
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePursuing theranostics: a multimodal architecture approach†
Aidan A.
Bender
 a,
Connor K.
Holiski
a,
Mary
Embree
b,
Heather M.
Hennkens
bc,
John R.
Klaehn
d,
Ellie
Lundgreen
a,
Andrew G.
Roberts
e,
Peter R.
Zalupski
f and
Tara
Mastren
*a
a,
Connor K.
Holiski
a,
Mary
Embree
b,
Heather M.
Hennkens
bc,
John R.
Klaehn
d,
Ellie
Lundgreen
a,
Andrew G.
Roberts
e,
Peter R.
Zalupski
f and
Tara
Mastren
*a
aNuclear Engineering Program, University of Utah, 110 Central Campus Dr. Suite 2000B, Salt Lake City, UT 84112, USA. E-mail: tara.mastren@utah.edu
bUniversity of Missouri Research Reactor, Columbia, MO 65211, USA
cDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
dBiological and Chemical Process Sciences, Idaho National Laboratory, Idaho Falls, ID 83415, USA
eDepartment of Chemistry, University of Utah, Salt Lake City, UT 84112, USA
fAqueous Separations and Radiochemistry, Idaho National Laboratory, Idaho Falls, ID 83415, USA
First published on 24th October 2024
Abstract
Theranostics is a field of nuclear medicine which uses the same targeting vector and chelating system for both a diagnostic and therapeutic radionuclide, allowing for uniformity in imaging and treatment. This growing field requires the development of more flexible chelate systems that permit novel targeting strategies. Toward this end, a multimodal architecture has been realized, making use of a phosphazene-based core and click chemistry to achieve a flexible and customizable scaffold. The six arm phosphazene-based core can scaffold six DTPA chelating motifs or a mixed set of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 DTPA:DFO chelates resulting in two multimodal compounds, pDbDt and pDbDtDf, respectively. Terbium complexes displayed strong luminescence, supporting that the structures act as an organic antenna for luminescence. Metal displacement titration studies confirmed the desired structures as well as the capability for heterometallic labeling of the structures. These structures were found to have high thermal and biological stability in vitro. Radiolabeling of each compound resulted in high molar activity labeling of each compound: 169 MBq nmol−1: [161Tb]Tb–pDbDt, 170 MBq nmol−1: [89Zr]Zr–pDbDtDf, and the mixed radiolabeling illustrated chelation of both radionuclides in a 1:1 ratio. This multimodal architecture is promising as a heterometallic structure for coupling of both a diagnostic and a therapeutic radionuclide with a highly customizable core structure.
:3 DTPA:DFO chelates resulting in two multimodal compounds, pDbDt and pDbDtDf, respectively. Terbium complexes displayed strong luminescence, supporting that the structures act as an organic antenna for luminescence. Metal displacement titration studies confirmed the desired structures as well as the capability for heterometallic labeling of the structures. These structures were found to have high thermal and biological stability in vitro. Radiolabeling of each compound resulted in high molar activity labeling of each compound: 169 MBq nmol−1: [161Tb]Tb–pDbDt, 170 MBq nmol−1: [89Zr]Zr–pDbDtDf, and the mixed radiolabeling illustrated chelation of both radionuclides in a 1:1 ratio. This multimodal architecture is promising as a heterometallic structure for coupling of both a diagnostic and a therapeutic radionuclide with a highly customizable core structure.
Introduction
Theranostics
As the field of nuclear medicine continues to expand each year,1 the need for compatibility between diagnostic and therapeutic techniques continues to escalate. Additionally, patient-specific dosimetry is a rising field with its own challenges and unique considerations.2 In order to combine these three distinct disciplines, a class of radiopharmaceuticals known as theranostics has been proposed.3–5 Theranostics make use of the bifunctional radiolabeling of biomolecules (Fig. 1) to deliver both an imaging radionuclide and a therapeutic radionuclide using the same targeting moiety. This approach minimizes any difference in biological behavior, allowing for more accurate modeling of treatment behavior. However, there are limitations to this approach, stemming from the fundamental differences in chemistry of certain radionuclides for radiolabeling, as well as disparate half-lives (e.g., 18F and 177Lu have incompatible chemical differences including drastically different half-lives).6 To eliminate these differences, there are ongoing searches for “theranostic pairs”, radionuclides of the same element, such as 64Cu/67Cu and 86Y/90Y.7 Despite these advancements in theranostics, there is still a need for a system that satisfies the need for patient-specific dosimetry. | ||
| Fig. 1 A bifunctional chelate system employing either a diagnostic isotope for PET/SPECT imaging or a therapeutic radionuclide. The bifunctional chelator may be attached to a biological vector before or after radiolabeling. | ||
Out of this need arises the expanding potential of multimodal systems. Already, multimodal imaging makes use of simultaneous imaging modalities such as PET/MRI and PET/CT to generate strong corroborating images of the target and surrounding tissue.8 Such techniques allow for greater certainty in diagnosis and better accuracy in determining a therapeutic route. If these multimodal imaging systems could be combined with the theranostic pair systems highlighted above, a clear path toward patient-specific dosimetry and treatment monitoring can be imagined.
However, theranostics is not contained to nuclear medicine alone. The concept of a theranostic system finds a home in chemotherapy as well, where an antibody–drug conjugate can be monitored by a PET or SPECT agent using the same antibody.9–11 Surgical resection of tumors uses brightfield microscopy for analyzing and spotting tumors on the exterior of tissue, but is often paired with chemiluminescent sensors for elucidating the presence of tumors buried within tissue.12–14 Fluorescent probes require careful synthesis of an organic scaffold which itself fluoresces or which enhances the fluorescence of a lanthanide metal.15 Imaging for surgical and pre-surgical work is a growing area of research and has need for an expanding library of probes.16 These theranostic applications also require flexible and novel approaches to imaging and therapy.
A multimodal chelate architecture
The more flexible a multimodal system is, the more viable it will be in the theranostic field. Therefore, the search for this system began with establishing a central ring that could be easily modified multiple times. Due to its stability and favorable solubility, a phosphazene ring was selected.17 This ring can be modified with a variety of pendent arms. The first pass at this system was aimed at using click chemistry18 across all six arms for very simple modification (Fig. 2). To these arms a variety of moieties could be added using Cu-catalyzed or Cu-free click chemistry, from bifunctional chemotherapy ligands, targeting antibodies or peptides,19 as well as chelates for radionuclides for imaging (PET/SPECT) and therapeutic (targeted alpha therapy/targeted radiotherapy) applications,20–22 lanthanides for chemiluminescence23,24 or MRI contrast25,26 and high Z elements for radio-sensitization.27,28 The simplest version of this ring creates a dendrimer-like structure with six identical arms. Here, this simple system was developed with diethylenetriaminepentaacetic acid (DTPA) chelates. The flexibility of this system was demonstrated by modification to a system split evenly between DTPA and deferoxamine (DFO) for the heterobimetallic labeling with a lanthanide and zirconium. We hypothesized that these two systems would demonstrate heterobimetallic labeling for the purpose of pursuing theranostic systems, which could be used for real-time imaging of a therapeutic agent. The feasibility of these heterobimetallic systems would illustrate the flexibility of a phosphazene core for targeted radiotherapy.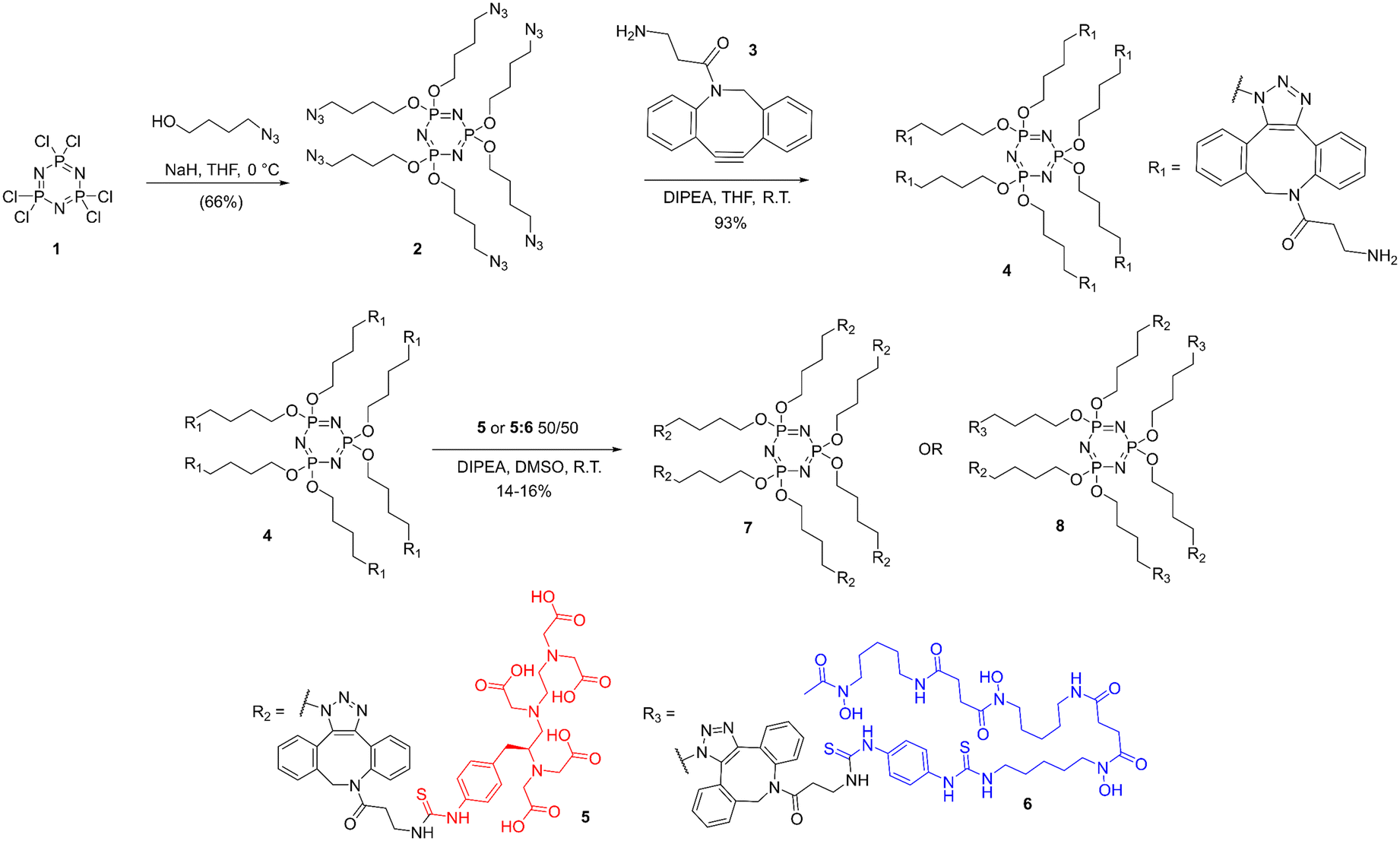 | ||
| Fig. 2 Synthesis of (7) and its derivative (8) making use of commercially available SCN-Bn-DTPA (5) and SCN-Bn-deferoxamine (6). | ||
Experimental procedures
Reagents
p-SCN-Bn-DTPA (5) and p-SCN-Bn-DFO (6) were purchased from Macrocyclics, Inc. DBCO-amine was purchased from Click Chemistry Tools. All solvents were purchased from Fisher Scientific and VWR. All other chemicals were purchased from Thermo Fisher Scientific. H5DTPA was recrystallized from hot water, characterized and standardized by titration with HCl. Gadolinium and terbium oxides (>99.99% Pangea Intl, China) and bismuth oxide (99.9%, Aldrich) were used to prepare the stock solutions by dissolving 1 equivalent metal oxide in 5.9 equivalents of corresponding acid (HClO4 for Gd and Tb and HNO3 for Bi, TraceSELECT, Sigma Aldrich). The undissolved oxide residue was filtered out, and the stock Tb(ClO4)3 solution was adjusted to contain 0.001 M HClO4 and Bi(NO3)3 to contain 1 M HNO3. The concentration of Gd and Tb was determined by complexometric titration with tetrasodium ethylenediamine-N,N,N′,N′-tetraacetic acid (Na4EDTA) to a xylenol orange endpoint in 1 mM N,N′-diethylpiperazine buffer solution at pH 5.00. A DP-5 phototrode (Mettler Toledo) operated by a Mettler Toledo T5 graphix autotitrator was used to identify the endpoint. The concentration of Gd and Tb was confirmed using ICP-MS analysis. The concentration of Bi3+ ion (0.04857 ± 0.00006 moles per kg) was determined using Na2EDTA stock and xylenol orange indicator. Zirconium oxynitrate dihydrate (Aldrich) was boiled in appropriate amount of 8 M HNO3 to ensure dissolution of the solid and yield targeted aqueous acidity of 1 M. The Zr(NO3)4 solutions were filtered using 0.2 μm PVDF filter membranes. The concentration of Zr was determined via a back-titration with Bi stock solution using xylenol orange indicator. The concentration of HNO3 in Bi and Zr stocks was determined using potassium oxalate to complex the metals and titrate the acid with standard NaOH to phenolphthalein endpoint. All other chemicals were used without further purification.General chemistry remarks
NMR spectra were collected using a 500 MHz Bruker Ascend at room temperature (rt), using tetramethylsilane (TMS) as an internal standard, from which all chemical shifts (δ) are reported in ppm. NMR spectra were obtained by dissolution of samples in DMSO-d6. LRMS Mass spectra were acquired using an Advion Expression compact mass spectrometer (ESI), HRMS were acquired using an Agilent Triple Quad GC (ESI). Analytical HPLC was performed using a Thermo Scientific Ultimate 3000 system, with the mobile phase H2O/CH3CN (solvent A/solvent B, each with 0.1% TFA) being delivered at 1.5 mL min−1, passed through a Thermo Scientific Hypersil GOLD C18 250 mm column, λ = 360 nm. Preparative HPLC purification was performed using an ECOM separations system, with the mobile phase (solvent A/solvent B) being delivered at 15.0 mL min−1, pass through an XTerra MS C18 250 mm prep column, λ = 360 nm. For both HPLC systems, the solvent gradient was as follows: 5% solvent B from 0 to 5 minutes; 5% to 50% B from 5–20 minutes; 50% B from 20–25 minutes. Fluorimetry data was collected using a Jobin Yvon IBH FluoroLog-3 fluorometer (HORIBA).Production and purification of terbium-161
Terbium-161 was produced at the University of Missouri Research Reactor (MURR) by irradiating 1.75 mg of enriched gadolinium(III) oxide (160Gd enrichment of 99.96 ± 0.003%) in the flux trap (neutron flux: nominal, thermal ≈3.8 × 1014 n cm2 s−1 for 140.85 hours. An aliquot of 161Tb, equivalent to approximately 175 μg of 160Gd metal, was first isolated via cation exchange chromatography (AG 50W-X8, 200–400 mesh) utilizing 2-hydroxyisobutyric acid (70 mM, pH 4.75). Subsequently, it was concentrated using bDGA (50–100 μM) extraction chromatography and subjected to a secondary separation and concentration using LN (100–150 μM) and bDGA (50–100 μM) resins.29,30Synthesis of pDbDt (7) and pDbDtDf (8)
The full synthetic route is shown in Fig. 2. Alkylation of the hexachloro-phosphazene trimer was carried out with high yield (66%), resulting the hexa-azide product 2. This product was purified by column chromatography and characterized using NMR spectroscopy and GCMS. To modify the ring further in preparation for reaction with isothiocyanate-functionalized chelates, intermediary 4 was achieved via copper-free click chemistry with DBCO-amine 3 in very high yields (92%). This product was purified by preparative HPLC and characterized by NMR and HR-LCMS. Functionalizing the ring with chelates took advantage of the well-established reaction between primary amines and isothiocyanates to attach either DTPA alone or a mixture of DTPA and DFO. Each reaction was purified with preparative HPLC (yields 16% and 14%, respectively) and characterized by NMR spectroscopy, GCMS, and lanthanide luminescence. Full synthetic procedures and analysis are available in the ESI.†Fluoroscopy of terbium
Tb3+ solutions were prepared in 20 mM HClO4. DTPA was dissolved in water by minimal addition of concentrated NaOH and standardized using a Mettler Toledo T5 titrator. Compounds 7 and 8 were dissolved in MES buffer with minimal addition of 50% NaOH (<0.1% v/v final concentration). All solutions were prepared by weight and all metal–chelate solutions were buffered with MES buffer (pH = 5.5) and ionic strength was held constant at 0.16 M.Tb3+ solutions were observed by fluoroscopy to determine optimal excitation wavelength, emission bandwidth, fluorimeter excitation and emission slit widths and integration times. These values were confirmed by observation of [Tb]–DTPA. Excitation studies of [Tb]–7 were used to select the ideal excitation wavelength of 488 nm for chelated terbium.
Competition assays between Tb3+ and Bi3+ were carried out with both DTPA and 7 in MES buffer (pH = 5.5). Samples were mixed to allow for equilibration of species in solution before fluorimetry. Equilibration was confirmed by analysis after 4 hours, 24 hours, and 7 days. Similar competition assays were carried out with Tb3+ and Gd3+ as well as Tb3+ and Zr3+. Maximum emission values were fit with sigmoidal curves to determine the number of metal ions per complex (in the case of DTPA, 1 metal per complex, and in the case of 7, multiple metal ions per complex) using GraphPad Prism. In the case of 8, only the Tb3+ and Zr3+ competition was carried out.
Lifetime measurements of 7 were carried out using the same fluorescence system at 22 ms. Time-dependent fluorescence spectra were collected on 4 samples in both D2O and H2O and lifetimes determined from a single exponential fit. The number of inner sphere water molecules, q, was determined using the equation  , where A = 4.2 ms and B = 0.06 ms.31
, where A = 4.2 ms and B = 0.06 ms.31
Thermal stability of [Tb]6–7 was determined by fluorescence spectra of samples at 20 °C and 37 °C taken daily for 5 weeks as compared to Tb3+ samples incubated at the same temperatures. Biological stability was determined by HPLC analysis of samples incubated in fetal bovine serum (FBS) at 20 °C and 37 °C for 2 weeks.
Radiolabeling of pDbDt and pDbDtDf
Radiolabeling of both 7 and 8 was carried out in MES buffer (pH = 6.0). 0.25 mM of each complex was incubated with 0.185 MBq of 161Tb or 0.185 MBq 89Zr at 37 °C for 20 minutes. Samples were analyzed by radio-iTLC and radio-HPLC to confirm radiolabeling. To determine apparent molar activity, a serial dilution of 7 (1 mM–1 pM) were incubated with 0.185 MBq 161Tb at 37 °C for 20 minutes and analyzed by radio-iTLC. Heteroradionuclide samples were prepared and analyzed by the same method, with the addition of 0.0925 MBq of 89Zr and 0.0925 MBq of 161Tb to a serial dilution of 8 (1 mM–1 pM) at 37 °C for 20 minutes. Samples were analyzed by radio-iTLC. Ratios of bound radionuclides were determined by HPGe analysis of a radio-HPLC-purified sample of 0.0925 MBq [89Zr/161Tb]–8 before and after decay of 89Zr.Results and discussion
Synthesis
Each synthetic target was produced in high purity (see HPLC traces in ESI†). The intermediate 4 was collected in high yield, and the products 7 and 8 were collected in sufficient yields for this work (∼15%), though methods of establishing higher yields could be studied for further expansion on this work. While mass spectrometry and NMR spectroscopy are the standard analyses for confirmation of molecular structure, the lanthanide luminescence compliments the complex nature of the spectra of these more traditional methods. While the conformational complexity of the full structures creates more complex 1H and 13C NMR shifts along with rotameric effects, the phosphorus in the central phosphazene allows for a simple method (and very simple spectra, with its single peak) of confirming the presence of the central ring in each sample, and the lack of significant shift of that single peak suggests uniformity of that ring (ESI† Fig. S7, S10 and S16). While there are very minor shifts in 1H environments between each compound, the presence of 1H shifts of each component strongly suggests the corresponding products (ESI† Fig. S5–S16), especially when paired with the shifting retention time of HPLC peaks (and the singular nature of those peaks) (ESI† Fig. S1–S4). Mass spectroscopic analyses reveal complex, peptide-like ionization signatures. When these traditional analyses are combined with the luminescence studies, confidence in the proposed structures can be well established. Compound 8 was isolated with significant purity by prep-HPLC in the primarily 3:3 DTPA:DFO ratio as shown, but inevitably includes other ratios of DTPA:DFO. Based on the luminescence studies, the overall ratio is still overwhelmingly 3:3.
Luminescence
The excitation spectrum of terbium as compared to the excitation spectra of 7 display some of the immediate challenges with this system. First and foremost, the classic excitation values for europium and terbium are obvious (Fig. 3A), but thoroughly overwhelmed by the excitation spectra of 7. We hypothesize that this is caused by the pi-conjugated systems of the DBCO moiety. Unfortunately, this very strongly interferes with the emission spectra of both europium and terbium when using the more traditional excitation wavelengths. Instead, the lower (but still significant) excitation wavelength at 488 nm for terbium presents itself as an excitation wavelength that interacts much less significantly with the pi-conjugated systems while still exciting terbium. As can be seen, not only does the 488 nm excitation create terbium emission spectra very consistent with those expected of terbium (Fig. 3B), but 7 actually enhances these spectra, in keeping with the organic “antenna” approach toward pi-conjugated organic moieties in other systems.32 The ability to track conjugation of lanthanides by luminescence is underutilized in development of radiotracers to date; these results indicate the field could benefit from taking advantage of these tools. | ||
| Fig. 3 A) Excitation spectra of europium and terbium complexes, as well as the excitation spectra of 7. B) Emission spectra of terbium complexes. C) Emission spectra of [Tb]–7 at decreasing solution concentrations. | ||
Between the expected emission spectra, the low limit of detection (6 μM) (Fig. 3C), and the similar luminescence lifetime of both DTPA alone and 7 (Table 1), the system behaves similarly to simpler systems while exhibiting enhanced terbium emissions.
| Compound | q (#H2O) |
|---|---|
| [Tb]–DTPA | 0.86 ± 0.5 |
| [Tb]–7 | 0.79 ± 0.5 |
Heterobimetallic studies
Of the three heterobimetallic systems studied, the bismuth-terbium system is the simplest. Due to bismuth having a significantly higher stability constant compared to terbium for DTPA,33 it was expected that bismuth would displace terbium quickly and with high stability. This is demonstrated by the decrease in terbium luminescence and can be seen at very low concentrations of bismuth (Fig. 4A). As bismuth concentrations reach equivalence to terbium, the luminescence dropped to the same level as full uncoordinated terbium ions, as expected. Further analysis resulted in a sigmoidal curve fitting revealing that 0.075 mM 7 coordinates all bismuth in solution when [Bi] = 0.42 ± 0.03 mM, suggesting that there are 5.6 ± 0.6 chelates per molecule of 7, consistent with the hexa-chelate structural configuration of 7. Based on measurements of this system at 4 hours, 24 hours, and 7 days, the bismuth–terbium system reached equilibrium within the first 4 hours and does not appreciably change over time. This simple system provides a clear-cut example of a displacement titration for creation of a heterometallic complex. | ||
| Fig. 4 Competition assays of Bi/Tb for A) DTPA and B) compound 7, as well as the competition assay for Gd/Tb for C) compound 7. | ||
While a gadolinium–terbium system introduces more complexity, the actual results are remarkably similar to those of the bismuth–terbium system, with the exception that more gadolinium was required in solution to displace all terbium from the chelate (approximately twice as much Gd, relative to Bi, Fig. 4C). The stabilities of DTPA complexes with gadolinium and terbium are very similar, resulting in a much slower metal exchange equilibrium. The luminescence spectra of terbium stabilized within 48 hours of mixing. However, once equilibrium was reached, the system remained stable for over a week.
In the case of 7, a competition between zirconium and terbium gave very similar results to the bismuth–terbium competition, due to the difference between the metals' stability constants. However, it is known that DTPA does not stably chelate zirconium in vivo, and as such, the alternative 8 was used to study this competition. Fig. 5 shows that terbium is coordinated by both DTPA and DFO moieties and, as zirconium is introduced into the system, it is partially displaced before reaching a hetero-metal chelate environment, containing both zirconium and terbium. We hypothesize this is caused by zirconium preferentially displacing terbium in the DFO chelates, leaving the DTPA chelates intact until Zr content is further increased.34 This metal exchange titration yields 2.8 ± 0.3 DFO chelates and 5.7 ± 0.4 total chelates (and thereby ∼3 DTPA chelates) as the molecular construct of 8, matching the MS results.
 | ||
| Fig. 5 Competition assay between zirconium and terbium with compound 8, showing two clear displacement curves, with curve 1 occurring at lower concentrations, likely displacement of Tb out of DFO by Zr, and curve 2 at higher concentrations, the displacement of Tb out of DTPA by Zr. | ||
The thermal stability of 7 was measured over 5 weeks by terbium fluorescence and the system was shown to have over 90% stability for over 4 weeks, and over 80% stability at 5 weeks at both 20 and 37 °C (Fig. 6A). Additionally, the biological stability in FBS was found to be over 90% for over 8 days and over 80% stability at 2 weeks by HPLC (Fig. 6B).
 | ||
| Fig. 6 A) Thermal stability of [Tb]–7. B) Stability of [161Tb]Tb–7 in FBS. | ||
Radiolabeling
7 was successfully radiolabeled with 161Tb, and 8 was radiolabeled with 89Zr with high apparent molar activity (Table 2) (Fig. 7A–D). 8 was radiolabeled with both radionuclides simultaneously in good apparent molar activity as well (Table 2) (Fig. 7E and F). The HPGe analysis of the HPLC-purified sample suggests binding of both radionuclides simultaneously (Table 3) in a 1:1 ratio, as hypothesized. A solvent-front peak is located at 2 minutes (where unlabeled radioactivity would appear) in the UV-vis spectra. [161Tb]–7 appears to have a minor degradation peak at 14 minutes (Fig. 7A), but this component is not radiolabeled and the chemical purity is >95%.
| Compound | AMA (MBq nmol−1) |
|---|---|
| [161Tb]–7 | 169 ± 7 |
| [89Zr]–8 | 170 ± 6 |
| [89Zr/161Tb]–8 | Tb = 91 ± 5Zr = 105 ± 4 |
 | ||
| Fig. 7 A and B) radio-HPLC trace and apparent molar activity of [161Tb]–7. C and D) radio-HPLC trace and apparent molar activity of [89Zr]–8. E and F) radio-HPLC trace and apparent molar activity of [161Tb/89Zr]–8. | ||
:1 coordination of Tb/Zr to 8
| Radionuclide | Activity (Bq) | Atoms | Atoms/molecules pDbDtDf |
|---|---|---|---|
| [161Tb] | 3.22 × 105 | 4.57 × 1011 | 2.05 |
| [89Zr] | 3.70 × 105 | 4.35 × 1011 | 1.95 |
Lanthanide luminescence with this system has a limiting concentration of 6 μM, which is significantly higher than the concentrations of radionuclides at standard working conditions (nM or pM magnitudes). However, the luminescence provided an excellent companion study to the heterobimetallic labeling with 89Zr and 161Tb with near-identical complexation ratios as the displacement titration would suggest.
Limitations and future work
Both 7 and 8 make use of all six of their pendent arms for chelation, which is not desired in a targeted approach. Ideally, one arm is either attached to a targeting molecule prior to functionalization with chelates, or one arm is left open after chelates are attached. In this case, the targeting molecule is likely an antibody, based on the size of the system, such that adding the phosphazene core to the antibody first is a reasonable method. The orthogonal protection of one arm could enable this goal. The overall reaction conditions are mild and could be translated to an aqueous methodology to further protect the antibodies. Once the full architecture is attached to an antibody, full toxicity and biodistribution studies would need to be performed.The flexibility of the phosphazene-azide system is demonstrated here. We hypothesize that this flexibility could be further expanded by introducing other functional arms to the phosphazene core. By differentiating the arms, greater control over the number of each modification could be exerted.
As demonstrated in the heterobimetallic labeling of 7 and 8, this system shows potential for use in theranostic applications. It can already be labeled with a PET imaging agent such as 89Zr and a therapeutic agent such as 161Tb, but further customization could be easily imagined. By interchanging the chelates used (such as using DOTA or HEHA), an analogous system could be labeled simultaneously with a SPECT radionuclide such as 99mTc and a therapeutic radionuclide such as 177Lu and 225Ac. In addition, a heterometallic system with gadolinium may prove to be a good contrast agent for complimentary MRI. With high specific activity and good thermal and biological stability, this system demonstrates the flexibility required for biological applications. If one of the pendent arms is left unmodified, it could be conjugated via click chemistry to a biomolecule of choice. These in vitro and in vivo experiments will be pursued in the future.
Based on the synthesis of 8, the system could be further diversified without changing the pendent arms. If yield was sacrificed, close stoichiometric control of functionalized arms could be achieved. If maintaining good yield is desired, then differentiating the arms prior to functionalization should be targeted.
Conclusions
In conclusion, a multimodal chelate system was synthesized using a phosphazene core and the principles of click chemistry. A hexa-DTPA system (pDbDt) and a tri-DTPA, tri-DFO system (pDbDtDf) were used to generate heterometallic complexes with Tb–Bi, Tb–Gd, and Tb–Zr as monitored by lanthanide luminescence. Both 7 and 8 were radiolabelled with 161Tb and 89Zr alone or with a mixture of the two, resulting in simultaneous radiolabelling with a therapeutic and diagnostic isotope. The radiolabelling occurred in high molar activity.A highly flexible system for the simultaneous conjugation of multiple radionuclides to a phosphazene core in pursuit of a theranostic system for targeted radiotherapy (TRT) was demonstrated. This work suggests the need for more investigation into multimodal systems for TRT and highlights the usefulness of lanthanide luminescence in developing these probes.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Travis Grimes and Dean Peterman for their advice on Terbium luminescence, and Paul Oblad for advice and assistance with NMR spectra acquisition. This research was supported by the Laboratory Directed Research and Development Program, U.S. Department of Energy, under DOE Idaho Operations Office contract DE-AC07-05ID14517 with Idaho National Laboratory, managed by Battelle Energy Alliance, LLC, and by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program. The SCGSR program is administered by the Oak Ridge Institute for Science and Education for the DOE under contract number DE-SC0014664. We are grateful for financial support of this research from the University of Utah Department of Chemistry, and the University of Utah Department of Civil and Environmental Engineering. The NMR spectroscopy results included in this report were recorded at the David M. Grant NMR Center, a University of Utah Core Facility. Funds for construction of the Center and the helium recovery system were obtained from the University of Utah and the National Institutes of Health awards 1C06RR017539-01A1 and 3R01GM063540-17W1, respectively. NMR spectroscopy instruments were purchased with support of the University of Utah and the National Institutes of Health award 1S10OD25241-01. Research was partially funded by the NRC Graduate Research Fellowship under award number 31310019M0042.References
- C. A. Kunos, D. A. Mankoff, M. K. Schultz, S. A. Graves and D. A. Pryma, Semin. Radiat. Oncol., 2021, 31, 3–11 CrossRef PubMed.
- O. D. Joe, Z. Pat, H. John and K. Adam, J. Nucl. Med., 2022, 63, 1467 CrossRef.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- I. F. Chaple and S. E. Lapi, J. Nucl. Med., 2018, 59, 1655–1659 CrossRef CAS PubMed.
- R. Hernandez, H. F. Valdovinos, Y. Yang, R. Chakravarty, H. Hong, T. E. Barnhart and W. Cai, Mol. Pharmaceutics, 2014, 11, 2954–2961 CrossRef CAS.
- E. Hindie, P. Zanotti-Fregonara, M. A. Quinto, C. Morgat and C. Champion, J. Nucl. Med., 2016, 57, 759–764 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS.
- M. R. Brandt, C. Vanasschen, J. Ermert, H. H. Coenen and B. Neumaier, Dalton Trans., 2019, 48, 3003–3008 RSC.
- S. Ponziani, G. Di Vittorio, G. Pitari, A. M. Cimini, M. Ardini, R. Gentile, S. Iacobelli, G. Sala, E. Capone, D. J. Flavell, R. Ippoliti and F. Giansanti, Int. J. Mol. Sci., 2020, 21, 5510 CrossRef CAS.
- Y. Hiroshima, T. M. Lwin, T. Murakami, A. A. Mawy, T. Kuniya, T. Chishima, I. Endo, B. M. Clary, R. M. Hoffman and M. Bouvet, J. Surg. Oncol., 2016, 114, 951–958 CrossRef CAS.
- O. Jacobson, Q. Li, H. Chen, G. Niu, D. O. Kiesewetter, L. Xu, K. Cook, G. Yang, W. Dall'Acqua, P. Tsui, L. Peng and X. Chen, J. Nucl. Med., 2017, 58, 1838 CrossRef CAS PubMed.
- T. Nagaya, Y. A. Nakamura, P. L. Choyke and H. Kobayashi, Front. Oncol., 2017, 7, 314 CrossRef.
- L. Privitera, I. Paraboschi, D. Dixit, O. J. Arthurs and S. Giuliani, Innov. Surg. Sci., 2021, 6, 161–172 Search PubMed.
- K. Wang, Y. Du, Z. Zhang, K. He, Z. Cheng, L. Yin, D. Dong, C. Li, W. Li, Z. Hu, C. Zhang, H. Hui, C. Chi and J. Tian, Nat. Rev. Bioeng., 2023, 1, 161–179 CrossRef CAS.
- Q. Zhang, S. O'Brien and J. Grimm, Nanotheranostics, 2022, 6, 184–194 CrossRef PubMed.
- Y. Zhang, G. Zhang, Z. Zeng and K. Pu, Chem. Soc. Rev., 2022, 51, 566–593 RSC.
- A. Medici, G. Fantin, P. Pedrini, M. Gleria and F. Minto, Macromolecules, 1992, 25, 2569–2574 CrossRef CAS.
- L. S. Campbell-Verduyn, A. K. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 11117–11120 CrossRef CAS PubMed.
- C. Müller, C. A. Umbricht, N. Gracheva, V. J. Tschan, G. Pellegrini, P. Bernhardt, J. R. Zeevaart, U. Köster, R. Schibli and N. P. van der Meulen, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1919–1930 CrossRef PubMed.
- A. J. Amoroso, I. A. Fallis and S. J. A. Pope, Coord. Chem. Rev., 2017, 340, 198–219 CrossRef CAS.
- E. Birnbaum, M. Fassbender, M. Ferrier, K. John and T. Mastren, in The Heaviest Metals: Science and Technology of the Actinides and Beyond, John Wiley & Sons, LTD, 2018, ch. 26, pp. 445–466 Search PubMed.
- S. Ahenkorah, I. Cassells, C. M. Deroose, T. Cardinaels, A. R. Burgoyne, G. Bormans, M. Ooms and F. Cleeren, Pharmaceutics, 2021, 13, 599 CrossRef CAS.
- M. Tropiano, C. J. Record, E. Morris, H. S. Rai, C. Allain and S. Faulkner, Organometallics, 2012, 31, 5673–5676 CrossRef CAS.
- G. Tian, L. R. Martin, Z. Zhang and L. Rao, Inorg. Chem., 2011, 50, 3087–3096 CrossRef CAS PubMed.
- J. Zhu, E. M. Gale, I. Atanasova, T. A. Rietz and P. Caravan, Chemistry, 2014, 20, 14507–14513 CrossRef CAS PubMed.
- L. C. Silvio Aime, C. Cavallotti, E. Gianolio, G. B. Giovenzana, A. M. Pietro Losi, G. Palmisano and M. Sisti, Inorg. Chem., 2004, 43, 7588–7590 CrossRef PubMed.
- J. Choi, G. Kim, S. B. Cho and H.-J. Im, J. Nanobiotechnol., 2020, 18, 122 CrossRef.
- J. Xie, L. Gong, S. Zhu, Y. Yong, Z. Gu and Y. Zhao, Adv. Mater., 2019, 31, e1802244 CrossRef PubMed.
- I. Da Silva, T. R. Johnson, J. C. Mixdorf, E. Aluicio-Sarduy, T. E. Barnhart, R. J. Nickles, J. W. Engle and P. A. Ellison, Molecules, 2021, 26, 7513 CrossRef CAS PubMed.
- S. Lehenberger, C. Barkhausen, S. Cohrs, E. Fischer, J. Grünberg, A. Hohn, U. Köster, R. Schibli, A. Türler and K. Zhernosekov, Nucl. Med. Biol., 2011, 38, 917–924 CrossRef CAS PubMed.
- A. G. Cosby, S. H. Ahn and E. Boros, Angew. Chem., Int. Ed., 2018, 57, 15496–15499 CrossRef CAS.
- C. Xie, Y. Wu, H. F. Chau, T. Zhang, C. T. Wong, Y. H. Yeung, P. L. Lam, J. C. G. Bünzli, L. J. Charbonnière, G. L. Law and K. L. Wong, Adv. Opt. Mater., 2023, 11, 2203057 CrossRef CAS.
- T. S. Grimes and K. L. Nash, J. Solution Chem., 2014, 43, 298–313 CrossRef CAS.
- M. T. Friend and N. A. Wall, Inorg. Chim. Acta, 2019, 484, 357–367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00221k |
| This journal is © The Royal Society of Chemistry 2025 |