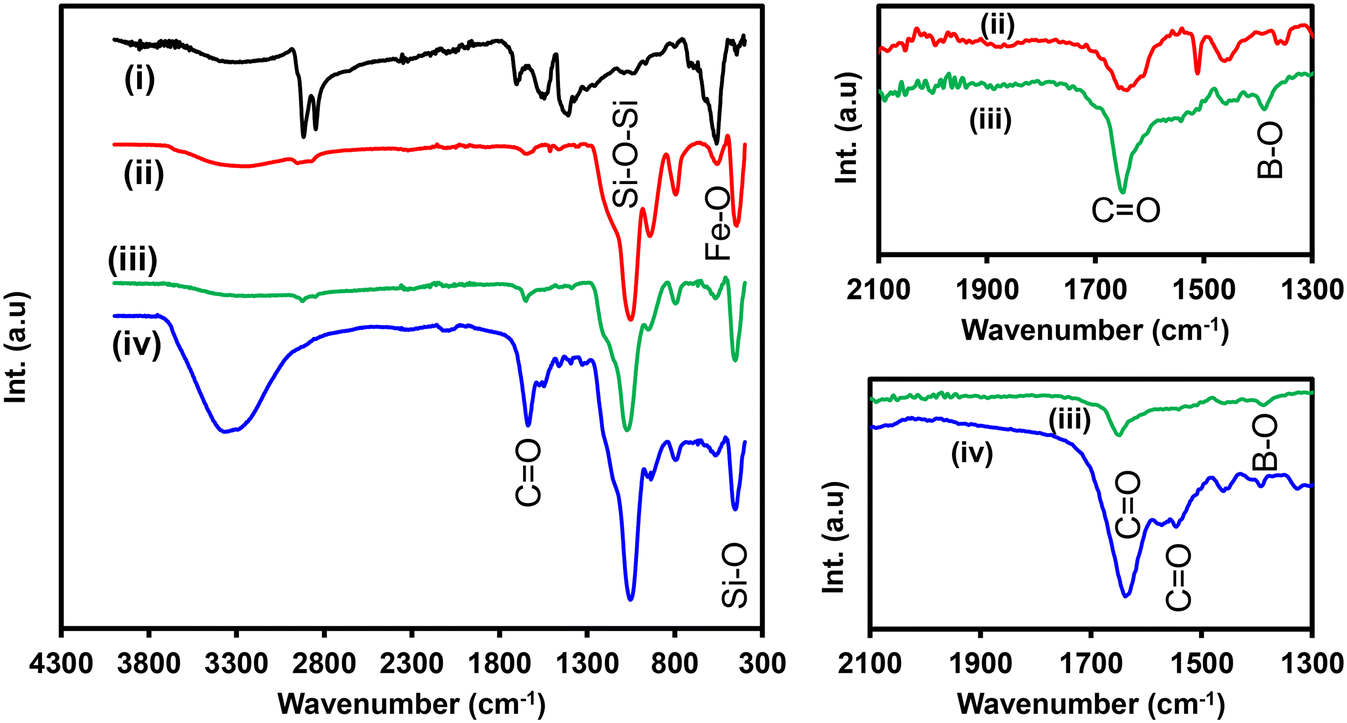 | ||
| Fig. 4 FT-IR spectra of (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, (iii) Fe3O4@SiO2-PBANPs, and (iv) Fe3O4@SiO2-PBA-Ab1 (enlarged 1300–2100 cm−1 for B–O, CO). | ||
Fig. 5 shows the zeta potential (ζ, mV) plot obtained at various pH values ranging from 2 to 10 in PBS, 10 mM for (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, (iii) Fe3O4@SiO2-PBANPs, and (iv) Fe3O4@SiO2-PBA-Ab1. Fig. 5(i) shows zeta potential changes on Fe3O4NPs and a negative increasing trend of zeta potential values up to −30 mV as the pH values increase. The negative increase in the zeta potential value is due to the deprotonation of the surface hydroxyl (OH) and negative surface charge. The zeta potential was −23.8 mV at pH 7.0. After coating with the silica shell as shown in Fig. 5(ii) for Fe3O4@SiO2NPs, the zeta potential value started at +2.0 mV at pH 2. As the pH increased, the zeta potential values became negative and increased negatively up to −43.2 mV at pH 10. The isoelectric point (IEP) was observed at pH 2.3. At pH 7.0, the zeta potential was −32.2 mV. The negative increase in the zeta potential was due to the deprotonation of the silanol (Si–OH) groups of the silica shell. After surface functionalization with TES-PBA as shown in Fig. 5(iii), a negative increase in the zeta potential charge from +24.0 mV at pH 3 to −40.0 mV at pH 10 was observed. The isoelectric point was observed at pH 5.4. The zeta potential charge was due to the deprotonation of the (B–OH) hydroxyl groups from TES-PBA. A zeta potential of −30.0 mV was obtained at pH 7.0. Fig. 5(iv) shows the pH effect on Fe3O4@SiO2-PBA-Ab1. A negative increase in the zeta potential from +25.0 mV at pH 2.0 to −23 mV at pH 10 was observed. An isoelectric point was obtained at pH 6.4 and the zeta potential value of −3.7 mV was observed at pH 7.0. The observed changes in zeta potential values at different pH solutions confirm the successful step-by-step modification and bioconjugation of the Ab1 to form Fe3O4@SiO2-PBA-Ab1.
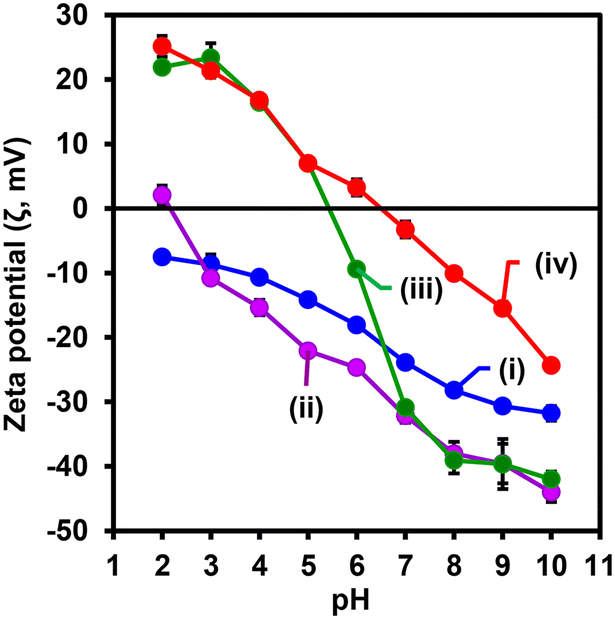 | ||
| Fig. 5 Zeta potential (ζ, mV) vs. pH (2–10, PBS, 10 mM) for (i) Fe3O4NPs, (ii) Fe3O4@SiO2NPs, (iii) Fe3O4@SiO2-PBANPs, and (iv) Fe3O4@SiO2-PBA-Ab1. | ||
The Bradford assay was used for protein quantification of Ab1 bioconjugated onto Fe3O4@SiO2−PBANPs. The calibration curve at different BSA concentrations was used to measure the protein solution before and after conjugation. The UV-vis absorbance measurements were performed at 595 nm for BSA and the same wavelength was used for determining the protein (Ab1) concentration. The protein amount bioconjugated onto Fe3O4@SiO2-PBANPs to form Fe3O4@SiO2-PBA-Ab1 was calculated by subtracting the determined concentration of the supernatant from the initial concentration in solution of Ab1. After bioconjugation, the percentage of the conjugation efficiency (%CE) was calculated using eqn (1) below:
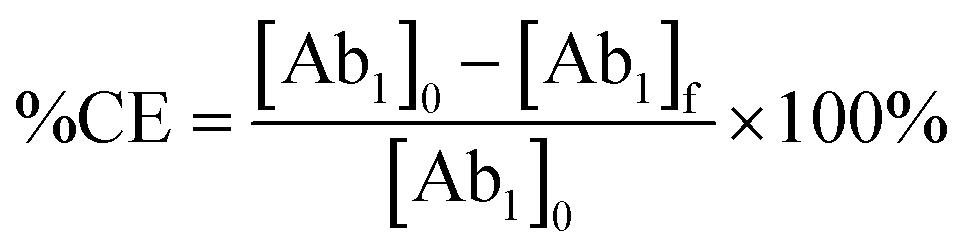 | (1) |
3.2 Evaluation and optimization of PSA capture and fluorescence detection
In the proposed immunosensor, Fe3O4@SiO2-PBA-Ab1/glucose nanobioconjugates were used to capture PSA from the sample solution as the first immunoreaction. Then, FITC@SiO2-PBA-Ab2/glucose fluorescence nanobioprobes were added to recognize the captured PSA by the second immunoreaction, forming the sandwich immunocomplex, Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA@SiO2@FITC. Scheme 2 shows the capture and fluorescence detection of PSA. | ||
| Scheme 2 Capture of PSA by Fe3O4@SiO2-PBA-Ab1/glucose and fluorescence detection using FITC@SiO2-PBA-Ab2/glucose. | ||
Fig. 6 shows the fluorescence emission spectra of the magnetic nanoparticles for (i) Fe3O4@SiO2-PBA-Ab1<PSA and (ii) Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC. In Fig. 6(i), no fluorescence absorption was observed for Fe3O4@SiO2-PBA-Ab1<PSA (after the PSA was captured). The same spectrum was observed in the absence of PSA. Absorption and fluorescence emission intensities were observed when PSA and FITC@SiO2-PBA-Ab2 were introduced in the system as shown in Fig. 6(ii). This confirmed that the sandwich immunoassay occured. The use of a magnet allowed for the separation of a sandwich immunoassay (Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC).
 | ||
| Fig. 6 (a) UV-vis absorption and (b) fluorescence emission spectra of (i) Fe3O4@SiO2-PBA-Ab1<PSA, and (ii) Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PSA-PBA-SiO2@FITC sandwich immunoassay. | ||
The concentration of the Fe3O4@SiO2-PBA-Ab1/glucose capture and FITC@SiO2-PBA-Ab1/glucose sensing nanobioprobes and the effect of the incubation time after PSA antigen capture were optimized. Both the concentration and incubation time parameters affect the sensitivity of the immunoassay. Fig. S2† shows the fluorescence intensity of the Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC immunoassay at (a) different concentrations of Fe3O4@SiO2-PBA-Ab1/glucose and (b) the effects of the incubation time on the immunosensor response during the detection of PSA. The concentration of PSA was 50 ng mL−1 and FITC@SiO2-PBA-Ab1/glucose was 200 μg mL−1. From Fig. S2(a),† the fluorescence emission intensity increased with increasing amounts of Fe3O4@SiO2-PBA-Ab1/glucose up to 100 μg mL−1. Similarly, in Fig. S2(b)† the fluorescence emission intensity increased with increasing incubation time up to 50 minutes. The optimum incubation time after the immunoassay for the detection of FITC@SiO2-PBA-Ab1/glucose in 10 mM NaOH was 20 minutes at 37 °C. The high intensity was achieved after exposing the sandwich assay to 10 mM NaOH that hydrolyses the silica and releases the FITC molecules into the solution.
3.3 Detection of PSA
Using the proposed fluorescence immunosensor, PSA was detected using fluorescence measurements as shown in Scheme 2. The dissolution fluorescence-linked immunosorbent assay (dFLISA) method was employed for the detection of PSA. The sandwich immunoassay Fe3O4@SiO2-PBA-Ab1<PSA>Ab2-PBA-SiO2@FITC was exposed to a 10 mM NaOH (20 μL) solution for 20 minutes to hydrolyze the silica and release the FITC molecules. The solution was subjected to fluorescence measurements at an excitation of 480 nm. Fig. 7(a) shows the fluorescence emissions of varying PSA concentrations from 2.0 pg mL−1 – 100 ng mL−1. The fluorescence emission intensity increased with increasing PSA concentrations. Fig. 7(b) shows the PSA dose–response curve for the correlation of the fluorescence intensity with an increase in the PSA concentrations. Fig. 7(c) shows the semi-log plot calibration curve of the proposed fluorescence immunosensor. The calibration curve of the change in fluorescence intensity Δ(Fluo. Int.)max = (Fluo. Int.i – Fluo. Int.o) vs. the ln[PSA] was obtained over the linear concentration range from 2.0 pg mL−1 to 100 ng mL−1. A linear regression equation with a regression coefficient (R2) = 0.978 is shown in Fig. 7(c) and below eqn (2):| Δ(Fluo. Int.)max = 415ln[PSA] + 4461 | (2) |
 | ||
| Fig. 7 (a) Bar graph of the fluorescence emission signal for different concentrations of PSA ranging from 2.0 pg mL−1 to 100 ng mL−1. (b) Dose–response curve of relative fluorescence intensity (ΔFImax = FIi − Flo). (c) Linear relation of relative fluorescence intensity (ΔFImax) against PSA concentrations, and (d) selectivity and specificity studies of the immunoassay sensor in the presence of PSA and other antibodies (n = 3). | ||
3.4 Specificity and selectivity of the immunosensor
The specificity and selectivity of the proposed immunosensor were investigated by determining the assay responses to PSA and other interfering analytes. Further, the real life application of the immunosensor was evaluated in newborn calf serum samples spiked with BSA, IgG antibody solutions and the mixture of IgG and PSA. Negative serum samples were used as blanks or negative samples with [PSA] = 0. The corresponding fluorescence response of the immunosensor, after removing the background interferences, are shown in Fig. 7(d). Fig. 7(d) shows the specificity and selectivity studies of the fluorescence immunosensor in the presence of PSA and other analytes like BSA and IgG, and a mixture of IgG and PSA. The IgG used is human IgG and not specific to PSA. From Fig. 7(d), the BSA and IgG antibody samples show very weak fluorescence response from trapped nanobioprobes during magnetic separation. The PSA and IgG + PSA spiked newborn calf serum samples gave high fluorescence intensity response. The immunosensor demonstrated excellent specificity and selectivity towards PSA detection. Furthermore, the comparison of the study was done with previously reported methods for detection for PSA as summarized in Table 1. The LOD of the study was found to be 0.81 pg mL−1 with an LOQ of 2.46 pg mL−1. Compared to other previously reported methods for PSA detection, their LODs of 10 pg mL−1,36 3.0 pg mL−1,8 83 pg mL−1,37 27 pg mL−1,38 and 30 pg mL−1.39 The previously reported methods employed the use of detection probes such as multi-CAT-AuNPs-Ab2, HRP-Ab2-SiO2NSs, CdTe@SiO2-Ab2, Ag/SiO2@RuBpy@SiO2-Ab2, and rGO-Ca:CdSe-Ab2, respectively. The proposed dFLISA immunosensor gave a lower LOD of 0.81 pg mL−1 for PSA detection. The proposed dFLISA in this study offered the advantage of using magnetic-silica capture nanobioconjugates for PSA concentration enrichment in the serum matrix and the highly fluorescent FITC-doped silica sensing nanobioprobes. The amplification of fluorescence signals was observed upon dissolution of the fluorescein–silica nanoparticles. The proposed method has many advantages especially considering very low concentrations of detected PSA by the proposed system and the scourge of prostate cancer worldwide. Future investigations will study the threshold and Gleason score of the modified system for early prostate cancer diagnosis. The only limitation of the system is the shelf life and long-term stability of the antibodies for selectivity and specificity of the immunosensor. These have limited shelf life (which is according to the supplier one year after the purchase). The storage and shelf life studies are part of our ongoing research activities.| Detection probe | Capture substrate | Signal monitored | Linear concentration range (LCR) | LOD |
|---|---|---|---|---|
| TW: this work; multi-CAT-AuNP-Ab2: polyclonal goat anti-human PSA/catalase-labelled gold nanoparticles; HRP-Ab2-SiO2NSs: horseradish peroxidase-labelled anti-PSA antibody/silicon dioxide nanospheres; CdTe@SiO2-Ab2: PSA-labelled cadmium telluride@silica core–shell nanoparticles. HRP-anti-PSA-IgG: horseradish peroxidase-labelled anti-PSA antibody. Ag/SiO2@RuBpy@SiO2-Ab2: a three layered silver core–shell, outer-layered RuBpy doped-silica fluorescent composite surface functionalized with the PSA antibody. IMN: immunomagnets. | ||||
| FITC@SiO2-PBA-Ab2/glucoseTW | Fe3O4@SiO2-PBA-Ab1/glucose | Fluorescence | 2.0 pg mL−1 – 100 ng mL−1 | 0.81 pg mL−1 |
| CdTe@SiO2-Ab28 |
Fe3O4–Ab1 | Fluorescence | 10 pg mL−1 – 5.0 ng mL−1 | 3.0 pg mL−1 |
| HRP-Ab2-SiO2NSs36 | Well/Ab1/BSA | Fluorescence | 30 pg mL−1 – 100 ng mL−1 | 10 pg mL−1 |
| HRP-anti-PSA-IgG37 | Well-anti-PSA-mIgY | Fluorescence | 0.10–3.38 ng mL−1 | 83 pg mL−1 |
| Ag/SiO2@RuBpy@SiO2-Ab238 |
IMN-Ab1 | Photo-luminescence | 0.10–100 ng mL−1 | 27 pg mL−1 |
| Multi-CAT-AuNPs-Ab239 |
MB-Ab1 | Colorimetric | 50 pg mL−1 – 20 ng mL−1 | 30 pg mL−1 |
4. Conclusions
In this work, we have developed a highly sensitive and selective fluorescence immunoassay for rapid detection of PSA using anti-PSA polyclonal antibody fluorescent-silica nanobioconjugates for fluorescence sensing and magnetic-silica antibody conjugates for PSA capture. The step-by-step preparation and characterization of the magnetic-silica antibody conjugates were successfully performed using microscopic and spectroscopic techniques. The preparation was successful as spherical magnetic-silica antibody capture nanobioconjugates were obtained. A sandwich immunocomplex of the fluorescein–silica sensing nanobioprobes and magnetic-silica capture nanobioconjugates formed through two immunoreactions used for the accurate detection of PSA. Compared with the ELISA method, the magnetic-silica capture nanobioconjugates assisted in PSA enrichment using a magnet to separate the captured PSA from the matrix. In addition, the fluorescent silica nanobioprobes enhanced the fluorescence intensity upon dissolution, using NaOH. This resulted in an amplified fluorescence signal due to the FITC molecules being released, enhancing the sensitivity of the immunoassay. The combined effects of the low background noise and increased fluorescence intensity resulted in a low limit of detection and limit of quantification in pg mL−1. Moreover, the oriented bioconjugation of monoclonal and polyclonal PSA antibodies promoted specificity and selectivity to PSA. Potential interfering proteins could not be detected using this system confirming its specificity and selectivity even at higher concentrations. The presented immunoassay was simple, specific, and ultrasensitive for PSA detection in newborn calf serum samples (representing real samples). The specificity of the proposed method was due to the use of anti-PSA specific antibodies for PSA detection. Other immunosensors for specific biochemical targets or molecules can be adapted to this method.Data availability
Data will be made available on request.Author contributions
Tumelo Msutu: conceptualization, methodology, formal analysis, validation, data curation, investigation, writing – original draft, visualization. Philani Mashazi: conceptualization, methodology, validation, data curation, visualization, supervision, writing – review & editing, resources, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank the National Research Foundation (NRF) for research funds through the NRF-STINT Bilateral (UID 118725) and NRF CPRR Grant supported this work. The authors also thank Rhodes University for research funding through Researcher Development Grant (RDG P5/17/2015), Rated Research Grant (RRG) and Sandisa Imbewu. TM thanks Gauteng City Regional Academy (GCRA) for an MSc Scholarship.References
- E. A. Stura, B. H. Muller, M. Bossus, S. Michel, C. Jolivet-Reynaud and F. Ducancel, J. Mol. Biol., 2011, 414, 530–544 CrossRef CAS PubMed.
- D. Damborska, T. Bertok, E. Dosekova, A. Holazova, L. Lorencova, P. Kasak and J. Tkac, Microchim. Acta, 2017, 184, 3049–3067 CrossRef CAS.
- D. C. Pérez-Ibave, C. H. Burciaga-Flores and M.-Á. Elizondo-Riojas, Cancer Epidemiol., 2018, 54, 48–55 CrossRef.
- H. Van Poppel, M. J. Roobol, C. R. Chapple, J. W. F. Catto, J. N'Dow, J. Sønksen, A. Stenzl and M. Wirth, Eur. Urol., 2021, 80, 703–711 CrossRef PubMed.
- L. Yang, H. Zhao, G. Deng, X. Ran, Y. Li, X. Xie and C.-P. Li, RSC Adv., 2015, 5, 74046–74053 RSC.
- P. Song, B. Yang, Z. Peng, J. Zhou, Z. Ren, K. Fang, L. Yang, L. Wang and Q. Dong, Int. J. Surg., 2020, 76, 64–68 CrossRef PubMed.
- G. S. M. Kammeijer, J. Nouta, J. J. M. C. H. de la Rosette, T. M. de Reijke and M. Wuhrer, Anal. Chem., 2018, 90, 4414–4421 CrossRef CAS PubMed.
- Y. Zhao, W. Gao, X. Ge, S. Li, D. Du and H. Yang, Anal. Chim. Acta, 2019, 1057, 44–50 CAS.
- S. Veeranarayanan, A. Cheruvathoor Poulose, S. Mohamed, A. Aravind, Y. Nagaoka, Y. Yoshida, T. Maekawa and D. S. Kumar, J. Fluoresc., 2012, 22, 537–548 CrossRef CAS.
- M. D. Nguyen, H.-V. Tran, S. Xu and T. R. Lee, Appl. Sci., 2021, 11, 11301 CrossRef CAS PubMed.
- L. Zhang, H. Shao, H. Zheng, T. Lin and Z. Guo, Int. J. Miner., Metall. Mater., 2016, 23, 1112–1118 CrossRef CAS.
- R. C. Popescu, E. Andronescu and B. S. Vasile, Nanomaterials, 2019, 9, 1791 CrossRef CAS PubMed.
- A. S. Teja and P.-Y. Koh, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22–45 CrossRef CAS.
- L. Zhang, R. He and H.-C. Gu, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef CAS.
- F. Ahangaran, A. Hassanzadeh and S. Nouri, Int. Nano Lett., 2013, 3, 23 CrossRef.
- S. Mohapatra, N. Panda and P. Pramanik, Mater. Sci. Eng., C, 2009, 29, 2254–2260 CrossRef CAS.
- C. Y. Wang, J. M. Hong, G. Chen, Y. Zhang and N. Gu, Chin. Chem. Lett., 2010, 21, 179–182 CrossRef CAS.
- N. V. Jadhav, A. I. Prasad, A. Kumar, R. Mishra, S. Dhara, K. R. Babu, C. L. Prajapat, N. L. Misra, R. S. Ningthoujam, B. N. Pandey and R. K. Vatsa, Colloids Surf., B, 2013, 108, 158–168 CrossRef CAS PubMed.
- M. Bloemen, W. Brullot, T. T. Luong, N. Geukens, A. Gils and T. Verbiest, J. Nanopart. Res., 2012, 14, 1100 CrossRef PubMed.
- Q. Fan, Y. Guan, Z. Zhang, G. Xu, Y. Yang and C. Guo, Chem. Phys. Lett., 2019, 715, 7–13 CrossRef CAS.
- S. Wang, J. Tang, H. Zhao, J. Wan and K. Chen, J. Colloid Interface Sci., 2014, 432, 43–46 CrossRef CAS PubMed.
- Z. Lu, J. Dai, X. Song, G. Wang and W. Yang, Colloids Surf., A, 2008, 317, 450–456 CrossRef CAS.
- O. Adeniyi, S. Sicwetsha and P. Mashazi, ACS Appl. Mater. Interfaces, 2020, 12, 1973–1987 Search PubMed.
- G. Asab, E. A. Zereffa and T. Abdo Seghne, Int. J. Biomater., 2020, 2020, 1–11 Search PubMed.
- A. Adesina, O. Adeniyi and P. Mashazi, Electroanalysis, 2023, 35, 120–130 CrossRef.
- S. Kralj, M. Drofenik and D. Makovec, J. Nanopart. Res., 2011, 13, 2829–2841 CrossRef CAS.
- P.-C. Lin, S.-H. Chen, K.-Y. Wang, M.-L. Chen, A. K. Adak, J.-R. R. Hwu, Y.-J. Chen and C.-C. Lin, Anal. Chem., 2009, 81, 8774–8782 CrossRef CAS.
- C. Bi, S. Zhang, Y. Li, X. He, L. Chen and Y. Zhang, New J. Chem., 2018, 42, 17331–17338 RSC.
- T. Msutu, O. Adeniyi and P. Mashazi, Sens. Diagn., 2024, 3, 1167–1176 RSC.
- R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC.
- D. K. Kim and J. W. Lee, J. Korean Ceram. Soc., 2018, 55, 625–634 CrossRef CAS.
- J. M. Xu, Y. X. Zhang, G. H. Li, S. Wang, H. L. Ding and S. C. Xu, Chem. Mater., 2012, 24, 4572–4580 CrossRef.
- R. M. Patil, P. B. Shete, N. D. Thorat, S. V. Otari, K. C. Barick, A. Prasad, R. S. Ningthoujam, B. M. Tiwale and S. H. Pawar, RSC Adv., 2014, 4, 4515–4522 RSC.
- J. Nayeem, M. A. A. Al-Bari, M. Mahiuddin, M. A. Rahman, O. T. Mefford, H. Ahmad and M. M. Rahman, Colloids Surf., A, 2021, 611, 125857 CrossRef CAS.
- C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H.-J. Gao, Nanoscale, 2011, 3, 701–705 RSC.
- L. Li, W. Zhang, Y. Wei, L. Yu and D. Feng, J. Anal. Methods Chem., 2022, 2022, 1–9 Search PubMed.
- A. Łupicka-Słowik, R. Grzywa, E. Leporowska, D. Procyk, J. Oleksyszyn and M. Sieńczyk, Ann. Lab. Med., 2019, 39, 373–380 CrossRef.
- D.-D. Xu, Y.-L. Deng, C.-Y. Li, Y. Lin and H.-W. Tang, Biosens. Bioelectron., 2017, 87, 881–887 CrossRef CAS.
- Z. Gao, M. Xu, L. Hou, G. Chen and D. Tang, Anal. Chem., 2013, 85, 6945–6952 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The materials, characterization and methods are further discussed in the ESI. See DOI: https://doi.org/10.1039/d4sd00292j |
| This journal is © The Royal Society of Chemistry 2025 |