
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maleic anhydride derived diphosphines: adaptable chelators for receptor-targeted 99mTc, 64Cu and 188Re radiotracers
Rachel E.
Nuttall
 *a,
Ingebjørg N.
Hungnes
a,
Truc T.
Pham
a,
Oliver W. L.
Carter
a,
Alex
Rigby
a,
Natasha
Patel
a,
Zilin
Yu
a,
Julie
Cleaver
b,
Jennifer D.
Young
ab,
Gary J. R.
Cook
a,
Lefteris
Livieratos
ac,
Jane
Sosabowski
b,
Hong Hoi
Ting
d,
Nicholas
Vetter
d,
Paul G.
Pringle
e and
Michelle T.
Ma
*a
*a,
Ingebjørg N.
Hungnes
a,
Truc T.
Pham
a,
Oliver W. L.
Carter
a,
Alex
Rigby
a,
Natasha
Patel
a,
Zilin
Yu
a,
Julie
Cleaver
b,
Jennifer D.
Young
ab,
Gary J. R.
Cook
a,
Lefteris
Livieratos
ac,
Jane
Sosabowski
b,
Hong Hoi
Ting
d,
Nicholas
Vetter
d,
Paul G.
Pringle
e and
Michelle T.
Ma
*a
aSchool of Bioengineering and Imaging Sciences, King's College London, 4th Floor Lambeth Wing, St Thomas' Hospital, London, SE1 7EH, UK. E-mail: rachel.nuttall@kcl.ac.uk; michelle.ma@kcl.ac.uk
bCentre for Cancer Biomarkers and Biotherapeutics, Barts Cancer Institute, Queen Mary University of London, John Vane Science Centre, Charterhouse Square, London, EC1M 6BQ, UK
cDepartment of Nuclear Medicine, Guy's and St Thomas' Hospitals NHS Foundation Trust, Guy's Hospital, London, SE1 9RT, UK
dOncobeta GmbH, 85748 Garching, Munich, Germany
eSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 26th August 2025
Abstract
The diagnostic imaging radionuclide, 99mTc, used in Single Photon Emission Computed Tomography (SPECT), has extraordinary potential for enabling economical molecular receptor imaging in oncology, provided suitable chelators are available to enable kit-based radiolabelling. We report the development of two new bis(phosphino)maleic anhydrides, DPAn and DPMEP, that exhibit increased electron donor capacity and concomitant increased radiochemical yields compared to their first-generation diphosphine analogues. Both DPAn and DPMEP can be reacted with a wide range of biological targeting vectors, including receptor-targeted peptides, carbohydrates, vitamins and small-molecule inhibitors. Exemplar diphosphine-peptide bioconjugates, DPAn-PSMAt and DPMEP-PSMAt (which target the prostate-specific membrane antigen, PSMA), can be formulated into kits to enable near-quantitative, one-pot radiosynthesis of new 99mTc radiotracers, cis/trans-[99mTcO2(DPAn-PSMAt)2]+ and cis/trans-[99mTcO2(DPMEP-PSMAt)2]+, respectively. We demonstrate that the two exemplar 99mTc radiotracers, cis/trans-[99mTcO2(DPAn-PSMAt)2]+ and cis/trans-[99mTcO2(DPMEP-PSMAt)2]+, display favourable SPECT imaging properties in murine prostate cancer models, including high tumour uptake, fast clearance from circulation, excretion via a renal pathway and high metabolic stability. The same diphosphine-peptide bioconjugates can also be radiolabelled with the Positron Emission Tomography (PET) isotope, 64Cu, and the radiotherapeutic β−-emitting isotope, 188Re, in high radiochemical yields. The new DPAn and DPMEP chelator platforms thus enable development of novel molecular imaging radiopharmaceuticals for 99mTc SPECT, 64Cu PET and 188Re systemic radiotherapy.
Introduction
In oncology, receptor-targeted molecular imaging using Single Photon Emission Computed Tomography (SPECT) or γ-scintigraphy has utility in diagnosis, disease staging and clinical decision-making. One class of radiotracer used for this purpose consists of a peptide attached to a chelator, which in turn is complexed to a radioactive metal ion. Two of the most prominent SPECT radiotracers employed for this purpose are 111In-DTPA-octreotide and 99mTc-EDDA/HYNIC-octreotide: both target the somatostatin receptor 2 that is overexpressed in neuroendocrine cancers.1 Whilst SPECT/γ-scintigraphy procedures with these radiotracers have been superseded in some healthcare settings by more sensitive Positron Emission Tomography (PET) imaging coupled with the PET radiotracer 68Ga-DOTA-octreotate,2 they are still widely used in many clinics where PET is not available.There are several factors that have led to the prevalence of SPECT/γ-scintigraphy imaging procedures. First, worldwide, there is simply more SPECT and γ-scintigraphy infrastructure than PET infrastructure, including in lower and middle income countries.3–6 Second, 99mTc (t½ = 6 h; 90% γ, 140 keV) is widely distributed and available from bench-top 99Mo/99mTc generators in the form of 99mTcO4− in aqueous saline solution.5,6 Third, 99mTc radiotracers, which are routinely used in 30–40 million procedures globally each year, are produced using simple one- or two-step kits in near-quantitative radiochemical yields.5–7
Kits for the preparation of 99mTc radiopharmaceuticals typically contain buffering salts, a reducing agent to reduce 99mTcVIIO4−, a chelator derivative that ultimately complexes the 99mTc metal ion to form the desired radiopharmaceutical, and other reagents including weak chelators, which serve to stabilise 99mTc intermediates.5,7 The majority of 99mTc radiopharmaceuticals are for imaging perfusion or anatomical processes.5,7 Molecular imaging using 99mTc-EDDA/HYNIC-octreotide is not as prevalent, in part because of the low incidence of neuroendocrine cancer.
However, recognising the utility and availability of molecular SPECT/γ-scintigraphy infrastructure, in recent years, several new 99mTc-labelled peptides have been clinically evaluated for receptor-targeted molecular imaging of the prostate-specific membrane antigen (PSMA) overexpressed in prostate cancer. These include 99mTc-MIP-1404 (also known as 99mTc-Trofolastat),8,999mTc-PSMA-I&S10,11 and 99mTc-EDDA/HYNIC-iPSMA,12,13 which have been shown to be viable alternatives to efficacious PET diagnostic agents that similarly target PSMA.
There is an additional incentive for development of 99mTc radiotracers: in some cases, chemically analogous Re complexes are accessible, providing access to pairs of “theranostic” 99mTc and 188Re radiopharmaceuticals for diagnosis and therapy, respectively. The rhenium radionuclide, 188Re (t½ = 17 h; 100% β−, Emax = 2.12 MeV; 15% γ, 155 keV), which can also be produced from a bench-top generator like 99mTc, emits cytotoxic β− particles. 188Re radiopharmaceuticals can be effective therapeutics. For example, the lipophilic 188Re-labelled radiopharmaceutical 188Re-lipiodol is not only clinically efficacious for treatment of inoperable liver cancer,14 but is also economically viable in lower and middle income countries where supplies of other β−-emitting radiopharmaceuticals are limited due to economical and/or geographical barriers.15,16 Indeed, a newly developed pair of 99mTc/188Re-labelled PSMA-GCK01 theranostic agents has shown favourable properties in preclinical and initial first-in-human studies,17 demonstrating the feasibility of molecular imaging and therapeutic 99mTc/188Re pairs.
We have recently explored diphosphine derivatives as potential platforms for radiolabelling receptor-targeted biomolecules.18–21 These diphosphines include 2,3-bis(diphenylphosphino)maleic anhydride (DPPh)22,23 and 2,3-bis(di-p-tolylphosphino)maleic anhydride (DPTol) (Fig. 1) which react with the primary amine groups of peptides to furnish diphosphine-peptide (DP-peptide) conjugates.18–20† The DP-peptide derivatives coordinate [TcO2]+ or [ReO2]+ motifs to yield complexes of the type cis/trans-[MO2(DP-peptide)2]+ (M = Tc, Re); radiolabelled 99mTc and 188Re isotopologues are also synthetically accessible. We have also demonstrated that the resulting 99mTc and 188Re radiotracers retain affinity for their cognate target receptors in vitro and in vivo, and have favourable biodistribution pathways including rapid clearance from the bloodstream via a renal pathway.18,20 We note that these derivatives also coordinate the PET imaging isotope, 64Cu (t½ = 12.7 h; 18% β+, Emax = 653 keV), rapidly at room temperature, furnishing radiotracers of formula [Cu(DP-peptide)2]+ that show high stability in serum.19,22,23
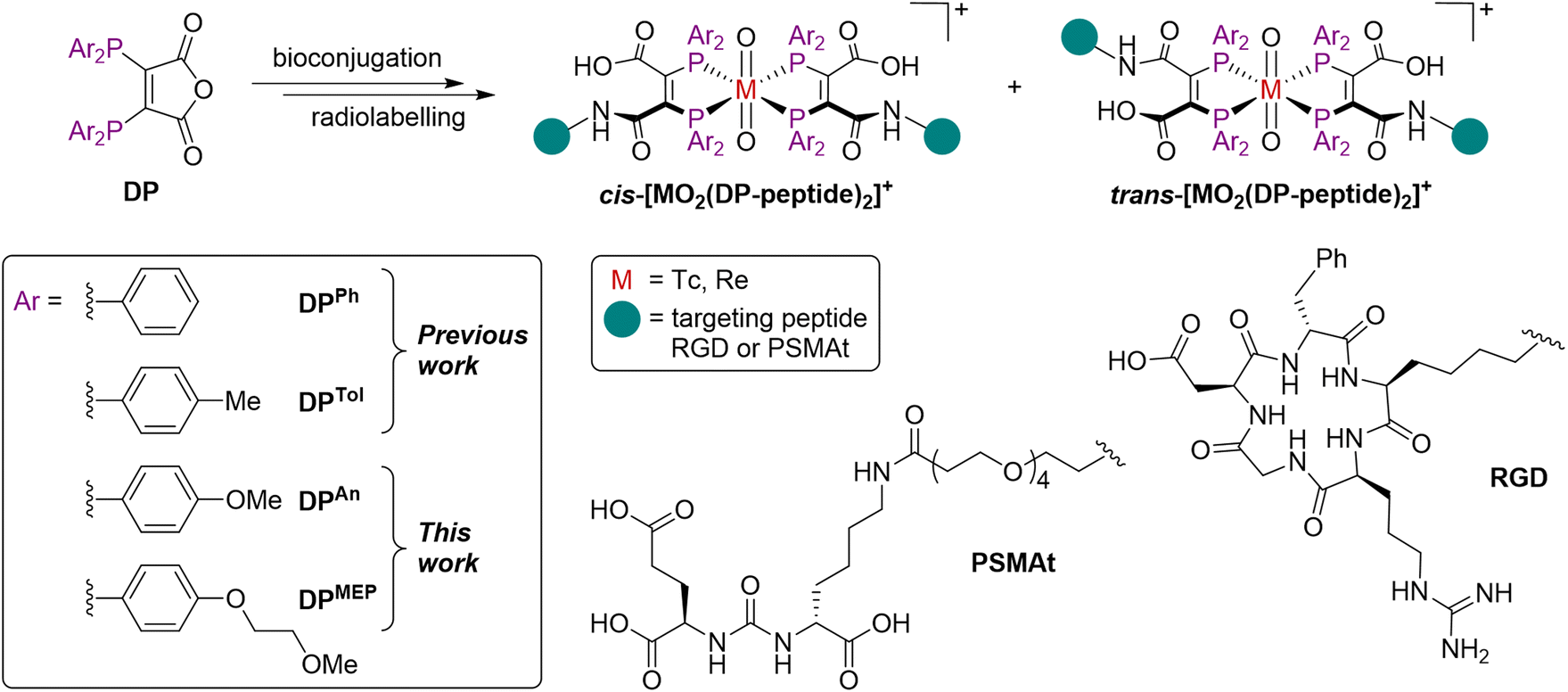 | ||
| Fig. 1 Structures of cis/trans-[MO2(DP-peptide)2]+ complexes, where M = Tc or Re.18–20 | ||
However, DP-peptide derivatives of DPPh do not provide 99mTc radiotracers in sufficiently high radiochemical yield to enable preparation using a one-step kit without subsequent purification to remove unreacted 99mTc precursors.19 Furthermore, radiochemical yields of 188Re derivatives are relatively low (<50%).20DP-peptide derivatives of DPTol (which contains p-tolyl substituents in place of phenyl groups) have shown increased electron donor capacity and concomitant increased radiochemical yields compared to DPPh derivatives.19 However, even with the improved reactivity of DPTol, we have not been able to obtain radiochemical yields of [99mTcO2(DP-peptide)2]+ compounds above ∼85–90%. This is a barrier to clinical translation.
To address this, we have synthesised two novel diphosphine maleic anhydrides, DPAn and DPMEP (Fig. 1), which possess either p-anisyl or p-(2-methoxyethoxy)phenyl groups on the phosphines, respectively. Bioconjugates of these diphosphine maleic anhydride platforms enable near-quantitative radiochemical syntheses of 99mTc radiotracers. In our most comprehensive report yet, we have explored the scope of possible bioconjugation reactions with both DPAn and DPMEP using a range of biological targeting vectors. We have attached DPAn and DPMEP to a PSMA-targeted dipeptide (PSMAt), enabling comparison of DPAn-PSMAt and DPMEP-PSMAt with our prior conjugate, DPPh-PSMAt, including formation of 99mTc, 188Re and 64Cu complexes. Lastly, our novel 99mTc radiotracers have been assessed in murine models of prostate cancer using SPECT/CT imaging. We have therefore demonstrated the full utility of our novel and improved DPAn and DPMEP platforms for enabling economical and accessible production of pairs of receptor-targeted theranostic radiotracers.
Experimental
Details of experimental procedures are included in the SI. All animal experiments and procedures were ethically reviewed and approved by the Animal Welfare & Ethical Review Boards at either King's College London or Barts Cancer Institute. All animal experiments and procedures were then carried out in accordance with approvals from these committees and review boards, alongside the Animals (Scientific Procedures) Act 1986 UK Home Office regulations governing animal experimentation.Results
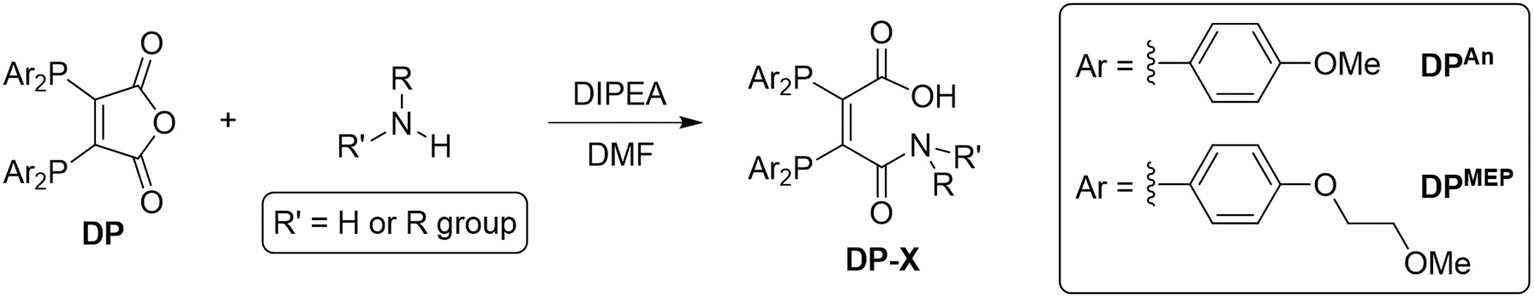
Synthesis of DPAn and DPMEP and their PSMAt conjugates
Two novel bis(phosphino)maleic anhydrides were synthesised bearing either p-anisyl (DPAn) or p-(2-methoxyethoxy)phenyl (DPMEP) substituents. By reaction of the required Ar2PH with 2,3-dichloromaleic anhydride in the presence of triethylamine base, DPAn and DPMEP were synthesised in 89% and 61% yield, respectively (Scheme 1). The syntheses of all precursors, including both diarylphosphines, are described in the SI.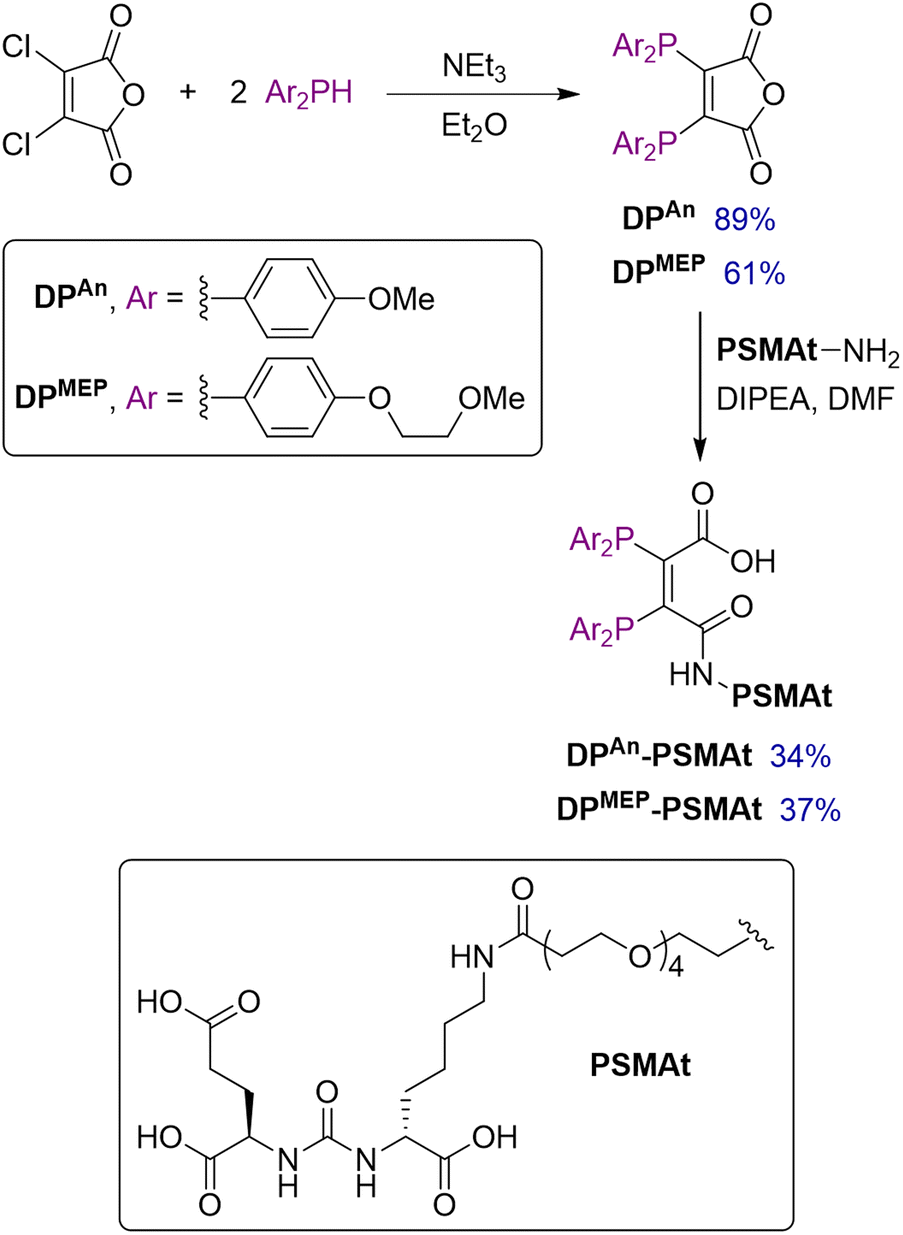 | ||
| Scheme 1 Synthesis of bis(phosphino)maleic anhydrides, DPAn and DPMEP. | ||
Next, both diphosphines, DPAn and DPMEP, were conjugated to a PSMA-targeting dipeptide, PSMAt-NH2. Using the pendant primary amine in PSMAt-NH2 under mild basic conditions, the maleic anhydrides readily ring-opened to form diphosphine bioconjugates, DPAn-PSMAt and DPMEP-PSMAt in 34% and 37% isolated yield, respectively (Scheme 1). As previously reported for this type of ligand,18,19DPAn-PSMAt and DPMEP-PSMAt were stable in the solid state; however, they both slowly oxidised in solution under atmospheric conditions. Under acidic conditions, a reverse reaction occurred to reform DPAn/MEP and PSMAt-NH2.
Bioconjugation reactions of DPAn and DPMEP with bioactive small molecules and peptides
To investigate the versatility of this DP platform, the reaction of each of DPAn and DPMEP with a range of biologically active amines (Fig. 2) was assessed, including: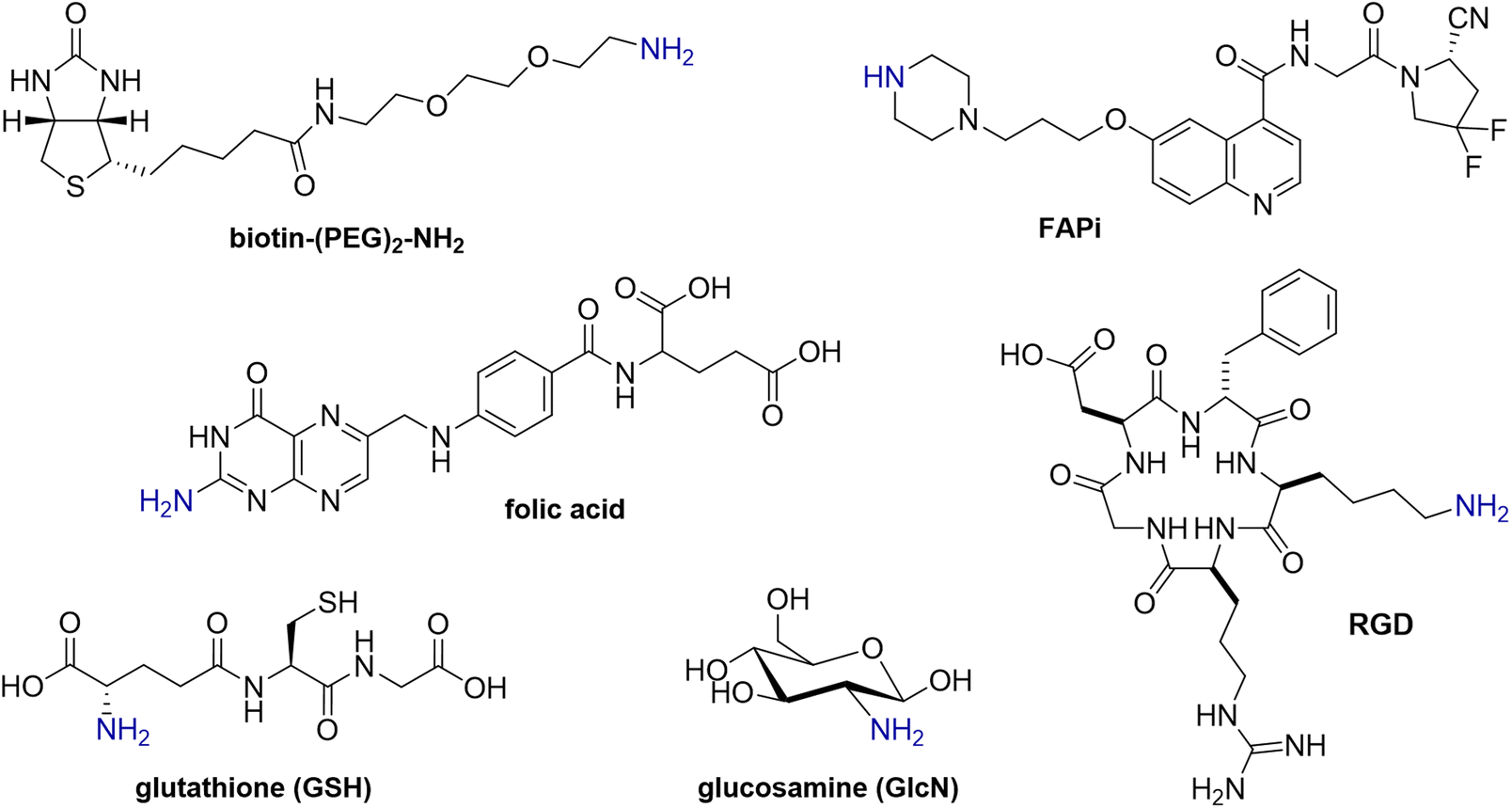 | ||
| Fig. 2 Structures of R–NH2 and RR′–NH amines. | ||
• Glucosamine (GlcN), a monosaccharide, as an exemplar carbohydrate;
• Biotin, a vitamin, which binds strongly to streptavidin protein; the biotin/streptavidin pair is used for purification and detection in biotechnology applications;24,25
• Folic acid, a vitamin which targets the folate receptor, overexpressed in ovarian, lung, breast and kidney cancers;26,27
• RGD, a cyclic pentapeptide, which targets the αvβ3-integrin receptor overexpressed in inflammation, neovasculature and many cancers;28
• Glutathione (GSH), a thiol-containing antioxidant tripeptide;
• FAPi, a small molecule receptor-targeted inhibitor, which targets the fibroblast activation protein of fibroblastic cells, associated with hard-to-treat cancers (e.g. pancreatic, lung, ovarian cancers).29 Notably, the FAPi motif is a secondary amine (R2NH), whereas all other amine derivatives above contain primary amines (RNH2).
The crude reaction mixtures were analysed by LC-MS (Table 1, see SI for full chromatograms). All reactions underwent >95% consumption of the respective DP and/or amine (except for folic acid which showed no reaction). The lack of reactivity of folic acid with either DPAn or DPMEP is presumably due to the low nucleophilicity of the primary amine on the pteridine ring. While both reactions with GSH showed full consumption of DPAn/MEP, multiple species were detected by LC-MS. Analysis by 31P{1H} NMR spectroscopy indicated that although the desired amide derivative was formed, additional reactions also occurred, which we attribute to the reactive thiol group of GSH (see SI). When 1-propanethiol was separately reacted with DPAn, multiple 31P{1H} NMR signals with similar chemical shifts were observed. In short, both new DPAn and DPMEP compounds react with thiols to give multiple products: consideration should be taken when a biomolecule with a free thiol group is reacted with these diphosphine compounds.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | DP | R–NH2 or RR′–NHa | DP-X | Expected m/z for [M + H]+ | Observed m/z | Conversionb |
| a RR′–NH only applies to the reaction with FAPi, which contains a secondary amine. b Approximate conversion was calculated using the UV signal (280 nm) in HPLC chromatogram(s). See SI for full chromatograms. c NR = no reaction. d Mixture of products. | ||||||
| 1 | DPAn | Biotin-(PEG)2-NH2 | DPAn-biotin | 961.3 | 961.7 | >95% |
| 2 | FAPi | DPAn-FAPi | 1073.4 | 1072.9 | >95% | |
| 3 | Folic acid | DPAn-folic acid | 1028.3 | — | NRc | |
| 4 | Glucosamine (GlcN) | DPAn-GlcN | 766.2 | 766.3 | >95% | |
| 5 | Glutathione (GSH) | DPAn-GSH | 894.2 | 894.4 | ∼83%d | |
| 6 | RGD | DPAn-RGD | 1190.5 | 1190.8 | >95% | |
| 7 | DPMEP | Biotin-(PEG)2-NH2 | DPMEP-biotin | 1138.4 | 1138.0 | >95% |
| 8 | FAPi | DPMEP-FAPi | 1249.5 | 1249.8 | >95% | |
| 9 | Folic acid | DPMEP-folic acid | 1204.4 | — | NRc | |
| 10 | Glucosamine (GlcN) | DPMEP-GlcN | 942.3 | 942.6 | >95% | |
| 11 | Glutathione (GSH) | DPMEP-GSH | 1070.3 | 1071.1 | ∼86%d | |
| 12 | RGD | DPMEP-RGD | 1366.6 | 1366.9 | >95% | |
Evaluating the donor properties of DPAn, DPMEP and derivatives: IR spectra of Mo complexes
The IR stretching frequencies of CO ligands (νCO) in metal complexes can be used to assess the binding properties of ligands, and complexes of the type cis-[Mo(CO)4L2] have previously been utilised for this purpose.30 Strong σ-donors, such as phosphines, increase π-back-bonding from Mo to CO ligands, resulting in lower CO stretching frequencies.Ligands DPAn and DPMEP, alongside the progenitor DPPh (for comparison) were reacted with cis-[Mo(CO)4(nbd)] to give cis-[Mo(CO)4(DP)] complexes (Scheme 2). Additionally, each of these complexes was reacted with (2-methoxyethyl)amine (MOE-NH2) to yield ring-opened compounds of the type [Mo(CO)4(DP-NH-MOE)]−,19 which all contained an amide bond, to model DP-peptide conjugate species (Scheme 2). These complexes were isolated as salts of MOE-NH3+.
 | ||
| Scheme 2 Mo(0) complexation of DP ligands to give cis-[Mo(CO)4(DP)] and subsequent ring-opening to give cis-[Mo(CO)4(DP-NH-MOE)]−. | ||
The IR νCO data for these cis-[Mo(CO)4L2] complexes (alongside 31P{1H} NMR and selected 13C{1H} NMR data) are shown in Table 2. There was a decrease in νCO in the order [Mo(CO)4(nbd)] > [Mo(CO)4(DPPh)] > [Mo(CO)4(DPAn)] ∼ [Mo(CO)4(DPMEP)] > [Mo(CO)4(DPPh-NH-MOE)]− > [Mo(CO)4(DPAn-NH-MOE)]− > [Mo(CO)4(DPMEP-NH-MOE)]−. Importantly, complexes of DPAn and DPMEP and their amide conjugates exhibited lower νCO values compared with analogous DPPh complexes, indicating that the new DPAn and DPMEP ligands possess increased electron donor capacities compared to DPPh derivatives.
| Compound | v CO (cm−1) | 31P{1H} NMR (ppm) | 13C{1H} NMR (ppm) of M–CO ligandsd |
|---|---|---|---|
| a CH2Cl2, 1 mg mL−1, “∼” denotes shoulder peak. b 162 MHz, CDCl3. c 162 MHz, CD2Cl2. d 126 MHz CD2Cl2. | |||
| DPPh | — | −20.5 (s)b | — |
| DPAn | — | −22.3 (s)b | — |
| DPMEP | — | −22.4 (s)b | — |
| [Mo(CO)4(nbd)] | 2041, 1951, 1888 | — | — |
| [Mo(CO)4(DPPh)] | 2031, ∼1939, 1920 | 51.7 (s)b | 215.2 (m, COeq), 208.9 (t, 2JP,C = 8.5 Hz, COax)19 |
| [Mo(CO)4(DPAn)] | 2028, ∼1932, 1916 | 49.0 (s)b | 215.9–215.5 (m, COeq), 209.1 (t, 2JP,C = 8.6 Hz, COax) |
| [Mo(CO)4(DPMEP)] | 2028, ∼1932, 1915 | 48.9 (s)b | 215.8–215.4 (m, COeq), 209.0 (t, 2JP,C = 8.6 Hz, COax) |
| [MOE-NH3][Mo(CO)4(DPPh-NH-MOE)] | 2024, ∼1931, 1902 | 72.5 (d, J = 3.3 Hz), 70.2 (d, J = 3.3 Hz)c | 217.9 (m, COeq), 210.0 (t, 2JP,C = 8.6 Hz, COax)19 |
| [MOE-NH3][Mo(CO)4(DPAn-NH-MOE)] | 2023, ∼1928, 1899 | 69.3 (s), 67.4 (s)c | 218.0–217.5 (m, COeq), 209.7 (t, 2JP,C = 8.5 Hz, COax) |
| [MOE-NH3][Mo(CO)4(DPMEP-NH-MOE)] | 2021, 1927, 1897 | 69.1 (d, J = 2.7 Hz), 66.4 (d, J = 2.7 Hz)c | 218.3–217.8 (m, COeq), 209.8 (t, 2JP,C = 8.5 Hz, COax) |
The νCO stretches were all lower for the amide conjugates, [Mo(CO)4(DPX-NH-MOE)]− (X = Ph, An, MEP), compared with the precursor species, [Mo(CO)4(DPX)] (X = Ph, An, MEP), indicating that DP-NHR ligands are significantly better electron donating ligands than the highly electron withdrawing bis(phosphino)maleic anhydride precursor ligands.
99mTc and 188Re radiolabelling using generator-produced eluate
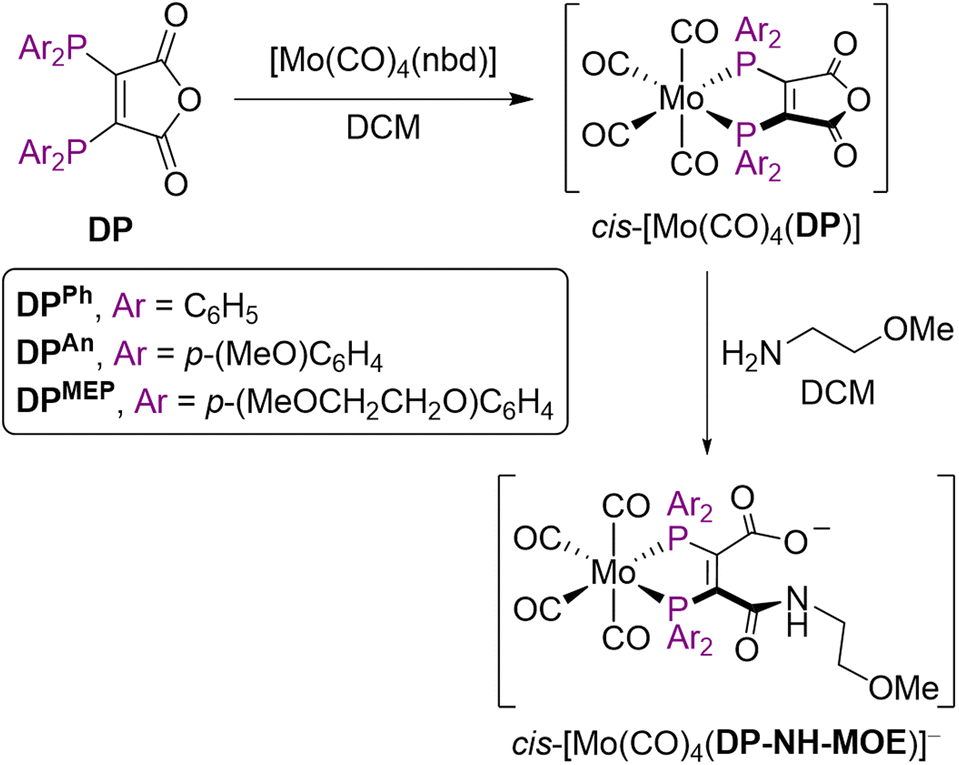
We next elected to study the reactions of DPAn-PSMAt and DPMEP-PSMAt derivatives with generator-produced 99mTc and 188Re. For this, the DPAn-PSMAt and DPMEP-PSMAt conjugates were incorporated into lyophilised kits containing all components required for labelling: reducing agent (SnCl2), stabilising chelator (sodium tartrate) and buffer (sodium bicarbonate) (Table S1).For 99mTc radiosyntheses, the kits were reconstituted using generator produced 99mTcO4− (200 MBq) in saline solution (500 μL) alongside an analogous DPPh-PSMAt kit for comparison. The mixtures were then heated at 100 °C for 10 min before radio-iTLC and radio-HPLC analyses. Both [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ were consistently formed in ≥95% RCY (Table 3), whilst side-by-side radiolabelling reactions of DPPh-PSMAt yielded <80% RCY. The major radiochemical impurity in these reactions was 99mTc colloidal species. Analytical reverse-phase radio-HPLC chromatograms of reactions containing either [99mTcO2(DPAn-PSMAt)2]+ or [99mTcO2(DPMEP-PSMAt)2]+ showed only one major radioactive species, which we attributed to a mixture of cis- and trans-[99mTcO2(DPAn-PSMAt)2]+ or cis- and trans-[99mTcO2(DPMEP-PSMAt)2]+ (Fig. 3). Under these HPLC conditions, cis and trans isomers were not resolved, however using alternative HPLC conditions (vide infra), cis and trans isomers for [99mTcO2(DPAn-PSMAt)2]+ could be discerned (see Fig. 5).
| Complex | RCY (%) | Mean difference compared to [99mTcO2(DPPh-PSMAt)2]+ |
|---|---|---|
| [99mTcO2(DPPh-PSMAt)2]+ | 76.9 ± 1.9 (n = 4) | Comparator |
| [99mTcO2(DPAn-PSMAt)2]+ | 95.1 ± 0.7 (n = 6) | Mean difference = 18.2, 95% confidence interval = 15.3 to 21.1%, p = 0.0001 |
| [99mTcO2(DPMEP-PSMAt)2]+ | 95.3 ± 1.3 (n = 7) | Mean difference = 18.9, 95% confidence interval = 15.9 to 21.8%, p = 0.0002 |
| [188ReO2(DPAn-PSMAt)2]+ | 84.7 ± 7.1% (n = 4) | — |
| [188ReO2(DPMEP-PSMAt)2]+ | 83.1 ± 6.8% (n = 4) | — |
 | ||
| Fig. 3 Radiochromatograms of reactions containing (a) [99mTcO2(DPAn-PSMAt)2]+ and (b) [99mTcO2(DPMEP-PSMAt)2]+, including radiochromatograms of 99mTc samples after either incubation in human serum at 37 °C, or after administration to a mouse, followed by collection of urine 2 hours post-administration. Radio-HPLC analyses of serum and urine samples indicate that 99mTc radiotracers possess high stability in the presence of serum proteins and in vivo. | ||
For 188Re radiosyntheses, generator-produced 188ReO4− was first reduced to a 188ReV-citrate precursor, using a mixture of sodium citrate and SnCl2, as previously described.31,32 Then, aqueous solutions of 188ReV-citrate (49–130 MBq, 85 μL) were added to the contents of two kits containing DPAn-PSMAt or DPMEP-PSMAt (Table S1), and the mixtures heated to 90 °C for 30 min. The radiochemical yield of [188ReO2(DPAn-PSMAt)2]+ measured 84.7% ± 7.1% (n = 4), and [188ReO2(DPMEP-PSMAt)2]+ measured 83.1% ± 6.8% (n = 4). Analytical reverse-phase radio-HPLC chromatograms (Fig. 4) showed that the reaction products, [188ReO2(DPAn-PSMAt)2]+ and [188ReO2(DPMEP-PSMAt)2]+, formed one major radiochemical species, with the species observed at 2–3 min attributed to unreacted 188ReO4−/188ReV-citrate precursor(s). Signals attributed to the desired 188Re-radiolabelled products were broadened at the baseline, and it is possible that there are minor 188Re-radiolabelled side products present in this final kit-based reaction solution.
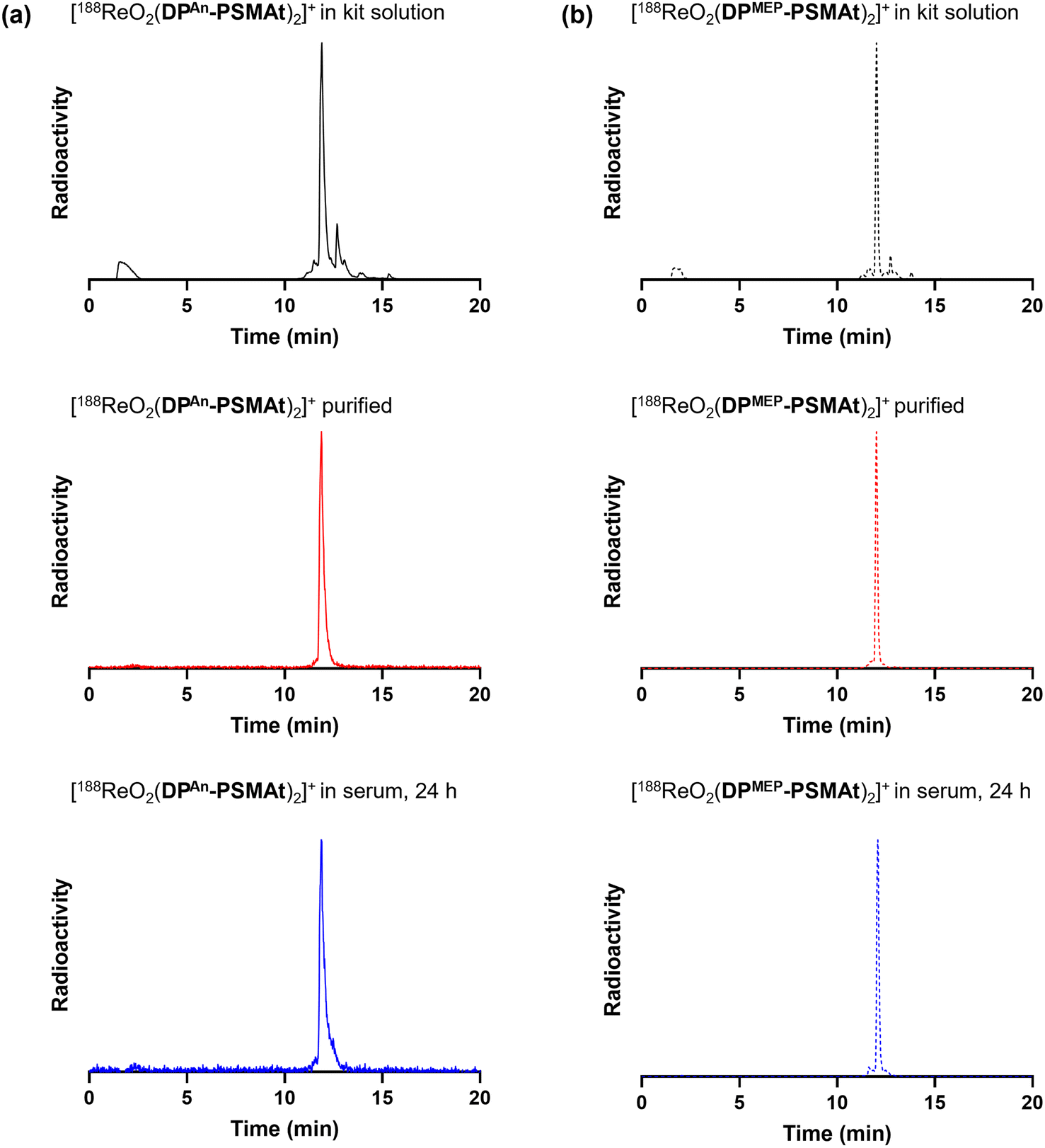 | ||
| Fig. 4 Radiochromatograms of reactions containing (a) [188ReO2(DPAn-PSMAt)2]+ and (b) [188ReO2(DPMEP-PSMAt)2]+, including radiochromatograms of 188Re-labelled samples after purification and then incubation in human serum at 37 °C. Radio-HPLC analyses of serum samples indicate that 188Re radiotracers possess high stability in the presence of serum proteins. | ||
The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DOCT/PBS of the new 99mTc and 188Re radiotracers was measured (Table 4), with radiotracers isolated from unreacted 99mTc/188Re precursor where required, prior to partition coefficient studies. The logDOCT/PBS values of all four new radiotracers were less than −3.0, indicating that all are highly hydrophilic.
DOCT/PBS of the new 99mTc and 188Re radiotracers was measured (Table 4), with radiotracers isolated from unreacted 99mTc/188Re precursor where required, prior to partition coefficient studies. The logDOCT/PBS values of all four new radiotracers were less than −3.0, indicating that all are highly hydrophilic.
DOCT/PBS values of [MO2(DP-PSMAt)2]+ complexes, where M = 99mTc or 188Re
| logDOCT/PBS |
||
|---|---|---|
| 99mTc | 188Re | |
| [MO2(DPAn-PSMAt)2]+ | −3.22 ± 0.04 | −3.47 ± 0.05 |
| [MO2(DPMEP-PSMAt)2]+ | −3.35 ± 0.12 | −3.91 ± 0.10 |
99gTc radiolabelling
To verify that DPAn-PSMAt and DPMEP-PSMAt form analogous complexes, the long-lived Tc radionuclide, 99gTc (t½ = 2.1 × 105 years), was utilised.99gTcO4− (0.06 kBq) was reacted with the contents of either a DPAn-PSMAt or DPMEP-PSMAt kit (Table S1), and analysed by LC-MS. Two prominent signals were observed by UV at 330 nm for each kit-based reaction containing either [99gTcO2(DPAn-PSMAt)2]+ or [99gTcO2(DPMEP-PSMAt)2]+ (Fig. 5). The first signal corresponded to the desired product, as evidenced by the appearance of a signal in the associated mass spectrum corresponding to species with a formula of either [99gTcO2(DPAn-PSMAt)2]+ ([M + H]2+: obs m/z = 1218.8, calc m/z = 1218.9) or [99gTcO2(DPMEP-PSMAt)2]+ ([M + H]2+: obs m/z = 1394.9, calc m/z = 1394.5). The second signal corresponded to that of unreacted DPAn-PSMAt or DPMEP-PSMAt.
 | ||
| Fig. 5 (a-i) and (a-ii) UV chromatogram and radiochromatograms of reactions containing [MO2(DPAn-PSMAt)2]+ (M = 99gTc, 99mTc or 188Re); (a-iii) mass spectrum showing signal corresponding to [99gTcO2(DPAn-PSMAt)2]+. (b-i and b-ii) UV chromatogram and radiochromatograms of reactions containing [MO2(DPMEP-PSMAt)2]+ (M = 99gTc, 99mTc or 188Re); (b-iii) mass spectrum showing signal corresponding to [99gTcO2(DPMEP-PSMAt)2]+. | ||
Each of [99mTcO2(DPAn-PSMAt)2]+, [99mTcO2(DPMEP-PSMAt)2]+, [188ReO2(DPAn-PSMAt)2]+ and [188ReO2(DPMEP-PSMAt)2]+ was also analysed using the same analytical reverse-phase radio-HPLC (Fig. 5). Analogous 99gTc, 99mTc and 188Re compounds exhibited highly similar HPLC retention times, with tracers based on DPAn-PSMAt giving rise to two closely eluting peaks (for [99mTcO2(DPAn-PSMAt)2]+tR = 18.18 and 18.23 min; for [188ReO2(DPAn-PSMAt)2]+tR = 17.98 and 18.05 min). We attribute the observation of these two radioactive signals to the formation of cis/trans isomers (Fig. 1). Tc and Re complexes of DPMEP-PSMAt also likely result in formation of cis/trans isomers, but under these HPLC conditions, these isomers cannot be resolved ([99mTcO2(DPMEP-PSMAt)2]+tR = 17.82 min; for [188ReO2(DPMEP-PSMAt)2]+tR = 17.75 min). The similar HPLC behaviours of analogous 99mTc and 188Re radiotracers is consistent with analogous pairs of 99mTc/188Re compounds being isostructural.
64Cu radiolabelling
We and others have previously demonstrated that diphosphines can both (i) reduce 64Cu2+ to 64Cu+ in aqueous solution, and (ii) coordinate to the resulting 64Cu+ radionuclide to yield [64Cu(diphosphine)2]+.19,22,23 In these radiochemical reactions, the diphosphine is in large excess compared to the 64Cu radiometal.To demonstrate the utility of these new compounds for radiolabelling with 64Cu for PET, each of DPAn-PSMAt (50.0 μg, 43.3 μmol) and DPMEP-PSMAt (57.7 μg, 43.3 μmol) were reacted with 64Cu2+ (7–8 MBq) in an aqueous solution (0.1 M ammonium acetate, pH 7) at ambient temperature for 10 min. For each reaction, analyses by analytical reverse-phase radio-HPLC (Fig. 6) showed the formation of a single major product, which we attributed to [64Cu(DPAn-PSMAt)2]+ (tR = 12.78 min, 79% RCY), and [64Cu(DPMEP-PSMAt)2]+ (tR = 12.67 min, 87% RCY). The radioactive signals of these products were coincident with the UV signal of the non-radioactive species, [natCu(DPAn-PSMAt)2]+ (tR = 12.71 min), and [natCu(DPMEP-PSMAt)2]+ (tR = 12.52 min), prepared and characterised as described in the SI.19
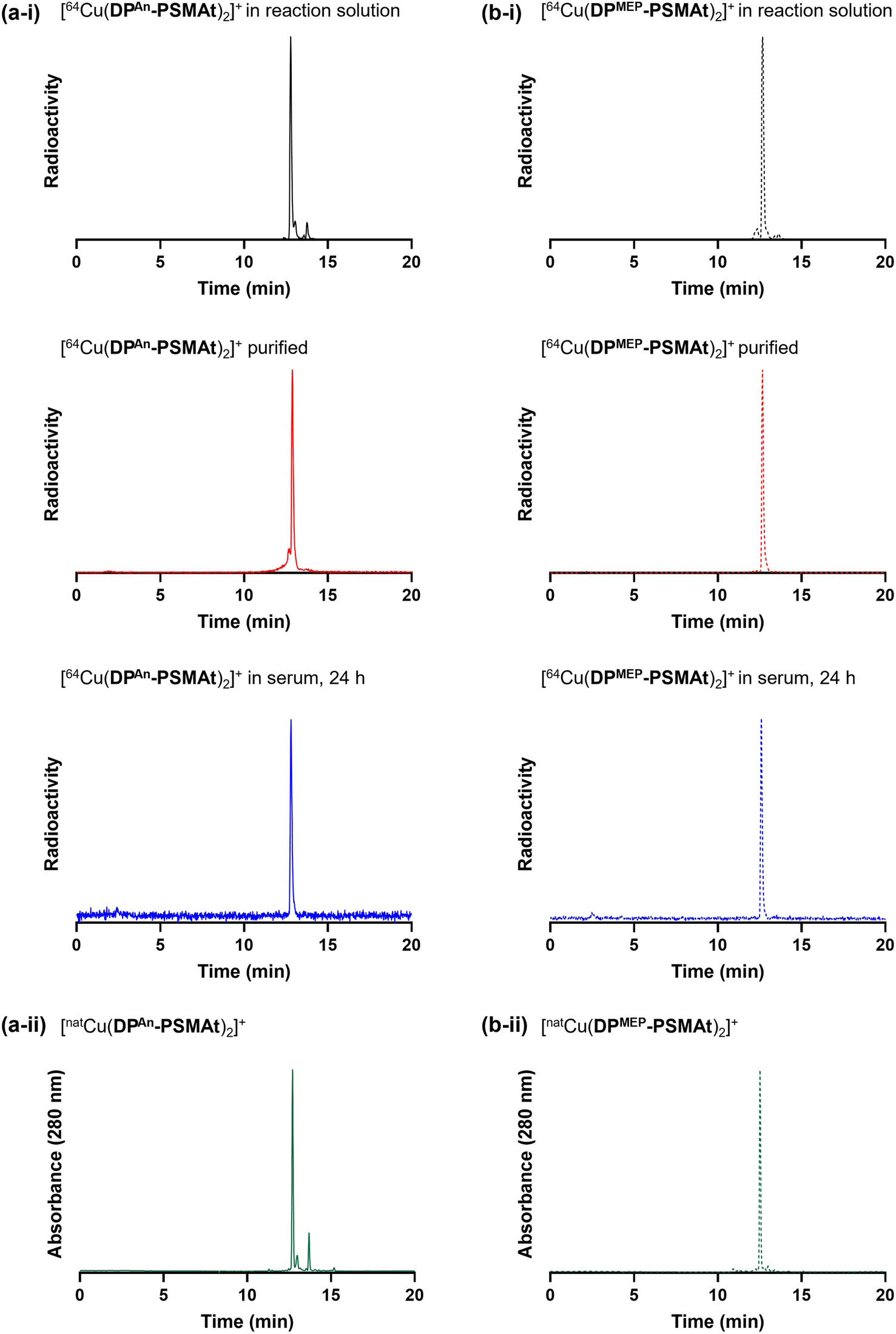 | ||
| Fig. 6 Radiochromatograms of (a-i) [64Cu(DPAn-PSMAt)2]+ and (b-i) [64Cu(DPMEP-PSMAt)2]+, including radiochromatograms of 64Cu samples after purification and then incubation in human serum at 37 °C. Radio-HPLC analyses of serum samples indicate that 64Cu radiotracers possess high stability in the presence of serum proteins. UV chromatograms of (a-ii) [natCu(DPAn-PSMAt)2]+ and (b-ii) [natCu(DPMEP-PSMAt)2]+. | ||
The new radiotracers, [64Cu(DPAn-PSMAt)2]+ and [64Cu(DPMEP-PSMAt)2]+, were purified using reverse-phase HPLC, reformulated in aqueous PBS (phosphate buffered saline) solution, and incubated with human serum at 37 °C. Radio-HPLC (Fig. 6, and SI) indicated that both [64Cu(DPAn-PSMAt)2]+ and [64Cu(DPMEP-PSMAt)2]+ were present after a 24 h incubation period, with <4.1% dissociated 64Cu detected. Both 64Cu radiotracers were also stable in aqueous PBS solution (see SI).
In vitro uptake of 99mTc tracers in prostate cancer cells
To compare the relative uptake and specificity of the new radiotracers for PSMA, [99mTcO2(DPPh-PSMAt)2]+, [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ (50 kBq) were purified from unreacted 99mTc precursors and excess DP-PSMAt peptide, and each incubated with PSMA-expressing DU145-PSMA+ prostate cancer cells33 or LNCaP prostate cancer cells (5 × 105 cells). After 1 h incubation, uptake of each radiotracer was quantified. Each radiotracer was also (i) co-incubated with the PSMA-inhibitor, PMPA (2-phosphonomethyl pentanedioic acid), in the presence of either DU145-PSMA+ cells or LNCaP cancer cells and (ii) incubated with parental DU145 cells that do not express PSMA. All three radiotracers exhibited uptake in PSMA-expressing prostate cancer cells (Fig. 7). This uptake was specific: DU145-PSMA+ and LNCaP cell uptake of each tracer could be blocked with PMPA, and there was negligible uptake in parental DU145 cells.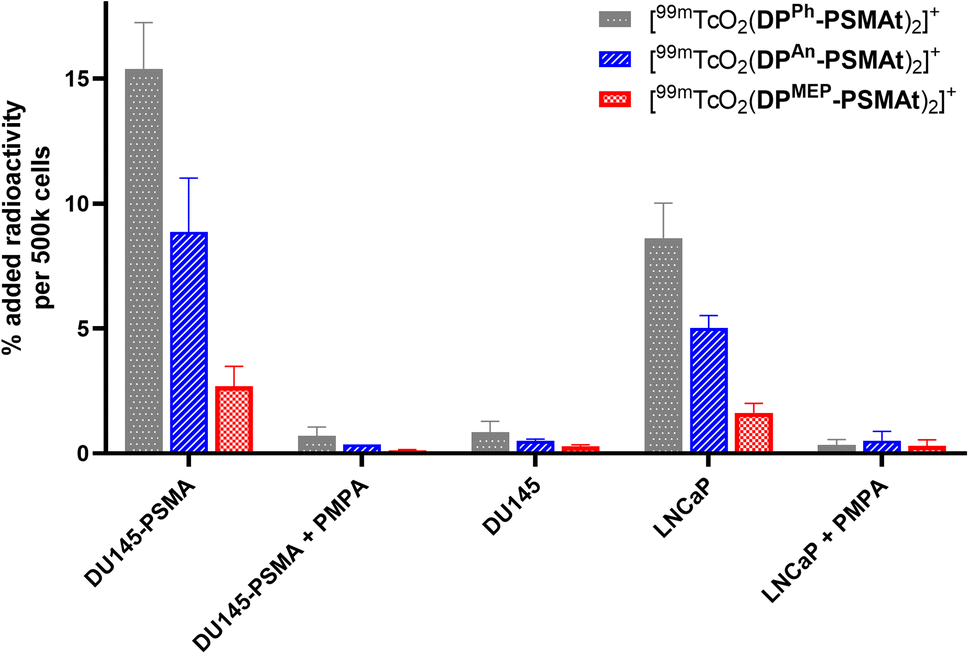 | ||
| Fig. 7 In vitro uptake of [99mTcO2(DPPh-PSMAt)2]+, [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ radiotracers in PSMA-expressing cell lines (DU145-PSMA+ and LNCaP), in the presence of an excess of PSMA inhibitor (PMPA) in PSMA-expressing cell lines, and in a cell line that does not express PSMA (DU145). | ||
The PSMA-specific uptake of [99mTcO2(DPPh-PSMAt)2]+ was significantly higher than that of either [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ in both DU145-PSMA+ cells and LNCaP cancer cells. For example, in DU145-PSMA+ cells, [99mTcO2(DPPh-PSMAt)2]+ measured 15.4 ± 1.9 %AR (% added radioactivity), [99mTcO2(DPAn-PSMAt)2]+ measured 8.9 ± 2.1 %AR (mean difference = 6.5 %AR, p = 0.02) and [99mTcO2(DPMEP-PSMAt)2]+ measured 2.7 ± 0.8 %AR (mean difference = 12.7 %AR, p = 0.003). The PSMA-specific uptake of [99mTcO2(DPAn-PSMAt)2]+ was significantly higher than that of [99mTcO2(DPMEP-PSMAt)2]+ in both DU145-PSMA+ cells and LNCaP cancer cells. For example, in DU145-PSMA+ cells, mean difference = 6.2 %AR (p = 0.03).
In vivo SPECT/CT and biodistribution studies of 99mTc tracers in mice bearing prostate cancer xenografts
The biodistribution of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ was studied using SPECT/CT imaging and ex vivo biodistribution methods. In these studies, [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ were used directly from kit-based formulations, without further purification, mimicking procedures used in clinical radiopharmacies and nuclear medicine departments. Mice were injected with [99mTcO2(DPAn-PSMAt)2]+ or [99mTcO2(DPMEP-PSMAt)2]+ (4–5 MBq), containing 3 μg (2.6 nmol) of DPAn-PSMAt or 3.5 μg (2.6 nmol) of (DPMEP-PSMAt) via the tail-vein, and culled 2 h post-injection, followed by dissection, organ/tissue weighing and counting.SPECT/CT studies were undertaken in male SCID/Beige mice bearing DU145-PSMA+ prostate cancer xenografts administered [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+. SPECT images were acquired from 15 to 120 min post-injection of radiotracer. In SPECT/CT scans of mice administered either [99mTcO2(DPAn-PSMAt)2]+ or [99mTcO2(DPMEP-PSMAt)2]+, tumours (T) could be delineated, with higher tumour to background ratios at later time points (Fig. 8). The kidneys (K) and bladder (B) were also clearly visible across all these timepoints with low background levels, indicating that both the radiotracers are rapidly cleared from the blood pool via a renal pathway.
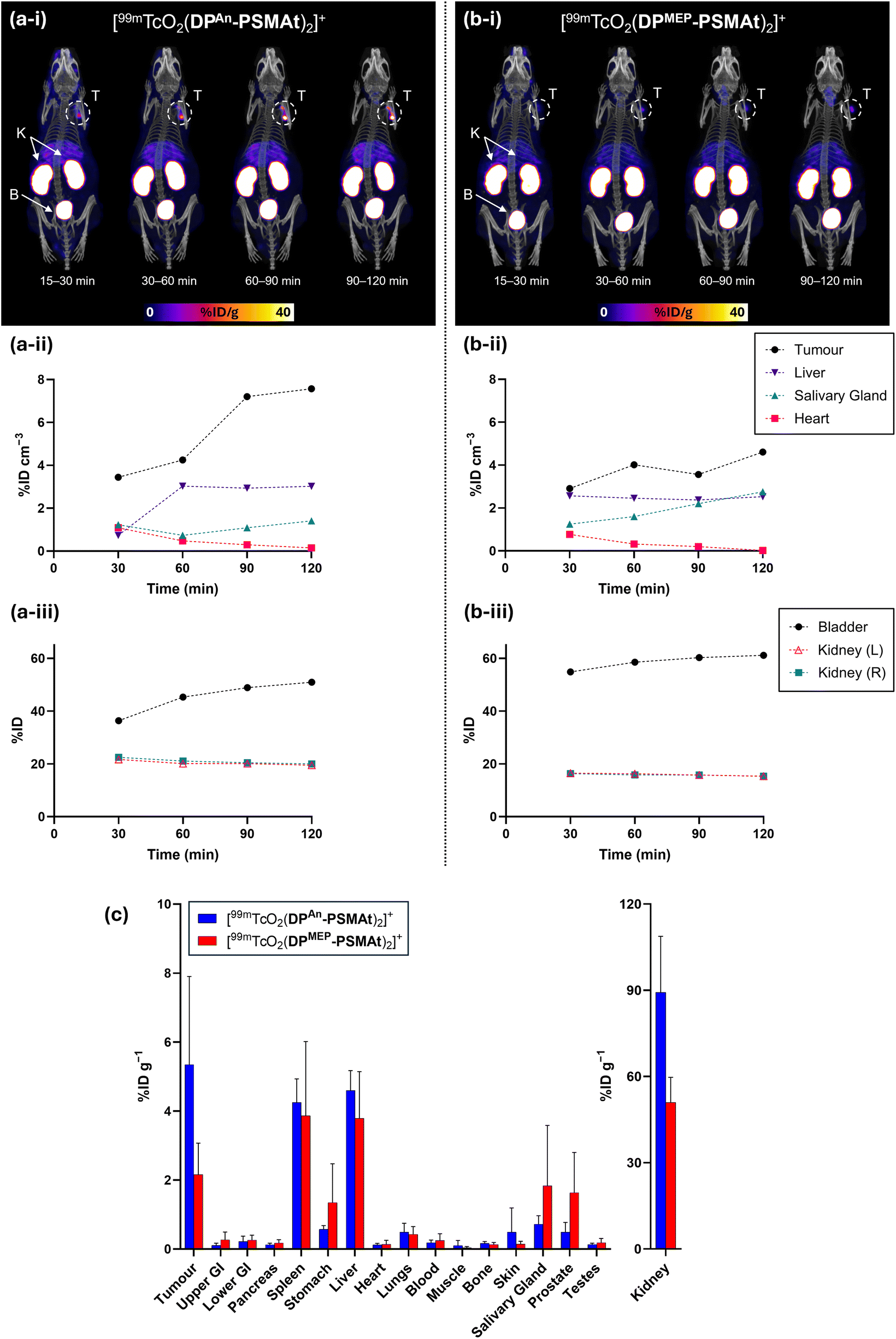 | ||
| Fig. 8 Whole body SPECT/CT maximum intensity projections and image quantification of SCID/Beige mice bearing DU145-PSMA+ tumours administered either (a) [99mTcO2(DPAn-PSMAt)2]+ or (b) [99mTcO2(DPMEP-PSMAt)2]+. T = tumour, K = kidneys, B = bladder. (c) Biodistribution (2 h post-injection) of [99mTcO2(DPAn-PSMAt)2]+ or [99mTcO2(DPMEP-PSMAt)2]+ in SCID/Beige mice bearing DU145-PSMA+ prostate cancer xenografts (mean ± SD, n = 3–4). | ||
Quantification of SPECT/CT images (acquired at 15–30 min, 30–60 min, 60–90 min and 90–120 min post-injection, Fig. 8) indicated that tumour uptake of [99mTcO2(DPAn-PSMAt)2]+ was higher than that of [99mTcO2(DPMEP-PSMAt)2]+, consistent with in vitro uptake studies and ex vivo biodistribution data (vide infra). Additionally, for the mouse administered [99mTcO2(DPAn-PSMAt)2]+, the 99mTc tumour concentration noticeably increased from 30 min until 2 h post-injection.
SPECT/CT indicated that 99mTc residualised in salivary glands for animals administered either [99mTcO2(DPAn-PSMAt)2]+ or [99mTcO2(DPMEP-PSMAt)2]+. Two well-documented mechanisms of radiotracer uptake likely account for salivary gland uptake:
(i) PSMA is expressed at low levels in the salivary glands, kidneys and small intestine, with PSMA-targeted radiotracers showing uptake in the salivary glands, in both mice and human patients.34–36 Indeed, the salivary glands are considered dose-limiting organs, and uptake of radiotherapeutic PSMA-targeted radiopharmaceuticals can lead to xerostomia in prostate cancer patients.36
(ii) TcO4−, bearing a single negative charge and with a similar polyatomic radius to that of the iodide anion, acts as a substrate for the sodium iodide symporter in vivo,37 and therefore accumulates in organs expressing the sodium iodide symporter – in mice, this includes the thyroid, salivary glands and stomach.38 SPECT imaging quantification (consistent with biodistribution data, vide infra) indicated that 99mTc concentrations were higher in the salivary glands for the mouse administered [99mTcO2(DPMEP-PSMAt)2]+ compared to the mouse administered [99mTcO2(DPAn-PSMAt)2]+. Indeed, 99mTc concentrations in the salivary glands increased over time for the former subject. This suggests that [99mTcO2(DPMEP-PSMAt)2]+ could possess lower in vivo stability than [99mTcO2(DPAn-PSMAt)2]+, with complex dissociation and oxidation of the [99mTcVO2]+ motif resulting in formation of 99mTcO4−in vivo, and increased 99mTc accumulation in the salivary glands for animals administered [99mTcO2(DPMEP-PSMAt)2]+.
The biodistribution of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ was further studied in (i) male SCID/Beige mice bearing DU145-PSMA+ prostate cancer xenografts (Fig. 8) and (ii) male nude mice bearing PSMA-expressing LNCaP prostate cancer xenografts (see SI).
For all animals, significant concentrations of 99mTc radioactivity (2–5 %ID g−1) were measured in PSMA-expressing tumours 2 h post-injection, and although 99mTc tumour concentrations were consistently higher for animals administered [99mTcO2(DPAn-PSMAt)2]+, there were no statistically significant differences between [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ in either mouse model. For both tracers, there were also significant concentrations of 99mTc radioactivity in the spleen, which is known to express low levels of PSMA and accumulate PSMA-targeted radiotracers,9,10 and the liver. Importantly, clearance from the blood pool and subsequent excretion was largely via a renal route, with concentrations of 40–90 %ID g−1 measured in kidneys.
We also noted that for both SCID/beige and nude mice studies, higher 99mTc concentrations were observed in the stomach and salivary glands for animals administered [99mTcO2(DPMEP-PSMAt)2]+ compared to animals administered [99mTcO2(DPAn-PSMAt)2]+, although these differences were not statistically significant. This observation is consistent with observations from SPECT/CT imaging studies, and further supports our conjecture that [99mTcO2(DPMEP-PSMAt)2]+ has lower in vivo stability than [99mTcO2(DPAn-PSMAt)2]+, with in vivo formation of 99mTcO4− from dissociation of [99mTcO2(DPMEP-PSMAt)2]+ resulting in higher levels of 99mTc activity in organs expressing the sodium iodide symporter.
Overall, the in vivo biodistribution demonstrates that both [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ are useful SPECT or γ-scintigraphy imaging agents for PSMA expression in prostate cancer.
Stability of [MO2(DPAn-PSMAt)2]+ and [MO2(DPMEP-PSMAt)2]+ (M = 99mTc, 188Re)
Both [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+, prepared using kits, were incubated at 37 °C in human serum for up to 24 h. Analytical reverse-phase radio-HPLC (Fig. 3) demonstrated that >98% of both radiotracers remained intact in serum. [188ReO2(DPAn-PSMAt)2]+ and [188ReO2(DPMEP-PSMAt)2]+, prepared using kits and then isolated using reverse-phase HPLC in >98% radiochemical purity, were also incubated in human serum under the same conditions. Radio-HPLC (Fig. 4) similarly showed that 188Re-labelled radiotracers were stable in serum over 24 h, with <1.5% of 188Re dissociating over this time. These 99mTc and 188Re radiotracers were also stable when left to stand in kit-based reaction solutions, or in PBS solution (see SI for full chromatograms).Urine was also collected in in vivo99mTc biodistribution experiments (from male SCID/Beige mice bearing DU145-PSMA+ prostate cancer xenografts at 2 h post-injection) and analysed using reverse-phase radio-HPLC (Fig. 3). Radio-chromatograms indicated that both [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ were excreted in urine intact, indicating that both radiotracers have high stability in vivo.
Discussion
We have recently reported DPPh and DPTol derivatives of the 2,3-bis(diphenylphosphino)maleic anhydride platform, and shown that the resulting 99mTc and 188Re radiotracers have desirable properties as molecular theranostic agents.19,20 Here, we have optimised this platform technology through development of DPAn and DPMEP, and demonstrated that these compounds can be attached to a variety of amine-containing bioactive molecules, including peptides, carbohydrates, vitamins and small molecule inhibitors. The resulting diphosphine conjugates can be radiolabelled with 99mTc, 188Re and 64Cu, thus enabling formation of radiotracers for molecular SPECT imaging, radiotherapy and molecular PET imaging, respectively.Significantly, as hypothesised, the increased σ-donating ability of DPAn and DPMEP derivatives, due to para substituents on aryl rings of the phosphines, leads to further increased RCYs within this family of diphosphine chelators (DPAn ∼ DPMEP > DPTol > DPPh). The kit formulations of DPAn and DPMEP conjugates developed here use relatively small amounts of DPAn-PSMAt and DPMEP-PSMAt diphosphine bioconjugates (110 nmol, 113–145 μg), minimising competitive target receptor binding of unreacted ligand present in formulations administered to in vivo subjects. The high RCYs (≥95%) of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ achieved using our one-step kit formulation negate the need for time-intensive and often complicated purification procedures to remove unreacted 99mTc precursors. In a clinical context, rapid, one-step radiolabelling and formulation protocols using kits are desirable: this simplicity of producing a radiopharmaceutical for immediate patient injection makes widespread clinical adoption feasible. We are yet to optimise 188Re radiolabelling protocols including kit formulations for new 188Re radiotracers, however, we note that the radiochemical yields (>83%) are significantly improved compared to RCYs of prior DPPh and DPTol conjugate derivatives.20 In these previous reactions, 188Re radiotracer yields were often highly variable, and commonly less than 30%.
The logDOCT/PBS values observed here are all lower than those previously reported for [99mTcO2(DPPh/Tol-PSMAt)2]+ derivatives, consistent with the more hydrophilic methoxy and methyl ethylene glycol substituents of aryl rings, compared to DPPh and DPTol analogues. This also poses potential advantages for receptor-targeted biomolecular vectors that are significantly more hydrophobic than the PSMAt pharmacophore tested here.
We observe decreasing uptake of [99mTcO2(DPX-PSMAt)2]+ (X = Ph, An, MEP) radiotracer analogues in PSMA-positive prostate cancer cells in the order Ph > An > MEP and we note that this corresponds with increasing size and hydrophilicity of the para substituents: H < OCH3 < OCH2CH2OCH3. It is well documented that aromatic groups are important in PSMA affinity, due to the hydrophobic binding pocket of PSMA.39,40 We postulate that the para substituents interfere with binding of the radiotracer to PSMA.
In the biodistribution experiments undertaken for this work, prostate cancer tumour uptake of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ was lower than previously reported for [99mTcO2(DPPh-PSMAt)2]+ and [99mTcO2(DPTol-PSMAt)2]+ in the same murine tumour models.20 This was expected: here, kit-based formulations of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ were not purified from unreacted, excess DPAn-PSMAt and DPMEP-PSMAt respectively, prior to injection. It is likely that DPAn-PSMAt and DPMEP-PSMAt compete for binding to PSMA receptor in vivo.
Liver concentrations of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ were observed to be higher than that previously measured for first-generation [99mTcO2(DPPh-PSMAt)2]+ and [99mTcO2(DPTol-PSMAt)2]+ radiotracers in the same murine models. An increased liver accumulation of a tracer can correlate with increased hydrophobicity of compounds, but we note that logDOCT/PBS values indicate that [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ are both more hydrophilic than their predecessors. We therefore attribute the higher liver accumulation of [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ to either their increased size, or alternatively, the different formulation of the radiotracer dose, which includes unlabelled bioconjugate precursor, which could affect the biodistribution.
Compared to animals administered [99mTcO2(DPAn-PSMAt)2]+, animals administered [99mTcO2(DPMEP-PSMAt)2]+ presented higher 99mTc concentrations in salivary glands and the stomach. This could indicate that [99mTcO2(DPMEP-PSMAt)2]+ exhibits a degree of instability in vivo, as both the salivary glands and the stomach express the sodium iodide symporter and are known to take up 99mTcO4−.38 It is also possible that some salivary gland uptake is due to known salivary gland PSMA expression.34,35 However, except for the possible formation of small amounts of 99mTcO4−in vivo, both [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ demonstrated high metabolic stability, as evidenced by their excretion intact in urine, and their high stability in serum (ex vivo) over 24 h.
Our new diphosphine chelator technology has advantages over other 99mTc and 188Re radiolabelling platforms. For instance, the radiosynthesis and purification of 99mTc-MIP-1404, which is based on the [99mTc(CO)3]+ synthon, is time-consuming. It requires (i) formation of an intermediate fac-[99mTc(CO)3(H2O)3]+ precursor prior to (ii) chelation with the tridentate MIP-1404 chelator-peptide, and finally (iii) purification and formulation before administration.9 Other molecular 99mTc-radiopharmaceuticals, including 99mTc-EDDA/HYNIC-octreotide1 and 99mTc-EDDA/HYNIC-iPSMA,8,9 utilise the 6-hydrazinopyridine-3-carboxylic acid (HYNIC) platform, which coordinates to 99mTc and acts as the attachment point for the bioactive receptor-targeting vectors. The ethylenediamine co-ligand chelates to the remaining coordination sites on the Tc metal centre. Whilst some of these HYNIC-based radiopharmaceuticals can be prepared from a single kit,13 their structures remain ill-defined:41,42 it is unknown whether HYNIC coordinates to Tc via the hydrazine group only, or as a bidentate ligand, via the hydrazine and pyridyl groups. Importantly, HYNIC-based bioconjugates are not known to provide isostructural Tc and Re derivatives,41,42 and therefore cannot be used to develop dual 99mTc/188Re theranostic tracers.
Compared with the existing 99mTc-radiolabeled PSMA-targeted tracers, 99mTc-MIP-1404, 99mTc-PSMA-I&S, and 99mTc-EDDA/HYNIC-iPSMA, the new [99mTcO2(DPAn-PSMAt)2]+ and [99mTcO2(DPMEP-PSMAt)2]+ radiotracers demonstrate either increased or comparable blood clearance at 1–2 h after administration (Table 5). [99mTcO2(DPAn-PSMAt)2]+ exhibits either decreased or comparable residualisation in murine liver compared to these other PSMA-targeted tracers.
| Radiotracer (time post-injection) | Blooda | Livera | Kidneya |
|---|---|---|---|
| a % ID g−1, ± standard deviation. | |||
| [99mTcO2(DPAn-PSMAt)2]+ (athymic nude, 2 h) | 0.11 ± 0.04 | 0.90 ± 0.15 | 87.6 ± 13.3 |
| [99mTcO2(DPMEP-PSMAt)2]+ (athymic nude, 2 h) | 0.17 ± 0.10 | 7.38 ± 3.98 | 46.4 ± 23.2 |
| [99mTcO2(DPPh-PSMAt)2]+ (athymic nude, 2 h)20 | 0.23 ± 0.05 | 0.35 ± 0.05 | 203.56 ± 13.67 |
| [99mTcO2(DPTol-PSMAt)2]+ (athymic nude, 2 h)20 | 0.28 ± 0.05 | 0.52 ± 0.05 | 212.23 ± 29.49 |
| 99mTc-MIP-1404 (athymic nude, 1 h)9 | 0.13 ± 0.03 | 0.14 ± 0.03 | 105 ± 37 |
| 99mTc-PSMA-I&S (SCID, 1 h)10 | 1.73 ± 0.50 | 1.58 ± 0.24 | 186 ± 23 |
| 99mTc-EDDA/HYNIC-iPSMA (athymic, 1 h)13 | 0.18 ± 0.08 | 2.18 ± 0.19 | 23.63 ± 3.56 |
These diphosphine chelators also enable simple, one-pot preparation of stable molecular radiotracers based on radioactive copper isotopes, such as 64Cu for PET imaging, and its “theranostic” companion, 67Cu (t½ = 61.9 h; 100% β−, Emax = 561 keV; 44% γ, 185 keV), which emits β− particles and has utility for systemic radiotherapy.43 Critically, this allows application of the same diphosphine-peptide bioconjugate for theranostic radiopharmaceuticals that span both SPECT and PET imaging.
Concluding remarks
In summary, our new and highly versatile bis(phosphino) maleic anhydride platforms, DPAn and DPMEP, enable facile preparation of diphosphine bioconjugates that can be simply radiolabelled with 99mTc for SPECT imaging, 64Cu for PET imaging and 188Re for systemic radiotherapy, leading to the possibility of theranostic radiotracers. Importantly, these 99mTc radiotracers can be prepared in high radiochemical yields (≥95%) using a single step kit. This is a critical advance upon our prior DPPh and DPTol technology, as it allows for simple, one-step formulation of peptide-based 99mTc radiotracers, and presents new opportunities for economical molecular imaging using widely available 99mTc production infrastructure and γ-scintigraphy/SPECT cameras. We are currently expanding the application of DPAn and DPMEP chemistry to other therapeutically relevant receptor targets, to develop a versatile suite of molecular SPECT, PET and radiotherapeutic tracers.Author contributions
REN conceived research, designed experiments, undertook experimental work and drafted the manuscript; MTM and PGP conceived research, designed experiments and drafted the manuscript; INH, OWLC, AR, NP, ZY, JC and JDY undertook experimental work; TTP conceived research and undertook experimental work; JS, GJRC and LL contributed to experimental design; NV and HHT provided a 188W/188Re Oncobeta generator and provided expertise on its use and potential clinical applications.Conflicts of interest
Some of the authors have submitted a patent application describing the intellectual property described herein. Nicholas Vetter is CEO of OncoBeta, who provide 188Re generators, including those used in this study. No other potential conflicts of interest relevant to this article exist.Data availability
The data supporting this article, including abbreviations, experimental procedures, characterisation data, additional figures, chromatograms and spectra have been included as part of the SI. See DOI: https://doi.org/10.1039/d5sc02110c.Acknowledgements
This research was supported by a Cancer Research UK Career Establishment Award (C63178/A24959), the EPSRC (EP/S032789/1), the Cancer Research UK National Cancer Imaging Translational Accelerator Award (C4278/A27066), the EPSRC programme for Next Generation Molecular Imaging and Therapy with Radionuclides (EP/S019901/1), ‘MITHRAS’, the Wellcome Multiuser Equipment Radioanalytical Facility funded by Wellcome Trust (212885/Z/18/Z), and the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/L015366/1). SPECT/CT scanning equipment at KCL was funded by an equipment grant from the Medical Research Council (MR/X011992/1). The authors are grateful for research assistance with animal work from James Cormack, Harmony Blythin and Hagen Schmidt. We would like to thank Dr Paul J. Gates and the mass spectrometry facility of the University of Bristol, UK and Oscar Ayrton and the mass spectrometry facility of King's College London, UK for obtaining high-resolution mass spectra.References
- M. Gabriel, C. Decristoforo, E. Donnemiller, H. Ulmer, C. W. Rychlinski, S. J. Mather and R. Moncayo, J. Nucl. Med., 2003, 44, 708–716 CAS.
- M. S. Hofman, G. Kong, O. C. Neels, P. Eu, E. Hong and R. J. Hicks, J. Med. Imaging Radiat. Oncol., 2012, 56, 40–47 CrossRef PubMed.
- A. Shinto, Curr. Trends Clin. Med. Imaging., 2017, 1, 91–94 Search PubMed.
- J. A. Jackson, I. N. Hungnes, M. T. Ma and C. Rivas, Bioconjugate Chem., 2020, 31, 483–491 CrossRef CAS PubMed.
- C. Rivas, J. A. Jackson, I. N. Hungnes and M. T. Ma, Compr. Coord. Chem. III, 2021, 9, 706–740 Search PubMed.
- M. Riondato, D. Rigamonti, P. Martini, C. Cittanti, A. Boschi, L. Urso and L. Uccelli, J. Med. Chem., 2023, 66, 4532–4547 CrossRef CAS PubMed.
- S. Liu and S. Chakraborty, Dalton Trans., 2011, 40, 6077–6086 RSC.
- C. Schmidkonz, C. Hollweg, M. Beck, J. Reinfelder, T. I. Goetz, J. C. Sanders, D. Schmidt, O. Prante, T. Bäuerle, A. Cavallaro, M. Uder, B. Wullich, P. Goebell, T. Kuwert and P. Ritt, Prostate, 2018, 78, 54–63 CrossRef CAS PubMed.
- S. M. Hillier, K. P. Maresca, G. Lu, R. D. Merkin, J. C. Marquis, C. N. Zimmerman, W. C. Eckelman, J. L. Joyal and J. W. Babich, J. Nucl. Med., 2013, 54, 1369–1376 CrossRef CAS PubMed.
- S. Robu, M. Schottelius, M. Eiber, T. Maurer, J. Gschwend, M. Schwaiger and H. J. Wester, J. Nucl. Med., 2017, 58, 235–242 CrossRef CAS PubMed.
- T. Maurer, S. Robu, M. Schottelius, K. Schwamborn, I. Rauscher, N. S. van den Berg, F. W. B. van Leeuwen, B. Haller, T. Horn, M. M. Heck, J. E. Gschwend, M. Schwaiger, H. J. Wester and M. Eiber, Eur. Urol., 2019, 75, 659–666 CrossRef PubMed.
- I. O. Lawal, A. O. Ankrah, N. P. Mokgoro, M. Vorster, A. Maes and M. M. Sathekge, Prostate, 2017, 77, 1205–1212 CrossRef CAS PubMed.
- G. Ferro-Flores, M. Luna-Gutiérrez, B. Ocampo-García, C. Santos-Cuevas, E. Azorín-Vega, N. Jiménez-Mancilla, E. Orocio-Rodríguez, J. Davanzo and F. O. García-Pérez, Nucl. Med. Biol., 2017, 48, 36–44 CrossRef CAS PubMed.
- P. Bernal, J. L. Raoul, J. Stare, E. Sereegotov, F. X. Sundram, A. Kumar, J. M. Jeong, P. Pusuwan, C. Divgi, P. Zanzonico, G. Vidmar, J. Buscombe, T. T. M. Chau, M. M. Saw, S. Chen, R. Ogbac, M. Dondi and A. K. Padhy, Semin. Nucl. Med., 2008, 38, S40–S45 CrossRef PubMed.
- A. Shinto, World J. Nucl. Med., 2017, 16, 1–2 CrossRef PubMed.
- A. Shinto, M. Mallia, M. Kameswaran, K. Kamaleshwaran, J. Joseph, E. Radhakrishnan, I. Upadhyay, R. Subramaniam, M. Sairam, S. Banerjee and A. Dash, World J. Nucl. Med., 2018, 17, 228–235 CrossRef PubMed.
- J. Cardinale, F. L. Giesel, C. Wensky, H. G. Rathke, U. Haberkorn and C. Kratochwil, J. Nucl. Med., 2023, 64, 1069–1075 CrossRef CAS PubMed.
- I. N. Hungnes, F. Al-Salemee, P. J. Gawne, T. Eykyn, R. A. Atkinson, S. Y. A. Terry, F. Clarke, P. J. Blower, P. G. Pringle and M. T. Ma, Dalton Trans., 2021, 50, 16156–16165 RSC.
- I. N. Hungnes, T. T. Pham, C. Rivas, J. A. Jarvis, R. E. Nuttall, S. M. Cooper, J. D. Young, P. J. Blower, P. G. Pringle and M. T. Ma, Inorg. Chem., 2023, 62, 20608–20620 CrossRef CAS PubMed.
- T. T. Pham, I. N. Hungnes, C. Rivas, J. Cleaver, G. Firth, P. J. Blower, J. Sosabowski, G. J. R. Cook, L. Livieratos, J. D. Young, P. G. Pringle and M. T. Ma, J. Nucl. Med., 2024, 65, 1087–1094 CrossRef CAS PubMed.
- R. E. Nuttall, T. T. Pham, A. C. Chadwick, I. N. Hungnes, G. Firth, M. A. Heckenast, H. A. Sparkes, M. C. Galan, M. T. Ma and P. G. Pringle, Inorg. Chem., 2023, 62, 20582–20592 CrossRef CAS PubMed.
- J. S. Lewis, J. Zweit, J. L. J. Dearling, B. C. Rooney and P. J. Blower, Chem. Commun., 1996, 1093–1094 RSC.
- J. S. Lewis, S. L. Heath, A. K. Powell, J. Zweit and P. J. Blower, J. Chem. Soc., Dalton Trans., 1997, 855–861 RSC.
- C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed.
- S. Bongarzone, T. Sementa, J. Dunn, J. Bordoloi, K. Sunassee, P. J. Blower and A. Gee, J. Med. Chem., 2020, 63, 8265–8275 CrossRef CAS PubMed.
- C. Müller and R. Schibli, J. Nucl. Med., 2011, 52, 1–4 CrossRef PubMed.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- C. Imberti, S. Y. A. Terry, C. Cullinane, F. Clarke, G. H. Cornish, N. K. Ramakrishnan, P. Roselt, A. P. Cope, R. J. Hicks, P. J. Blower and M. T. Ma, Bioconjugate Chem., 2017, 28, 481–495 CrossRef CAS PubMed.
- C. Kratochwil, P. Flechsig, T. Lindner, L. Abderrahim, A. Altmann, W. Mier, S. Adeberg, H. Rathke, M. Röhrich, H. Winter, P. K. Plinkert, F. Marme, M. Lang, H. U. Kauczor, D. Jäger, J. Debus, U. Haberkorn and F. L. Giesel, J. Nucl. Med., 2019, 60, 801–805 CrossRef CAS PubMed.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 621–627 CrossRef CAS.
- H. Gali, T. J. Hoffman, G. L. Sieckman, N. K. Owen, K. V. Katti and W. A. Volkert, Bioconjugate Chem., 2001, 12, 354–363 CrossRef CAS PubMed.
- K. K. Kothari, H. Gali, K. R. Prabhu, N. Pillarsetty, N. K. Owen, K. V. Katti, T. J. Hoffman and W. A. Volkert, Nucl. Med. Biol., 2002, 29, 83–89 CrossRef CAS PubMed.
- F. Kampmeier, J. D. Williams, J. Maher, G. E. Mullen and P. J. Blower, EJNMMI Res., 2014, 4, 1–10 CrossRef CAS PubMed.
- J. Roy, B. M. Warner, F. Basuli, X. Zhang, K. Wong, T. Pranzatelli, A. T. Ton, J. A. Chiorini, P. L. Choyke, F. I. Lin and E. M. Jagoda, Cancer Biother. Radiopharm., 2020, 35, 284–291 CrossRef CAS PubMed.
- S. Piron, J. Verhoeven, E. De Coster, B. Descamps, K. Kersemans, L. Pieters, A. Vral, C. Vanhove and F. De Vos, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- N. Heynickx, K. Herrmann, K. Vermeulen, S. Baatout and A. Aerts, Nucl. Med. Biol., 2021, 98–99, 30–39 CrossRef CAS PubMed.
- J.-K. Chung, J. Nucl. Med., 2002, 43, 1188–1200 CAS.
- F. Man, L. Lim, A. Volpe, A. Gabizon, H. Shmeeda, B. Draper, A. C. Parente-Pereira, J. Maher, P. J. Blower, G. O. Fruhwirth and R. T. M. de Rosales, Mol. Ther., 2019, 27, 219–229 CrossRef CAS PubMed.
- M. Benešová, U. Bauder-Wüst, M. Schäfer, K. D. Klika, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Med. Chem., 2016, 59, 1761–1775 CrossRef PubMed.
- M. Benesová, M. Schäfer, U. Bauder-Wüst, A. Afshar-Oromieh, C. Kratochwil, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Nucl. Med., 2015, 56, 914–920 CrossRef PubMed.
- R. C. King, M. B. U. Surfraz, S. C. G. Biagini, P. J. Blower and S. J. Mather, Dalton Trans., 2007, 4998–5007 RSC.
- L. K. Meszaros, A. Dose, S. C. G. Biagini and P. J. Blower, Inorg. Chim. Acta, 2010, 363, 1059–1069 CrossRef CAS.
- S. E. Rudd, J. Van Zuylekom, C. Cullinane, B. J. Blyth and P. S. Donnelly, Chem. Sci., 2025, 16, 3998–4005 RSC.
Footnote |
| † DPPh and DPTol and their derivatives have alternatively been abbreviated to “DP1” and “DP2”, respectively in our prior report in a medical journal.20 |
| This journal is © The Royal Society of Chemistry 2025 |