
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayer-by-layer assembly: an emerging, tailored and robust platform for solar water splitting
Wen-Cheng
Liu†
a,
Yan-Qing
Cai†
c and
Fang-Xing
Xiao
 *ab
*ab
aCollege of Materials Science and Engineering, Fuzhou University, New Campus, Minhou, Fujian Province 350108, China. E-mail: fxxiao@fzu.edu.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, PR China
cSchool of Advanced Manufacturing, Fuzhou University, Jinjiang, 362200, PR China
First published on 29th August 2025
Abstract
Photoelectrochemical (PEC) water splitting represents a highly promising technology to convert solar energy into clean and renewable chemical fuels. Among the various strategies utilized for customizing photoelectrodes, layer-by-layer (LbL) assembly has emerged as a green, simple, and easily accessible technique for rationally constructing multilayered heterostructures in terms of versatility, flexibility, and atomic-level interface configuration modulation. However, precise design of robust photoelectrodes based on LbL assembly still remains in the exploratory stage. This review comprehensively summarizes the recent advancements in the fabrication of composite multilayer photoanodes via LbL assembly, highlighting the LbL assembly construction with diverse substrates (e.g., metal oxides, transition metal sulfides) and building blocks of varying sizes and dimensions (e.g., quantum dots, nanoclusters, nanoparticles, nanosheets). Furthermore, this review underscores the role of these building blocks in extending the light absorption and improving the solar water oxidation performance. Most importantly, the latest endeavors devoted to mediating directional charge transfer routes in artificial PEC systems are specifically summarized. Finally, prospects and challenges of LbL assembly technology in photoelectrode engineering for PEC water splitting are outlined, aiming to inspire innovative strategies for the smart design of composite nanostructured photoelectrodes towards solar energy conversion.
1. Introduction
In recent decades, the technology of converting water into green hydrogen fuel using abundant, green, and sustainable solar energy has become a crucial solution to address the fossil fuel depletion crisis.1 Currently, the predominant solar-driven water-splitting methods include photocatalysis, photothermal catalysis, and photoelectrochemical (PEC) catalysis. However, photocatalytic hydrogen production is hindered by rapid charge recombination (<1 ns), low solar-to-hydrogen (STH) efficiency (<3%), and poor long-term stability.2 Photothermal catalysis consumes a large amount of additional energy. In contrast, a PEC cell leverages the photo-electron coupling mechanism to achieve STH conversion efficiencies exceeding 10% (e.g., copper indium gallium selenide and halide perovskite-based cells), signifying its emerging potential for industrial-scale hydrogen production.3 Compared with heterogeneous photocatalysis, PEC technology can employ a broader scope of interface engineering optimization strategies to enhance the charge separation and corrosion resistance of photoelectrodes, thereby improving the stability of hydrogen evolution. Moreover, PEC cells generally operate under mild conditions, significantly reducing the emission of harmful byproducts and providing a more cost-effective solution for solar-driven hydrogen production.4 Consequently, PEC water splitting has emerged as a key technology for solar-to-hydrogen conversion in the pursuit of sustainable energy solutions.In order to improve the STH conversion efficiency of PEC systems, previous endeavors have been mainly devoted to doping with impurity atoms or ions, surface passivation and modification, and designing Type-II or Z-scheme heterojunctions.5 Although these methods can improve the efficiency of photoelectrocatalysis to some extent, current research frontiers are mainly centered on exploring new composite photoelectrodes, but have met with limited success. The emergence of nanotechnology provides a feasible and effective way to readily construct high-efficiency composite artificial photosystems.6 Currently, bottom-up thin film fabrication methods primarily include Langmuir–Blodgett (LB), self-assembled monolayers (SAMs), chemical vapor deposition (CVD), hydrothermal techniques, and electrodeposition,7 but they inevitably encounter obstacles in practical applications. For example, the LB method typically requires semiconductor substrates with high mechanical strength. The SAM method faces issues of insufficient stability and a complex molecular design process when preparing a multilayer structure. The CVD method incurs high production costs in the actual fabrication of thin films and generates toxic byproducts that are harmful to the environment.8 Therefore, there is an urgent need to develop precise, efficient, and cost-effective thin film fabrication methods to integrate nanoscale building blocks with semiconductors for crafting novel composite photoelectrodes.
Thus far, the layer-by-layer (LbL) assembly approach has been developed to address the limitations of conventional bottom-up thin-film fabrication techniques. It is suitable for the composite assembly of a wide range of materials, including polyelectrolytes, nanoparticles, biomacromolecules, and organic small molecules, and facilitates the preparation of both organic and inorganic thin films.9 In addition, LbL assembly provides tunable multilayer nanoarchitectures, enabling precise control of key parameters such as film thickness, morphology, and properties at the nanoscale, with the preparation process being environmentally friendly and cost-efficient. Furthermore, LbL assembly improves the stability of composite nanomaterials through orderly interface design via molecular interactions such as electrostatic adsorption and covalent cross-linking.10 Consequently, LbL assembly provides a versatile platform for fabricating novel heterostructured photoelectrodes for solar energy conversion. Although some reviews have summarized the progress in the fabrication of spatially multilayered architectures via LbL assembly, they lack a systematic and insightful elaboration on the correlation between the structure of building blocks and PEC water splitting performances. Furthermore, charge transport characteristics mediated by the unique interface configuration endowed by LbL assembly have not yet been elucidated.
Motivated by these considerations, the current review aims to provide a detailed overview of the up-to-date advancements in PEC water splitting systems constructed by LbL assembly under ambient conditions. This review initially elucidates the essential principles of LbL assembly and PEC water splitting, followed by a comprehensive discussion of heterostructured photoelectrodes assembled with diverse types of substrates (e.g., metal oxides, transition metal sulfides, etc.) and functional building blocks of various sizes and dimensions (e.g., quantum dots, nanoclusters, nanoparticles, nanosheets, etc.). In addition, the present review summarizes the mechanistic correlation between the spatially hierarchical structures of multilayered photoelectrodes and their PEC water splitting performances. This work provides valuable information on the exploration of a large variety of LbL-assembled PEC systems with fine tuning of charge transport and separation for solar-to-hydrogen conversion.
2. A brief overview of LbL assembly
Layer-by-layer (LbL) assembly represents a nanoscale fabrication technique that exploits complementary interactions, such as electrostatic force and hydrogen bonding, to sequentially deposit materials onto interfaces with high precision.11 The concept of LbL assembly was first introduced in 1966 by Iler et al., who systematically studied the adsorption behavior of small particles, multivalent ions, and water-soluble polymers, laying the foundation for its interfacial mechanism.12 A major milestone in LbL assembly came in 1991 with the work of Decher and colleagues, who successfully fabricated and systematically characterized 100-layer polyelectrolyte films.13 This pioneering work established LbL assembly as a robust and adaptable platform for fabricating nanostructured thin films with diverse functional applications. Building on its core principle of integrating multiple functional building blocks, LbL assembly offers precise control over critical parameters, including film thickness, composition, and architecture, thereby enabling the rational design of spatially organized multilayered nanostructures on a wide range of substrates.14 Compared with conventional thin film fabrication techniques, LbL assembly provides superior precision, enhanced stability, and improved fabrication efficiency, addressing limitations such as poor interfacial control and material compatibility. These unique advantages have enabled LbL assembly to be widely adopted in various disciplines, including energy storage (e.g., supercapacitors and lithium-ion batteries), catalysis, biomedical drug delivery, electronics, and photonic devices.7aIn order to adapt to the assembly of building blocks with different geometric shapes and specific functional requirements, LbL assembly technology has developed assembly modes such as immersion assembly, spin assembly, spray assembly, fluidic assembly and electromagnetic assembly (Fig. 1a).15 Among these methods, spray assembly or spin assembly is generally applicable to planar substrates and requires complex equipment (e.g., high-pressure spray guns or centrifuges).10 In contrast, immersion assembly, performed within the solution phase, exhibits broad adaptability to substrates with diverse geometric characteristics, including porous structures, curved surfaces, and three-dimensional (3D) architectures. This method promotes the uniform deposition of functional building blocks, thereby offering advantages for the fabrication of structurally complex PEC devices. Furthermore, unlike fluidic assembly, which requires stringent control of hydrodynamic conditions, and electromagnetic assembly, which is limited to magnetically responsive materials,16 immersion assembly enables the formation of stable multilayer structures without the need for elaborate assembly protocols.17 Therefore, immersion assembly represents the most versatile approach for LbL assembly of photoelectrodes, owing to its broad material compatibility, operational simplicity, and precise controllability.
 | ||
| Fig. 1 (a) LbL assembly techniques for different assembly modes (reprinted with permission from ref. 16, Copyright 2016 American Chemical Society). (b) Schematic illustration of the immersion LbL assembly process (reprinted with permission from ref. 10, Copyright 2015, American Association for the Advancement of Science). (c) Molecular interactions between functional building blocks in LbL assembly (reprinted with permission from ref. 26, Copyright 2018 Multidisciplinary Digital Publishing Institute). (d) Actual photograph of the automated LbL assembly equipment designed by our group, used for LbL assembly of spatially multilayered heterostructure photoelectrodes. | ||
Beyond the different assembly techniques, the molecular interactions underlying the LbL assembly process include electrostatic interaction, hydrogen bonding interaction, covalent bonding interaction, host–guest interaction, and coordination chemistry interaction.18 Notably, electrostatic interaction represents the predominant driving force in LbL assembly, owing to its wide applicability, straightforward processing, and excellent interface stability.19 As illustrated in Fig. 1b, the initial step of electrostatically driven immersion LbL assembly involves immersing a positively charged substrate in an anionic building block solution containing functional substances, such as polymers or nanoparticles. This process results in the formation of a single-layer film on the substrate surface. Following each adsorption step, the substrate is thoroughly rinsed with ultrapure water to eliminate physically adsorbed substances while preserving the chemically bonded monolayer. Subsequently, the single-layer film is immersed in a cationic solution, facilitating the formation of a double-layer film structure through charge attraction.20 By repeating this cycle, a desired n-layer film structure can be achieved, allowing for precise control of composition and concentration at the nanoscale by adjusting the number of layers.21
In addition to the core electrostatic interaction mechanism, the key parameters regulating the LbL assembly process such as the charge density of the polyelectrolyte (PE), reaction temperature, pH value, ionic strength, and adsorption time are essential in determining the thickness, internal structure, and stability of the multilayer film.22 Importantly, precise adjustment of these parameters to optimize interfacial connection between the different building blocks remains an area of ongoing investigation. Establishing standardized and systematic assembly schemes is critical for enhancing the structural robustness and functional efficiency of LbL-assembled architectures. For example, in the MOs/(PDDA/Aux NCs)n multilayer heterostructure photoanodes with efficient cascade charge transfer characteristic, the oppositely charged PDDA and Aux NCs serve as building blocks for LbL assembly construction.23 It is essential to precisely adjust the concentration of building blocks and assembly cycle, as excessive polyelectrolyte and functional building block deposition can both lead to adverse effects. In addition, the assembly sequence between different building blocks is also crucial, as it is closely related to fine-tuning of electron transfer in the multilayer structure. For example, as demonstrated by Li et al., multilayer photoanodes prepared with different assembly sequences of PDDA and MXene quantum dots show significantly different photocurrent densities, implying that the cascade charge transfer pathway can be rationally designed via structure modulation.24 In general, the rational design of the LbL-assembled multilayer heterostructure needs to consider the following key points: (1) band alignment between building blocks and semiconductor materials such as Type-II and Z-scheme band structure, (2) the roles played by polyelectrolytes between building blocks such as promoting charge transfer, serving as molecular glue, and forming an interfacial dipole, and (3) stability of the connection between functional building blocks. The LbL assembly principle based on molecular interaction has been widely accepted and will provide inspiring guidance for the future design and optimization of LbL-assembled multilayer photoelectrodes.
As shown in Fig. 1d, our group has independently developed an automated LbL assembly device for fabrication of spatially multilayered heterostructure photoelectrodes. This equipment effectively addresses the issues of low efficiency, non-uniform coating, and poor quality in the traditional manual LbL process, demonstrating high automation levels with superior coating uniformity and quality. Specifically, the device enables precise control of critical parameters including assembly sequence, coating duration, deposition rate, and cycle number through an intelligent control system, significantly reducing manual operation time while overcoming the technical limitations of conventional methods in terms of efficiency and uniformity. Furthermore, the integrated N2 atmosphere protection and temperature control modules substantially enhance coating quality and film stability, offering considerable practical value. Notably, this LbL assembly system exhibits distinct advantages over conventional multilayer fabrication techniques such as CVD, electrodeposition, hydrothermal, LB, and SAM methods. Specifically, our LbL device operates even under ambient conditions with minimal material consumption (90% utilization rate) and broad compatibility with diverse inorganic or organic materials, demonstrating remarkable advantages in operational simplicity, safety, and cost-effectiveness.25 Consequently, leveraging our team's proprietary high-precision automated control system, LbL assembly technology is poised to overcome traditional process limitations, enabling efficient and stable large-scale production while providing robust technical support for industrial manufacturing of next-generation functional composite photoelectrodes.
3. Principle of PEC water splitting
PEC water splitting, which utilizes semiconductor materials to absorb solar energy and drive the redox reactions of water enabling the sustainable production of hydrogen, is regarded as a crucial strategy to alleviate the global energy crisis.27 The origins of PEC water splitting can be traced back to A. E. Becquerel's pioneering work in 1839, when he first observed the photocurrent in silver halide materials.28 This foundational discovery laid the groundwork for subsequent advancements in solar energy conversion. However, the true breakthrough in integrating PEC systems into solar energy conversion was propelled by the seminal contributions of Fujishima and Honda.29 As illustrated in Fig. 2a–c, the fundamental principle of PEC water splitting involves photoinduced redox reactions at semiconductor electrodes under light irradiation, resulting in the production of hydrogen (H2) and oxygen (O2).30 PEC reactions usually occur in electrochemical cells; in a typical PEC system, an electron-rich n-type semiconductor photoanode (e.g., TiO2, Fe2O3, or BiVO4) absorbs incident photons, exciting electrons from the valence band (VB) to the conduction band (CB), thereby generating electron–hole pairs. The photogenerated electrons migrate through an external circuit to the hole-rich p-type semiconductor (e.g., Cu2O, NiO, or CoO), where they reduce protons (H+) to H2. Simultaneously, the holes accumulated in the VB oxidize water molecules at the photoanode, releasing O2.31 Nevertheless, the rapid recombination of photogenerated electron–hole pairs critically impedes charge separation, representing a major barrier to achieving high-efficiency PEC water splitting. To address this issue, an external bias is typically applied in PEC systems to facilitate charge separation and effectively overcome the inherent energy barrier of semiconductors. Furthermore, co-catalysts (e.g., Pt, graphene, NiFe-layered double hydroxides) can be introduced in the fabrication of photoelectrodes to reduce the overpotential and enhance the reaction kinetics.32 By synergistically enhancing light absorption, facilitating charge separation, and optimizing surface catalytic activity, PEC water splitting enables highly efficient solar-to-hydrogen energy conversion. This technology provides a promising pathway for solar-driven hydrogen production, offering a critical solution to the efficient storage and utilization of renewable energy. | ||
| Fig. 2 Schematic illustration of PEC water splitting using (a) a photoanode, (b) photocathode, and (c) a tandem configuration combining both a photoanode and photocathode (reprinted with permission from ref. 30, Copyright 2014 Wiley-VCH Verlag). (d) CB and VB edges of semiconductor photocatalysts, along with their corresponding redox potentials in aqueous media (reprinted with permission from ref. 33, Copyright 2024 Royal Society of Chemistry). | ||
Specifically, the chemical reactions of hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) involved in the PEC water splitting reaction are as follows:
Reaction at the anode:
| 2H2O + 4h+ → O2 + 4H+ | (1) |
Reaction at the cathode:
| 4H+ + 4e− → 2H2 | (2) |
Net Reaction:
| 2H2O → 2H2 + O2 | (3) |
To comprehensively evaluate PEC system performance, several critical efficiency metrics are utilized. These metrics include the standard solar-to-hydrogen conversion efficiency (ηSTH), applied bias photon-to-current efficiency (ABPE), incident photon-to-current conversion efficiency (IPCE), charge transfer lifetime (τ), faradaic efficiency (FE), stability, and electrochemical active surface area (ECSA).34 Each parameter provides distinct insights into the catalytic characteristics of PEC systems. Among these indicators, ηSTH represents the ultimate performance metric for all solar conversion photosystems. The ηSTH is quantitatively defined as the ratio between the energy content of evolved hydrogen (calculated via Gibbs free energy ΔG = 237 kJ mol−1) and the incident solar energy input:35
 | (4) |
In PEC photosystems, ABPE is essential for quantifying the STH conversion efficiency under applied bias conditions. It quantifies the efficiency with which absorbed solar photons generate photocurrent at a specified applied bias. The ABPE is calculated using the following equation:36
 | (5) |
 | (6) |
 | (7) |
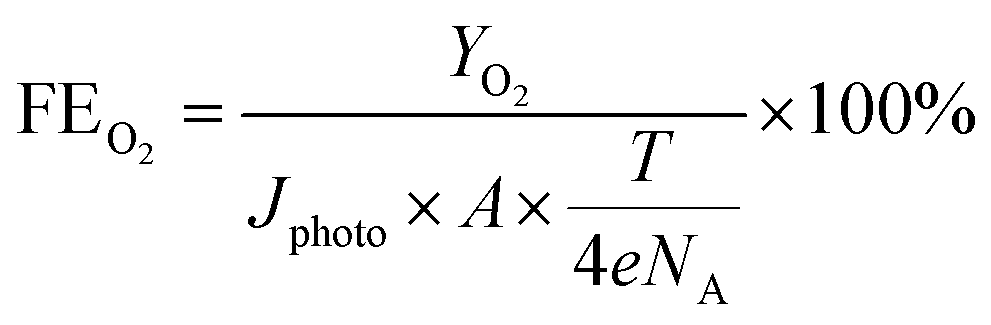 | (8) |
 | (9) |
Electrochemical active surface area (ECSA) refers to the actual surface area of electrode materials that participates in electrochemical reactions, reflecting effective catalyst utilization. Unlike geometric area, ECSA considers microstructural features such as surface roughness, porosity, and degree of active site exposure, making it a critical parameter for evaluating photoelectrode performance. The electrochemical double-layer capacitance (Cdl) at catalyst surfaces is typically determined by measuring non-faradaic capacitive currents from cyclic voltammetry curves at multiple scan rates. The ECSA calculation formula is as follows:38
 | (10) |
Efficient PEC water splitting holds potential to economically compete with conventional fossil fuel-based hydrogen production technologies, provided that its STH conversion efficiency approaches or exceeds approximately 10% (comparable to an efficiency threshold dictated by fossil fuel economics).39 The energy band structure of semiconductor materials primarily determines the achievable efficiency of PEC water splitting. For spontaneous PEC water splitting without an external bias, the semiconductor must have a minimum bandgap energy exceeding the thermodynamic requirement of 1.23 eV, corresponding to the Gibbs free energy for water splitting.40 Furthermore, the CB edge must lie at a more negative potential, and the VB edge must be more positive, relative to the hydrogen evolution (0 V vs. RHE) and oxygen evolution potentials (1.23 V vs. RHE), respectively. Fig. 2d summarizes the CB and VB positions of representative semiconductor photocatalysts, which are essential for rational adjustment and optimization of the energy-level structures of catalyst in designing photoelectrodes.41 A catalyst with a bandgap of 1.6–2.4 eV is generally considered optimal as it can maximize the absorption of sunlight while overcoming the overpotential in charge transfer between the photoanode and photocathode and minimizing energy loss. Beyond band structure considerations, PEC water splitting efficiency significantly depends on the interfacial charge transfer kinetics at the semiconductor–electrolyte interface. Achieving efficient charge separation and transport is essential for suppressing charge recombination and maximizing the overall photocurrent generation efficiency.42 Consequently, designing high-performance PEC systems necessitates a coordinated approach that integrates semiconductor band engineering with optimization strategies for enhancing charge separation, to enable efficient solar energy conversion.
4. Novel LbL-assembled photoelectrodes for PEC water splitting
In this section, we introduce the design and application of novel LbL-assembled heterostructures for PEC water splitting, focusing particularly on employing various substrate materials and assembling functional building blocks. We first categorize various semiconductors, including TiO2, WO3, ZnO, BiVO4, and CdS, as substrates for LbL assembly, leveraging their unique light absorption properties and energy band structures to enhance solar water oxidation efficiency. Additionally, we elucidate the rational integration of quantum dots (QDs) (e.g., transition metal chalcogenide QDs, graphene QDs, and MXene QDs), nanoclusters (NCs) (e.g., metal NCs and polyoxometalate NCs), and nanoparticles (NPs) (e.g., metal NPs and up-conversion NPs) as functional building blocks. These components exhibit a remarkable ability to promote charge separation or extend light absorption, which are the key factors in improving PEC performances. Furthermore, we investigate the LbL assembly of composite multilayer films utilizing graphene nanosheets and polyelectrolyte multilayers as the building blocks, where the constructed high-efficiency charge transfer pathways effectively enhance the PEC efficiency of photoelectrodes. By summarizing these assembly strategies, this section will elucidate the structure–performance relationship of spatially multilayered photoelectrodes fabricated by the LbL assembly technique.4.1. LbL assembly with different semiconductors as substrates
 | ||
| Fig. 3 (a) Top-view scanning electron microscopy (SEM) images, (b) high-resolution transmission electron microscopy (HR-TEM) images, and (c) transient photocurrent (I–t) responses of TNRAs/(PDDA/Agx NCs)4 multilayer heterostructures. (d) Schematic representation of the energy band structure of TNRA and Ag NCs [Ag9 (SG)6NCs, Ag16 (SG)9NCs, Ag31 (SG)19NCs] (reprinted with permission from ref. 47, Copyright 2022 Royal Society of Chemistry). (e) Schematic illustration for LbL assembly of TNTA/(BPEI/CdSe QDs)4 photoanodes. (f) Cross-sectional SEM images of (I) TNTAs and (II) TNTA/(BPEI/CdSe QDs)4 (reprinted with permission from ref. 48, Copyright 2019 Royal Society of Chemistry). | ||
Compared with rod-shaped TNRAs, tubular porous TiO2 nanotube arrays (TNTAs) have a larger specific surface area and more reactive sites, which can effectively improve the PEC performance.49 For example, Xiao et al. used a facile and easily accessible LbL assembly strategy to uniformly deposit metal (Au, Ag, Pt) colloidal NPs on the TNTAs and investigated the effect of the number of deposition cycles on the PEC performances of TNTAs/metal NP heterostructures.50 Specifically, PEC performances of the Au/TNTA heterostructure increase with the assembly cycle (n = 1, 2, 4, 6…), reach an optimal value at n = 8, and decrease with increasing deposition layer to 10 layers. This result is because excessive deposition of metal NPs leads to unfavorable agglomeration, which hinders the active sites of TNTAs. Therefore, this work demonstrates that, for the LbL assembly system of metal NPs and TiO2, an appropriate number of assembly cycles is the key factor affecting the PEC performances of the multilayer heterostructure photoanodes. As shown in Fig. 3e and f, in an analogous work, Hou et al. alternately deposited negatively charged transition metal chalcogenide (TMC) QDs and positively charged ultra-thin branched polyethyleneimine (BPEI) on the TNTA substrate, resulting in the TNTAs/(BPEI/CdX QDs)n (X = Se, Te, S) photoanodes.48 The UV-visible diffuse reflectance spectroscopy (DRS) results indicate that, compared to the original TNTAs, the ternary heterostructures with different assembly cycles all exhibit enhanced light absorption, with the TNTAs/(BPEI/CdX QDs)8 photoanode showing the strongest absorption. In addition, in a series of PEC tests, the TNTAs/(BPEI/CdX QDs)8 multilayer photoelectrode exhibited significantly enhanced water oxidation activity under simulated solar irradiation. This enhanced solar energy conversion efficiency is attributed to the tight interfacial bonding mode between TMC QDs and TNTAs enabled by the LbL assembly technique, which maximizes the strong photosensitizing effects of TMC QDs. In addition to TNRAs and TNTAs, one-dimensional TiO2 nanobelt (TNB) structures have also been utilized for LbL assembly construction. Specifically, Zhang et al. prepared the TNB substrate by a hydrothermal method, and then WO3 nanowires (WO3 NRs) were deposited on the TNBs by electrostatic attraction, and finally graphene oxide (GO) was attached on the TNBs/(WO3 NRs) framework to obtain the TNBs/(WO3 NRs/GO)n ternary nanohybrid heterostructures.51 This study reveal that the TNBs/(WO3 NRs/GO)n ternary composites exhibit the highest photocurrent density under visible light irradiation, which is attributed to the forming well-aligned Type-II energy level configuration between TNBs, WO3, and GO afforded by LbL assembly. The LbL assembly technique enables close integration of WO3 NRs and GR on the TNB scaffold, endowing the spatially multilayered nano-architecture with a cascade electron transfer channel that significantly accelerates charge transport. Consequently, rational selection of applicable functional building blocks in LbL assembly construction is of paramount importance.
 | ||
| Fig. 4 (a) Schematic illustration of the LbL assembly process for WO3 NPAs/(CdSe QDs/PSS)8 photoelectrodes. (b) Transient kinetic curves of WO3 NPAs/(CdSe QDs/PSS)8 multilayer photoanodes (reprinted with permission from ref. 54, Copyright 2022 Wiley-VCH Verlag). (c) Schematic diagram illustrating the LbL assembly of N-GQD/ZnO NW photoelectrodes. (d) Hydrogen production performance of N-GQD/ZnO NWs under simulated sunlight (100 mW cm−2). (e) Photostability of GQDs/ZnO NW and N-GQDs/ZnO NW heterostructures with zero bias vs. Ag/AgCl (reprinted with permission from ref. 57, Copyright 2016 Royal Society of Chemistry). Cross-sectional SEM images of (f) BiVO4 and (g) BiVO4/(Agx NCs/PAH)8 heterostructured photoanodes. (h) LSV curves of BiVO4/(Agx NCs/PAH)8 photoelectrodes (reprinted with permission from ref. 58, Copyright 2024 American Chemical Society). | ||
In another work on sheet-like WO3, Li et al. used an electrostatic LbL assembly strategy to deposit multilayers of PDDA and CdSe NCs on WO3 substrates to obtain WO3/(PDDA/CdSe)n heterostructured photoanodes.55 The results manifest that the synergistic interaction between PDDA as an efficient electron transfer medium and CdSe NCs as a photosensitizer contributes to the cascade charge transfer channels, notably accelerating the interfacial charge transfer kinetics and boosting charge separation, definitively endowing the WO3/(PDDA/CdSe)4 multilayer photoanodes with exceptional PEC water oxidation performance under simulated and visible light irradiation. Furthermore, the formation of this cascade charge transfer channels originates from the LbL assembly strategy, which endows the tightly bound WO3 and CdSe NCs with good energy level alignment. Specifically, PDDA and CdSe MSCs form highly ordered cascading electron transfer pathways, further accelerating the spontaneous transfer of photogenerated electrons from the LUMO of CdSe NCs to the CB energy level of WO3 NPA, thereby enhancing charge transfer and separation efficiency. Besides the sheet-like WO3 materials, Lin et al. assembled Au NPs on the WO3 nanorods (NRs) via a facile LbL assembly approach to obtain the Au/WO3 multilayer structure.56 In this work, alternate deposition of PSS and PDDA imparts a positive charge to the WO3 NR surface, enabling the LbL assembly with negatively charged Au NPs. The role of Au NPs as electron traps rather than plasmonic photosensitizers accelerates interfacial electron transfer, thereby extending the lifetime of photoinduced electron–hole pairs. Based on the above, considering the representative multilayer photoanodes fabricated via LbL assembly incorporating WO3 and CdSe, where WO3 serves as an ideal substrate model with excellent stability, future research would greatly enhance this ideal model by introducing more controllable building blocks.
4.2. LbL assembly with quantum dots as building blocks
As 0D nanomaterials, QDs exhibit unique PEC properties closely related to their size.72 The size of QDs is comparable to or smaller than the exciton Bohr radius of their bulk materials, showing a pronounced quantum size effect and allowing precise bandgap tuning by controlling the particle size. Therefore, in LbL assembly, QDs enable forming suitable energy level alignment with the substrate materials, thereby enhancing the charge transport between different building blocks and thus improving the PEC water splitting performances. Additionally, QDs possess a high surface-to-volume ratio and exhibit a quantum tunneling effect, making them ideal building blocks for constructing multilayered photoanodes.22a This section provides a comprehensive overview of high-efficiency photoelectrodes fabricated using diverse QDs including TMC QDs, graphene QDs, and MXene QDs as primary building blocks for LbL assembly. | ||
| Fig. 5 (a) Schematic illustration for LbL assembly of TNTAs/(CdX@AET QDs/CdX@MAA QDs)n multilayered photoanodes. (b) I–t curves of TNTAs/(CdX@AET QDs/CdX@MAA QDs)n photoanodes (reprinted with permission from ref. 78, Copyright 2021 American Chemical Society). (c) HR-TEM images of TNTAs/(TMC QDs/PSS)2 photoanodes. (d) I–t curves and (e) EIS results of TNTAs/(TMC QDs/PSS)2 photoanodes under visible light irradiation (reprinted with permission from ref. 79, Copyright 2020 American Chemical Society). (f) TEM image of MoS2 QD aqueous solution and histogram of size distribution. (g) UV-Vis absorption spectra of MoS2 QD aqueous solutions, as well as photographs and bandgap energy measurements. (h) PL spectra of MoS2 QD aqueous solution at different excitation wavelengths (reprinted with permission from ref. 80, Copyright 2020 Royal Society of Chemistry). | ||
 | ||
| Fig. 6 (a) Schematic illustration of the LbL assembly process for the fabrication of (M/GQDs)n (M = Au, Ag, Pt) multilayer thin films. (b) Atomic force microscopy (AFM) image and (c) LSV curves of the (Au/GQDs)10 multilayer film (reprinted with permission from ref. 84, Copyright 2017 Royal Society of Chemistry). (d) Charge injection efficiency of TiO2/(PDDA/MQDs)6 and TiO2 photoanodes. (e) Photocurrents of TiO2/(PDDA/MQDs)6 and TiO2 photoanodes under continuous simulated sunlight irradiation (AM 1.5G) (reprinted with permission from ref. 24, Copyright 2023 Wiley-VCH Verlag). (f) Schematic illustration depicting the PEC water oxidation mechanism of TNRAs/(TMC QDs/MQDs)8 photoanodes. (g) Carrier densities of TNRAs, TNRAs/(TMC QDs)8 and TNRAs/(TMC QDs/MQDs)8 photoanodes (reprinted with permission from ref. 86, Copyright 2024 Wiley-VCH Verlag). | ||
4.3. LbL assembly with nanoclusters as building blocks
 | ||
| Fig. 7 (a) Ball-and-stick and (b) space-filling models of Au25(SCH2CH2Ph)18 NCs (reprinted with permission from ref. 98, Copyright 2013 American Chemical Society). (c) X-ray crystal structure and (d) methyl group orientations of [Ag24Au(SPhMe2)18]− (reprinted with permission from ref. 99, Copyright 2015 Wiley-VCH Verlag). (e) Schematic diagram of the PEC water splitting mechanism for TNTAs/(BPEI–AuxNCs)2 photoanodes. (f) HRTEM image of the TNTAs/(BPEI/AuxNCs)2 heterostructure photoanode (reprinted with permission from ref. 95, Copyright 2023 Wiley-VCH Verlag). (g) EIS results and (h) Bode plots of WO3/(PDDA/Aux)2 (reprinted with permission from ref. 23, Copyright 2020 American Chemical Society). (i) I–t curves (bias: 1.0 V vs. RHE) of TNRAs/(PDDA/Au1−xAgx NCs)4 multilayer photoanodes (reprinted with permission from ref. 97, Copyright 2023 Wiley-VCH Verlag). | ||
In addition to Au NCs, Tang et al. also comprehensively explored the efficient role of multilayer Ag NCs [Agx@GSH, Ag9@GSH, Ag16@GSH, and Ag31@GSH NCs] and PDDA deposition on the TNRA substrate.47 Compared with bare TNRAs, the LbL-assembled TNRAs/(PDDA/Ag NCs)4 heterostructure exhibits significantly enhanced carrier density, which is attributed to the role of PDDA as an efficient electron-trapping mediator that accelerates the charge transfer between the adjacent Ag NCs. Compared with monometallic NCs, alloy NCs composed of multiple metal atoms exhibit more intriguing PEC properties. For instance, Su et al. utilized a LbL assembly technique to alternately deposit Au1−xAgx@GSH NCs and PDDA on the TNRA substrate.97 As shown in Fig. 7i, the TNRAs/(PDDA/Au1−xAgx NCs)4 photoanodes exhibited significantly elevated photocurrent compared with the unexcited TNRA photoanodes under visible light irradiation. Furthermore, the PEC water splitting performances of TNRAs/(PDDA/Au1−xAgx NCs)n photoanodes with varied LbL assembly sequences were also systematically investigated. The results suggested that the LbL assembly sequence could profoundly influence the electron transfer pathway and thereby enable precise control over charge separation and transfer between MNCs and metal oxides. However, it should be considered that TNRAs/(PDDA/Au1−xAgx)4 demonstrated limited photostability due to the deactivation of Au1−xAgx@GSH NCs caused by superoxide radical oxidation. Therefore, for various alloy NCs (AuPt, AuCu), how to design multilayer structures that can protect the active metal atoms remains an important area for further investigation, similar to the specific protective effect of BPEI on Aux NCs. This series of studies on MNCs highlights the universal significance of constructing a directional charge transfer route via LbL assembly, providing valuable insights for the rational design of MNC-based photoelectrodes. It is worth emphasizing that these MNCs with well-defined energy level structures and atomic configurations demonstrate universal compatibility with MOs. The assembly model of “MOs/non-conjugated polymer/MNCs”, which establishes a well-oriented charge transfer pathway, can provide systematic theoretical guidance for the future design and optimization of LbL-assembled PEC devices.
 | ||
| Fig. 8 (a) Model diagrams of classic Keggin anion [XM12O40]3− (X = P, Si, Bi, etc., M = Mo, W, V, Nb, etc.) (reprinted with permission from ref. 101, Copyright 2018 Springer Nature). (b) Diagram depicting the LbL assembly of Mn6SiW and g-C3N4 composite films (reprinted with permission from ref. 104, Copyright 2021 Multidisciplinary Digital Publishing Institute). (c) Schematic diagram of photoanodes prepared using different polyelectrolytes and metal oxides (reprinted with permission from ref. 107, Copyright 2017 American Chemical Society). (d) OER performance of an Fe2O3/(Co-POM/GO)n multilayer photoanode (reprinted with permission from ref. 108, Copyright 2018 American Chemical Society). (e) LSV curves and (f) stability of Cu2O and BiVO4 PEC cells (reprinted with permission from ref. 109, Copyright 2018 Royal Society of Chemistry). | ||
Inspired by these studies, Ryu's research group further conducted a comprehensive investigation of POM NCs as primary building blocks for fabricating spatially multilayered photoanodes. As shown in Fig. 8c, a LbL assembly strategy was employed to deposit various cationic polyelectrolytes including BPEI, PDDA, PAH, or linear-poly(ethylenimine) (l-PEI), along with tetracobalt-substituted polyoxometalate [Co4(H2O)2(PW9O34)2]10− (Co-POM) on different metal oxide substrates (Fe2O3, BiVO4, TiO2).107 Interestingly, regardless of the type of PE used, the Fe2O3/(PE/Co-POM)n photoanodes exhibit significant PEC performance improvement compared with bare Fe2O3. This is attributed to the alternating cationic and anionic material layers formed by LbL assembly, which function as interfacial dipoles and help reduce charge transfer resistance. Furthermore, when the same deposition was applied to BiVO4 and TiO2 substrates, the resulting photoanodes show notable enhancements in both stability and PEC performances. To further demonstrate the advantages of the LbL assembly, a randomly mixed film compromising b-PEI and POM was prepared on Fe2O3via a drop-casting method. However, compared with the LbL-assembled Fe2O3/(b-PEI/Co-POM)10 multilayer heterostructure photoanode, it exhibits negligible photocurrent density. This indicates that well-defined interface structure and integration mode enabled by LbL assembly are crucial for enhancing the PEC performances. Moreover, Choi et al. deposited positively charged graphene oxide (GO) and negatively charged Co-POM [Co4(H2O)2(PW9O34)2]10− on Fe2O3 through LbL assembly technology.108 The results demonstrate that compared with bare Fe2O3, the Fe2O3/(Co-POM/GO)n spatially multilayered photoanode exhibits significantly enhanced OER performance (Fig. 8d) while maintaining good stability during prolonged PEC reactions. This enhancement stems from GO serving as an efficient hole transport material and Co-POM acting as an effective water oxidation catalyst, with the tightly integrated multilayer film enabled by LbL assembly maximizing their synergistic effects on electron transfer. In another study, Kim et al. used a LbL assembly strategy to deposit Co-POM [Co4(H2O)2(PW9O34)2]10− and l-PEI on BiVO4, resulting in BiVO4/(l-PEI/Co-POM)n multilayer photoanodes, while nickel-containing POM [Ni4(H2O)2(VW9O34)2]10− (Ni-POM) and PEI were assembled on the Cu2O, forming the Cu2O/(l-PEI/Ni-POM)n photocathodes.109 The results revealed that the PEC cell achieved unassisted water splitting under zero external bias, with its photocurrent density demonstrating remarkable stability for at least 2 h (Fig. 8e and f). This significant enhancement can be attributed to: (1) improved charge transfer due to catalyst deposition; (2) enhanced charge transport to the catalyst through the formation of interfacial dipoles; (3) increased stability resulting from the protective coating formation. Another noteworthy aspect is that conventional functionalization methods for photoelectrodes are typically material-specific and require harsh processing conditions (e.g., vacuum/high-temperature treatments) along with complex chemical syntheses, often involving toxic/hazardous chemicals (such as HF and H2SO4). In contrast, the LbL assembly strategy developed in this study enables the fabrication of PEC devices entirely utilizing aqueous building block solutions under ambient conditions, representing an environmentally benign approach. This simple, universally applicable, and eco-friendly methodology demonstrates significant practical value for real-world applications. Therefore, the deposition of highly catalytically active POMs using LbL assembly technology demonstrates definitive and universal significance for enhancing the PEC performance of MOs. In particular, [Co4(H2O)2(PW9O34)2]10− serves as an ideal building block that will provide guiding principles for the design of future multilayer spatial structures.
4.4. LbL assembly with nanoparticles as building blocks
 | ||
| Fig. 9 (a) Schematic illustration of electron transfer pathways in TNTAs/Au NPs/Aux NCs multilayer nanostructured photoanodes (reprinted with permission from ref. 115, Copyright 2015 American Chemical Society). (b) Faradaic efficiency of Fe2O3/(Co-POM)10/(Ag NPs)5 photoanodes. (c) A diagram showing the LbL assembly of UCNPs, Ag NPs, and POM. (d) PL spectra of (UCNP/PSS/Ag/PSS)3. (e) LSV curves of (Ag NPs/UCNPs)5/Fe2O3/(Co-POM)5 (reprinted with permission from ref. 116, Copyright 2019 Wiley-VCH Verlag). (f) Flowcharts for LbL assembly of GNs/CdS QDs, GNs, and CdS QDs multilayers (reprinted with permission from ref. 120, Copyright 2014 American Chemical Society). (g) Schematic illustration of LbL-assembled polyelectrolyte multilayers and charge carrier transport pathways during PEC water oxidation (reprinted with permission from ref. 121, Copyright 2019 Wiley-VCH Verlag). | ||
4.5. LbL assembly with layered materials as building blocks
5. Structure–performance mechanism analysis
In recent years, significant progress has been made in fabricating spatially multilayered heterostructure photoelectrodes by integrating diverse functional building blocks using the LbL assembly technique. However, due to the wide scope of building blocks and the lack of precise characterization tools, the structure–performance mechanism underlying LbL-assembled PEC devices remains unclear. In this section, we summarize the several classic models of functional building blocks (e.g., TMC QDs, Ag NCs, and Au NCs) combined with metal oxides (MOs) to construct the multilayer heterostructure photoelectrodes. We further elucidate the correlation between the cascade charge transfer pathways enabled by these multilayer structures and their PEC performances. From the perspective of band structure, applicable energy level alignment between the building blocks and semiconductor substrate is fundamental to optimizing charge migration and separation. As shown in Fig. 10a, the energy level alignment between CdX QDs (X = Se, Te, S) modified with different ligands [2-aminoethanethiol (AET) and mercaptoacetic acid (MAA)] and the classical n-type semiconductor TiO2 illustrates this optimization.78 In such a carrier transfer model, photoexcited electrons are transferred from the CB of CdX QDs to the CB of TiO2, participating in the reduction reaction. Similarly, in the multilayer heterostructure photoelectrode constructed with Ag NCs or Au NCs, photoexcited electrons migrate from the LUMO of metal NCs to the CB of TiO2 (Fig. 10b and c).90b,130 | ||
| Fig. 10 (a) Schematic diagram of energy level matching between different CdX QDs (X = Se, Te, S) capped with AET and MAA ligands and TiO2 (reprinted with permission from ref. 78, Copyright 2021 American Chemical Society). (b) Schematic diagram of energy level matching between different Ag NCs and TiO2 (reprinted with permission from ref. 130, Copyright 2023 American Chemical Society). (c) Schematic diagram of energy level matching between different Au NCs and TiO2 (reprinted with permission from ref. 90b, Copyright 2023 Royal Society of Chemistry). (d) Schematic illustration of the tandem charge transfer pathway of MOs/(PDDA/Aux NCs)n heterostructures (reprinted with permission from ref. 23, Copyright 2021 American Chemical Society). (e) Schematic diagram depicting the charge transport characteristic of TiO2/(PDDA/MQDs)n photoanodes (reprinted with permission from ref. 24, Copyright 2023 Wiley-VCH Verlag). | ||
On the other hand, it is important to note that polyelectrolyte (PE) thin films are a critical component in constructing multilayer heterostructure photoelectrodes, as exemplified by the classic MOs/(PE/metal NCs)n photosystems. During the LbL assembly process, PE thin films provide a strong electrostatic force to stabilize the integration of functional building blocks with different charge properties. Furthermore, the critical role of PE interlayers in facilitating charge transfer within multilayer architectures has been experimentally demonstrated. For example, the WO3/(PDDA)2 photoanode shows significantly enhanced photocurrent compared with WO3/SiO2/(PDDA)2 with an interim insulating layer, highlighting the crucial role of PDDA in accelerating interfacial charge transfer.23 In summary, the key roles of PE in LbL-assembled multilayer photoelectrodes are as follows: (1) acting as electrostatic adhesives for combining functional building blocks with opposite charge properties, (2) serving as an essential medium to improve charge migration and separation between the semiconductor substrate and functional building blocks, (3) forming interfacial dipoles that improve the flat-band potential of photoanodes and create more favorable thermodynamic conditions for water splitting reactions, and (4) functioning as an effective protective coating to reduce corrosion of multilayer photoelectrodes. Therefore, with modulating the favorable energy level alignment and harnessing the critical roles of PEs, coupled with the face-to-face intimate integration interface enabled by LbL assembly, a directional charge transfer mechanism is stimulated that significantly enhanced the charge transport and separation efficiency.
As illustrated in Fig. 10d and e, schematic diagrams depicting the cascade charge transfer mechanisms of MOs/(PDDA/Aux NCs)n and TiO2/(PDDA/MQDs)n multilayer heterostructure photoanodes are presented.23,24 Specifically, in the MOs/(PDDA/Aux NCs)n multilayer heterostructure, ultrathin PDDA layers are alternately and tightly intercalated at the interface of functional building block layers, acting as an efficient electron transfer mediator. Therefore, the non-conjugated polymer of PDDA accelerates the unidirectional electron transfer from the adjacent Aux NC layer to the CB of MOs (WO3, TiO2, Fe2O3). Through this configuration design, the spatially stacked ultrathin PDDA layers serve as relays for cascade electron transfer, effectively suppressing the charge recombination over Aux NCs. Hence, the synergistic interaction of the alternately deposited ultrathin PDDA layer and Aux@GSH NCs endowed by LbL assembly as a charge transfer mediator and light harvesting antennae for constructing tandem charge transfer channel leads to the substantially enhanced PEC water oxidation activities of MOs/(PDDA/Aux)2 heterostructures under simulated solar and visible light irradiation. Moreover, extensive experimental evidence demonstrates that this cascade charge transfer pathway is universally applicable to a wide variety of building blocks and substrate materials. Therefore, spatially multilayer photoelectrodes with cascade charge transfer pathways fabricated via the LbL assembly strategy provide critical paradigms for the future design of functionalized high-performance PEC devices.
6. Outlook and perspectives
The LbL assembly technique has become a widely adopted strategy for constructing spatially multilayer heterostructure photoelectrodes with an optimized charge transfer pathway, owing to its precise control over interfacial structure at the atomic scale. While the LbL assembly technique exhibits remarkable potential for constructing efficient PEC water splitting systems, significant challenges remain in the rational integration of advanced building blocks at the microstructural level to further improve STH conversion efficiency, which includes the following aspects:(1) Structural stability under complex conditions: the long-term operational efficiency and durability of multilayered nanostructured photoelectrodes fabricated by LbL assembly, especially under intense light irradiation and in harsh aqueous environments, have not yet been fully achieved. Currently, functional layers are primarily connected by simple electrostatic interactions, which are not enough to maintain long-term structural stability. Future research should focus on the development of PEC devices incorporating stronger intermolecular interactions. For example, rational design of multilayer nanostructures supported by a combination of molecular interactions (e.g., electrostatic forces, hydrogen bonds, covalent bonds, and biospecific interactions) could fundamentally enhance the stability between the functional layers. Moreover, integrating LbL assembly technology with scalable manufacturing techniques such as photolithography, inkjet printing, or 3D printing, while maintaining its inherent flexibility and precision, could significantly accelerate the design and development of next-generation PEC devices.
(2) Advanced functional building blocks: the exploration of novel materials remains a primary driving force for improving the PEC water splitting performance. Although this review summarizes several photosensitizers and co-catalysts used in LbL assembly, many promising candidate materials in the PEC field remain underexplored. For instance, perovskite materials with high photoelectric conversion efficiency, biomimetic catalysts with superior light-harvesting properties, and single-atom catalysts with exceptional catalytic activity possess significant potential for further development. It is anticipated that scientific integration of these powerful building blocks via the LbL assembly strategy could enable unprecedented STH conversion efficiencies.
(3) Mechanistic elucidation of LbL-assembled PEC devices and PEC reaction: due to the limitations of conventional characterization tools, the current understanding of the growth process and PEC water splitting reaction mechanism of LbL-assembled multilayer nanostructures is mainly at the theoretical level. Specifically, multilayer thin-film photoelectrodes prepared from ultrathin polyelectrolytes possess an ultralow deposition amount, making it challenging to directly observe their internal structures using conventional characterization tools such as SEM, TEM, or atomic force microscopy (AFM). Utilizing more advanced characterization techniques like aberration-corrected transmission electron microscopy (AC-TEM), in situ TEM, and focused ion beam scanning electron microscopy (FIB-SEM) is crucial for confirming the integrated architectures of various functional building blocks. Moreover, unraveling the rapid and complex charge separation and transfer process in PEC water splitting reactions is critical for improving the STH conversion efficiency of PEC devices. In the future, more advanced in situ characterization methods such as transient absorption spectroscopy (TAS), ambient pressure X-ray photoelectron spectroscopy (AP-XPS), and secondary ion mass spectrometry (SIMS) can provide deeper insights into these unexplored processes. In addition, introduction of theoretical computational models in LbL-assembled multilayer heterostructures will provide valuable insights into the interaction between different building blocks. For example, Yu et al. employed first principles density functional theory (DFT) to investigate the binding modes between different components in LbL-assembled multilayer structures.131 Specifically, the Multiwfn software based on efficient algorithms can be used to evaluate the strength of electrostatic interaction in diverse binding configurations, while VMD (Visual Molecular Dynamics) can generate color-coded van der Waals surface diagrams of various binding modes and calculate their corresponding binding energies, visually demonstrating the steric effects. By constructing theoretical models of different building block materials, parameters such as binding energies, charge densities, oxygen vacancies, and free energy changes can be computed, thereby providing more definitive explanations for reaction mechanisms.132 These computational models establish a rigorous theoretical framework for elucidating interfacial interaction in LbL-assembled systems, whose deeper implementation should be prioritized in future studies.
(4) Intelligent AI design: the integration of artificial intelligence (AI) and machine learning technologies into LbL assembly and PEC reactions undoubtedly represents a critical future research direction. By leveraging AI algorithms, researchers can rapidly screen potential high-performance building block materials based on existing data such as physicochemical properties, energy level alignment, and interfacial interaction.133 For instance, combining high-throughput computational and experimental databases, AI can predict optimal material combinations and assembly strategies, thereby significantly reducing experimental exploration time and costs. Furthermore, deep learning techniques can analyze how various parameters including deposition sequence, duration, and concentration influence the final PEC performances, establishing the comprehensive parameter-performance mapping models. Additionally, AI-based predictive models can forecast performance degradation in multilayer photoelectrodes during prolonged operation, identifying key failure mechanisms such as material decomposition or building block deactivation and proposing targeted optimization strategies.134 Following this developmental roadmap, future AI-embedded LbL and PEC systems will achieve fully automated deposition control and intelligent experimental analysis through real-time data acquisition and processing, ultimately advancing intelligent, modular, and highly efficient operation.
However, this intelligent transformation faces significant challenges. For example, the LbL assembly process involves diverse materials and complex interfacial interactions, yet currently available high-quality data such as precise energy level alignment and intermolecular forces remain limited, constraining AI model accuracy. Addressing this requires collaborative efforts to generate large-scale, high-precision databases through high-throughput experimental platforms and advanced in situ characterization techniques such as in situ XPS and Raman spectroscopy. Moreover, designing multilayer photoelectrodes for PEC applications involves intricate physicochemical processes that current AI models struggle to fundamentally comprehend and explain. Developing AI algorithms that intrinsically incorporate physicochemical principles while integrating empirical data from techniques like Kelvin probe force microscopy and EIS may provide deeper mechanistic insights.135 Most crucially, realizing this vision demands sophisticated hardware support, including precision sensors, real-time data acquisition systems, and high-speed computing capabilities, all of which will undoubtedly mature alongside advancements in computing and energy technologies. Therefore, establishing comprehensive databases to support reliable benchmark AI models has emerged as an urgent priority, necessitating coordinated efforts across the global LbL research community.
7. Conclusions
In summary, this work comprehensively summarized the latest developments of PEC water oxidation mediated by LbL assembly. The principles of LbL assembly and PEC water splitting are concisely introduced, and then a large variety of spatially multilayer heterostructure photoelectrodes fabricated by LbL assembly are elucidated, followed by a presentation of the emerging vision in this booming research field. The present review will provide fresh insights and practical guidance for the strategic design of advanced PEC devices via the LbL assembly strategy, thereby offering strong technical support for the large-scale development of green hydrogen energy in the future.Author contributions
Wen-Cheng Liu and Yan-Qing Cai drafted the manuscript. Fang-Xing Xiao guided this work and corrected the manuscript. All the authors contributed to a critical discussion on the data and the manuscript.Conflicts of interest
The authors declare no conflict of interest.Abbreviations
| LbL | Layer-by-layer |
| STH | Solar-to-hydrogen |
| PEC | Photoelectrochemical |
| LB | Langmuir Blodgett |
| SAMs | Self-assembled monolayers |
| CVD | Chemical vapor deposition |
| 0D | Zero-dimensional |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| TMC QDs | Transition metal chalcogenide quantum dots |
| GR | Graphene |
| GO | Graphene oxide |
| VB | Valence band |
| CB | Conduction band |
| RHE | Reversible hydrogen electrode |
| HER | Hydrogen evolution reaction |
| OER | Oxygen evolution reaction |
| LSV | Linear sweep voltammetry |
| OCVD | Open-circuit photovoltage decay |
| ABPE | Applied bias photon-to-current efficiency |
| IPCE | Incident photon-to-current conversion efficiency |
| FE | Faradaic efficiency |
| PL | Photoluminescence |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| AFM | Atomic force microscopy |
| PSS | Poly(4-styrenesulfonic acid) |
| FTO | Fluorine-doped tin oxide |
| ITO | Indium tin oxide |
| NPAs | Nanoplate arrays |
| NRs | Nanorods |
| MOs | Metal oxides |
| TNRAs | TiO2 nanorods |
| MOF | Metal–organic framework |
| TNTAs | TiO2 nanotube arrays |
| GSH | L-Glutathione |
| PDDA | Poly(diallyl-dimethylammonium chloride) |
| BPEI | Branched poly-ethyleneimine |
| l-PEI | linear-poly(ethylenimine) |
| TNBs | TiO2 nanobelts |
| NPs | Nanoparticles |
| NWs | Nanowires |
| N-GQDs | Nitrogen-doped graphene quantum dots |
| PAH | Poly(allylamine hydrochloride) |
| QDs | Quantum dots |
| AET | 2-Aminoethanethiol |
| MAA | Mercaptoacetic acid |
| NCs | Nanocrystals |
| MNCs | Metal nanoclusters |
| POMs | Polyoxometalates |
| MNPs | Metal nanoparticles |
| SPR | Surface plasmon resonance |
| UCNPs | Up-conversion nanoparticles |
| NIR | Near-infrared |
| GNs | Graphene nanosheets |
| rGO | Reduced graphene oxide |
| AI | Artificial intelligence |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
The support by the award Program for Minjiang scholar professorship is gratefully acknowledged. This work was financially supported by the National Natural Science Foundation of China (No. 21703038, 22072025) and Fujian Institute of Research on the Structure of Matter, Chinese Academy of Science acknowledged (No. 20240018).References
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- H. Wu, H. L. Tan, C. Y. Toe, J. Scott, L. Z. Wang, R. Amal and Y. H. Ng, Adv. Mater., 2019, 32, 1904717 CrossRef PubMed.
- (a) W. Chen, D. H. Wang, W. Y. Wang, X. Liu, Y. Y. Liu, C. Wang, Y. Kang, S. Fang, X. D. Yang, W. G. Gu, D. Y. Luo, Y. M. Luo, Z. T. Qu, C. J. Zuo, Y. Kang, L. Cheng, W. S. Yan, W. Hu, R. Long, J. H. He, K. Liang, S. Liu, Y. J. Xiong and H. D. Sun, Nat. Commun., 2025, 16, 879 CrossRef CAS PubMed; (b) J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- F. Safari and I. Dincer, Energy Convers. Manage., 2020, 205, 112182 CrossRef CAS.
- B. Alfakes, C. Garlisi, J. Villegas, A. Al-Hagri, S. Tamalampudi, N. S. Rajput, J. Y. Lu, E. Lewin, J. Sá, I. Almansouri, G. Palmisano and M. Chiesa, Surf. Coat. Technol., 2020, 385, 125352 CrossRef CAS.
- C. H. Kwon, Y. Ko, D. Shin, M. Kwon, J. Park, W. K. Bae, S. W. Lee and J. Cho, Nat. Commun., 2018, 9, 4479 CrossRef PubMed.
- (a) A. Wichmann, G. Schnurpfeil, J. Backenköhler, L. Kolke, V. A. Azov, D. Wöhrle, M. Bäumer and A. Wittstock, Tetrahedron, 2014, 70, 6127–6133 CrossRef CAS; (b) Y. Paz, Beilstein J. Nanotechnol., 2011, 2, 845–861 CrossRef CAS PubMed.
- B. J. Liu, Q. Chen, Q. L. Mo and F. X. Xiao, Coord. Chem. Rev., 2023, 493, 215285 CrossRef CAS.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707–718 RSC.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, 2491 CrossRef PubMed.
- F. X. Xiao, M. Pagliaro, Y. J. Xu and B. Liu, Chem. Soc. Rev., 2016, 45, 3088–3121 RSC.
- R. K. Iler, J. Colloid Interface Sci., 1966, 21, 569–594 Search PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210, 831–835 CrossRef.
- B. Y. Ning, S. H. He, X. Lin and F. X. Xiao, Inorg. Chem., 2024, 63, 23742–23748 CrossRef CAS PubMed.
- I. M. Thomas, Appl. Opt., 1987, 26, 4688–4691 CrossRef CAS PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- S. Dey, K. Mohanta and A. J. Pal, Langmuir, 2010, 26, 9627–9631 CrossRef CAS PubMed.
- Y. J. Zhang, S. G. Yang, Y. Guan, W. X. Cao and J. Xu, Macromolecules, 2003, 36, 4238–4240 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831–1841 Search PubMed.
- S. Hou, M. H. Huang, Y. B. Li, S. Xu, X. Lin, X. Y. Fu and F. X. Xiao, Inorg. Chem., 2020, 59, 16654–16664 CrossRef CAS PubMed.
- (a) S. T. Dubas, T. R. Farhat and J. B. Schlenoff, J. Am. Chem. Soc., 2001, 123, 5368–5369 CrossRef CAS PubMed; (b) P. Bieker and M. Schönhoff, Macromolecules, 2010, 43, 5052–5059 CrossRef CAS.
- Z. Q. Wei, S. Hou, X. Lin, S. Xu, X. C. Dai, Y. H. Li, J. Y. Li, F. X. Xiao and Y. J. Xu, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed.
- S. Li, Q. L. Mo, S. C. Zhu, Z. Q. Wei, B. Tang, B. J. Liu, H. Liang, Y. Xiao, G. Wu, X. Z. Ge and F. X. Xiao, Adv. Funct. Mater., 2022, 32, 2110848 CrossRef CAS.
- B. J. Starr, V. V. Tarabara, M. Herrera-Robledo, M. Zhou, S. Roualdès and A. Ayral, J. Membr. Sci., 2016, 514, 340–349 CrossRef CAS.
- S. C. Zhang, M. Xing and B. Y. Li, Int. J. Mol. Sci., 2018, 19, 1641 CrossRef PubMed.
- S. Li, Q. L. Mo, Y. Xiao and F. X. Xiao, Coord. Chem. Rev., 2023, 477, 214948 CrossRef CAS.
- A. E. Becquerel, C. R. Acad. Sci., 1839, 9, 145–149 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238(5358), 37–38 CrossRef CAS PubMed.
- J. Lin, W. Wang and G. Li, Adv. Funct. Mater., 2020, 30, 2005677 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- C. M. Ding, J. Y. Shi, Z. L. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- L. Y. Yang, F. Li and Q. J. Xiang, Mater. Horiz., 2024, 11, 1638–1657 RSC.
- S. Hou, H. W. Xie and F. X. Xiao, Inorg. Chem., 2024, 63, 8970–8976 CrossRef CAS PubMed.
- C. R. Jiang, S. J. Moniz, A. Q. Wang, T. Zhang and J. W. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- H. J. Huang, D. S. Yu, F. Hu, S. C. Huang, J. N. Song, H. Y. Chen, L. L. Li and S. J. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CrossRef CAS PubMed.
- Y. Yoon, B. Yan and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 2397–2400 CrossRef CAS PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 RSC.
- J. T. Li and N. Q. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- X. Yan, J. N. Yuan, F. X. Xiao and B. Liu, Mater. Horiz., 2024, 11, 5136–5140 RSC.
- X. Zhang, Y. Liu, S. T. Lee, S. Yang and Z. Kang, Energy Environ. Sci., 2014, 7, 1409–1419 RSC.
- H. Yang, J. Bright, S. Kasani, P. Zheng, T. Musho, B. Chen, L. Huang and N. Wu, Nano Res., 2019, 12, 643–650 CrossRef CAS.
- B. Tang, S. C. Zhu, H. Liang, S. Li, B. J. Liu and F. X. Xiao, J. Mater. Chem. A, 2022, 10, 4032 RSC.
- S. Hou, X. C. Dai, Y. B. Li, M. H. Huang, T. Li, Z. Q. Wei, Y. He, G. Xiao and F. X. Xiao, J. Mater. Chem. A, 2019, 7, 22487 RSC.
- Z. Y. Li, Y. H. Chen, J. R. Zhu, Q. Chen, S. J. Lu and F. X. Xiao, Inorg. Chem., 2023, 62, 16965–16973 CrossRef CAS PubMed.
- F. X. Xiao, Chem. Commun., 2012, 48, 6538–6540 RSC.
- J. Y. Zhang and F. X. Xiao, J. Mater. Chem. A, 2017, 5, 23681–23693 RSC.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- S. Gonçalves, P. Quitério, J. Freitas, D. Ivanou, T. Lopes, A. Mendes, J. P. Araújo, C. T. Sousa and A. Apolinário, ACS Appl. Mater. Interfaces, 2024, 16, 64389–64409 CrossRef PubMed.
- Z. Q. Wei, S. Hou, S. C. Zhu, Y. Xiao, G. Wu and F. X. Xiao, Adv. Funct. Mater., 2022, 32, 2106338 CrossRef CAS.
- Z. Y. Li, M. Yuan and F. X. Xiao, Small, 2025, 21, 2409513 CrossRef CAS PubMed.
- H. J. Lin, T. Li, M. H. Huang, X. C. Dai, Y. B. Li and F. X. Xiao, J. Phys. Chem. C, 2019, 123, 28066–28080 CrossRef CAS.
- Z. P. Zeng, F. X. Xiao, X. C. Gui, R. Wang, B. Liu, Y. Tan and T. Thatt, J. Mater. Chem. A, 2016, 4, 16383–16393 RSC.
- Q. Chen, Y. Xiao and F. X. Xiao, Inorg. Chem., 2024, 63, 1471–1479 CrossRef CAS PubMed.
- K. J. Kim and Y. R. Park, Appl. Phys. Lett., 2001, 78, 475–477 CrossRef CAS.
- L. Zhang, Y. Huang, C. H. Dai, Q. M. Liang, P. Yang, H. H. Yang and J. H. Yan, J. Electron. Mater., 2019, 48, 1724–1729 CrossRef CAS.
- Y. Q. Sun, B. Xu, Q. Shen, L. F. Hang, D. D. Men, T. Zhang, H. L. Li, C. C. Li and Y. Li, ACS Appl. Mater. Interfaces, 2017, 9, 31897–31906 CrossRef CAS PubMed.
- S. J. Yuan, J. K. Mu, R. Y. Mao, Y. G. Li, Q. H. Zhang and H. Z. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5719–5725 CrossRef CAS PubMed.
- X. Yan, H. W. Xie, G. Wu and F. X. Xiao, Chin. Chem. Lett., 2025, 36, 110279 CrossRef CAS.
- X. Y. Zhong, Y. L. Li, H. W. Wu and R. S. Xie, Mater. Sci. Eng., B, 2023, 289, 116278 CrossRef CAS.
- G. Wu, Q. L. Mo, Y. Xiao, K. Wang, X. Z. Ge, S. R. Xu, J. L. Li, Y. Q. Shao and F. X. Xiao, Inorg. Chem., 2023, 62, 520–529 CrossRef CAS PubMed.
- Y. Liang, T. Tsubota, L. P. Mooij and R. van de Krol, J. Phys. Chem. C, 2011, 115, 17594–17598 CrossRef CAS.
- G. L. Chang, D. G. Wang, Y. Y. Zhang, A. Aldalbahi, L. H. Wang, Q. Li and K. Wang, Nucl. Sci. Tech., 2016, 27, 108 CrossRef.
- Y. B. Li and F. X. Xiao, Inorg. Chem., 2025, 64, 3608–3615 CrossRef CAS PubMed.
- (a) L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC; (b) Q. Chen, X. Z. Ge, L. H. Yu and F. X. Xiao, Inorg. Chem., 2023, 62, 19358–19365 CrossRef CAS PubMed.
- (a) W. Q. Fan, Q. H. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC; (b) P. Su, X. Yan and F. X. Xiao, Chem. Sci., 2024, 15, 14778–14790 RSC.
- Y. B. Li, T. Li, X. C. Dai, M. H. Huang, S. Hou, X. Y. Fu, Z. Q. Wei, Y. He, G. Xiao and F. X. Xiao, ACS Appl. Mater. Interfaces, 2020, 12, 4373 CrossRef CAS PubMed.
- Y. X. Chen, M. Cheng, C. Lai, Z. Wei, G. X. Zhang, L. Li, C. S. Tang, L. Du, G. F. Wang and H. D. Liu, Small, 2023, 19, 2205902 CrossRef CAS PubMed.
- (a) Q. L. Mo, J. L. Li, S. R. Xu, K. Wang, X. Z. Ge, Y. Xiao, G. Wu and F. X. Xiao, Adv. Funct. Mater., 2023, 33, 2210332 CrossRef CAS; (b) Q. Chen, Y. H. Chen, J. R. Zhu, Z. Y. Li and F. X. Xiao, Adv. Funct. Mater., 2025, 35, 2417139 CrossRef CAS.
- (a) X. Z. Ge, K. Wang, Q. L. Mo, Y. Xiao, J. L. Li, G. Wu, S. R. Xu and F. X. Xiao, Catal. Sci. Technol., 2023, 13, 479–489 RSC; (b) Q. L. Mo, S. R. Xu, J. L. Li, X. Q. Shi, Y. Wu and F. X. Xiao, Small, 2023, 19, 2300804 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- (a) S. C. Zhu and F. X. Xiao, ACS Catal., 2023, 13, 7269–7309 CrossRef CAS; (b) X. Yan, K. Wang and F. X. Xiao, Inorg. Chem., 2023, 62, 17454–17463 CrossRef CAS PubMed.
- (a) X. Yan, X. Y. Fu and F. X. Xiao, Adv. Funct. Mater., 2023, 33, 2303737 CrossRef CAS; (b) M. Yuan, X. Yan, J. N. Yuan, P. Su, Q. Chen and F. X. Xiao, J. Catal., 2024, 437, 115667 CrossRef CAS.
- S. Hou, Q. L. Mo, S. C. Zhu, S. Li, G. G. Xiao and F. X. Xiao, Inorg. Chem., 2021, 61, 1188–1194 CrossRef PubMed.
- S. Hou, Z. Q. Wei, X. C. Dai, M. H. Huang and F. X. Xiao, Inorg. Chem., 2020, 59, 7325–7334 CrossRef CAS PubMed.
- Z. Q. Wei, X. C. Dai, S. Hou, Y. B. Li, M. H. Huang, T. Li, S. Xu and F. X. Xiao, J. Mater. Chem. A, 2020, 8, 177 RSC.
- (a) X. Li, J. Yu, S. Wageh, A. A. Al-Ghamdi and J. Xie, Small, 2016, 12, 6640–6696 CrossRef CAS PubMed; (b) Q. Chen, J. L. Li, Q. L. Mo and F. X. Xiao, Chem. Eng. J., 2024, 497, 154584 CrossRef CAS.
- (a) K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS; (b) X. Yan, B. X. Zheng, J. R. Zhu, Y. B. Li and F. X. Xiao, Inorg. Chem., 2025, 64, 3572–3581 CrossRef CAS PubMed.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef CAS PubMed.
- Z. P. Zeng, F. X. Xiao, H. Phan, S. F. Chen, Z. Z. Yu, R. Wang, T. Q. Nguyen and T. T. Y. Tan, J. Mater. Chem. A, 2018, 6, 1700–1713 RSC.
- Z. Zeng, T. Li, Y. B. Li, X. C. Dai, M. H. Huang, Y. He, G. Xiao and F. X. Xiao, J. Mater. Chem. A, 2018, 6, 24686 RSC.
- P. Su, S. Li and F. X. Xiao, Small, 2024, 20, 2400958 CrossRef CAS PubMed.
- L. X. Yin, G. Y. Xu, P. Nie, H. Dou and X. G. Zhang, Chem. Eng. J., 2018, 352, 695–703 CrossRef CAS.
- (a) X. J. Chen, X. D. Ren, Z. L. Liu, L. Zhuang and J. T. Lu, Electrochem. Commun., 2013, 27, 148–151 CrossRef CAS; (b) Y. S. Cai, J. Q. Chen, P. Su, X. Yan, Q. Chen, Y. Wu and F. X. Xiao, Chem. Sci., 2024, 15, 13495–13505 RSC.
- D. J. Yan, X. C. Fu, Z. C. Shang, J. K. Liu and H. A. Luo, Chem. Eng. J., 2019, 361, 853–861 CrossRef CAS.
- (a) Y. B. Li and F. X. Xiao, J. Mater. Chem. A, 2023, 11, 589 RSC; (b) H. Liang, Q. Chen, Q. L. Mo, Y. Wu and F. X. Xiao, J. Mater. Chem. A, 2023, 11, 9401–9426 RSC.
- (a) Q. L. Mo, X. C. Dai, Y. Xiao and F. X. Xiao, Chin. Chem. Lett., 2023, 34, 107901 CrossRef CAS; (b) B. X. Zheng, J. N. Yuan, P. Su, X. Yan, Q. Chen, M. Yuan and F. X. Xiao, J. Mater. Chem. A, 2025, 13, 4908–4920 RSC.
- Y. B. Li and F. X. Xiao, Chem. Sci., 2025, 16, 2661–2672 RSC.
- (a) L. A. Peyser, A. E. Vinson, A. P. Bartko and R. M. Dickson, Science, 2001, 291, 103–106 CrossRef CAS PubMed; (b) X. Yan, M. Yuan, Y. L. Yuan, P. Su, Q. Chen and F. X. Xiao, Chem. Sci., 2024, 15, 10625–10637 RSC.
- Y. S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075 CrossRef CAS PubMed.
- Q. L. Mo, X. C. Dai and F. X. Xiao, Small, 2023, 19, 2302372 CrossRef CAS PubMed.
- Y. Xiao, Q. L. Mo, G. Wu, K. Wang, X. Ge, S. R. Xu, J. L. Li, Y. Wu and F. X. Xiao, J. Mater. Chem. A, 2023, 11, 2402 RSC.
- P. Su, B. Tang and F. X. Xiao, Small, 2024, 20, 2307619 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed.
- J. H. Zhou, W. C. Zhao, Z. H. Miao, J. G. Wang, Y. Ma, H. T. Wu, T. D. Sun, H. S. Qian and Z. B. Zha, ACS Nano, 2020, 14, 2126–2136 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- K. V. Grzhegorzhevskii, P. S. Zelenovskiy, O. V. Koryakova and A. A. Ostroushko, Inorg. Chim. Acta, 2019, 489, 287–300 CrossRef CAS.
- A. Ostroushko, I. Gette, I. Danilova, E. Mukhlynina, M. Tonkushina and K. Grzhegorzhevskii, Nanotechnol. Russ., 2014, 9, 577–582 CrossRef CAS.
- Y. Wu, X. X. Yu, Z. J. Fu, J. Y. Pei and L. H. Bi, Catalysts, 2021, 11, 856 CrossRef CAS.
- J. Pei and L. Bi, Catalysts, 2022, 12, 696 CrossRef CAS.
- Q. Zhou, Y. Du, Z. Y. Qu and L. H. Bi, J. Electroanal. Chem., 2021, 894, 115339 CrossRef CAS.
- D. Jeon, H. Kim, C. Lee, Y. Han, M. Gu, B. S. Kim and J. Ryu, ACS Appl. Mater. Interfaces, 2017, 9, 40151–40161 CrossRef CAS PubMed.
- Y. Choi, D. Jeon, Y. Choi, D. Kim, N. Kim, M. Gu, S. Bae, T. Lee, H. W. Lee and B. S. Kim, ACS Nano, 2018, 13, 467–475 CrossRef PubMed.
- H. Kim, S. Bae, D. Jeon and J. Ryu, Green Chem., 2018, 20, 3732–3742 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- N. Zhang, C. Han, Y. J. Xu, J. J. Foley Iv, D. Zhang, J. Codrington, S. K. Gray and Y. G. Sun, Nat. Photonics, 2016, 10, 473–482 CrossRef CAS.
- P. K. Ngumbi, S. W. Mugo and J. M. Ngaruiya, IOSR J. Appl. Chem., 2018, 11, 25–29 CAS.
- L. Liu, X. Zhang, L. Yang, L. Ren, D. Wang and J. Ye, Natl. Sci. Rev., 2017, 4, 761–780 CrossRef CAS.
- Z. Cao, Y. L. Yin, P. Fu, D. Li, Y. L. Zhou, Z. J. Wen, Y. H. Peng, W. K. Wang, W. C. Zhou and D. S. Tang, J. Electrochem. Soc., 2020, 167, 026509 CrossRef CAS.
- F. X. Xiao, Z. P. Zeng and B. Liu, J. Am. Chem. Soc., 2015, 137, 10735–10744 CrossRef CAS PubMed.
- H. Kim, Y. You, D. Kang, D. Jeon, S. Bae, Y. Shin, J. Lee, J. Lee and J. Ryu, Adv. Funct. Mater., 2019, 29, 1906407 CrossRef CAS.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- (a) B. Zhou, B. Y. Shi, D. Y. Jin and X. G. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed; (b) F. Wang, Y. Han, C. S. Lim, Y. H. Lu, J. Wang, J. Xu, H. Y. Chen, C. Zhang, M. H. Hong and X. G. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- Y. Lim, S. Y. Lee, D. Kim, M. Han, H. S. Han, S. H. Kang, J. K. Kim, U. Sim and Y. I. Park, Chem. Eng. J., 2022, 438, 135503 CrossRef CAS.
- F. X. Xiao, J. W. Miao and B. Liu, J. Am. Chem. Soc., 2014, 136, 1559–1569 CrossRef CAS PubMed.
- S. Bae, D. Kim, H. Kim, M. Gu, J. Ryu and B. S. Kim, Adv. Funct. Mater., 2020, 30, 1908492 CrossRef CAS.
- (a) M. Faraji, M. Yousefi, S. Yousefzadeh, M. Zirak, N. Naseri, T. H. Jeon, W. Choi and A. Z. Moshfegh, Energy Environ. Sci., 2019, 12, 59–95 RSC; (b) J. R. Zhu, Y. H. Chen, Z. Y. Li, Q. Chen and F. X. Xiao, Inorg. Chem., 2023, 62, 18649–18659 CrossRef CAS PubMed.
- M. Kavitha, P. Gopinath and H. John, Phys. Chem. Chem. Phys., 2015, 17, 14647–14655 RSC.
- S. J. Teh, C. W. Lai and S. B. A. Hamid, J. Energy Chem., 2016, 25, 336–344 CrossRef.
- D. Kim, M. Gu, Y. Choi, H. Kim, J. Ryu and B. S. Kim, ACS Appl. Energy Mater., 2020, 3, 7103–7112 CrossRef CAS.
- (a) P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS; (b) X. Yan, J. H. Dong, J. Y. Zheng, Y. Wu and F. X. Xiao, Chem. Sci., 2024, 15, 2898–2913 RSC.
- P. Bian, N. Li, G. Li, S. Zhang, X. Liu, J. Gu, B. Liu and T. Jiao, Colloids Surf., A, 2023, 656, 130460 CrossRef CAS.
- J. L. Liu, X. Yan, J. N. Yuan, Y. Wu, X. Wang and F. X. Xiao, Small, 2024, 20, 2405514 CrossRef CAS PubMed.
- J. N. Yuan, X. Yan, B. X. Zheng, J. Q. Chen and F. X. Xiao, Inorg. Chem. Front., 2025, 12, 1553–1567 RSC.
- Z. Q. Wei and F. X. Xiao, Inorg. Chem., 2023, 62, 6138–6146 CrossRef CAS PubMed.
- Y. Yu, J. Y. Liu, S. S. Chen, Y. Song, R. R. Chen, J. Yu, J. H. Zhu, Y. Li, Q. Liu and J. Wang, Chem. Eng. J., 2024, 486, 149783 CrossRef CAS.
- B. Huang, G. F. von Rudorff and O. A. von Lilienfeld, science, 2023, 381, 170–175 CrossRef CAS PubMed.
- A. Maqsood, C. Chen and T. J. Jacobsson, Adv. Sci., 2024, 11, 2401401 CrossRef CAS PubMed.
- X. P. Bai and X. C. Zhang, Nano-Micro Lett., 2025, 17, 135 CrossRef PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |