
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePaper sensors for the measurement of nitric oxide release from endothelial cells
Syed Hassan
Ali
a and
Raphaël
Trouillon
 *abc
*abc
aDepartment of Electrical Engineering, Polytechnique Montréal, Montréal, QC, Canada. E-mail: raphael.trouillon@polymtl.ca
bTransMedTech Institute, Montréal, QC, Canada
cSNC research Group, Montréal, QC, Canada
First published on 24th January 2025
Abstract
Nitric oxide (NO) is a ubiquitous and important biological mediator. However, its detection and chemical analysis are challenging due to its short lifetime in biological conditions. Paper-based NO sensors combining the ease of fabrication and affordability of paper with the quantitative capabilities of electrochemical methods are presented for the detection and quantification of NO in cultured cells. Nafion-coated and eugenol-functionalized paper devices were built and characterized using a NO donor. The electrochemical interferences from nitrite, a common interferent for NO sensing, were successfully screened out. Finally, preliminary data were obtained from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 endothelial cells cultured directly, in an extracellular matrix, on the paper device. In response to vascular endothelial growth factor exposure, NO secretion was detected and quantified.
000 endothelial cells cultured directly, in an extracellular matrix, on the paper device. In response to vascular endothelial growth factor exposure, NO secretion was detected and quantified.
1. Introduction
Nitric oxide (NO) is a ubiquitous biological mediator, a compound released by endothelial, neuronal, immune, and other cells to allow them to communicate with one another.1–5 It is a key component of efficient immune response3,6 and a critical agent in the regulation of blood pressure and angiogenesis and the growth of new blood vessels.7,8 However, extremely high rates of NO synthesis are associated with adverse health conditions including cancer.2,9 Tumors are typically highly angiogenic structures containing high levels of growth factors.10,11 One of these families of molecules, vascular endothelial growth factors (VEGFs), activates endothelial cells into an angiogenic state and stimulates the secretion of NO.7 VEGF is thus a clinical target, and anti-VEGF therapies are being used clinically.12 Therefore, the quantification of NO release may be useful in the diagnosis of cancer as well as determining the efficacy of different cancer treatments.Unfortunately, the quantification of NO is challenging, especially in a biological context, owing to its short lifetime in physiological conditions as it is quickly oxidized to nitrites.13 This rapid scavenging has complicated the accurate measurement of NO levels and thus impeded its precise study. Amongst the most common methods for NO detection and quantitation, the Griess test is a colorimetric assay.14 This method oxidizes NO to nitrites and measures the nitrite levels. This two-step strategy limits the selectivity of the assay, as endogenous nitrites are included in the readout and are not compatible with fast in situ analyses. Similarly, diaminofluoresceins are fluorescent compounds for NO detection.15 They are suitable for fast microscopic assays but are also prone to limited selectivity.16
Electrochemical methods are attractive solutions for biosensors owing to their adaptability to a wide range of samples and measurement scenarios, sensitivity, low cost and rapid response time.17–19 As NO is electroactive, it can be readily oxidized on a wide range of substrates and several NO electrochemical sensors have been proposed. Malinski's work highlighted the usefulness of porphyrinic compounds,20,21 and eugenol-based electrodes have been proposed for the measurement of NO released by neurons.22 There have been significant improvements in the past decade in the detection of NO with significant improvements in the selectivity, sensitivity, sensocompatibility, and cytocompatibility.17 Limits of detection (LoDs) of a few nM have been reported with modified carbon microelectrodes.22 However, most of these techniques are specialist solutions that limit the dissemination of the technology.
Paper-based biochips have attracted significant interest over the past decade.23–27 Paper is here used not only as a substrate for handling liquid passively, taking advantage of the capillary action of porous paper, but also for adding sensors, electronic or mechanical components. The wicking action of paper is particularly useful as it allows for pumpless microfluidics. Paper is easy to handle, cut and functionalize and is highly amenable to a wide range of bioassays.23,28,29 The integration of electrochemical sensors has been demonstrated on several types of paper substrate.30–34 These electrodes are usually made from conductive inks or polymers (e.g. PEDOT-PSS solutions, carbon nanotubes (CNTs) or graphite suspensions, etc.) deposited or printed directly on the paper.35,36
However, there are several limitations to paper-based electroanalytical devices.37 The opacity of paper hinders the use of microscopy. Fluorescence microscopy or openings in the paper can help circumvent this issue.38 The size of the features deposited on the paper is limited by the characteristics of the paper surface, i.e. the size of the pores (in the 10 to 100 μm range, depending on the paper). This sets a limit to miniaturization of features on paper substrates, even though specialized papers for printed electronics are available. The random nature of paper, especially the filter paper used here, can also impact the reproducibility of the devices. Finally, as the conductive structures are often printed and/or deposited as a thin layer on the cellulose substrate, these thin-film electrodes can typically show a lower conductivity than traditional ones. It has been found that maintaining small electrode dimensions can help limit the drop in potential in the device.32 Electrode designs with low aspect ratios are better than ones with large aspect ratios, probably as they improve conductivity, but overall keeping all the dimensions as small as reasonably possible (typically 1 to 2 mm) led to the best electrode response, possibly by lowering the electrode capacitance.32 The smallest electrode dimension is set by the typical pore size (the device must be significantly larger than this distance to overcome the paper surface randomness) and the specifications of the fabrication systems. It also reduces the amount of reactant needed to functionalize the device. Taking these limitations into account, paper can nevertheless be used to build efficient sensors, benefiting from ease of use and fabrication as well as passive fluid handling via capillary action.
Importantly, cell culture on paper substrates has been well described.39 Here, cells are suspended in a gel of extracellular matrix (ECM) and the solution is then spotted on paper. As the gel solidifies, an ECM matrix containing cells is maintained in the pores of the paper (typically a filter or chromatography paper), which acts as a scaffold providing mechanical strength to the fragile gel. The resulting construct is a 3D cell structure that can be used for studying the cellular response to hypoxia or for testing drugs.40,41 It has been reported that these samples can be easily integrated into microfluidic chips and describe well the neurochemical response of the cells to stimulation,42 or the ability of muscle cells to capture glucose when exposed to insulin.43 The possibility of combining electrodes on paper with cell culture in the same paper device has recently been reported and used to test different neurosecretion altering drugs.38
In this study, paper-based sensors are fabricated and tested in varying concentrations of NO. The devices were also characterized with scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). The final devices were calibrated and tested against common interferences. Finally, preliminary results showing the possibility of seeding endothelial cells onto the paper devices and observing their secretion of nitric oxide in response to VEGF exposure are reported.
2. Methodology
2.1. Chemicals and solutions
Unless stated otherwise, all the reactants were purchased from Millipore-Sigma (Canada). Deionized water was used throughout the experiments (resistivity 18 MΩ cm). The CNT ink is a commercial aqueous suspension of single-walled CNTs (0.2 mg ml−1, ref. 791490). The solution contains sodium dodecyl sulfate (SDS) to prevent aggregation of the nanotubes.2.2. Electrochemical setup
All the electrochemical measurements were run with a CHI 1040C potentiostat (CH Instruments, TX, USA). A 3-electrode setup was used, with a Pt counter electrode and an Ag|AgCl (3 M NaCl) reference electrode. All the electrode potentials are reported vs. Ag|AgCl (3 M NaCl). In some experiments, 3-mm glassy carbon (GC) electrodes were used after polishing them with 0.03 μm alumina slurry.2.3. Paper electrode fabrication
Whatman filter paper was laser cut using a Trotec laser cutter to create paper electrodes (Fig. 1A). The paper electrodes were coated with the CNT ink.31 The electrodes were dip coated in the CNTs three times and air-dried after each coat by placing the paper electrodes on a glass slide. The electrodes were then placed in an oven for 5 min at 90 °C. 3–4 mm-wide strips of parafilm were melted onto the paper electrodes by placing them on the paper and then placing the paper electrodes into the oven at 100 °C until the parafilm has melted. This section of melted paraffin is used to limit the active site of the electrode44,45 and prevent the capillary wicking of the test solution along the length of the paper electrode as it could interfere with the connection to the potentiostat (see Fig. 1A). The active part of the electrode was cut to a length of 2 mm to complete the paper device (Fig. 1B). | ||
| Fig. 1 Fabrication of the NO sensors. (A) Schematic showing the preparation protocol: (1) cutting paper pieces, (2) coating in CNT ink, (3) blocking part of the paper with paraffin, (4) depositing Nafion and (5) electropolymerizing eugenol. (B) Photograph of the final device showing the different parts of the paper electrode (scale bar: 5 mm). | ||
2.4. NO sensor functionalization
The NO sensing functionalization was adapted from a published report.22 The paper electrodes were coated with Nafion by dipping them three times in 5% (v/v) Nafion in ethanol solution and air-drying after each coating. Afterwards, the electrodes were placed in an oven at 90 °C for 4 min. The electrodes were placed in 0.1 mM eugenol (see structure in Fig. 2A) in 0.1 M NaOH. Cyclic voltammograms (CVs) were run for 25 cycles at a scan rate (SR) = 100 mV s−1 between 0 V and 1.25 V (Fig. 2B).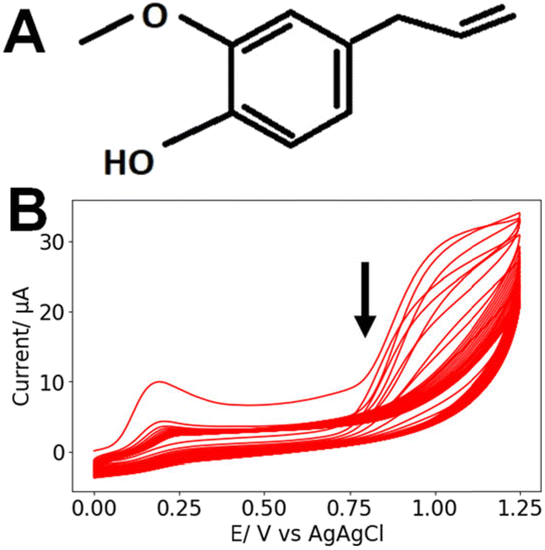 | ||
| Fig. 2 (A) Chemical structure of eugenol. (B) Typical CVs recorded during the electrodeposition of 0.1 mM eugenol in 0.1 M NaOH (25 cycles, SR = 100 mV s−1). The arrow indicates the order of the scans. | ||
2.5. SEM and XPS analyses
SEM images of the different samples were acquired with a Quattro ESEM microscope. The sensors were also analyzed using X-ray photoelectron spectroscopy (XPS). A VG ESCALAB 250Xi instrument was used to acquire XPS spectra. The spectra were analyzed using Avantage v6.5 with all high-resolution peaks fitted using symmetrical Lorentzian/Gaussian peak functions.For both SEM and XPS, the paper sensors were examined at each stage of sensor fabrication: blank Whatman filter paper, paper coated with CNT ink, paper coated with CNT and Nafion, and lastly, finished electrodes with CNT ink, Nafion, and electropolymerized eugenol.
2.6. NO electrochemistry
All the NO electrochemistry was performed in aerated PBS at room temperature. For the initial characterization of the system three repeats of differential pulse voltammetry (DPV) were run between 0 V and 1 V in PBS. Diethylamine NONOate (DEA-NONOate), a NO donor, was used as a NO source. Once exposed to a neutral pH, DEA-NONOate spontaneously releases NO molecules in a time-dependent manner.46 A 10 mM stock solution of DEA-NONOate was prepared in 0.01 M NaOH.46 Different aliquots of this stock were pipetted into 10 mL of PBS to reach 100 μM, 200 μM or 300 μM final concentrations of DEA-NONOate. Three DPV experiments were then run again under the same conditions to calibrate the sensor. The rate of the production and degradation of NO from DEA-NONOate was also analyzed with amperometry at 0.9 V with a runtime of 1800 s. The first 300 s were used to allow the system to stabilize in PBS. Afterwards the DEA-NONOate was injected into the PBS ([DEA-NONOate] = 100 μM, 200 μM or, 300 μM) and the current was measured for 1500 s. The baseline due to sensor stabilization was subtracted to isolate the contribution of the faradaic NO current.2.7. Simulation of NO release
In addition to the experimental measurements, the kinetics of NO release from DEA-NONOate were computed using MATLAB. The concentration profile of NO release was calculated using the differential equation provided by Griveau et al. for the kinetics of NO release from DEA-NONOate:46where
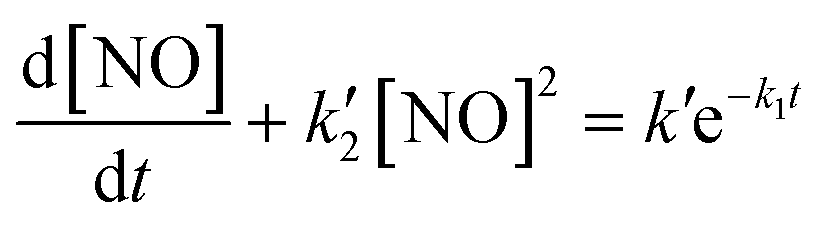
 , k2 = 2 × 106 M−2 s−1, [O2] = 1.3 × 10−3 atm−1, k′ = vNOk1[NONOate]0, vNO = 1.5, and k1 = 28.5 × 10−4 s−1.
, k2 = 2 × 106 M−2 s−1, [O2] = 1.3 × 10−3 atm−1, k′ = vNOk1[NONOate]0, vNO = 1.5, and k1 = 28.5 × 10−4 s−1.
2.8. Cell culture and measurements
The immortalized endothelial cell line EA.hy926 (from ATCC) was used as a vascular model. The cells were grown sub-confluently in DMEM supplemented with 10% fetal calf serum in a humid incubator, 5% CO2, 37 °C. The medium was changed every other day, and the cells were passaged weekly, up to 10 passages.To measure NO release in endothelial cells, 100000 cells in 2 μl ECM gel from Engelbreth–Holm–Swarm murine sarcoma were deposited on the sensing part of a paper NO sensor. The ECM was allowed to set for 5 min in an incubator, and the sensing extremity of the chip was then placed in warm medium for ∼1 h in the humid incubator.
To run the experiment, the device was placed in warm (37 °C) PBS, and an amperometric measurement was initiated at 0.9 V. After baseline stabilization (∼100 s), VEGF was added to obtain a final concentration of 20 ng ml−1.
2.9. Fluorescence microscopy
After allowing the ECM gel to set, the cells on the paper electrodes were fixed in 3.7% methanol-free formaldehyde solution in PBS for 15 min at room temperature. Next, the cell membranes were permeabilized in 0.1% Triton™ X-100 in PBS for 15 min, and the PBS containing the cells was next incubated for 40 min with Alexa Fluor™ 488 Phalloidin (Thermo Fisher). Between each step, the samples were washed twice in prewarmed (37 °C) PBS.After staining, the cells were imaged with a Nikon Eclipse TiE inverted C2 epifluorescence microscope fitted with a 20× objective, and z-stacks were acquired. ImageJ was used to process the micrographs. To better observe the paper geometry, differential interference contrast (DIC) was used to image the general topology of the sample. The DIC image was inverted and is here reported in blue to highlight the position of the paper fiber. The contrast of the images was modified to better show the position of the cells in the paper fibers.
2.10. Data analysis
The sensitivity of the electrodes is defined as the coefficient of the NO concentration in the linear fit. The LoD of the sensor was calculated as the concentration associated to thrice the standard deviation of the blank current. In the case of the NO amperometric measurement, the data were smoothed with a Savitzky–Golay filter, and the baseline was subtracted after a linear fit. Unless specified otherwise, the data are presented as mean ± SD for N measurements. Where applicable, datasets were compared using a two-tailed Student's t-test.3. Results and discussion
3.1. SEM imaging
Typical SEM images of the different samples are shown in Fig. 3: the paper/CNT, paper/CNT/Nafion and paper/CNT/Nafion/eugenol assemblies. In all cases, larger paper (cellulose) fibers can be observed, with a coating of CNT covering the cellulose and, in some cases, filling the pores. The addition of Nafion or eugenol does not lead to significant changes in surface texture or structure. Importantly, in all the samples observed, the paper devices retained a high level of porosity (as indicated by the arrows in Fig. 3). This is critical for cell culture as the cells are maintained in the bulk of the paper between the cellulose fibers.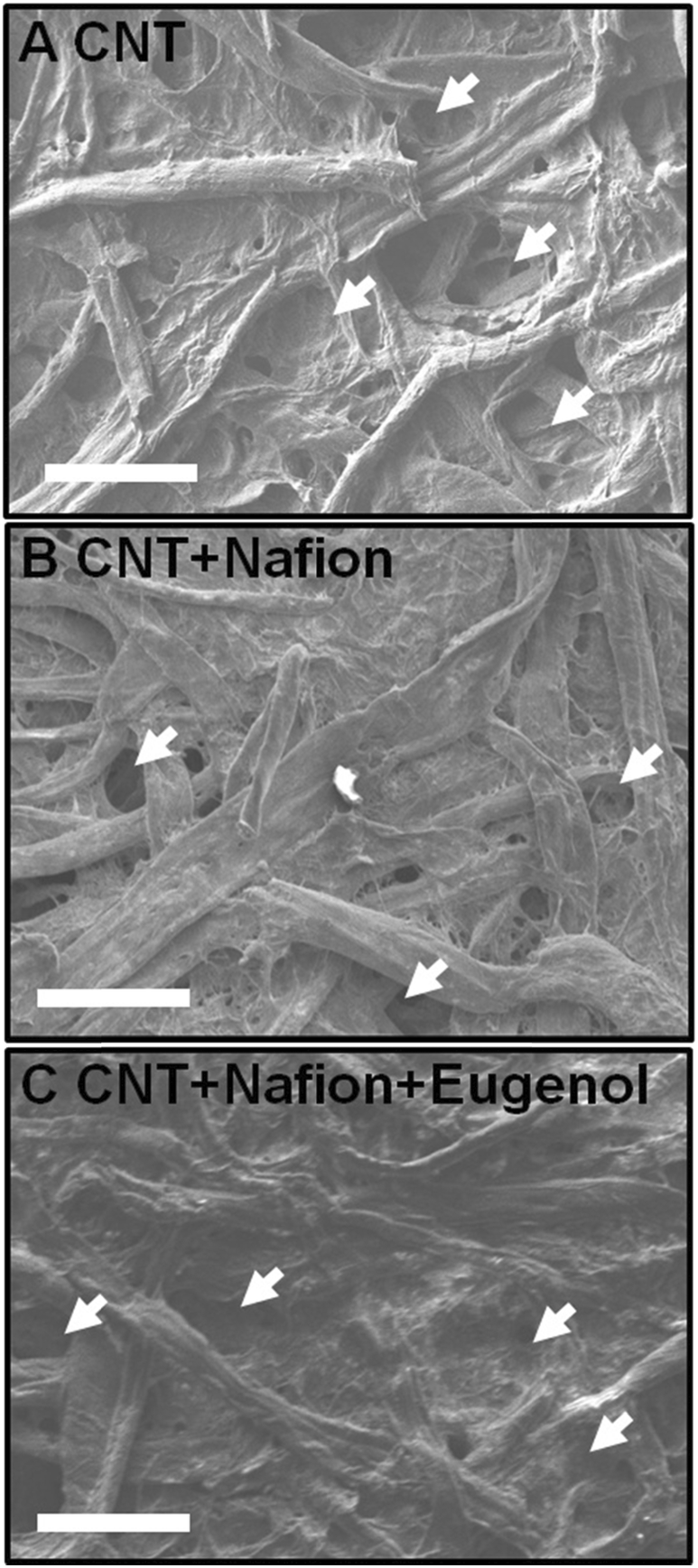 | ||
| Fig. 3 Typical SEM images for different paper preparations: (A) paper/CNT, (B) paper/CNT/Nafion and (C) paper/CNT/Nafion/eugenol. Scale bars: 100 μm. The arrows highlight the presence of large pores on all the micrographs. | ||
3.2. XPS analysis
The XPS data for different electrode preparations are shown in Fig. 4. As the CNT ink is deposited on the pristine paper, Na 1s (1071.2 eV) and S 2p (169.0 eV) peaks are observed. This is attributed to the presence of SDS in the CNT ink. The electrode is then encapsulated in Nafion, a fluorosulfonic acid, as revealed by the presence of an F 1s peak (688.8 eV). Finally, after eugenol deposition, the relative height of the F 1s peak vs. the O 1s peak is greatly reduced. The Si 2p (102.4 eV) and Si 2s (174.5 eV) peaks are likely contamination due to silicon leaking from the glassware containing the alkaline eugenol solution (in 0.1 M NaOH). This analysis confirms the successive deposition of the different layers of the NO paper sensor. | ||
| Fig. 4 XPS traces obtained for (i) pristine paper, (ii) paper with CNT, (iii) paper/CNT/Nafion and (iv) paper/CNT/Nafion/eugenol. | ||
3.3. Kinetics of NO release from DEA-NONOate
DEA-NONOate was used as an easily accessible source of NO. Each molecule theoretically releases 2 molecules of NO once at physiological pH, but previous studies highlighted that this release is incomplete and not instantaneous, as highlighted in Fig. 5A–C. To better understand these dynamics, the maximum [NO] released from different starting concentrations of DEA-NONOate was computed (Fig. 5D).46 As previously reported, the relationship between [DEA-NONOate] and the maximum [NO] is non-linear. Table 1 presents the maximum concentration [NO]max and the instant when that maximum is reached (tmax) computed from these simulations for the 3 concentrations used in the experiments.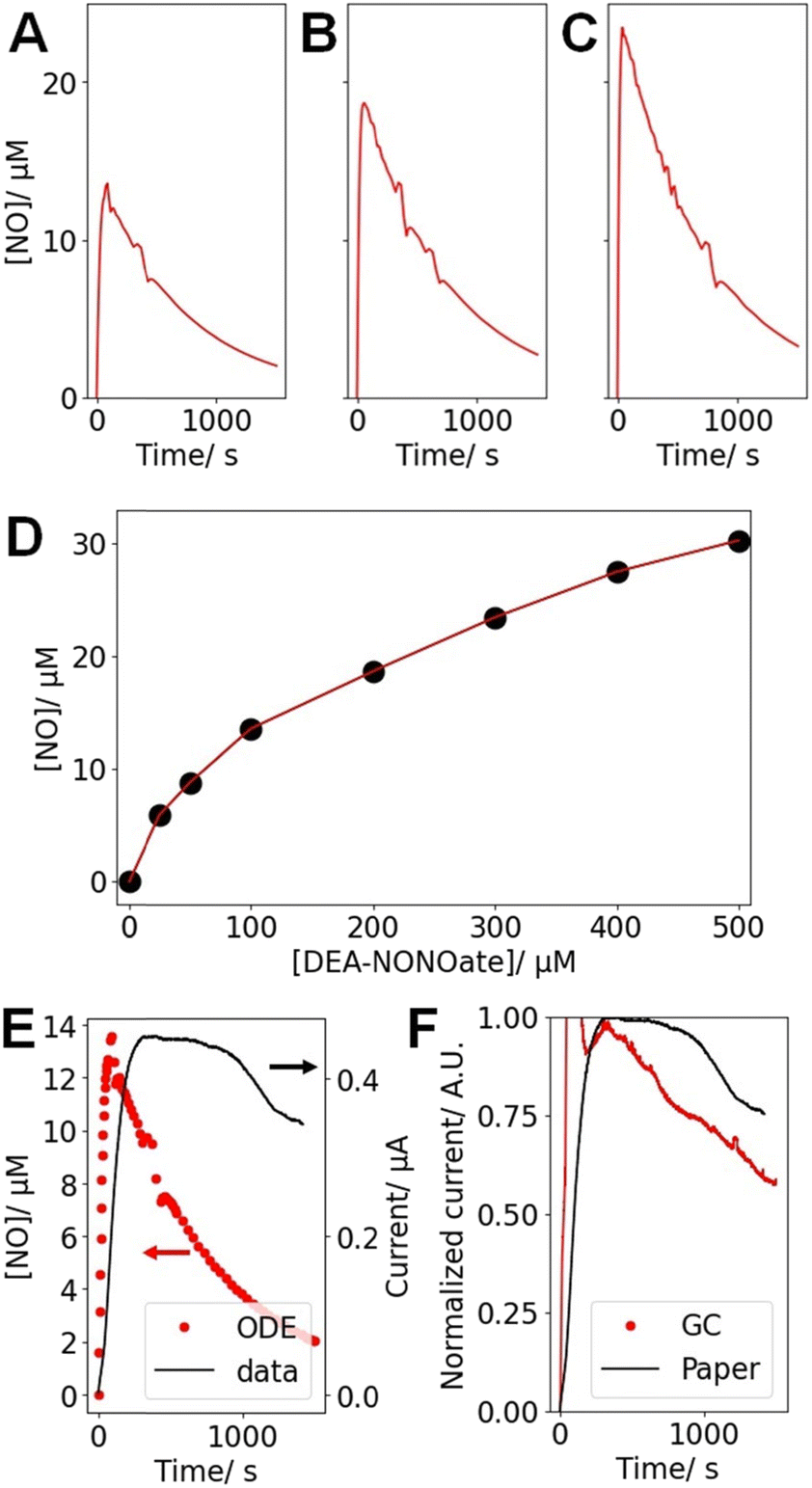 | ||
| Fig. 5 Results of the ODE describing the [NO] released by DEA-NONOate for starting concentrations of (A) 100 μM, (B) 200 μM and (C) 300 μM DEA-NONOate. (D) Peak NO concentration [NO]max for different [DEA-NONOate], as computed from the ODE described in ref. 46. (E) Comparison of the ODE for [DEA-NONOate] = 100 μM and the typical corresponding amperometric trace (at 0.9 V) recorded with a paper sensor after the addition of 100 μM DEA-NONOate to PBS over ∼1500 s. (F) Comparison of the amperometric data (at 0.9 V) recorded with a paper sensor after the addition of 100 μM DEA-NONOate to PBS over ∼1500 s and obtained with a GC and a paper electrode, both prepared as NO sensors following the reported protocol. For the GC data, an injection artifact is observed in the first ∼100 s and was not considered in the analysis. | ||
| [DEA-NONOate]/μM | 100 | 200 | 300 |
| [NO]max/μM | 13.6 | 18.7 | 24.5 |
| t max/s | 89.8 | 57.5 | 41.6 |
To confirm these results experimentally, the amperometric response of the paper sensors in the presence of varying [DEA-NONOate] was obtained. The amperometric current (Fig. 5E) recorded at 0.9 V increases rapidly, thus confirming the sensitivity of the paper devices towards NO. Fig. 5E presents the amperometric data recorded for 100 μM DEA-NONOate and the corresponding simulation. In both cases, a sharp rise in current or [NO] is observed followed by a slow decrease. However, these kinetics are much faster in computation in comparison to the experimental data. This could be evidence of complex hindered mass transport of the DEA-NONOate in the pores of the paper and the Nafion layer, leading to diffusive dispersion of the peak.
To investigate this possibility, the ODE solution for 100 μM DEA-NONOate was convoluted with a Gaussian function to simulate a hindered diffusion pathway. The width of the Gaussian was adjusted so that the maxima of the diffusively hindered ODE and the experimental amperometric trace match. The fit was optimal for a 340 s Gaussian width, which likely arises from tortuous diffusion in the paper matrix. This diffusion time tdiff can be associated to a diffusion distance  , where D is the diffusion coefficient for unidimensional diffusion. DEA-NONOate, whose structure is C4H10N3NaO2, has a molecular weight of 209.6 Da. Assuming a diffusion coefficient of 10−10 m2 s−1 for DEA-NONOate, tdiff = 340 s is associated to xdiff = 261 μm. As the Whatman 114 paper is 190 μm thick, this analysis hints that the peak broadening observed in Fig. 5E can be associated with slower diffusion of the NO donor in the bulk of the porous paper coated with the Nafion film. Slower diffusion due to higher tortuosity in partially blocked medium has been well described.47,48
, where D is the diffusion coefficient for unidimensional diffusion. DEA-NONOate, whose structure is C4H10N3NaO2, has a molecular weight of 209.6 Da. Assuming a diffusion coefficient of 10−10 m2 s−1 for DEA-NONOate, tdiff = 340 s is associated to xdiff = 261 μm. As the Whatman 114 paper is 190 μm thick, this analysis hints that the peak broadening observed in Fig. 5E can be associated with slower diffusion of the NO donor in the bulk of the porous paper coated with the Nafion film. Slower diffusion due to higher tortuosity in partially blocked medium has been well described.47,48
To investigate the impact of the porous structure of paper on the detection kinetics, a GC electrode was used to prepare a NO sensor using the same protocol. This results in a flat sensing surface with properties similar to the paper device. In this case, the current increase following DEA-NONOate addition is faster, as it reaches its maximum earlier and decays more rapidly, than for the paper device (Fig. 5F). However, the observed current for GC is still slower than the profile predicted by the ODE. This could indicate that (i) the structure of the paper and (ii) the Nafion layer both decrease NO detection kinetics, probably because of diffusional hindrances.
Despite these slower kinetics, DEA-NONOate provides an easy source of NO, as demonstrated by amperometry. Importantly, it was found that [NO], thanks to the peak broadening, was relatively stable over the course of the voltammetric tests described below (>600 s) and is therefore suitable for the calibration of the paper NO sensors.
3.4. Voltammetric characterization
The sensors were also characterized with DPV to better understand their detection mechanisms. As a reference, the NO-sensing layers (Nafion, then eugenol) were deposited on GC electrodes (Fig. 6A). For this preparation, and as reported by others, a clear peak appears at ∼0.7 V for NO, and its magnitude increases with [NO]. This confirms the NO sensing capabilities of eugenol on carbon electrodes. | ||
| Fig. 6 Typical DPV recorded in PBS alone or supplemented with 100, 200, and 300 μM DEA-NONOate for (A) glassy carbon and (B) a typical paper NO sensor. (C) DPV calibration curve for the data recorded from the typical NO sensor presented in (B). | ||
If GC is replaced by a paper device coated with CNT, a very slight peak can still be observed in the same region but is very hard to resolve and cannot be used for analysis (Fig. 6B). This is expected to be due to the higher electrode resistance of the paper devices in comparison to the traditional GC, leading to peak broadening, as observed with other paper electrodes31,32,38 or very thin layer electrodes. However, a consequence of this peak broadening is that the overall DPV trace appears to be shifted towards higher currents as [DEA-NONOate], and therefore [NO], increases. The current recorded at the higher potential limit of the DPV, here at 1 V, was found to be a reliable marker of [NO]. Calibration curves were therefore constructed for each paper device from the DPV current recorded at 1 V. A similar potential (0.9 V) was used for the amperometric measurement to overcome the electrode resistivity.32 An example of such a calibration curve for a single typical NO paper sensor is presented in Fig. 6C.
The same calibration procedure was repeated for n = 11 electrodes and the independent calibration curves were obtained. Overall, each of these 11 calibration curves displayed a linear behavior, making it possible to interpolate the electrode response between 0 and 13.8 μM from 4 calibration points. Using a low number of calibration points (1 to 5) to calibrate a sensor is a common strategy once the calibration function is known, especially if it is linear.49–52 This approach was chosen as paper devices are fragile once wet and repeated manipulations can compromise their performances. It is thus necessary to limit the number of calibration measurements for a single device, for instance, in our case with 4 calibration points. These datasets were pooled to extract the general analytical behavior of the paper NO sensors. This allows for overcoming the sensor-to-sensor variability due to the random nature of paper. Overall, a general calibration curve is built for this sensor fabrication protocol, allowing for then using new sensors without calibration before the cell measurements. The sensitivity and the LoD associated to each paper electrode were computed. The median, 1st and 3rd quartiles for this non-parametric set of results are presented in Table 2.
| Median | Quartile 1 | Quartile 3 | |
|---|---|---|---|
| Sensitivity/μA M−1 mm−2 | 488.3 | 366.8 | 776.9 |
| LoD/μM | 1.7 | 1.4 | 2.3 |
3.5. Interferences
The signals recorded from DPV in 13.8 μM solutions of potential interferents were collected and compared to those obtained for 100 μM DEA-NONOate, corresponding to 13.8 μM NO, as presented in Table 3.| Compound | % response |
|---|---|
| Nitrite | 2.7 |
| Nitrate | 2.4 |
| Hydrogen peroxide | 14.0 |
| Ascorbic acid | 6.7 |
| Dopamine | 60.0 |
| Serotonin | 13.3 |
A critical requirement for NO sensing is high selectivity, especially in the presence of other nitrogen-based compounds. Nitrite (NO2−) is an electroactive product of NO oxidation and should be considered as the primary potential interferent. Here, a 2.7% level of interference is recorded, hinting at good screening from the Nafion layer. Nafion is a negatively charged polymer and efficiently blocks anions such as NO2−. Similarly, nitrate or ascorbic acid, an important biological antioxidant, did not lead to significant interferences. Hydrogen peroxide was found to lead to a 14% response, which can compromise measurements. In the context of NO chemistry, this molecule is relevant to immune response, where it is produced from superoxide ions co-released with NO to create peroxynitrite, a potent cytotoxic agent.53 However, high levels of hydrogen peroxide are not expected in physiological conditions of angiogenic response, and the impact of this interferent on our assays is expected to be limited.
However, neurotransmitters were found to produce higher interferences. The molecules are typically found in the brain where they mediate neuronal communication and can be readily oxidized on carbon electrodes. Dopamine and serotonin, two important neurotransmitters, were associated with 60.0% and 13.3% levels of interference, respectively. However, neurotransmitters are more likely to be found in neuronal samples and in specific parts of the brain associated with their release. This tissular and spatial selectivity ensures that they are unlikely to interfere with NO measurements in vascular models. Overall, the interference results agree with previous reports.22
3.6. VEGF-stimulated NO release
For the cell measurement assay, 100000 EA.hy926 cells were cultured directly in the pores of the paper in ECM. These immortalized hybrid endothelial cells were reported to secrete NO in response to VEGF.54 The volume of the cells + ECM gel is 2 μl. SEM images were obtained for the paper electrodes combined with ECM and cells (Fig. 7A and B). The micrographs reveal that the fibrous structure of paper can still be observed, but the well-defined pores seen in Fig. 3 are now filled with gel. The ECM gel here appears as a smooth coating featuring several cracks and fractures (indicated by arrows in Fig. 7A and B). These are likely due to the gel drying and shrinking in the high vacuum environment of the SEM. The cells themselves are not visible in these images, as they are embedded in the gel. Overall, the SEM investigation shows a high integration of the gel in the paper, as the ECM adheres well to the cellulose fibers and fills the pores of the device. The porous nature of the paper is thus critical to produce a 3D cellular construct, maintained in the cellulose fiber lattice of the paper. This confirms that the cell-on-paper approach allows for full integration of the biological component in the paper matrix. The absence of gaps or spaces between the gel and the paper hints at fast diffusion of molecules secreted from the cells to the sensor.
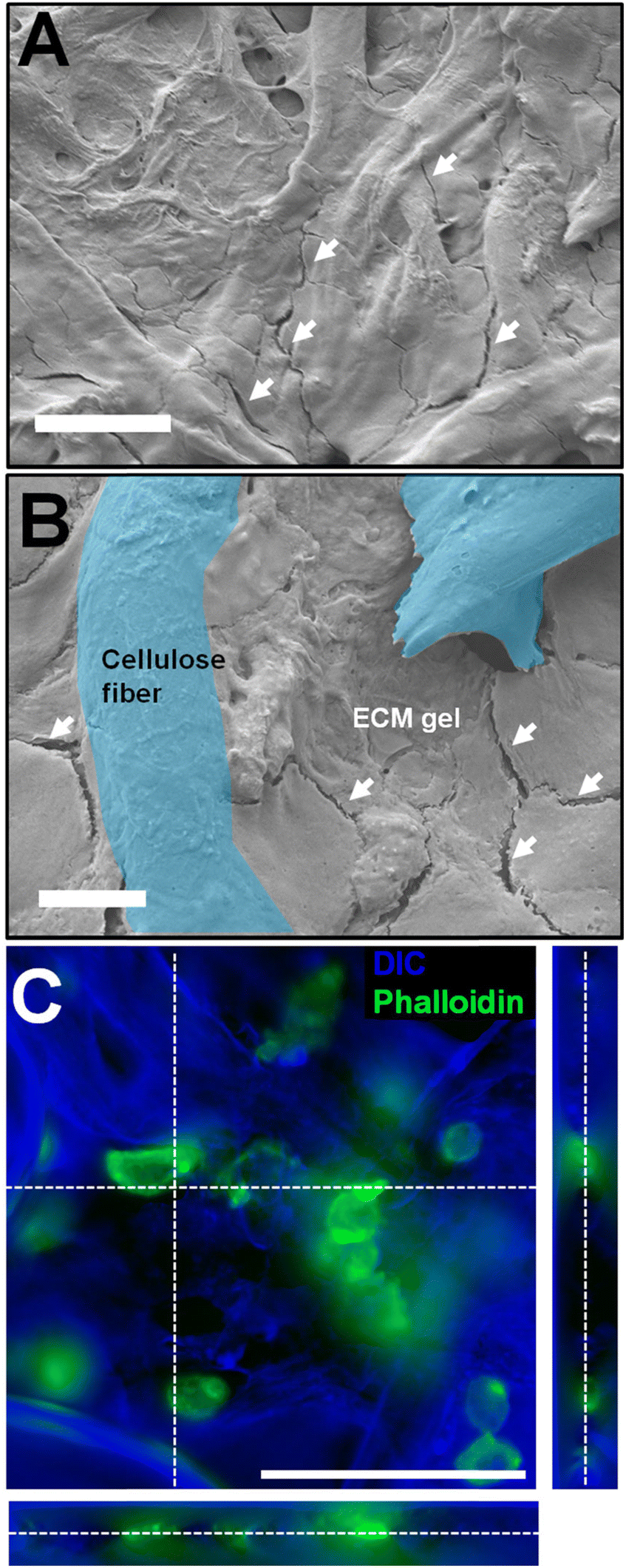 | ||
| Fig. 7 Microscopy of the paper devices prepared with EA.hy926 in ECM gel. (A and B) SEM images of the cell and gel-loaded paper chip (scale bars: (A) 50 μm and (B) 10 μm). On both panels, the white arrows highlight the presence of fissures that are an indication of the presence of the ECM gel coating the paper fibers. In (B), two cellulose fibers (paper) are highlighted in blue (false colors), underlining how the gel fills the pores and coats the paper fibers. (C) Fluorescence micrograph of EA.hy926 maintained on paper. The actin cytoskeleton appears in green (phalloidin staining) and the topology, mostly the paper fibers, appears in blue (inverted DIC). Scale bar: 100 μm; the stack is 50 μm thick. | ||
To directly observe the cells maintained in the gel, epifluorescence imaging was performed (Fig. 7C). The phalloidin stain marks the actin cytoskeleton, thus revealing the general shape of the cell. This image confirms that the endothelial cells are in the bulk of the paper (not only on the surface), between the fibers, in small clusters or as individual cells. This further stresses the importance of the porous structure allowing for cells to be cultured in a 3D matrix surrounded by electrochemical sensors deposited on the cellulose fibers.
Fig. 8A and B present averaged amperometric measurements (at 0.9 V, n = 3 for each case) for paper devices without and with 100000 EA.hy926 cells in ECM gel, following stimulation with 20 ng ml−1 VEGF at t = 0 s. In the absence of cells, the signal does not change and stays below the noise level, defined as thrice the SD of the noise over the baseline. However, in the presence of 100000 cells, an increase above this threshold (Fig. 8C, p < 0.001) in the current is recorded above the noise level of the device 13.8 ± 5.0 s after the addition of VEGF. The reported delayed release of NO after VEGF stimulation agrees with previously published reports.8,20,55
 | ||
| Fig. 8 NO analysis in cultured cells on the paper chip. Average amperometric traces for (A) no and (B) 100000 EA.hy926 cells maintained on the chips stimulated at t = 0 s with 20 ng ml−1 VEGF in PBS (for each case, n = 3 chips, mean ± SD). (C) Averages of the amperometric data (shown in A and B) recorded from 30 to 100 s for 0 and 100000 cells maintained in the paper device, flowing VEGF stimulation (mean ± SEM, n = 3 for each case, Student's t-test ***: p < 0.001). | ||
The current increase in the presence of EA.hy926 was thus attributed to the activity of cells only, and more specifically to VEGF-stimulated NO release. The increase in amperometric current of 0.55 μA indicates that [NO] = 3.75 μM after exposure to VEGF, using the DEA-NONOate amperometric calibration. This high concentration is expected from the densely seeded EA.hy926 cells. Assuming that NO is stable for 10 s in oxygenated buffers, and considering that there are 100000 cells in a 2 μl sample, this corresponds to a NO production rate for a single cell of 4.5 × 106 molecules per s.
3.7. Bioanalytical significance
For single endothelial or endocardial cells, [NO] following chemical or mechanical stimulation has been reported to be in the 100 nM to 1 μM range depending on the distance between the cell and the electrode and the type of cell.20,21,56 The instantaneous concentration of NO at the surface of single endothelial cells was measured as ∼1 μM for a sensed volume of 10−10 l,56 which corresponds to a rate of release of NO of 6 × 106 molecules per s per cell, in agreement with our data. The discrepancies can be largely attributed to the difficulties in measuring the sensed volume, but these preliminary data nevertheless support the use of paper NO sensors for the study of endothelial NO release.Endothelial cells are typically used in vitro to grow artificial vasculature57 owing to their capability to form microvessels in response to angiogenic factors like VEGF. This is not the case in the present assay, as the cells are tested a few hours after seeding. The fluorescence imaging clearly shows that the EA.hy926 cells are maintained as individual cells or in small clusters, not in large tubules. The assay is nevertheless significant as it provides quantitative information on the early steps of angiogenesis. Indeed, a preliminary angiogenic event is the release of NO, which promotes endothelial cell motility and proliferation, and measuring this response of significance to highlight the angiogenic role of compounds and growth factors, even in individual cells or monolayers.58–60 The VEGF concentration used here is also in agreement with the ones used for in vitro or in vivo assays.7,58,61–63
Finally, the cells are kept in a 3D matrix thanks to the capillary properties of paper, which increases the biomimicry of the assay. The use of porous paper as a device substrate allows for rapidly and easily building a 3D cell construct and sensing NO release in a 2 μl sample, thus providing easy access to quantitative measurements in an artificial tissue.38
4. Conclusion
NO paper-based sensors were fabricated and tested. Their selectivity against nitrite was tested, confirming their suitability for NO sensing. In comparison to carbon electrodes, the use of paper alters the analytical properties of the sensor but also provides an easy-to-use platform for cell biology. Furthermore, the sensor characteristics were found to be suitable for multicellular applications. Importantly, their applicability to cell measurements was investigated, and time dynamics of NO release following VEGF exposure were successfully resolved and compared to the literature. This is an early validation of the paper sensor approach on controlled, well-characterized samples, paving the way for application to more complex biological and pharmacological questions. This proof-of-concept study supports the validity of using paper devices for investigating NO biology in 3D cell models.Data availability
The data supporting this article have been included in the tables and figures.Author contributions
Conceptualization: R. T. Methodology: S. H. A. and R. T. Investigation: S. H. A. Formal analysis: S. H. A. Data curation: S. H. A. Validation: R. T. Writing – original draft: S. H. A. and R. T. Writing – review and editing: S. H. A. and R. T. Supervision: R. T. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The FRQNT, NSERC, NFRF, Polytechnique Montréal and the TransMedTech Institute are gratefully acknowledged for funding.References
- K.-D. Kröncke, K. Fehsel and V. Kolb-Bachofen, Nitric Oxide, 1997, 1, 107–120 CrossRef PubMed.
- S. Korde Choudhari, M. Chaudhary, S. Bagde, A. R. Gadbail and V. Joshi, World J. Surg. Oncol., 2013, 11, 118 CrossRef PubMed.
- J. R. Klinger and P. J. Kadowitz, Am. J. Cardiol., 2017, 120, S71–S79 CrossRef PubMed.
- S. Moncada and E. A. Higgs, Br. J. Pharmacol., 2006, 147, S193–S201 CrossRef CAS PubMed.
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524–526 Search PubMed.
- R. Trouillon, E. D. Williamson, R. J. Saint and D. O'Hare, Biosens. Bioelectron., 2012, 38, 138–144 CrossRef PubMed.
- A. Papapetropoulos, G. García-Cardeña, J. A. Madri and W. C. Sessa, J. Clin. Invest., 1997, 100, 3131–3139 Search PubMed.
- R. Trouillon, D.-K. Kang, H. Park, S.-I. Chang and D. O'Hare, Biochemistry, 2010, 49, 3282–3288 Search PubMed.
- Y. Nakamura, H. Yasuoka, M. Tsujimoto, K. Yoshidome, M. Nakahara, K. Nakao, M. Nakamura and K. Kakudo, Clin. Cancer Res., 2006, 12, 1201–1207 CrossRef PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef PubMed.
- H. T. Nguyen, L. N. Dupont, E. A. Cuttaz, A. M. Jean, R. Trouillon and M. A. M. Gijs, Microelectron. Eng., 2018, 189, 33–38 CrossRef.
- K. L. Meadows and H. I. Hurwitz, Cold Spring Harbor Perspect. Med., 2012, 2(10), a006577 Search PubMed.
- D. D. Thomas, X. Liu, S. P. Kantrow and J. R. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 355–360 Search PubMed.
- J. Sun, X. Zhang, M. Broderick and H. Fein, Sensors, 2003, 3, 276–284 Search PubMed.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef PubMed.
- X. Zhang, W.-S. Kim, N. Hatcher, K. Potgieter, L. L. Moroz, R. Gillette and J. V. Sweedler, J. Biol. Chem., 2002, 277, 48472–48478 CrossRef PubMed.
- M. D. Brown and M. H. Schoenfisch, Chem. Rev., 2019, 119, 11551–11575 CrossRef PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- L. Simonsson, M. E. Kurczy, R. Trouillon, F. Hook and A.-S. Cans, Sci. Rep., 2012, 2, 824 CrossRef PubMed.
- T. Malinski and Z. Taha, Nature, 1992, 358, 676–678 CrossRef PubMed.
- V. Brovkovych, E. Stolarczyk, J. Oman, P. Tomboulian and T. Malinski, J. Pharm. Biomed. Anal., 1999, 19, 135–143 Search PubMed.
- B. A. Patel, M. Arundell, K. H. Parker, M. S. Yeoman and D. O'Hare, Anal. Chem., 2006, 78, 7643–7648 CrossRef PubMed.
- T. Akyazi, L. Basabe-Desmonts and F. Benito-Lopez, Anal. Chim. Acta, 2018, 1001, 1–17 CrossRef PubMed.
- D. A. Bruzewicz, M. Reches and G. M. Whitesides, Anal. Chem., 2008, 80, 3387–3392 CrossRef PubMed.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- B. Deka, R. Kalita, D. Bhatia and A. Mishra, Sens. Int., 2020, 1, 100004 CrossRef PubMed.
- W. Chen, X. Fang, H. Li, H. Cao and J. Kong, Sci. Rep., 2016, 6, 1–7 CrossRef CAS.
- Z. Yao, P. Coatsworth, X. Shi, J. Zhi, L. Hu, R. Yan, F. Güder and H.-D. Yu, Sens. Diagn., 2022, 1, 312–342 RSC.
- J. Pelletier and R. Trouillon, Electrochim. Acta, 2024, 475, 143528 CrossRef CAS.
- R. Trouillon and M. A. M. Gijs, ChemPhysChem, 2018, 19, 1164–1172 CrossRef CAS PubMed.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chem., 2009, 81, 5821–5826 CrossRef CAS.
- J. Mettakoonpitak, K. Boehle, S. Nantaphol, P. Teengam, J. A. Adkins, M. Srisa-Art and C. S. Henry, Electroanalysis, 2016, 28, 1420–1436 CrossRef CAS.
- L. Bezinge, N. Tappauf, D. A. Richards, C.-J. Shih and A. J. deMello, ACS Sens., 2023, 8, 3964–3972 CrossRef CAS PubMed.
- E. Noviana and C. S. Henry, Curr. Opin. Electrochem., 2020, 23, 1–6 CrossRef CAS.
- A. T. Singh, D. Lantigua, A. Meka, S. Taing, M. Pandher and G. Camci-Unal, Sensors, 2018, 18, 2838 CrossRef.
- R. Trouillon and M. A. M. Gijs, Biosens. Bioelectron.:X, 2023, 14, 100327 CAS.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Y. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef.
- F. F. Tao, X. Xiao, K. F. Lei and I.-C. Lee, BioChip J., 2015, 9, 97–104 CrossRef.
- K. Ng, B. Gao, K. W. Yong, Y. Li, M. Shi, X. Zhao, Z. Li, X. Zhang, B. Pingguan-Murphy, H. Yang and F. Xu, Mater. Today, 2017, 20, 32–44 CrossRef.
- R. Trouillon and M. A. M. Gijs, RSC Adv., 2016, 6, 31069–31073 Search PubMed.
- R. Trouillon, M. C. Letizia, K. J. Menzies, L. Mouchiroud, J. Auwerx, K. Schoonjans and M. A. M. Gijs, Integr. Biol., 2017, 9, 810–819 CrossRef PubMed.
- S. Kasetsirikul, K. Clack, M. J. A. Shiddiky and N.-T. Nguyen, Micromachines, 2022, 13, 48 CrossRef.
- Y. S. Kim, Y. Yang and C. S. Henry, Sens. Actuators, B, 2018, 255, 3654–3661 Search PubMed.
- S. Griveau, C. Dumézy, P. Goldner and F. Bedioui, Electrochem. Commun., 2007, 9, 2551–2556 CrossRef.
- J. S. Mackie, P. Meares and E. K. Rideal, Proc. R. Soc. London, Ser. A, 1955, 232, 498–509 Search PubMed.
- R. Trouillon and A. G. Ewing, Anal. Chem., 2013, 85, 4822–4828 CrossRef PubMed.
- S. A. N. Gowers, V. F. Curto, C. A. Seneci, C. Wang, S. Anastasova, P. Vadgama, G.-Z. Yang and M. G. Boutelle, Anal. Chem., 2015, 87, 7763–7770 Search PubMed.
- S. A. N. Gowers, M. L. Rogers, M. A. Booth, C. L. Leong, I. C. Samper, T. Phairatana, S. L. Jewell, C. Pahl, A. J. Strong and M. G. Boutelle, Lab Chip, 2019, 19, 2537–2548 Search PubMed.
- Z. Mahmoudi, M. D. Johansen, J. S. Christiansen and O. Hejlesen, J. Diabetes Sci. Technol., 2014, 8, 709–719 CrossRef.
- A. U. Alam, D. Clyne, H. Jin, N.-X. Hu and M. J. Deen, ACS Sens., 2020, 5, 412–422 Search PubMed.
- C. Amatore, S. Arbault, D. Bruce, P. de Oliveira, M. Erard and M. Vuillaume, Chem. – Eur. J., 2001, 7, 4171–4179 CrossRef PubMed.
- M. K. Priya, G. Sahu, D. R. Soto-Pantoja, N. Goldy, A. M. Sundaresan, V. Jadhav, T. R. Barathkumar, U. Saran, B. M. Jaffar Ali, D. D. Roberts, A. K. Bera and S. Chatterjee, Angiogenesis, 2015, 18, 175–189 CrossRef PubMed.
- D. Feliers, X. Chen, N. Akis, G. G. Choudhury, M. Madaio and B. S. Kasinath, Kidney Int., 2005, 68, 1648–1659 CrossRef PubMed.
- D. J. Pinsky, S. Patton, S. Mesaros, V. Brovkovych, E. Kubaszewski, S. Grunfeld and T. Malinski, Circ. Res., 1997, 81, 372–379 Search PubMed.
- S. Kim, H. Lee, M. Chung and N. L. Jeon, Lab Chip, 2013, 13, 1489–1500 RSC.
- R. Trouillon, C. Cheung, B. A. Patel and D. O'Hare, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 929–936 Search PubMed.
- R. Trouillon, D.-K. Kang, S.-I. Chang and D. O'Hare, Chem. Commun., 2011, 47, 3421–3423 Search PubMed.
- R. Trouillon, D.-K. Kang, S.-I. Chang and D. O'Hare, Int. J. Mol. Sci., 2022, 23, 15277 CrossRef CAS.
- Y. Cao, P. Linden, J. Farnebo, R. Cao, A. Eriksson, V. Kumar, J.-H. Qi, L. Claesson-Welsh and K. Alitalo, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14389–14394 Search PubMed.
- D. Fukumura, T. Gohongi, A. Kadambi, Y. Izumi, J. Ang, C.-O. Yun, D. G. Buerk, P. L. Huang and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2604–2609 CrossRef CAS.
- S. A. Stacker, M. M. Halford, S. Roufail, C. Caesar and M. G. Achen, J. Visualized Exp., 2016, 53867 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |