DOI:
10.1039/D4SD00352G
(Paper)
Sens. Diagn., 2025,
4, 320-335
A new sequential dual flow lab-on-a-chip with a lyophilized one-component chemiluminescence substrate for high-sensitive microchannel lateral flow assay (mLFA)
Received
21st November 2024
, Accepted 10th February 2025
First published on 14th March 2025
Abstract
Recently, there has been a growing demand for the development of lab-on-a-chip (LOC) platforms with new assays and detection protocols for point-of-care-test (POCT) applications. So far, chemiluminescence (CL) detection-based immunoassays have shown promising performance for the high-sensitive POCT, but they require automated machines or multiple manual steps to perform the CL-based assay. In this work, a fully automated CL-based immunoassay was developed using a new sequential dual flow LOC with on-chip lyophilized CL substrate, and then a highly specific and sensitive immunoassay using a pair of single chain variable fragment (scFv) capture and detection antibodies was successfully performed. The concept of sequential and automatic control of dual flows, which was strongly desired for ensuring that the reconstituted detection antibody conjugated with horseradish peroxidase flowed first through the reaction zones and then the reconstituted CL substrate flowed, was newly developed and implemented on the LOC. In addition, a new one-component CL substrate in liquid format was introduced and lyophilized for the on-chip lyophilized substrate, developing a new lyophilization process. To evaluate the assay performance on the developed new LOC platform, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was chosen as a demonstration vehicle. The nucleocapsid (N) protein of SARS-CoV-2 was analyzed using the custom-developed scFv antibody pair from a phage display library system, which showed a better limit of detection (LoD) over the commercially available rapid diagnostic test (RDT) kits for detecting SARS-CoV-2. Finally, a portable reader for reading the CL signal from the CL-based microchannel lateral flow assay (CL-mLFA) was developed and used for evaluating the performance of the SARS-CoV-2 assay on the developed LOC platform. An LoD of approximately 1.6 ng mL−1 was achieved, which was acceptable for the early diagnosis of SARS-CoV-2 infection. The new CL-mLFA platform developed in this work, adopting the sequential dual flow LOC and the lyophilized one-component CL substrate, can be applied to other high-sensitive immunoassays in POCT for diagnosing various chronic or infectious diseases.
Introduction
The well-known criteria for the point-of-care-test (POCT), known as ASSURED, which stands for affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free, and delivered,1 were established by the World Health Organization (WHO) in 2004. To meet these criteria, over the past two decades, numerous efforts have been devoted to the development of biochemical analysis laboratories or systems on the micro-scale, resulting in lab-on-a-chip (LOC) and micro total analysis systems (μTAS).2 However, there is always a trade-off with at least one ASSURED criterion compared with the gold-standard clinical laboratory test. Among these criteria, sensitivity should be the most important criterion as it defines the proportion of true positive tests among the patients with a condition.3 A pre-requisite of this criterion is the limit of detection (LoD), known as analytical sensitivity, with respect to the upper reference limit (URL) of a healthy population.4 Benefits of microfluidic technologies, such as rapid response, high-sensitive reaction, and small reagent volume, make them appropriate for POCT applications.5 A category of POCT devices popularly known as rapid diagnostic tests (RDTs), based on the immunochromatographic technique6 is widely available, and it is primarily used for screening purposes only and provide qualitative results, which is not ideal for advanced POCT platforms.6,7
The commonly employed optical detection methods for immunoassays include absorbance, fluorescence, and chemiluminescence (CL). In recent years, CL-based immunoassays have gained attention owing to their superior sensitivity and simplicity for optical detection, but the inevitable use of fresh and liquid CL substrates makes the devices pseudo-POCTs.8–10 These platforms typically involve another loading step after sample introduction, as well as complications regarding the transport, storage, and handling of reagents. Thus, it is necessary to store reagents in a dry format to successfully develop POCT platforms with CL-based immunoassay. As a method to store CL reagents in a dry format on chips, thereby eliminating the need for an additional processing step, lyophilization has been reported.11,12 Especially for LOC application, our group reported the fabrication of a few disposable LOCs with self-contained assay reagents using lyophilization.12,13 The LOCs were capable of delivering the reconstituted lyophilized reagents to the reaction zones with a sample loading step but required an additional step to open a vent to proceed with the immunoassay,12,13 which significantly reduced the burden of liquid handling.
In the previous work, two components of the CL substrate, luminol and hydrogen peroxide, were lyophilized separately on the LOC. However, although the two lyophilized components showed a good performance for the CL-based assay, the result became unstable under various humidity conditions. Conversely, the sodium perborate-based CL substrate showed excellent stability after lyophilization for longer times, as reported by Deng et al.,11 and can potentially be incorporated into a POCT device. Sodium perborate enables the controlled and gradual release of hydrogen peroxide upon dissolution, which improves the stability and reproducibility of the chemiluminescence reaction in sensitive assays.14–16
One of the most challenging issues in the realization of CL-based microchannel lateral flow assays (CL-mLFAs) for POCT platforms is sequentially and timely delivering the immunoassay reagents to the reaction zones, which requires multiple flow paths and fluid controls. Some typical platforms developed by Lim et al.9 and Parandakh et al.17 required multiple additions of immunoassay reagents in liquid format at the sample loading port, which was unable to meet the desire for a sample-to-answer POCT feature. Thus, to automatically perform the desired CL-based immunoassay, it is necessary to have another branch of a microchannel with a delay line that can control the delivery time of the CL substrate by modifying the surface contact angle.
Another way to improve the immunoassay performance is by utilizing a biorecognition molecule with better affinity. A single-chain variable fragment (scFv) antibody provides high specificity to the nucleocapsid (N) protein and can be easily produced in mammalian expression systems, thus reducing the assay costs. Furthermore, due to its small size, an scFv antibody can be efficiently washed to increase the specificity.
Building on previous efforts, we report a one-component chemiluminescence-based, on-chip lyophilized mLFA with the sample-to-answer feature as a new POCT platform that addresses the major drawbacks of current POCT platforms. In this work, a new dual flow polymer LOC with a lyophilized sodium perborate-based one-component CL substrate with sequential dual flow control was fully developed and applied for detecting the N-protein of SARS-CoV-2. In addition, scFv antibodies specific to N-protein were developed and applied as a demonstration vehicle on the CL-mLFA. The N-protein is one of the four structural proteins of the SARS-CoV-2 virus and is the only structural protein present inside the virion, playing a crucial role in eliciting the host immune response.18 The N-protein is also found to be highly conservative in its structure19 across mutations, making it an efficient diagnostic biomarker for the detection of SARS-CoV-2.20 However, although several scFv antibodies specific to the SARS-CoV-2 N-protein are commercially available, a pair of capture and detection scFv antibodies for POCT has not been developed to date. Therefore, scFv antibodies specific to the N-protein using a phage display library system were generated in this work.
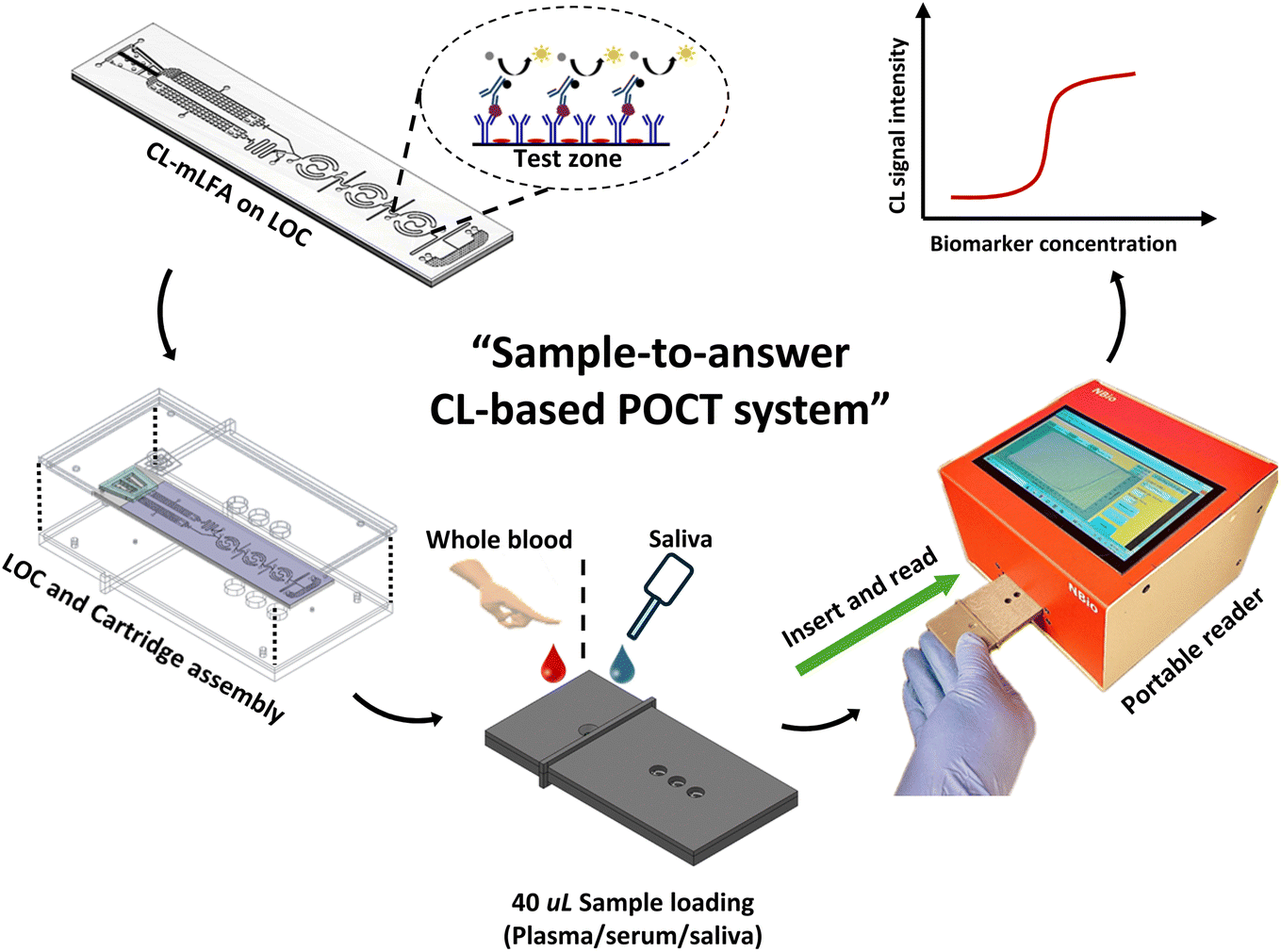
Fig. 1 shows an overview of the newly developed POCT platform, comprising a CL-mLFA on LOC, a functional cartridge, and a custom-designed portable reader. To perform the CL-mLFA, the developed LOC was assembled with the cartridge, and 40 μL of the patient's sample, such as saliva and plasma, was loaded on the sample loading port. Then, the cartridge was inserted into the slot of the portable reader to obtain the quantitative immunoassay signal.
 |
| | Fig. 1 Schematic illustration of the overall approach for sequential dual flow LOC with on-chip lyophilized one-component chemiluminescence substrate for high-sensitive mLFA, detected using a custom designed portable reader. | |
Design and fabrication of sequential dual flow polymer LOC
Concepts of sequential dual flow and assay protocol of the CL-mLFA on LOC
In this work, we developed a new CL-mLFA on LOC for a high-sensitive sandwich immunoassay using an on-chip lyophilized one-component CL substrate. Fig. 2(a) shows the schematic of the design of the CL-mLFA on LOC, with all the functional fluidic and sensing components labeled with outer dimensions of 67 × 15 × 1.2 mm. Fig. 2(b) shows a simplified and matched block diagram of the desired sequential dual flows. This diagram shows the major microfluidic elements, including a sample loading port, detection antibodies conjugated to horseradish peroxidase (DAb–HRP) and lyophilized one-component CL substrate chambers, two delay lines, reaction zones, and a waste reservoir. The sequential, time-controlled dual flow of reconstituted reagents to the reaction zones is depicted using the blue and red arrows, with the blue path first, followed by the red path with a time delay.
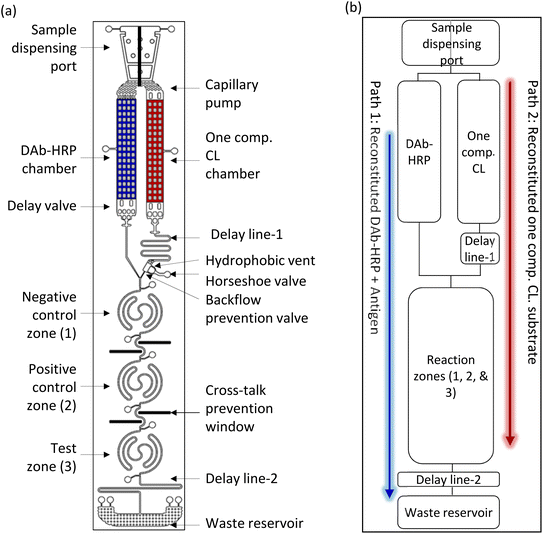 |
| | Fig. 2 Sequential dual flow LOC for CL-based mLFA. (a) Dual flow LOC for CL-mLFA with desired components and (b) simplified block diagram showing the sequential dual flow. | |
The assay protocol of CL-mLFA on LOC is described in Fig. 3. The CL-mLFA on LOC contains three reaction zones, each serving different purposes such as test, positive, and negative control. Sandwich immunoassay of the CL-mLFA was performed as follows: (a) the sample was loaded on the sample loading port; (b) the loaded sample flowed through both paths 1 and 2, simultaneously reconstituting the lyophilized or vacuum-dried assay reagents; (c) DAb–HRP, which was reconstituted in path 1, flowed first through the reaction zones pre-coated with capture antibodies (CAb) and blocking buffer, forming the solid-phase sandwich immunocomplex; (d) unbonded molecules were washed away by the remaining sample from path 1; and (e) the reconstituted CL substrate in path 2 after a desired time delay flowed through the reaction zones, reacting with HRP to generate a CL signal. The autonomous dual flow sequence after the sample loading is illustrated in Fig. 4, where path 1 represents the reconstituted DAb–HRP flow and path 2 represents the reconstituted one-component CL substrate flow. The flow timing was controlled by surface coatings, ensuring that the reconstituted DAb–HRP enters the reaction zones first. During this step, the DAb–HRP and N-protein complexes bind to the CAb on the test reaction zone. The flow was delayed in delay line-2, allowing sufficient incubation time in all the reaction zones. The delayed introduction of the reconstituted CL substrate flow was beneficial for ensuring the desired washing time. Therefore, the flow timing of path 2 was controlled to merge at the intersection point around 10 min after the sample loading, subsequently flowing through the reaction zones. Finally, the CL signals generated from reaction zones 1, 2, and 3 were read by both a conventional 96-well reader and a custom-designed portable reader.
 |
| | Fig. 3 Assay protocol for CL-based microchannel lateral flow assay (CL-mLFA). (a) Negative, positive and test zones after preparation for assay, (b) reaction zones filled with reactive molecules after path 1 reconstitution, (c) unbonded molecules washed away by remaining sample from path 1, and (d) reconstituted CL substrate in path 2 reacting with HRP in reaction zones to generate chemiluminescence signal. | |
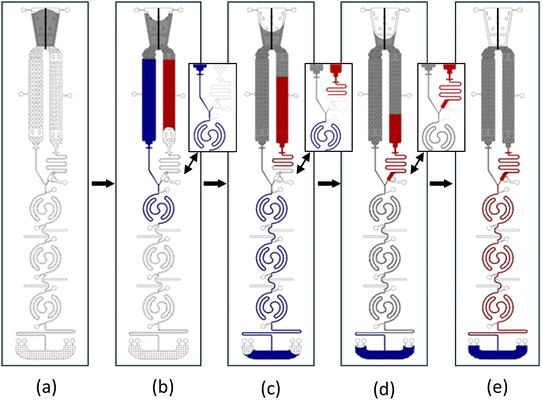 |
| | Fig. 4 Schematic flow of the LOC illustrating the autonomous two-path flow sequence after sample loading. (a) Sample loading and splitting, (b) DAb–HRP reconstitution and flow through reaction zones, (c) reaction zones washed by remaining sample in path 1, (d) reconstituted CL substrate arriving at the merge point after overcoming the delay line-1, and (e) flow of CL substrate through the reaction zones. | |
Design of LOC
The new CL-mLFA on LOC, as shown in Fig. 2(a), was designed to perform CL-based immunoassay on a chip with lyophilized reagents. The final goal was to develop a platform that can work as a sample-to-answer POCT with minimal user intervention other than a single sample loading and achieve sufficient analytical sensitivity for clinical use. In this case, the LOC was adopted to leverage the advantages of microfluidics.21,22 The entire volume of the LOC was designed to be 10 μL considering practical usage for POCT, consisting of elements including a sample loading port, DAb–HRP and one-component CL substrate lyophilization chambers, a horseshoe valve, a backflow prevention valve, delay lines, reaction zones, and a waste reservoir, as shown in Fig. 2(a). The overall concept of the flow to deliver the two lyophilized and reconstituted reagents to the reaction zones was based on the previous work by Ghosh et al.,13 but the full sample-to-answer feature was achieved by eliminating the necessity of the vent-opening step. Furthermore, the manufacturability of the LOC was also considered in this work, adopting a polymer-based LOC made of polystyrene and a new adhesive tape-based sealing method. The detailed dimensions of each component on the newly designed LOC are summarized in Table 1.
Table 1 Dimensions of the microfluidic elements on LOC (width, height and volume)
| Microfluidics elements on LOC |
Width (μm) |
Height (μm) |
Volume (μL) |
| Capillary pump |
N/A |
150 |
0.22 |
| DAb–HRP chamber |
2400 |
150 |
2.75 |
| CL chamber |
2100 |
150 |
2.75 |
| Delay line-1 |
150 |
150 |
0.18 |
| Backflow prevention valve |
75 |
75 |
0.05 |
| Reaction zone |
150 |
150 |
0.86 |
| Delay line-2 |
150 |
150 |
0.5 |
| Waste reservoir |
10500 |
150 |
2.17 |
Microfabrication of master mold and injection-molded polymer chips
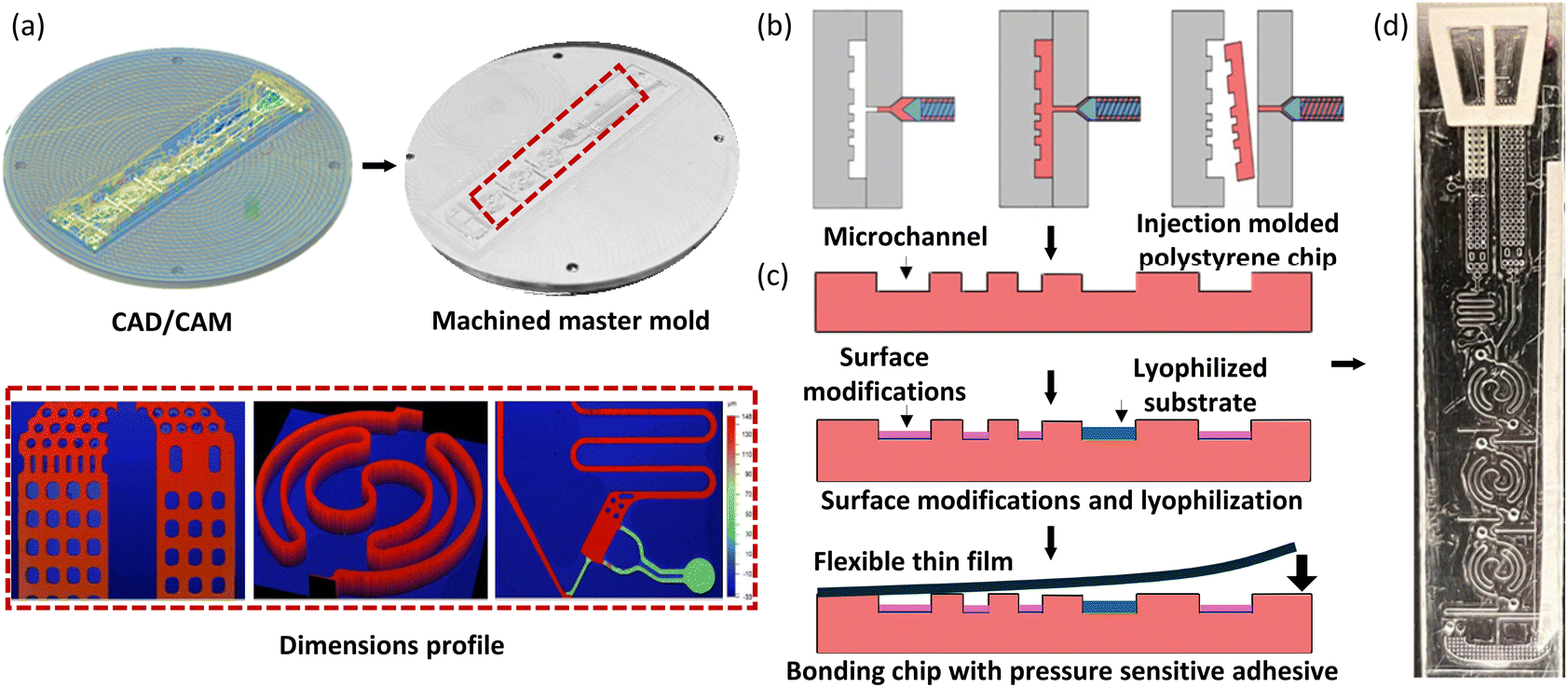
The LOC for CL-mLFA was injection-molded with polystyrene and sealed with adhesive tape using a pneumatic press for low-cost fabrication. Our lab developed and reported a new insertable disc-mold using an aluminium disc for injection molding, instead of the expensive and heavy mold.23 Thus, as shown in Fig. 5(a), the disc-mold of the polymer chip was first designed using the CAD/CAM software, Mastercam 2023 (Mastercam®, USA), according to the dimensions defined in Table 1. Machining a circular aluminium disc-mold was carried out using a 5-axis milling machine, Microlution 5100S (Microlution Inc., USA).13,24,25 The micro-machined aluminium disc-mold with a diameter of 3 inches is also shown in Fig. 5(a). After completing the fabrication, the dimensions of the micro-machined disc-mold were inspected using a contour microscope (Bruker, USA), as shown in Fig. 5(a) (lower). Then, the disc-mold was inserted in a BOY22 injection molding machine (BOY Machines Inc., Germany) and chips were injection-molded using polystyrene, 3100 (INEOS Styrolution, Germany), as illustrated in Fig. 5(b). The choice of polystyrene is because the conventional 96-well plates are mostly based on polystyrene due to its chemical inertness, hydrophobicity, non-reactivity, optical transparency, and majorly low cost over other polymers that have similar properties for LOC applications.24–26 The injection-molded polystyrene chips were processed by surface modification and lyophilization on the open chips, and then sealed using a pressure-sensitive adhesive tape, as shown in Fig. 5(c). A picture of the fabricated LOC for CL-based microchannel lateral flow assay (CL-mLFA) is shown in Fig. 5(d).
 |
| | Fig. 5 Design, microfabrication and assay preparation on the mLFA-LOC. (a) 3D CAD design of the master mold along with the toolpath for machining, the 3 inch aluminium micromachined master mold and the 3 inch aluminium master mold inspected using contour microscope Bruker GT-X to measure the machined dimensions (Table 1), (b) illustration of the injection molding process using polystyrene in BOY22, (c) chip released after injection molding, followed by surface modifications, lyophilization and sealed with a pressure sensitive adhesive tape, and (d) fully prepared polymer LOC to perform the test. | |
Development of ScFv antibody for N-protein of SARS-CoV-2
Hyperimmunization of mice
To develop the N-protein-specific scFv antibodies, the desired animal work was reviewed and approved by the Institutional Animal Care and Use Committee at Mississippi State University (IACUC). Two 6 week-old Balb/c mice (Jackson Laboratories, USA) were immunized via a footpad injection with a final volume of 15 μL containing 25 μg of recombinant N-protein of SARS-CoV-2 (YP_009724397.2, Sino Biological, China), emulsified in an equal volume of TiterMax adjuvant (Millipore-Sigma, USA). The animals were boosted 2 and 4 weeks later. A day before each immunization, mice sera were collected and tested for the detection of N-protein-specific antibodies by an indirect enzyme-linked immunosorbent assay (ELISA) using the recombinant N-protein.
Amplification and cloning of variable light (VL) and variable heavy (VH) chain genes
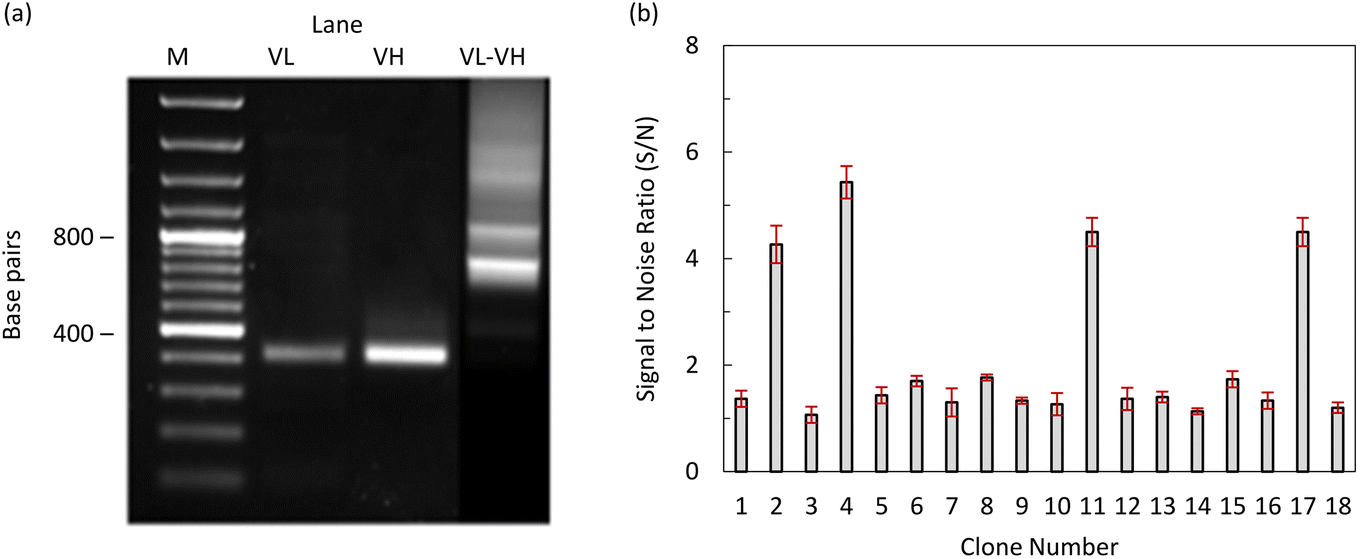
After verifying a robust N-protein-specific immunoglobulin G (IgG) response, the animals were humanely euthanized for spleen collection. Total ribonucleic acid (RNA) was extracted from the spleen cells using an RNeasy Mini Kit (Qiagen, USA). Complementary deoxyribonucleic acid (cDNA) was synthesized using a Superscript cDNA Synthesis Kit (ThermoFisher, USA) according to the manufacturer's instructions. The first polymerase chain reaction (PCR) was performed to amplify the VL and VH chain genes using cDNA and degenerative primer sets, as described previously by Lin et al.27 The PCR products were analyzed by agarose gel electrophoresis, and the approximately 400 bp and 350 bp bands corresponding to VH and VL genes, respectively, were excised from the gel and purified using a PureLink Quick Gel Extraction Kit (ThermoFisher, USA). A second PCR was performed to join the VH and VL genes with a flexible (G4S)3 linker (scFv), as described previously by Lin et al.27 The resulting approximately 750 bp PCR products were excised from the agarose gel and purified using a PureLink Quick Gel Extraction Kit (ThermoFisher, USA). The purified PCR products were digested with NotI and BglI restriction enzymes (Fermentas) and ligated into 50 ng of the digested pADL100 phagemid (Antibody Design Laboratories, USA) using T4 DNA ligase (ThermoFisher, USA). The ligated products were transformed into E. coli XL1-Blue competent cells, which were plated on Luria-Bertani (LB) agar supplemented with ampicillin (100 μg mL−1) and incubated overnight at 37 °C. Colonies from the LB agar plate were scraped and pooled in LB broth.
Construction of phage display ScFv library
E. coli XL-1 Blue transformed cells with the pADL100–scFv genes were grown in SOB-GAT medium (SOB broth supplemented with 100 mM glucose, 100 μg mL−1 ampicillin, and 10 μg mL−1 tetracycline at 37 °C overnight. The overnight culture was inoculated at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution in SOB-GAT medium and incubated at 37 °C until reaching an optical density of 0.3 at 600 nm. The cultures were infected with M13K07ΔPIII hyperphage (Progen, USA) at a multiplicity of infection of 20. The hyperphage-infected culture was incubated at 37 °C without shaking for 30 min, and then shaken at 200 rpm for 30 min. The cells were harvested by centrifugation at 3000 rpm for 10 min at room temperature and re-suspended in pre-warmed SOB-AKT medium (SOB broth supplemented with 100 μg mL−1 ampicillin, 50 μg mL−1 kanamycin, and 10 μg mL−1 tetracycline). The cultures were incubated overnight at 30 °C with shaking at 200 rpm. The overnight culture was centrifuged at 3000 rpm for 15 min at room temperature, and the supernatant was harvested into a fresh tube. Phage particles were precipitated by adding one-fifth volume of 20% polyethylene glycol and 2.5 M NaCl and incubated on ice for 1 h, followed by centrifugation at 4000 rpm/4 °C for 30 min. The pellet was re-suspended in phage dilution buffer (10 mM Tris-HCl pH 7.5, 20 mM NaCl, 2 mM EDTA) and stored at 4 °C until further use.
:1000 dilution in SOB-GAT medium and incubated at 37 °C until reaching an optical density of 0.3 at 600 nm. The cultures were infected with M13K07ΔPIII hyperphage (Progen, USA) at a multiplicity of infection of 20. The hyperphage-infected culture was incubated at 37 °C without shaking for 30 min, and then shaken at 200 rpm for 30 min. The cells were harvested by centrifugation at 3000 rpm for 10 min at room temperature and re-suspended in pre-warmed SOB-AKT medium (SOB broth supplemented with 100 μg mL−1 ampicillin, 50 μg mL−1 kanamycin, and 10 μg mL−1 tetracycline). The cultures were incubated overnight at 30 °C with shaking at 200 rpm. The overnight culture was centrifuged at 3000 rpm for 15 min at room temperature, and the supernatant was harvested into a fresh tube. Phage particles were precipitated by adding one-fifth volume of 20% polyethylene glycol and 2.5 M NaCl and incubated on ice for 1 h, followed by centrifugation at 4000 rpm/4 °C for 30 min. The pellet was re-suspended in phage dilution buffer (10 mM Tris-HCl pH 7.5, 20 mM NaCl, 2 mM EDTA) and stored at 4 °C until further use.
Bio-panning
To select N-protein-specific scFv antibodies, three rounds of phage rescue and bio-panning were performed. Briefly, ELISA plate wells were coated with the N-protein and blocked with 2% bovine serum albumin. The phage library was added to each well and incubated at room temperature for 1 h. After washing ten times with phosphate-buffered saline and Tween-20, the bound phages were eluted by adding 1 M glycine-HCl buffer (pH 2.2) and neutralized by adding 1 M Tris-HCl, pH 8.0. The eluted phage was amplified by infection with E. coli XL-1 Blue expressing PIII strain for the next round of bio-panning. After three rounds of bio-panning, the E. coli XL-1 Blue expressing PIII strain was infected with the eluted phages and plated on LB agar supplemented with ampicillin. Ninety-six colonies were randomly selected for further analysis.
Selection and expression of ScFv antibodies
To select high-affinity scFv antibodies to the N-protein antigen, soluble scFv antibodies were induced by IPTG from individual E. coli XL-1 Blue clones resulting from reinfection of the eluted phages from the third-round bio-panning, as described previously. A soluble scFv antibody was added to the wells of the ELISA plate coated with the N-protein, and the bound scFv antibody was detected with an anti-FLAG tag antibody (ThermoFisher, USA). Identified candidate clones were subjected to DNA sequencing. The identified scFv gene was synthesized in the pGenLenti vector (GenScript, USA) and co-transfected into HEK293T cells with pMD2.G (Addgene) and psPAX2 (Addgene) using TransIT-2020 Reagent (Mirus Bio, USA). After 72 h of culture, the culture supernatant containing lentivirus was harvested and passed through a 0.45 μm filter (Sartorius, USA) to remove the cell debris. Subsequently, the HEK293T cells cultured in a 6-well dish (approximately 50% confluent) were treated with 1 mL of lentivirus solution for 3 days. Then, the cultured cells were treated with 2 μg mL−1 puromycin and cultured for another 2 weeks to select the stable expression of scFv.
Construction and screening of phage display ScFv library
To generate an scFv antibody specific to the N-protein of SARS-CoV-2, a Balb/c mouse was immunized with recombinant N-protein. The VL and VH gene fragments, approximately 400 bp in length, were successfully amplified from cDNA prepared from the spleen of the immunized mouse. The VL and VH genes were joined by a (G4S)3 linker using SOE PCR, resulting in an scFv gene repertoire, as shown in Fig. 6(a). The scFv library was generated by cloning the scFv gene repertoires into pADL100 and transforming them into E. coli XL-1 Blue. Phage display scFv antibodies were generated by infection with the M13K07ΔPIII hyperphage. After three rounds of bio-panning, the reactivity of the candidate phage display scFv antibodies was assessed by ELISA using an anti-E tag antibody. Among the 20 candidates, 4 phage display scFv antibodies (2, 4, 11, and 17) showed positive reactions, defined as a signal-to-noise ratio (S/N) of >2, as shown in Fig. 6(b). These scFv antibodies were expressed in HEK293T cells using a lentiviral expression system and purified by nickel affinity chromatography.
 |
| | Fig. 6 (a) Amplification of the VL and VH genes from immunized mice and screening of phage display scFv antibodies generated from the third round of biopanning. Lane M: DNA Marker 100 bp (Fermentas); lane VL: PCR products of the VL gene; lane VH: PCR products of the VH gene; lane VL–VH: VL gene joined with VH gene by SOE-PCR, and (b) screening of high affinity phage display scFv antibody to the N-protein using an indirect ELISA. | |
Selection of ScFv antibody pair
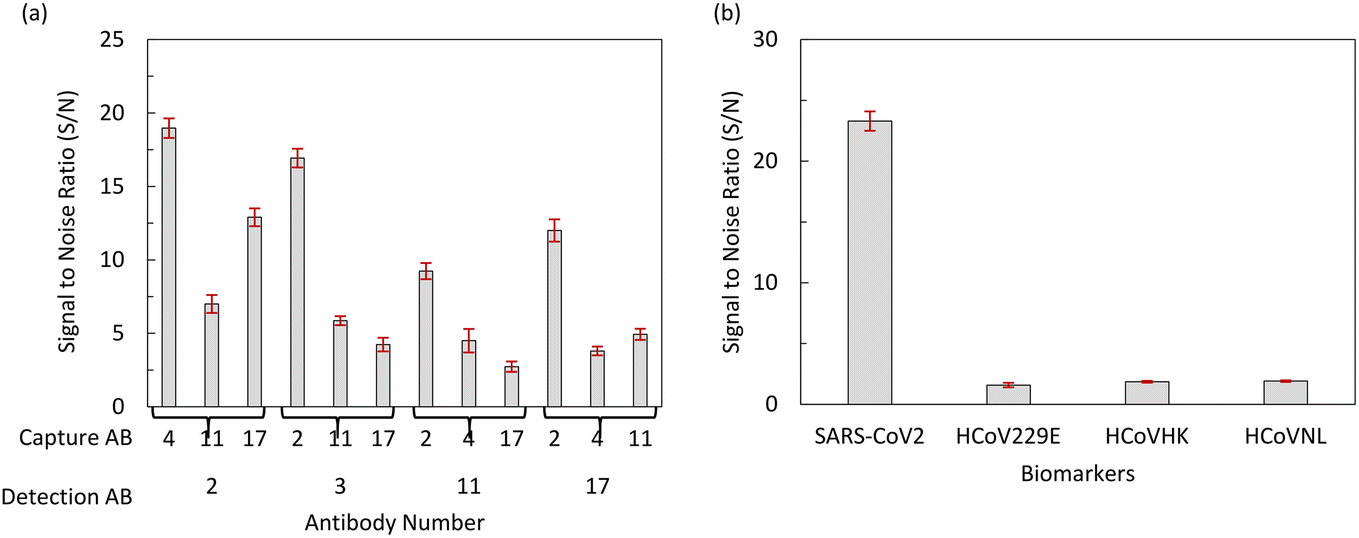
To select the best pair of scFv antibodies for detecting the N-protein of SARS-CoV-2, we performed a checkerboard sandwich ELISA using each unconjugated scFv antibody and each scFv antibody conjugated with HRP as capture and detection antibodies, respectively. A pair consisting of scFv4 as the capture antibody and scFv2 as the detection antibody showed an S/N ratio greater than 24, indicating the high affinity of these scFv antibodies against the N-protein of SARS-CoV-2, as shown in Fig. 7(a). The specificity of the scFv4 capture antibody and the scFv2 detection antibody was examined by sandwich ELISA using the N-proteins of various human coronaviruses, which showed S/N ratios of less than 2. This indicates that these scFv antibodies are highly specific to the N-protein of SARS-CoV-2, as shown in Fig. 7(b). Fig. 8 shows the amino acid sequences of the scFv2 and scFv4 antibodies, which were analyzed using the IMGT/V-QUEST program. Thus, we used the newly developed scFv antibodies to detect the N-protein of SARS-CoV-2.
 |
| | Fig. 7 Screening of a scFv antibody pair sensitive and specific to the N-protein of SARS-CoV-2. (a) A high-affinity pair of capture scFv and detection scFv conjugated with HRP to the N-protein of SARS-CoV-2 was screened by a sandwich ELISA, and (b) specificity of capture antibody #4 and detection antibody #2 was examined by a sandwich ELISA using the N-protein of SARS-CoV-2. | |
 |
| | Fig. 8 VL and VH amino acid sequence of scFv2 and scFv4 antibodies. (a) and (b) Show the amino acid sequences for scFv2, and (c) and (d) show the amino acid sequences for scFv4 respectively. The complementarity determining region (CDR) was analyzed using the IMGT/V-QUEST program version 3.2.20 of the international immunogenetics information system (https://www.imgt.org/HighV-QUEST/home.action). | |
Preparation of sequential dual flow polymer CL-mLFA
Immunoassay reagents for SARS-CoV-2 N-protein
The following reagents were procured to perform the N-protein immunoassay of the SARS-CoV-2 virus: rabbit capture antibody from Sino Biological, USA (68097-R150); detection antibody from Sino Biological, USA (40588-RC02) and phosphate-buffered saline (PBS) as CAb coating buffer and washing buffer from Abcam, USA (ab285410). The blocking buffer was used for blocking the reaction sites and as a diluent buffer from ThermoFisher, USA (StartingBlock Blocking Buffer™, 37578). The chemifluorescent substrate was from ThermoFisher, USA (QuantaRed, 15159). One component chemiluminescent substrate based on sodium perborate was from Cyanagen, Italy (Westar-One Extreme, XLSU180). Artificial serum (AS) was from CST Technologies Inc., USA (Serasub™). The HRP conjugation kit was from Abcam, USA (Lightning-Link®, ab102890), and the SARS-CoV-2 pseudovirus was from Virongy, USA (Ha-CoV-2). Specifically, the scFv antibodies utilized for the N-protein assay were produced by our collaborator, Dr. Seo's lab at Mississippi State University, who custom-developed scFv antibodies specific to the N-protein, as described in the previous section.
CL-mLFA assay preparation on polymer LOC
The assay preparation began with cutting and drilling vent holes in the injection-molded polystyrene chips, cleaning with isopropyl alcohol and deionized water, and drying with nitrogen gas. To modify the surface of the microchannels with the desired hydrophilicity or hydrophobicity, all channels and chambers of the chips were treated with O2 plasma (30 s, 1 SCFH, 100% power level) in the plasma-preen system (Plasmatic Systems Inc., USA), except the reaction zones, which were covered with adhesive tape, given that they should have a hydrophobic surface for the CAb immobilization and blocking step. Then, the surface of the microchannels was modified to realize the sequential dual flows of the two flow paths on the LOC. Surface modification coating solutions (X100 and A30, Joninn ApS, Denmark) were used to attain the desired hydrophilic and hydrophobic surfaces, as summarized in Table 2. Next, the reaction zones were prepared for CAb immobilization using the following protocol. In the test reaction zone, CAb was in the desired concentration, diluted in coating buffer, incubated for 20 min, and washed with PBS. Then, the StartingBlock blocking buffer was incubated for 20 min and washed with PBS. In the negative control reaction zone, the CAb incubation step was omitted, and thus only blocking proteins existed on the reaction zone surface. Once the reaction zones were incubated with the desired immunoassay reagents, they were vacuum-dried in a vacuum desiccator (SP Bel-Art, USA) at −0.1 MPa for 30 min.
Table 2 Coating conditions for the microfluidic elements on LOC to perform surface modification resulting in a sequential delivery of the reconstituted reagents to the reaction zone
| Microfluidic element |
Coating condition (%V) |
Contact angle (degrees) |
| Sample port |
100% X100 |
∼20 |
| DAb–HRP chamber |
| CL chamber |
| Delay line-1 |
30% X100 + 70% A30 |
∼80 |
| Horseshoe valve |
100% A30 |
∼100 |
| Reaction zone 1 |
Assay |
∼13 |
| Reaction zone 2 |
| Reaction zone 3 |
| Delay line-2 |
100% X100 |
∼20 |
| Waste reservoir |
In this work, we developed a new method to construct a flexible elastomer dam that can be easily prepared and removed after the lyophilization process, enabling the liquid to be held in the chamber constructed between the dams. To perform the lyophilization process of DAb–HRP and CL substrate, a rapid freezing step of these two liquids was required ahead of the drying step inside a liquid nitrogen dewar. A peelable, quick-dry silicone adhesive from World Precision Instruments, USA (Kwik-Cast low toxicity silicone sealant), that is not harmful to polystyrene was used to create the dam structures, as shown in Fig. 9(a). After lyophilization, the silicone adhesive dams were peeled off easily, and the areas still maintained hydrophilicity after the removal of the dams. 3 μL of DAb-HRP and 3 μL of one component CL substrate were dispensed, and the polymer chips containing liquid reagents were put in a custom-designed aluminium box, as shown in Fig. 9(b). An aluminium box with a diameter of 101.6 mm and thickness of 6.35 mm was used for achieving a stable temperature equilibrium during the freezing step and holding the temperature stability at the beginning of the drying step in the lyophilizer. The aluminium box was first located inside a liquid nitrogen dewar for stable freezing, such that the box was positioned at around 127 mm from the liquid nitrogen surface. The temperature during the entire freezing step was monitored through a thermocouple attached to the inside of the aluminium box, as shown in Fig. 9(c). Once the temperature reached −160 °C, the aluminium box was immediately transferred onto a lyophilizer rack (FreeZone 1 liter −54 °C benchtop freeze dryer, Labconco, USA) with the collector preset at −54 °C and 0.010 mbar of vacuum was applied for 10 h. As shown in Fig. 9(d), the substrate had a porous structure after lyophilization, which was critical for easy and quick reconstitution.13 The chips prepared with lyophilized reagents were bonded with pressure-sensitive adhesive tape, IST-121 QuickSeal, 60 μm in thickness (iST Scientific, UK), using a pneumatic press set to 60 psi for 30 s, as shown in Fig. 5(c), and Fig. 5(d) shows the fully prepared LOC for CL-mLFA.
 |
| | Fig. 9 Lyophilization of one-component CL substrate. (a) Construction of dam using peelable silicone adhesive Kwik-Cast and loading of the liquid CL substrate, (b) aluminium box that acts as a heat sink to enclose the chips and hold the temperature until the CL substrate lyophilizes, (c) aluminium box loaded with chips placed in the liquid nitrogen dewar for rapid freezing to −160 °C, and (d) aluminium box transferred to Labconco freeze dryer to perform lyophilization of the CL substrate. Post lyophilization, the silicone adhesive is easily peeled away, leaving behind the lyophilized substrate having a foam like consistency. | |
Delay time control on CL-mLFA LOC
The previous work by Ghosh et al.13 was based on vent opening for triggering the second flow, which was implemented manually once the flow from path 1 was complete. To eliminate this user intervention, we worked on generating automatic delay by coating the delay line-1 to have a delay of around 10 min, and thus the sample from path 2 could merge autonomously after the desired delay without any user interventions, such as opening another vent. Fig. 10 depicts the correlation between the contact angle and delay time in delay line-1. Based on this result, a contact angle of around 80°, giving the desired 10 min of delay time, was chosen. Table 3 shows how the appropriate delay was achieved by mixing the hydrophilic coating (X100) and hydrophobic coating (A30) at different ratios to get a range of delay line filling times and contact angles. According to Table 3, a mixture containing 30% of X100 (volume%) and 70% of A30 (volume%) was the appropriate choice for attaining the desired delay time of around 10 min in the developed LOC.
 |
| | Fig. 10 Delay line-1 flow time control: plot of contact angle vs. delay line-1 flow time. Each data point is the mean of triplicate and error bar represents the standard deviation. | |
Table 3 Delay line-1 flow time control: table shows the mixing of hydrophilic and hydrophobic coatings at different ratios and the resultant contact angle
| Hydrophilic X100 (V%) |
Hydrophobic A30 (V%) |
Delay line-1 fill timing (minutes) |
Contact angle (degree) |
| 100 |
0 |
<1 |
18 |
| 50 |
50 |
3–4 |
49 |
| 40 |
60 |
7–8 |
64 |
| 30 |
70 |
9–11 |
82 |
| 0 |
100 |
>30 |
103 |
Cartridge assembly with polymer LOC
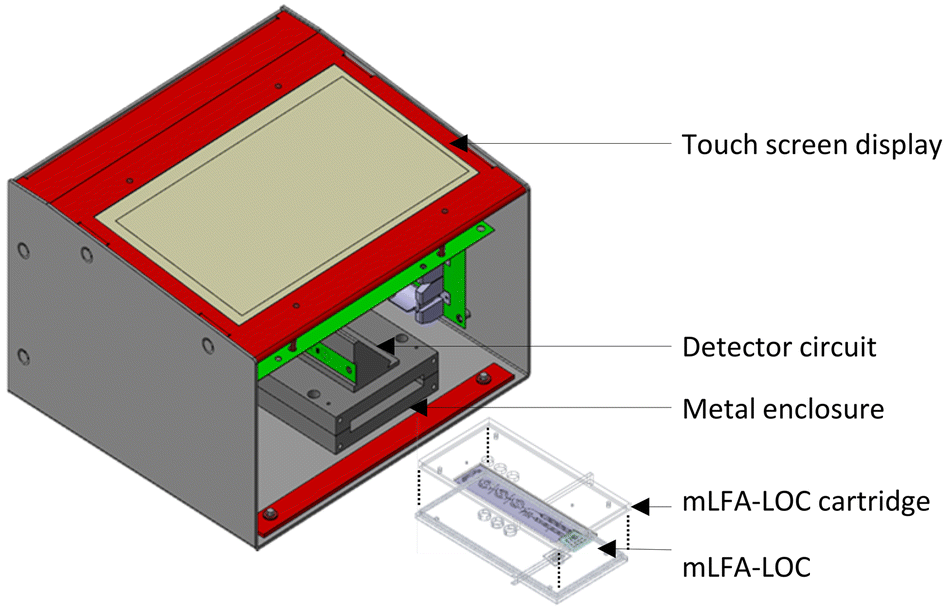
A cartridge is necessary for preventing ambient light exposure and acts as a means of interface for easy sample loading as well as equal sample splitting to each path. The cartridge was fabricated using a polymer 3D printer (UltiMaker, USA) to accommodate the developed LOC. In detail, the LOC was mounted inside the cartridge, and then 1 mm thick double-sided adhesive tape was used between the LOC and the sample loading port of the cartridge to prevent leakage during sample loading, as shown in Fig. 1 (LOC and cartridge assembly). Thus, the sample loading port allowed the loaded sample to split and flow to each path on the LOC. Thus, only when the cartridge was fully assembled with the LOC was it ready to perform the CL-mLFA. The cartridge had three opening holes of 6 mm in diameter, which aligned with the three reaction zones on the LOC, and thus the CL signals could reach the photodiodes and prevent crosstalk between the reaction zones.
Characterization of lyophilized one-component CL substrate
As mentioned, CL is a chemical reaction that produces energy in the form of light, which is commonly used as an optical detection method for quantifying biochemical assays such as ELISAs.28 Luminol-based CL is particularly popular due to its selectivity, simplicity, low cost, and high sensitivity.26,28 Typically, two-component-based CL substrates should be mixed right before their use to prevent the loss of their functionality, which indicates that these reagents are sensitive to light, temperature, and ambient humidity. The same concern should be considered for the on-chip lyophilization step, realized by the separated storage of CL substrate on the LOC.13 However, recently, one-component CL substrates have gained attention owing to their good stability as a proprietary mixture of a luminol derivative, an enhancer/electron mediator, and sodium perborate as an oxidizing agent.29 Thus, in this work, we transitioned from a two-component CL substrate to a one-component CL substrate, and this shift has benefits in terms of manufacturing and performance due to two primary reasons, as follows: (a) processing a one-component substrate is simpler than two components, eliminating the possibility of non-homogeneous reconstitution on-chip and (b) sodium perborate-based one-component CL substrates are more stable with a variation in ambient light and humidity.
The first step was to confirm the compatibility of the one-component CL substrate with the lyophilization process. The lyophilization process, as described in a previous section, was followed by characterization, with the results shown in Fig. 11(a). Here, an Optimiser™ microchannel-based microplate (MiCo BioMed Co., Ltd, South Korea) coated with streptavidin-HRP (Sav-HRP) at different concentrations was used to measure the CL signal intensity for both the fresh CL substrate in liquid form and the lyophilized and reconstituted CL substrate using the BioTek reader (Synergy HT, Agilent, USA). The sodium perborate-based, one-component CL substrate showed a matching trend in a wide range of Sav-HRP concentrations on the reaction surface, confirming its suitability for the lyophilization process and CL-mLFA on the LOC. To further investigate the chemical nature of the CL substrate during processing, we conducted ultraviolet-visible (UV-Vis) absorption spectroscopy using a NanoDrop™ Onec spectrophotometer (ThermoFisher, USA). Fig. 11(b) shows the absorbance spectrum of (i) the control (original), where the CL substrate was used in its liquid form as a standard, and (ii) the substrate solution obtained from the reconstitution of the lyophilized CL substrate. The recovery rate of the CL substrate was found to be over 85%, which indicated that the one-component CL substrate used in this work was acceptable for performing the CL-based mLFA on LOC after the lyophilization process.
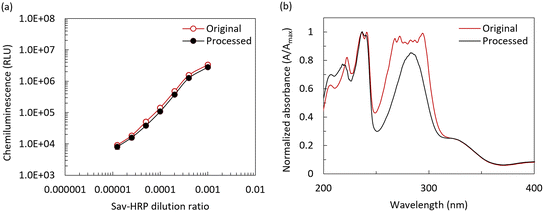 |
| | Fig. 11 Characterization of lyophilized one component CL substrate. (a) Comparison of one component CL intensity before (original) and after lyophilization (processed) with different dilutions of Sav-HRP coated on Optimiser™ microplate. Each data point is the mean of triplicate and error bar represents the standard deviation. Error bar is smaller than the data point, and (b) UV-vis absorption spectra of one component CL substrate before (original) and after lyophilization (processed) measured using NanoDrop™ One. | |
Development of a custom-designed portable reader for POCT
To achieve the quantification of the CL signal intensity from the CL-mLFA on LOC and analysis of the existence of biomarkers for the severity of a disease, a detection instrument is required.30–32 In this work, a custom-designed portable reader was developed to measure the CL signal from the CL-mLFA on LOC assembled in a cartridge. The cartridge helped to align the reaction zones on the LOC with the photodiodes in the portable reader, allowing the emitted CL signal to efficiently reach the respective photodiodes, preventing crosstalk between the photodiodes and avoiding exposure to ambient light as well. The reader had outer dimensions of 188 × 170 × 114 mm and housed optical photodiodes as part of the detector circuit, a microcontroller, a touch screen display to monitor the CL signals using a software application built with LabVIEW, and an enclosure to prevent ambient light exposure. The electrical and optical systems used in this work were based on the previous work by Ghosh et al.13 and modified to integrate them with the newly developed LOC.
Electronic and optical system design
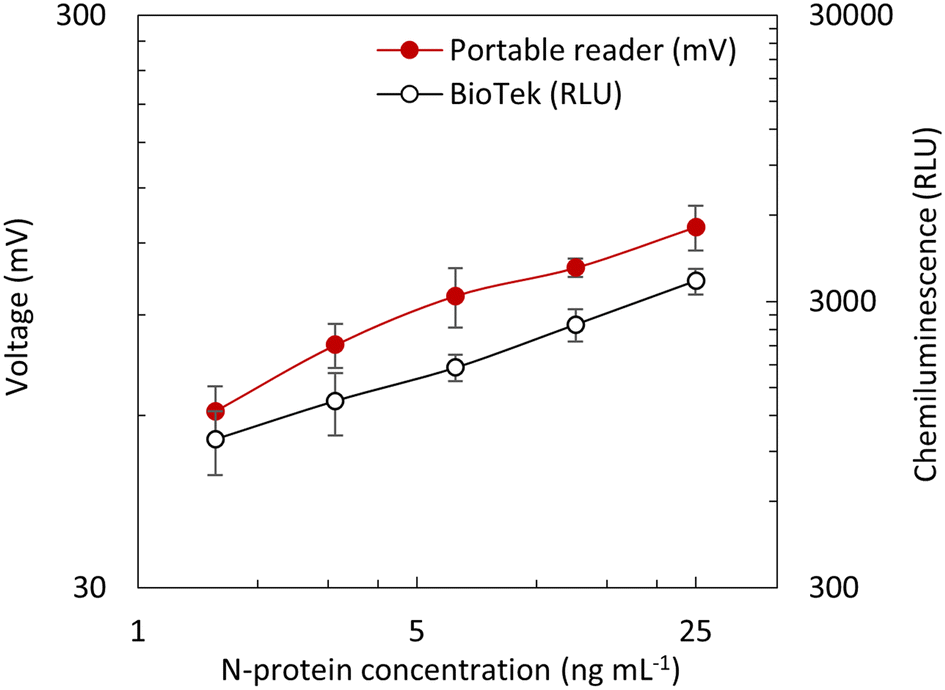
Fig. 12(a) shows a block diagram of the opto-electronic system, which was categorized into three subunits including the user interface, electronic control, and optical detection. The detection started with a single click, and the CL signal measurement began. The photodiodes (MicroFC-30035-SMT-C1, Onsemi, USA) had a peak detection wavelength of 420 nm, which is very close to the CL peak emission wavelength of 425 nm, acquiring the optical signal emitted from the reaction zones to current in the micro-ampere scale. The current was fed to an amplifier circuit built using an Op-Amp (OPA320, Texas Instruments, USA), and the trans-impedance amplifier converted this current to voltage and amplified the signal based on the feedback resistance, which could be controlled using a potentiometer. In this case, a feedback resistance of 100 kΩ showed a good performance in differentiating the signals for a given range of DAb–HRP (1 ng mL−1 to 1000 ng mL−1) present in the reaction zones. Then, the analog signal was fed to the analog-digital converter (ADS1118, Texas Instruments, USA), which converted the voltage signal to a 16-bit digital signal. The signal was transferred from ADS1118 to a microcontroller (Teensy 3.2, PJRC, USA) via a serial peripheral interface (SPI). Finally, the data from the microcontroller was transferred to the CPU (Intel® Compute Stick, Intel, USA) on request command via the serial interface. All the operations in the portable reader were controlled using the custom-developed LabVIEW software, which included the user interface for monitoring and analyzing the measured results as well as controlling the photodiode measurement via the CPU Intel Compute Stick. Fig. 12(b) shows a detailed flow of the opto-electronic signal in the portable reader. The setup to provide alignment of the LOC cartridge with the enclosure to prevent crosstalk, the photodiodes, and the photodiode detector circuit underneath the enclosure for CL signal detection is shown in Fig. 13. To validate the functioning of the portable reader, different concentrations of DAb-HRP were incubated in the reaction zones of the LOC, and a fresh CL substrate was introduced. Multiple sets of chips were measured using the portable reader and compared with the result from the BioTek reader. In both cases, the signal showed a similar trend in a given range of DAb–HRP, as shown in Fig. 14, and thus we could reliably proceed with measuring the assay performed on LOC with the portable reader.
 |
| | Fig. 12 (a) Block diagram of the opto-electronic system categorized into user interface, electronic control and optical detection, and (b) block diagram illustrating the flow of opto-electronic signals in the portable reader for quantifying the CL signal intensity. | |
 |
| | Fig. 13 Custom designed portable reader with an enclosure and a slot to insert the fully assembled mLFA-LOC device cartridge providing alignment with the photodiodes and prevents cross-talk. | |
 |
| | Fig. 14 Portable reader showing a trend for CL signal obtained from various concentrations of DAb–HRP in reaction zones of mLFA for a gain resistance of 100 KΩ, compared with BioTek reader. Each data point is the mean of triplicate and error bar represents the standard deviation. | |
Results and discussion
Immunoassay validation and optimization on Optimiser™
For diagnosing SARS-CoV-2 viral infection, a nasopharyngeal swab is commonly used as the sample. After sample collection, the swab is inserted in a tube containing an extraction buffer, which causes the viral cell to lyse, and the proteins present inside the virus are exposed and can be used for diagnosis, as illustrated in Fig. 15. The compatibility test of the extraction buffer was performed with SARS-CoV-2 pseudovirus, expressing all the proteins present in the actual virus due to its contagion and facility requirement for handling it.33 Given that pseudoviruses are non-replicative, they are allowed to be handled in a Biosafety Level 2 (BSL-2) facility.
 |
| | Fig. 15 Extraction scheme of N-protein from pseudovirus present in a nasopharyngeal swab sample using an extraction buffer. | |
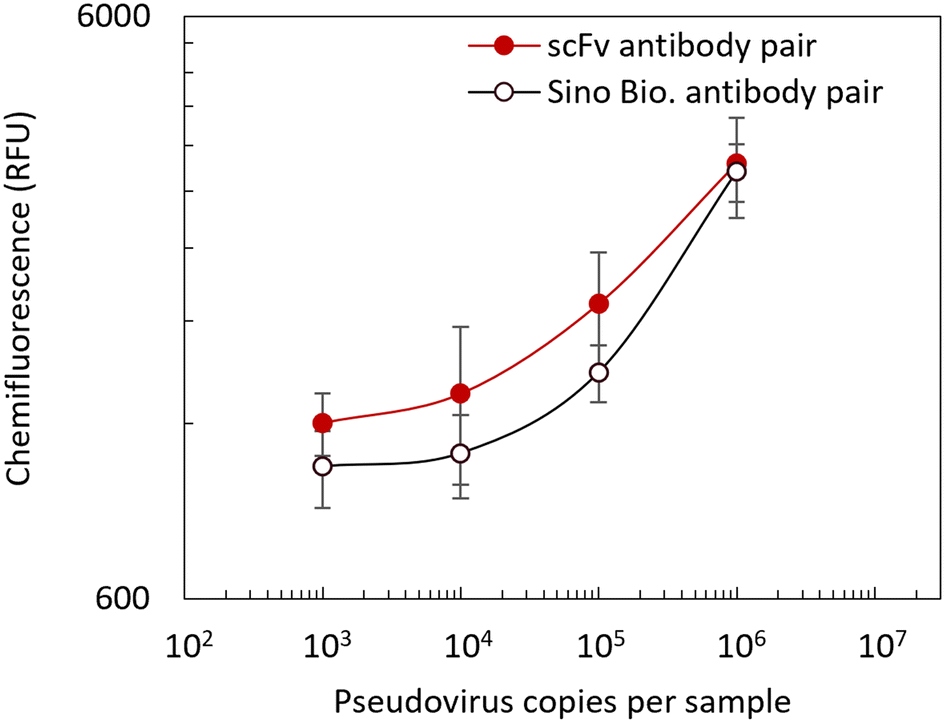
The immunoassay reagents were validated on a commercially available Optimiser™ microchannel-based microplate, which usually provides assay performance and sensitivity similar to the conventional 96-well assay by following the assay protocol based on the Optimiser™ Assay Transfer Guide.34 A standard curve was first obtained using the lysed sample in the extraction buffer described in the previous section to validate the assay reagents and assay protocol, including the extraction protocol. Fig. 16 shows the standard curves obtained from both the developed scFv antibody pair and commercially available antibody pair on the Optimiser™ platform for detecting N-protein, with a sample range of 103 pseudovirus copies per sample to 106 pseudovirus copies per sample. An LoD of 103 pseudovirus copies per sample was observed for the scFv antibody pair. The obtained results were compared with the N-protein-specific IgG antibody pair from Sino Biological and the LoD was found to be 105 pseudovirus copies per sample. The scFv antibody pair showed a 100 times better performance than the commercial Sino Biological antibody pair. Also, we performed the assay using a purified protein spiked sample with the same assay reagents and protocols. The standard curve obtained for the purified protein sample on the Optimiser™ platform is shown in Fig. 17. A detection range of 1.6 ng mL−1 to 100 ng mL−1 was tested, and it was observed that concentrations above 25 ng mL−1 showed saturation. An LoD of 1.6 ng mL−1 was achieved on this platform for the purified protein spiked sample.
 |
| | Fig. 16 Standard curve obtained for assay on the Optimiser microplate for N-protein extracted from pseudovirus, detected using the custom designed scFv antibody pair and compared with the commercially available IgG antibody pair from Sino Biological. Each data point is the mean of triplicate, and the error bar represents the standard deviation. The LoD of the developed scFv antibody pair was 103 copies per sample, while the LoD of the Sino Biological antibody pair was 105 copies per sample. | |
 |
| | Fig. 17 Standard curve obtained for assay on Optimiser™ microplate for N-protein in spiked sample. Each data point is the mean of triplicate and error bar represents the standard deviation. LoD was measured as 1.6 ng mL−1. | |
SARS-CoV-2 N-protein detection on LOC
According to the assay protocol in the Optimiser™ Assay Transfer Guide, at least three wash steps were required to wash away the unbound molecules to reduce background noise. Keeping this as a reference, we designed the flow protocol for CL-mLFA as follows: a 40 μL aliquot of the sample to be tested was loaded on the sample port of the cartridge. Due to the design of the cartridge, the sample naturally split into two equal parts of 20 μL, each following separate paths through the LOC chambers. The total volume of the three reaction zones and the DAb–HRP chamber was approximately 2.5 μL each. The reconstituted DAb–HRP, with the loaded sample, first passed through the reaction zones and started reacting there to form the sandwich immunocomplex (CAb – N-protein – DAb–HRP). The next 12.5 μL of the sample loaded initially passed through the reaction zones, which worked as washing buffer to remove the unbound molecules from the reaction zones. The remaining 5 μL was retained in the DAb–HRP chamber and reaction zones. Then, the flow from path 1 stopped as there was no sample remaining in the sample port of path 1, and the fluid front did not have enough capillary pressure to pull the sample. Fig. 18 shows the signal from the test reaction zone when there was no antigen present in the sample with one, two and three washing steps. According to this result, it can be clearly seen that at least three washing steps were required to remove a significant amount of the unbound DAb–HRP from the reaction zones given that the signal obtained after three washes was close to the signal from the blank reaction zone. Thus, while performing an assay on LOC, sufficient washing steps through the microchannel were guaranteed.
 |
| | Fig. 18 CL signal intensity obtained from a test reaction zone vs. the number of wash steps performed for that reaction zone. | |
After the validation of the assay reagent functionality and assay protocol on the Optimiser™ platform and optimizing the assay performance, which included the washing steps, we finally performed the CL-mLFA on LOC for N-protein extracted from the lysed sample of pseudovirus with both the custom-designed portable reader and the BioTek reader. The performed assay results are depicted in Fig. 19(a), showing that the trend was comparable for both readers, and hence the functionality of the custom-designed portable reader was well validated. Finally, the CL-mLFA for the pseudovirus of SARS-CoV-2, which was performed on the LOC platform with the protocol developed in this work, achieved a minimum detection of 103 copies per sample.
 |
| | Fig. 19 Performance comparison of the mLFA-LOC platform with the commercial RDT. (a) Standard curve obtained for the assay with N-protein extracted from pseudovirus on mLFA, quantified using portable reader and compared with BioTek reader. Each data point is the mean of triplicate, and the error bar represents the standard deviation, and (b) assay performed with N-protein extracted from pseudovirus on commercial RDT as per the kit’s instructions (Flowflex®, USA). LoD of the CL-mLFA on LOC was 103 copies per sample whereas the LoD of the commercial RDT was 105 copies per sample. | |
For a parallel comparison of various platforms using the same lysed pseudovirus test sample, we used commercially available rapid diagnostic test (RDT) kits (Flowflex® COVID-19 Antigen Home Test, USA) for COVID-19, following the kit's instruction manual. The photographs of the test results are shown in Fig. 19(b), where the commercially available RDTs could not show a positive result for pseudovirus copies below 105 copies per sample. Alternatively, the CL-mLFA showed an LoD of 103 copies per sample, which achieved 100 times lower LoD than the RDTs. This result clearly showed a high potential of the CL-mLFA on the LOC platform as a next-generation POCT. The CL-mLFA for the purified protein spiked sample on the LOC platform was tested and read using both the portable reader and the BioTek reader. The results are shown in Fig. 20. The achieved assay performances were similar to that of the Optimiser™ platform with an LoD of 1.6 ng mL−1. This result is another confirmation of the reliable assay performance achievable with the CL-mLFA on the LOC platform.
 |
| | Fig. 20 Standard curve obtained for N-protein assay with spiked sample on mLFA, quantified using the portable reader and compared with BioTek reader. Each data point is the mean of triplicate, and the error bar represents the standard deviation. LoD was measured as 1.6 ng mL−1. | |
Given that the reconstitution profile of the lyophilized DAb–HRP and the CL substrate was not explored enough, there might be room to further optimize the assay if the reconstitution profile can be controlled. The LoD actually decreased to 0.1 ng mL−1 when the assay was manually performed on the reaction zones of LOC with the purified protein spiked sample, which indicated the possibility for further improvement in sensitivity after further optimization. The CL-mLFA on the LOC platform with the portable reader has great potential to replace existing systems at POCT, providing an accurate result with better sensitivity and possibly competing with molecular diagnostics.
Since cost and scalability are also critical considerations for the real-world applications of POCT, we roughly estimated the cost to be around $15 per test and fabrication time to be around 15 h in batch processing mode. Due to the limitations of the current laboratory setting, we could achieve $15 per test, but we anticipate this cost to be reduced by 4–5 times if manufacturing with well-established facilities. The cost of one portable reader is estimated to be around $500, but if mass manufacturing is considered, its cost can significantly decrease.
Conclusion
In this work, a new CL-mLFA on an LOC platform for sample-to-answer POCT was successfully developed and characterized for the detection of SARS-CoV-2. The CL-mLFA on LOC was composed of an on-chip lyophilized one-component CL substrate based on sodium perborate, a sequential dual flow polymer LOC, and a portable reader for POCT applications. The developed platform could perform a high-sensitive and rapid immunoassay through the sequential dual flow control of assay reagents with minimal user intervention. The lyophilization process of the one-component CL substrate was developed and characterized well, confirming its functionality after reconstitution. The long-term stability of the lyophilized CL substrate needs to be further explored for the POCT applications. CL-mLFA of the N-protein assay with both pseudovirus and purified protein samples of SARS-CoV-2 was successfully performed on the LOC platform in response to the recent pandemic, achieving a limit of detection (LoD) of 103 copies per sample and 1.6 ng mL−1, respectively, using the scFv antibody pair developed in this work. Unlike commercially available antibodies, the scFv antibodies offered high specificity to the N-protein of SARS-CoV-2, which improved the performance of the N-protein assay. The results obtained support that the developed LOC platform with the portable reader has high potential as a promising POCT platform. The CL-mLFA on the LOC platform developed in this work is not only applicable for diagnosing infectious diseases but also other acute and chronic diseases.
Data availability
All the necessary data are within the main article in the form of graphs and tables.
Author contributions
Dr. Ahn's lab team members at the University of Cincinnati contributed equally to the overall concept of LOC for CL-mLFA in this work, and Supreeth Setty and Heeyeong Jang are considered the co-first authors. Team members of Dr. Seo's lab at Mississippi State University contributed equally to the custom-developed single-chain variable fragment (scFv) antibodies specific to the N-protein and related assay.
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Mississippi State University and approved by the Institutional Animal Care and Use Committee (22-525).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by MiCo BioMed Co., Ltd. with the University of Cincinnati's research contract # G402226 and partially by a grant from the Mississippi Center for Clinical and Translational Research, NIGMS, NIH (5U54GM115428). We'd like to thank Mr. Jeffrey Simkins in the Ohio Center for Microfluidic Innovation (OCMI) at the University of Cincinnati for assisting with the micro-milling of the aluminium disc-mold; Dr. Keun Seok Seo and the team at Mississippi State University for providing custom-designed scFv antibodies and guidance in developing the assay protocol; Dr. Lakshmi Narayan at NBio LLC for insights in the development of the portable reader; and Dr. Andrew Steckl and Dr. Daewoo Han at the University of Cincinnati for supporting the use of UV-vis spectrophotometer for the analysis of chemiluminescence (CL) substrate.
Notes and references
- H. Kettler, K. White and S. Hawkes, World Health Organization, 2004 Search PubMed.
- J. M. Cooper, Front. Lab. Chip. Technol., 2022, 1, 979398 CrossRef.
- J. Shreffler and M. R. Huecker, StatPearls, 2023 Search PubMed.
- K. E. Mullins and R. H. Christenson, Curr. Cardiol. Rep., 2020, 22, 1–10 CrossRef PubMed.
- L. Gervais and E. Delamarche, Lab Chip, 2009, 9, 3330–3337 RSC.
- J. Castillo-León, R. Trebbien, J. J. Castillo and W. E. Svendsen, Analyst, 2021, 146, 3750–3776 RSC.
- World Health Organization, https://www.who.int/news-room/questions-and-answers/item/simple-rapid-tests, (accessed June 2024).
- H. T. Kim, E. Jin and M. H. Lee, Biosensors, 2021, 11, 191 CrossRef CAS.
- G. S. Lim, S. M. Seo, S. H. Paek, S. W. Kim, J. W. Jeon, D. H. Kim, I. H. Cho and S. H. Paek, Sci. Rep., 2015, 5, 14848 CrossRef CAS PubMed.
- A. Xiang, F. Wei, X. Lei, Y. Liu, Y. Liu and Y. Guo, Eur. J. Clin. Microbiol. Infect. Dis., 2013, 32, 1557–1564 CrossRef CAS.
- J. Deng, M. Yang, J. Wu, W. Zhang and X. Jiang, Anal. Chem., 2018, 90, 9132–9137 CrossRef CAS PubMed.
- S. Ghosh and C. H. Ahn, Analyst, 2019, 144, 2109–2119 RSC.
- S. Ghosh, K. Aggarwal, V. Thiyagarajan Upaassana, T. Nguyen, J. Han and C. H. Ahn, Microsyst. Nanoeng., 2020, 6, 5 CrossRef CAS.
- U. Fritsche, Anal. Chim. Acta, 1980, 118, 179–183 CrossRef CAS.
- C. Y. Lee and C. W. Lien, U.S. Pat., No. 10711185, 2020 Search PubMed.
- X. Tao, W. Wang, Z. Wang, X. Cao, J. Zhu, L. Niu, X. Wu, H. Jiang and J. Shen, Luminescence, 2014, 29, 301–306 CrossRef CAS.
- A. Parandakh, O. Ymbern, W. Jogia, J. Renault, A. Ng and D. Juncker, Lab Chip, 2023, 23, 1547–1560 RSC.
- H. Yang and Z. Rao, Nat. Rev. Microbiol., 2021, 19, 685–700 CrossRef CAS.
- W. Feng, Y. Xiang, L. Wu, Z. Chen, Q. Li, J. Chen, Y. Guo, D. Xia, N. Chen, L. Zhang, S. Zhu and K. N. Zhao, J. Clin. Lab. Anal., 2022, 36, e24479 CrossRef CAS.
- Z. Bai, Y. Cao, W. Liu and J. Li, Viruses, 2021, 13, 1115 CrossRef CAS.
- J. Kai, A. Puntambekar, N. Santiago, S. H. Lee, D. W. Sehy, V. Moore, J. Han and C. H. Ahn, Lab Chip, 2012, 12, 4257–4262 RSC.
- L. Gervais, N. De Rooij and E. Delamarche, Adv. Mater., 2011, 23, H151–H176 CAS.
- C. C. Hong, J. W. Choi and C. H. Ahn, Lab Chip, 2004, 4, 109–113 RSC.
- C. Ahn, T. Lee, J. H. Shin, J. S. Lee, V. Thiyagarajan Upaassana, S. Ghosh and B. K. Ku, Microfluid. Nanofluid., 2023, 27, 72 CrossRef CAS.
- V. T. Upaassana, S. Ghosh, A. Chakraborty, M. E. Birch, P. Joseph, J. Han, B. K. Ku and C. H. Ahn, Anal. Chem., 2019, 91, 6652–6660 CrossRef CAS PubMed.
- A. Giussani, P. Farahani, D. Martínez-Muñoz, M. Lundberg, R. Lindh and D. Roca-Sanjuán, Chem. - Eur. J., 2019, 25, 5202–5213 CrossRef CAS PubMed.
- Z. Lin, P. Cao and H. Lei, Appl. Biochem. Biotechnol., 2008, 144, 15–26 CrossRef CAS PubMed.
- P. Khan, D. Idrees, M. A. Moxley, J. A. Corbett, F. Ahmad, G. von Figura, W. S. Sly, A. Waheed and M. I. Hassan, Appl. Biochem. Biotechnol., 2014, 173, 333–355 CrossRef CAS PubMed.
- L. J. Kricka, J. C. Voyta and I. Bronstein, Methods Enzymol., 2000, 305, 370–390 CAS.
- M. Zarei, TrAC, Trends Anal. Chem., 2017, 91, 26–41 CrossRef CAS.
- A. Shahzad, G. Köhler, M. Knapp, E. Gaubitzer, M. Puchinger and M. Edetsberger, J. Transl. Med., 2009, 7, 1–6 CrossRef PubMed.
- T. Tian, J. Li, Y. Song, L. Zhou, Z. Zhu and C. J. Yang, Lab Chip, 2016, 16, 1139–1151 RSC.
- J. A. Cruz-Cardenas, M. Gutierrez, A. López-Arredondo, J. E. Castañeda-Delgado, A. Rojas-Martinez, Y. Nakamura, J. A. Enciso-Moreno, L. A. Palomares and M. E. Brunck, Sci. Rep., 2022, 12, 17966 CrossRef CAS PubMed.
- MiCo BioMed Co., Ltd., https://micobiomed-usa.com/optimiser_platform_technology/optimiser-quick-reference-guide/, (accessed June 2024).
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Heeyeong Jang
a,
Heeyeong Jang