
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh throughput screening for the design of protein binding polymers†
Carolin
Bapp‡
 a,
Ahmed Z.
Mustafa‡
b,
Cheng
Cao
b,
Erica J.
Wanless
a,
Martina H.
Stenzel
*b and
Robert
Chapman
*a
a,
Ahmed Z.
Mustafa‡
b,
Cheng
Cao
b,
Erica J.
Wanless
a,
Martina H.
Stenzel
*b and
Robert
Chapman
*a
aSchool of Environmental and Life Sciences, University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: robert.chapman@newcastle.edu.au
bCentre for Advanced Macromolecular Design, School of Chemistry, UNSW Sydney, Kensington, NSW 2052, Australia. E-mail: m.stenzel@unsw.edu.au
First published on 1st July 2025
Abstract
Using polymers for protein encapsulation can enhance stability in processing environments and prolong activity and half-life in vivo. However, finding the best polymer structure for a target protein can be difficult, labour- and cost-intensive. In this study we introduce a high throughput screening approach to identify strong polymer–protein interactions by use of Förster Resonance Energy Transfer (FRET), enabling a rapid read out. We iteratively screened a total of 288 polymers containing varying hydrophilic, hydrophobic, anionic and cationic monomers against a panel of eight different enzymes (glucose oxidase, uricase, manganese peroxidase, bovine serum albumin, carbonic anhydrase, lysozyme, trypsin and casein). By optimisation of the assay conditions it was possible to read out strongly binding polymers at protein concentrations down to 0.1 μM. We were able to use the screening data to locate moderately selective polymer binders in most cases, and elucidate general trends in polymer design that lead to strong binding. Interestingly, these trends are not consistent across proteins, underscoring the value of a screening approach for identification of the best polymers. We applied this technique to identify lead polymers suitable for encapsulation of the important therapeutic protein TNF-related apoptosis-inducing ligand (TRAIL), at a concentration of 0.25 μM (5 μg mL−1). This approach should be valuable in the design of polymers for either selective protein binding, or for universal protein repulsion, particularly where the protein is too expensive to work with at high concentrations and large volumes.
Introduction
Directing the way in which proteins bind to synthetic polymers is key to unlocking their use in a wide range of applications from biomedical treatments,1,2 to industrial catalysis,3 and even the depolymerisation of plastic waste.4,5 In many cases, non-covalent encapsulation of a protein with a strongly binding polymer is advantageous, either to impart stability or direct activity.5–10 In others, it is important to minimise undesired binding as is the case in controlling the protein corona of nanoparticles for drug delivery, the make-up of which can significantly influence the in vivo response.11–13 Because of the unique surface landscape of any given protein, matching the design of polymeric systems to the surface properties is important to achieve the desired strength of binding without disruption to the protein's active or binding site.While we lack the tools to achieve the sequence control of proteins in a synthetic polymer, statistical populations of copolymers (termed random heteropolymers, RHPs) have been shown to very effectively and selectively encapsulate and stabilize proteins,5,6,14,15 and fold and mimic their function.16–18 Key to this approach is the insight that although a random copolymer lacks precise sequence definition, within any given population of appropriately designed copolymers there will exist some sequences that complement the protein surface well enough to encapsulate the protein, or which match the primary sequence well enough to fold and recapitulate the activity of the protein. The group of Ting Xu has shown that even using only four different monomers reflecting the four classes of amino acids (methyl methacrylate (MMA) and 2-ethylhexyl methacrylate (EHMA) for the hydrophobic residues, oligo(ethylene glycol) methyl ether methacrylate (OEGMA) for the hydrophilic residues and 3-sulfopropyl methacrylate potassium salt (SPMA) for the anionic residues), very effective mimics of membrane transport proteins, for example, can be found.18 In their study, proton transfer comparable to natural proton channels was achieved with purely synthetic polymers. Likewise the group has stabilised a range of common enzymes such as horseradish peroxidase,6 lipase and proteinase K.19 Encapsulation of lipase and proteinase K enabled dispersion into poly(caprolactone) and polylactic acid (PLA), respectively, and direct the activity of the enzyme to chain end depolymerisation instead of random chain scission.
While computational approaches can help refine the best RHP combinations to try for any given application,20,21 high throughput experimental screening approaches have been very helpful in refining the design of similar materials.22–28 Recent innovations in high throughput polymer synthesis have enabled screening of polymer structures in the open atmosphere,29 but applying these methods to designing polymers for single protein encapsulation requires efficient read out mechanisms for binding. Using activity assays as the readout, Gormley and coworkers recently applied a high throughput approach to designing polyelectrolytes for glucose oxidase, lipase, and horseradish peroxidase encapsulation.15 By screening a total of over 500 automatically synthesised polymers over five “learn-design-build-test” cycles, polymers able to improve the retained enzyme activity after thermal stress by up to 90% in some cases were identified. Importantly, most polymer combinations were largely inactive, and so this kind of high throughput screen was critical to guiding the polymer design. Unfortunately, easy activity assays are not always readily available for a given protein, particularly in the case of therapeutic proteins, and do not provide a direct read-out of binding. In many cases encapsulation would ‘switch off’ or modulate the activity of the underlying protein. Furthermore, many therapeutic and engineered proteins are very expensive, and this rules out techniques which require large amounts of material such as isothermal calorimetry (ITC), X-ray (SAXS) or light scattering (MALS).
To address this, we recently introduced the use of Förster resonance energy transfer (FRET) as an alternative and complementary method for screening protein–polymer binding.30 In these assays we attached cyanine 3 (Cy3) to our model protein (glucose oxidase) via amide coupling, and cyanine 5 (Cy5) to the polymer via a functionalised monomer and measured the FRET ratio upon excitation of Cy3. Using ITC, SAXS, we demonstrated that the FRET ratio correlates well with binding strength in a small library of positively charged polymers. In this work, we advanced this technique to screen a large polymer library prepared using automated synthesis, against a range of proteins varying in size and surface characteristics including glucose oxidase, uricase, manganese peroxidase, bovine serum albumin, carbonic anhydrase, lysozyme, trypsin and casein. Since the target polymers are soluble in water/alcohol mixtures, their synthesis is compatible with enzyme-assisted RAFT,29,31,32 and so can be prepared in high throughput. The ability to both synthesise polymers and measure protein binding in this way opens up the possibility of automating the design of polyelectrolytes for expensive proteins, as well as those which are hard to find a suitable binding (or non-binding) polymer for. We were interested to understand the limits of this approach to polymer design and to what extent the protein sequence could be used to predict polymer binding. We also sought to understand what general features of polymer design control binding strength (see Fig. 1) and performed complementary activity assays. With this information in hand, we then apply the method to design a polymer capable of encapsulating TNF-related apoptosis-inducing ligand (TRAIL),33–35 an expensive and poorly circulating chemotherapeutic protein.
 | ||
Fig. 1 Layout of the two polymer libraries synthesised. Each polymer was made at a degree of polymerisation (DP) 100 with randomly copolymerised hydrophobic, hydrophobic, cationic and anionic monomers. The libraries are organised according to charge in the vertical axis (red-blue scale) and have varying log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values (grey-purple scale). Across a row, the polymer compositions are identical, but the base hydrophilic monomer (Avs.H) is varied, as is the architecture (size and length of PEG block/graft). Full details of the polymers in each library are given in the ESI.† P values (grey-purple scale). Across a row, the polymer compositions are identical, but the base hydrophilic monomer (Avs.H) is varied, as is the architecture (size and length of PEG block/graft). Full details of the polymers in each library are given in the ESI.† | ||
Results and discussion
Polymer synthesis and enzyme labelling
Our donor dye of choice, cyanine 3 (Cy3-CO2H), was synthesised according to literature protocols,36 and conjugated to each protein using standard amide coupling conditions to the lysine residues (see further information on enzymes, Table S1†). The protein was then separated from free dye and coupling reagents using Sephadex G15. The extent of labelling was then measured spectroscopically and was found to be reproducible within replicates of a given protein (Table S2†). Naturally, the molar ratio of Cy3:protein after labelling varied with the molecular weight and number of lysines in the protein. Polyacrylamide libraries were synthesised by an oxygen tolerant RAFT method,31,32 in 200 μL PCR tubes with the help of a Beckmann Coulter NxP automated workstation. Polymerisations were conducted at 45 °C overnight in 10% DMSO/phosphate buffer mixtures, with 1 μM GOx to degas each polymerisation, sodium pyruvate to scavenge the peroxide generated, and VA-044 to act as the initiator. A summary of the polymers included in the library is shown in Fig. 1. All polymers contain a mixture of positively and negatively charged monomers, as well as hydrophilic and hydrophobic monomers, mimicking the amino acid library. Two different positively charged monomers were compared, one with a quarternised amine ([3-(acryloylamino)propyl] trimethylammonium, designated Q), and one with a tertiary amine expected to be protonated at neutral pH (N-[3-(dimethylamino)propyl] acrylamide, designated D). Likewise, two different negatively charged monomers were compared – a sulfonated monomer (2-acrylamido-2-methylpropane sulfonic acid, S) and carboxylic acid monomer (carboxyethyl acrylate, C). These acids allow comparison of anionic monomers of 2 vastly different pKa (<0 for S and +4.5 for C). Hydroxyethyl acrylate (H) and acrylamide (A) were compared as hydrophilic monomers, as these mimic the amino acid structures of glycine, and serine, and a range of hydrophobic monomers including benzyl acrylate (designated F, for its similarity to phenylalanine), methyl acrylate (M) and butyl acrylate (Y), covering a wide range of logP values (see calculations in Table S3†). We fixed the degree of polymerisation (DP) at 100 and varied the RAFT agent to incorporate a 2 kDa or 5 kDa polyethylene glycol (PEG) block, or no PEG block. All synthesized polymers were analysed with size exclusion chromatography (SEC), and the conversion of selected polymers was checked with 1H NMR spectroscopy, using an internal standard of 1,3,5-trioxine. The choice of DP is not very important to the results of the screen, as will be shown below. It is the composition of the polymer that matters for binding.
FRET screening on glucose oxidase
Initially, we sought to understand how varying the concentration of the protein and equivalents of polymer used would impact the FRET results. We began with a library of ∼192 polymers (library 1 in Fig. 1) and glucose oxidase (GOx), as this enzyme is (i) large (∼160 kDa), (ii) readily available and (iii) easy to label with our Cy3. After mixing the polymer and protein in a 384 well plate in 20 mM phosphate buffer (pH 7.4), the FRET intensity was calculated over the normalized fluorescence emission as described in eqn (1).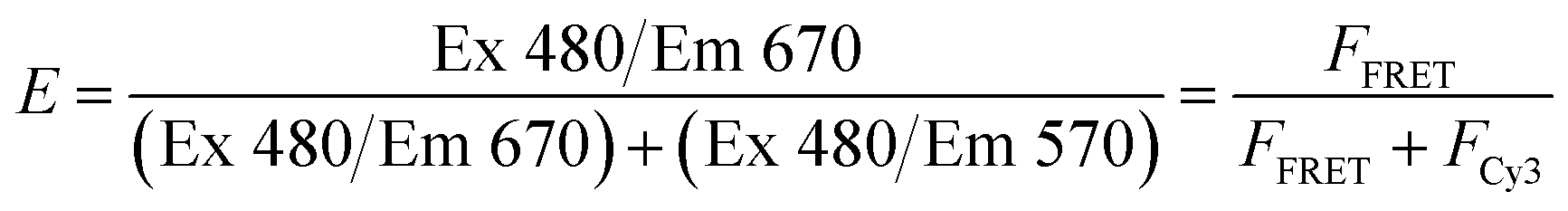 | (1) |
Any measurements that showed a standard deviation >0.1 in duplicate measurements were excluded, although this was very rare. In total, only 4% of data was excluded, which corresponds to less than 4 FRET ratios per 96 well plate. The entire database of FRET data is provided in csv form in the ESI.†Fig. 2 shows the FRET data for the polymer library at 146 eq. polymer to 0.1 μM GOx, 16 eq. polymer to 0.25 μM GOx, 16 and 32 eq. polymer to 0.5 μM GOx and 16 eq. polymer to 1 μM GOx. Reducing the overall protein concentration at a fixed equivalence of polymer (16 eq.) results in very little difference to the measured FRET ratio, until the GOx concentration drops to 0.25 μM, where the signal begins to fade. Likewise, doubling the polymer concentration from 6 to 32 eq. at a fixed GOx concentration (Fig. 2b) makes very little difference to the measured FRET ratios. This demonstrates the versatility of this readout mechanism. Provided the Cy3 and Cy5 signals are high enough to read, the background signals are generally low enough that the ratio of FRET signal to Cy3 emission doesn't greatly depend upon the assay conditions. At 0.1 μM GOx, it was still possible to read out strong binding polymers provided a large excess of the polymer was used, as shown in Fig. 2b for the 146 eq. polymer:GOx case. It is noteworthy that the high excess of polymer in this case does not contribute to any significant background, and the very low concentration of Cy3 can still be accurately measured. By diluting a subset of both strong and weak binding polymers we observed that strong binding polymers could be differentiated from weak binding polymers at even ∼25 nM protein (4 μg mL−1, Fig. S2†).
 | ||
| Fig. 2 FRET data presented as a heatmap. Averaged and normalized results of library 1 against GOx at (a) equal polymer to enzyme equivalents (16 eq.) and different concentrations (0.25, 0.50 and 1.00 μM) and (b) different polymer to enzyme equivalents (16 and 32 eq.) at the same enzyme concentration (0.50 μM), plus an additional screen at 0.10 μM and 146 eq. polymer to enzyme. Net charge of monomers as scale. | ||
As should be expected from the low isoelectric point of GOx (pI = 4.55), positively charged polymers were required for any binding. Using H or A as the hydrophilic component made little difference to binding, as can be seen by comparing the 1st and 2nd column of each data set in Fig. 2 which are identical in composition but for this difference. The 3rd and 4th columns in each data set show the H bearing polymers and A bearing polymers with a 5 kDa PEG block included and show somewhat stronger binding than the comparable PEG-free polymers in the first two columns. Most strikingly, however, incorporation of the quarternized amine (Q) over the tertiary amine D, and of ∼10% of the phenylalanine mimicking monomer F over other hydrophobic monomers lead to the strongest binding polymers to GOx (Fig. 3). Interestingly, the amino acid make-up of the protein (see the pie chart insert in Fig. 5) was not a very strong predictor of the composition of the strongest binding polymers, and some positively charged polymers which might be expected to bind well did not bind as well as others. We expect that this is in part because the total amino acid make-up of a protein is not necessarily representative of what is presented on the surface – hydrophobic amino acids will tend to be buried. However, it is also because even two different proteins with the same surface amino acid composition can display vastly different distributions. This can be seen by looking at the crystal structures of a panel of proteins used later in this study, obtained from the Protein Data Bank (PDB). Table S1† shows snapshots of the surface potential of these proteins, analysed using UCSF's ChimeraX program10 in order to calculate and visualise the protein's surface coulombic electrostatic potential (ESP) and hydrophobicity, also known as molecular lipophilicity potential (MLP). It is for this reason that screening methodologies are valuable – it is very difficult to predict from first principles which polymers will bind strongly and selectively to a given protein, but relatively easy to find by iterative screening.
 | ||
| Fig. 3 Detail from Fig. 2, showing the FRET hits (FRET ratio >0.375) of library 1 and corresponding polymers at a GOx concentration of 0.50 μM at 16 eq. polymer to enzyme. In each row the composition of monomers is the same, except for the base monomer m (H or A), plus PEG block for the last two columns. The results show high FRET binding for polymers with benzyl acrylate (F) and monomer Q. | ||
To test whether the length of the polymer is influencing the binding strength, a strongly binding polymer (P(H50-co-N40-co-Q10)) and a weakly binding polymer (P(H90-co-F10)) were synthesized at 5 different chain lengths (DP = 25, 50, 100, 200 and 400). We fixed the total Cy5 content in each polymerisation such that the concentration of dye is equivalent in each sample, but this means that longer polymers will contain more Cy5 monomers/chain. Because of this, we investigated the FRET from experiments in which the [Cy5] was held constant (resulting in different molar equivalences of the polymer) and from experiments in which the molar ratio of polymer was held constant (resulting in different concentrations of Cy5). Both were conducted at a GOx concentration of 1 μM. When the [Cy5] was held constant the FRET ratios were the same for all polymers (see Fig. S4, ESI†). Our results from the DP100 screens (Fig. 2) demonstrated that the molar equivalences of polymer has little influence on the FRET result, so the lack of increase in FRET as a function of chain length here demonstrates that this variable has little (if any) impact on binding. When the polymer concentration was fixed, the FRET ratio did increase with increasing chain length, but we believe this is due to the commensurate increase in Cy5 concentration and not due to any stronger binding. Longer polymers have a greater chance of bearing a Cy5 monomer than shorter polymers (as the ratio of Cy5:total monomer is fixed in our polymerisations, but not the ratio of Cy5:chains). This means that if the [Cy5] in the FRET experiment is fixed, there is a higher chance of the polymer bound to the protein bearing a Cy5 in the case of longer polymers. Further evidence for this is given by the fact that the increase in FRET ratio as a function of chain length in this experiment is greater for the more strongly binding polymer. The amount of Cy5 per chain in the weakly binding polymer doesn't influence the FRET result by as much, because fewer binding events occur.
SAXS analysis
Small angle X-ray scattering was used to gain additional information about the structural features of polymer–GOx composites in a small set of polymers. Samples were prepared by mixing 8 mol equivalents of polymer with GOx in PBS buffer. The fitting parameters of polymer–GOx composites were constrained by the size obtained from our previous DLS measurements on these polymers.30 The radius of the free GOx was determined through fitting Small-Angle X-ray Scattering (SAXS) data using a sphere model. The Scattering Length Density (SLD) of GOx was calculated using an SLD calculator, and the diameter obtained from Dynamic Light Scattering (DLS) measurements was employed to constrain the sphere fitting, resulting in a radius of 3 nm for free GOx. Within the core–shell model,37 the core radius and the calculated core SLD were fixed as those of the protein (see ESI, Tables S6–S8†). The SAXS data revealed a variation in shell thickness, despite minimal changes in the shell SLD. The shell thickness of the statistical cationic copolymers (Fig. 4 and Table S6†) increased with the polymer charge density for both acrylamide and hydroxyethyl acrylate polymer chains from 2.2 and 1.2 nm for polyacrylamide and poly(2-hydroxyethyl acrylate), respectively, to 4.6 nm for fully charged polymers. The shell thickness of PEG block copolymers (Fig. S3 and Table S7†) and PEG methyl ether acetate (PEGMEA) statistical cationic brush copolymers (Fig. S3 and Table S8†) increased proportionally to the charge density of the polymers. Although the SAXS shell thickness data cannot directly prove how much polymer is on the surface, the fact that the SLD remains constant suggests that thicker shells really do mean “more polymer”. In any event, the SAXS and FRET data are well correlated, supporting the conclusion that FRET signal is a good measure of polymer binding. Notably, SAXS data did not reveal a significant difference in particle size based on the architecture of the polymer. Although block copolymers may have stronger protein binding than statistical copolymers, some theoretical studies have demonstrated that both the charged block and the PEG block envelop the protein surface,38 consequently, there is unlikely to be a significant difference in shell thickness between statistical and block copolymers.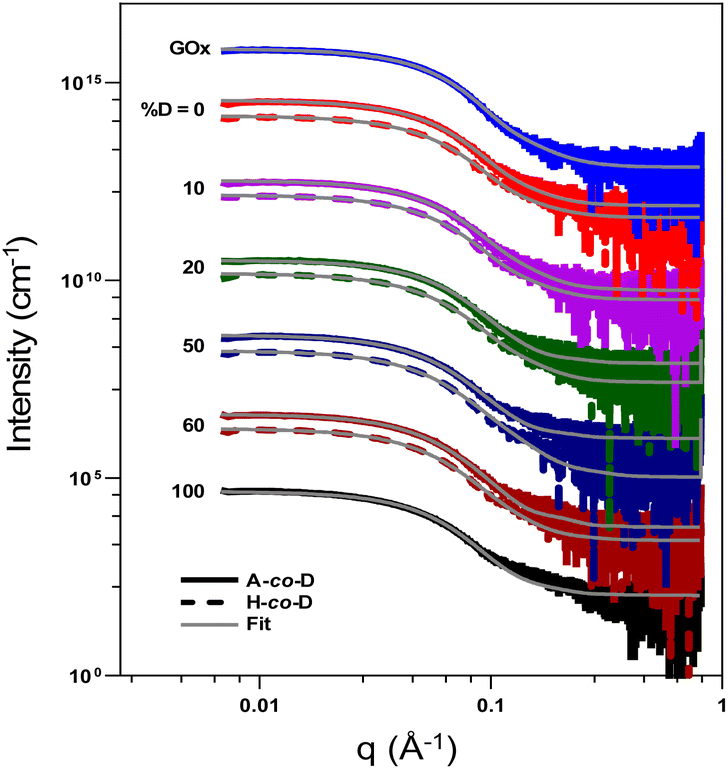 | ||
| Fig. 4 Small angle X-ray scattering (SAXS) data obtained upon mixing of polymers with GOx (1.0 mg mL−1) at poly/GOx = 8 with core–shell fits shown. Data is offset in the y-axis. Note: A = acrylamide, H = hydroxyethyl acrylate and D = N-[3-(dimethylamino) propyl]acrylamide. | ||
Relationship between activity and binding
In addition to estimating the binding strength, we were interested if we can find a correlation between the binding strength of a polymer and its ability to stabilize a protein. Each polymer was mixed with GOx ([GOx] = 40 μg mL−1, 16 eq. polymer) and incubated for 20 min in 60 °C, after which the sample was diluted and the activity of GOx at room temperature relative to a standard curve was measured using a standard HRP assay. Under such conditions, the activity of GOx with no polymer added decreased to 18% of its original activity (see Fig. S5, ESI†). The residual activity of the polymer/GOx mixtures after this heat treatment, expressed as a percentage of the GOx only control, are shown in Fig. 5. Results with an error >10% in duplicate measurements were excluded. The data for polymers bearing a thermoresponsive N-isopropyl acrylamide (NIPAM, N) monomer, as well as polymers with PEG architectures (copolymers, different lengths of block copolymers) are separated out in this analysis. Interestingly, despite significant variability in the residual GOx activity, no correlation to FRET ratio (binding strength) was observed in any of these families, and the R2 for a linear fit is low in all cases. Low stabilisation with high binding can be explained by denaturation or inhibition of the protein on binding, and high stabilisation with low binding might be explained by excipient effects or highly dynamic interactions. In any case, these results highlight that enzyme stabilisation and polymer binding are quite distinct from each other. If a polymer which stabilises the enzyme is desired, then screening for this directly is important, but stabilisation cannot be used as a proxy for binding strength. If binding strength is the readout that is desired, it must be measured directly, using FRET as we have here, or some other technique.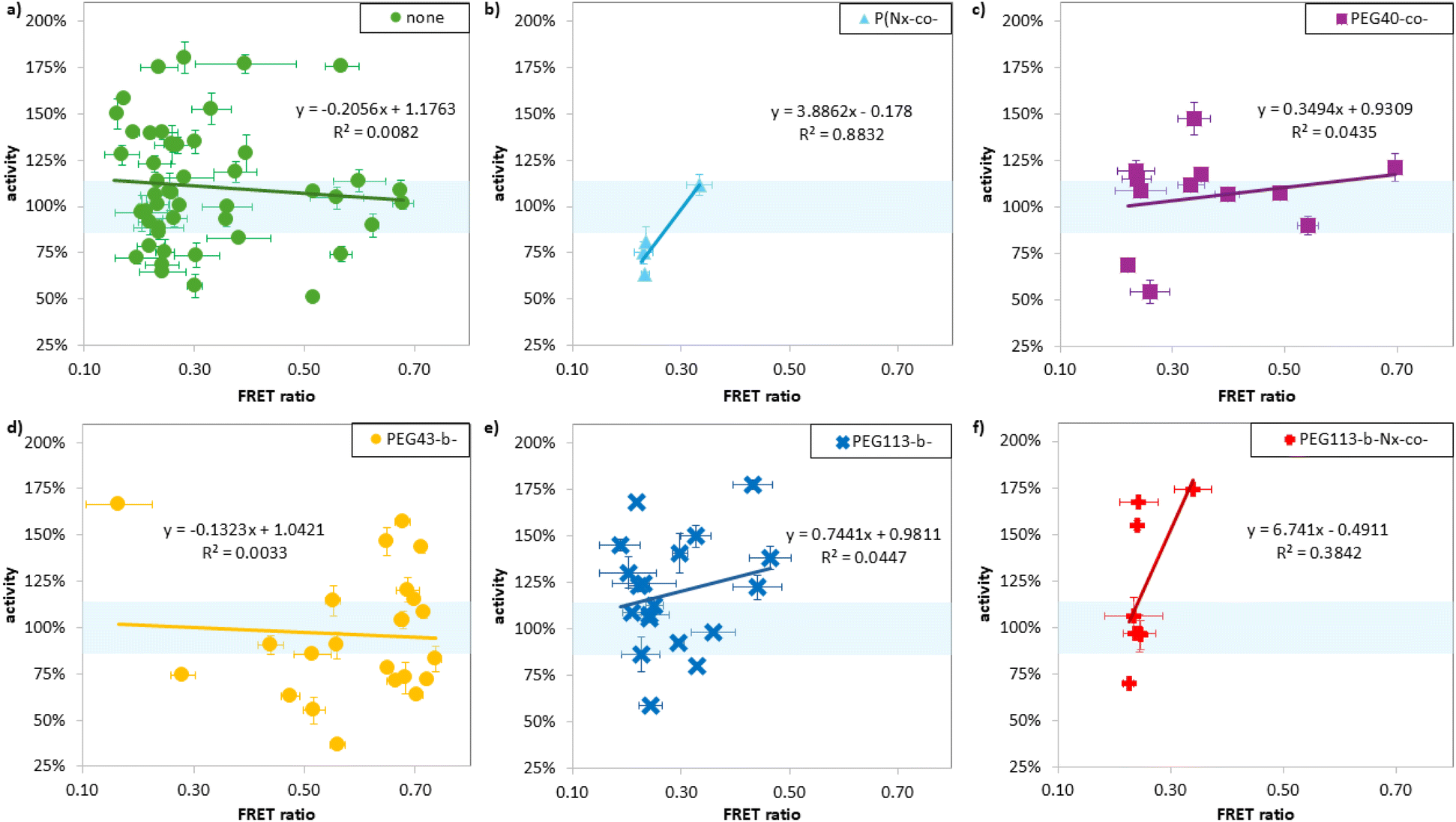 | ||
| Fig. 5 Activity of GOx in % after incubated with different polymers of library 1 and 2 for 20 min at 60 °C in 20 mM phosphate buffer vs. FRET ratio of given polymer. As FRET ratio was obtained at RT and the temperature for the activity was measured after incubation at 60 °C, the results were separated in different groups of polymers containing (a) no thermos-responsive behaviour or potential thermos-responsive behaviour given due to the monomers used (NIPAM, N monomer see (b)) or PEG co polymers (c) or block copolymers (d) and (e) or both (f). | ||
Screening against different protein substrates
The robust nature of this screening protocol, and its capacity to work at high dilution, should enable it to locate strong binding polymer compositions to any given protein. We were eager to understand (i) how the surface structure and size of a protein influences the best polymer to use in a given case, (ii) whether any general structural features of the polymers are predictors of strong binding, and (iii) to what extent the amino acid composition of the protein can be used to predict binding strength of a given polymer sequence. For this purpose, we prepared a second library (see Fig. 1) covering a broader range of hydrophobicities and structural complexity (including some amphiphilic polymers). This was screened against a panel of proteins varying in size, isoelectric point (pI) and charge distributions which included manganese peroxidase, casein, uricase, bovine serum albumin, trypsin, carbonic anhydrase and lysozyme (see crystal structures previously described in Table S1†).In most cases we screened at multiple polymer equivalences. While the overall trends did not change as the amount of polymer was varied, the optimised screens for each protein shown in Fig. 6 were all performed at 3 to 7 equivalents of polymer to protein. As can be seen in this figure, different proteins result in different ‘polymer hits’. A general trend corresponding to the pI of the enzymes is observed, with the best binding occurring at net lower polymer charge as the pI increased. This is seen most dramatically for the highly cationic protein lysozyme, where hits for good binding are only found for polymers with negative charge. However, while there are some common hits, screening in this way enables selection of a polymer in each case which is at least moderately selective towards the target protein. Such selectivity is still a long way from that which could be achieved by phage display for peptides,39 but is none the less notable given these polymers are simply copolymerised structures with no particular attention to sequence control. Interestingly different proteins seemed to prefer different polymer architectures, and this can be seen most clearly in the statistical analysis in Fig. 7a–c. This analysis compares the FRET ratio achieved for comparable polymers with and without PEG in the side chain (a), as a block (b) and comparing Hvs.A monomers (c) across all the proteins in the screen. Positive values in this analysis therefore represent an increase in binding strength results from each attribute in the polymer, while values close to zero indicate that this modification has negligible effect on the polymer binding strength. As expected from the GOx screen above, there is little effect of changing the hydrophilic monomer H to A in the polymer on binding to any protein. However, for carbonic anhydrase and uricase, binding is preferred for the PEG block copolymers, whereas GOx also shows good hits for polymers without any PEG components. By contrast, manganese peroxidase shows good binding with polymers with grafted PEG. As only a selection of polymers from the library has been screened against manganese peroxidase, it is unknown if PEG block copolymers would have resulted in even higher FRET ratios. These proteins have vastly different molecular weights, pI (Fig. 6) and surface characteristics (Table S1†), so these trends would be hard to predict from first principles.
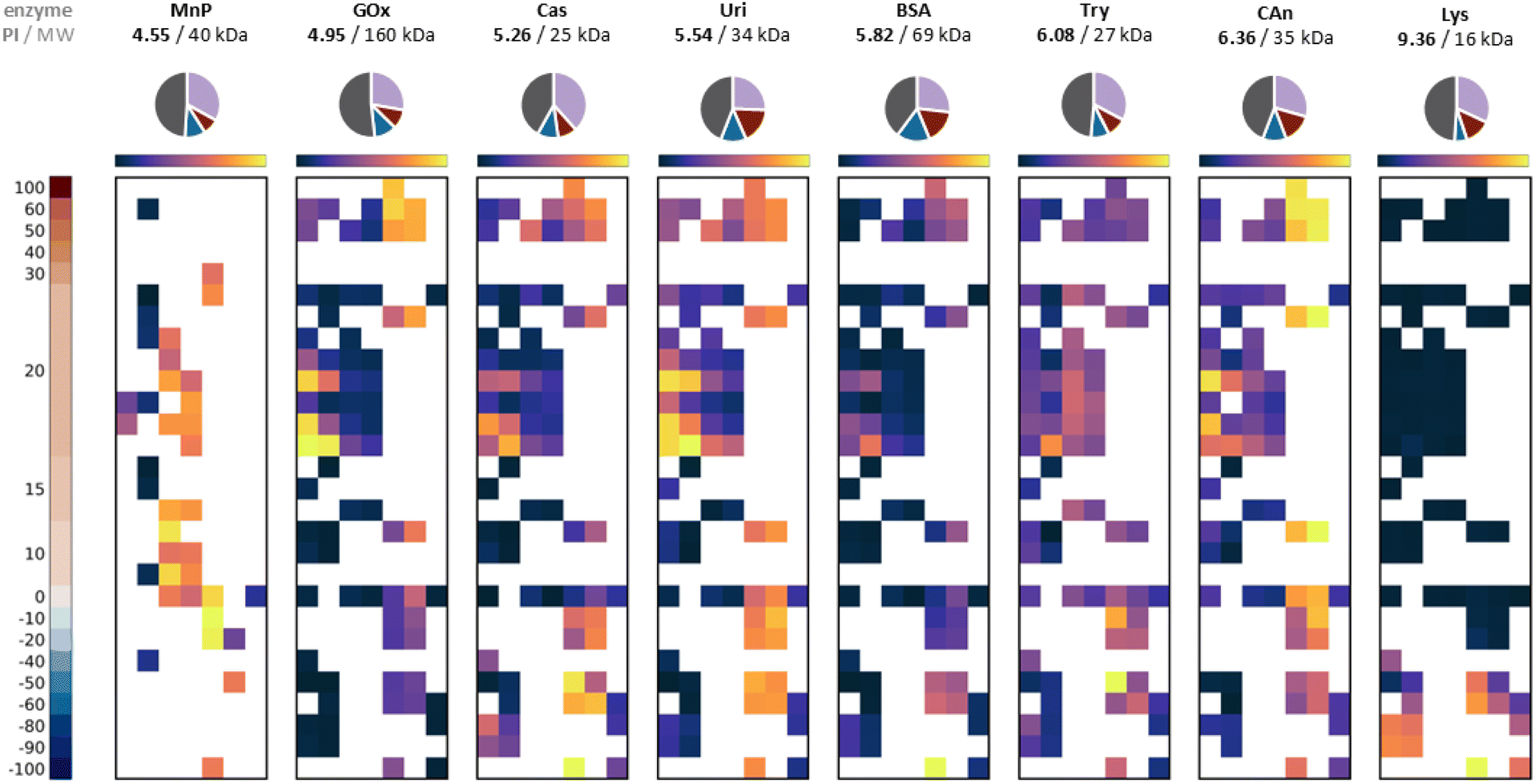 | ||
| Fig. 6 FRET ratio averaged results for library 3 against different enzymes: manganese peroxidase (MnP), GOx, bovine serum albumin (BSA), uricase (Uri), casein (Cas), trypsin (Try), carbonic anhydrase (CAn) and lysozyme (Lys). All at a concentration of 0.25 mg mL−1 and molecular equivalents of polymer ranging from 3 to 7 eq. Pie charts above each FRET map show the amino acid make-up of each protein structure (red = cationic amino acid, blue = anionic, purple = hydrophilic neutral, and grey = hydrophobic). | ||
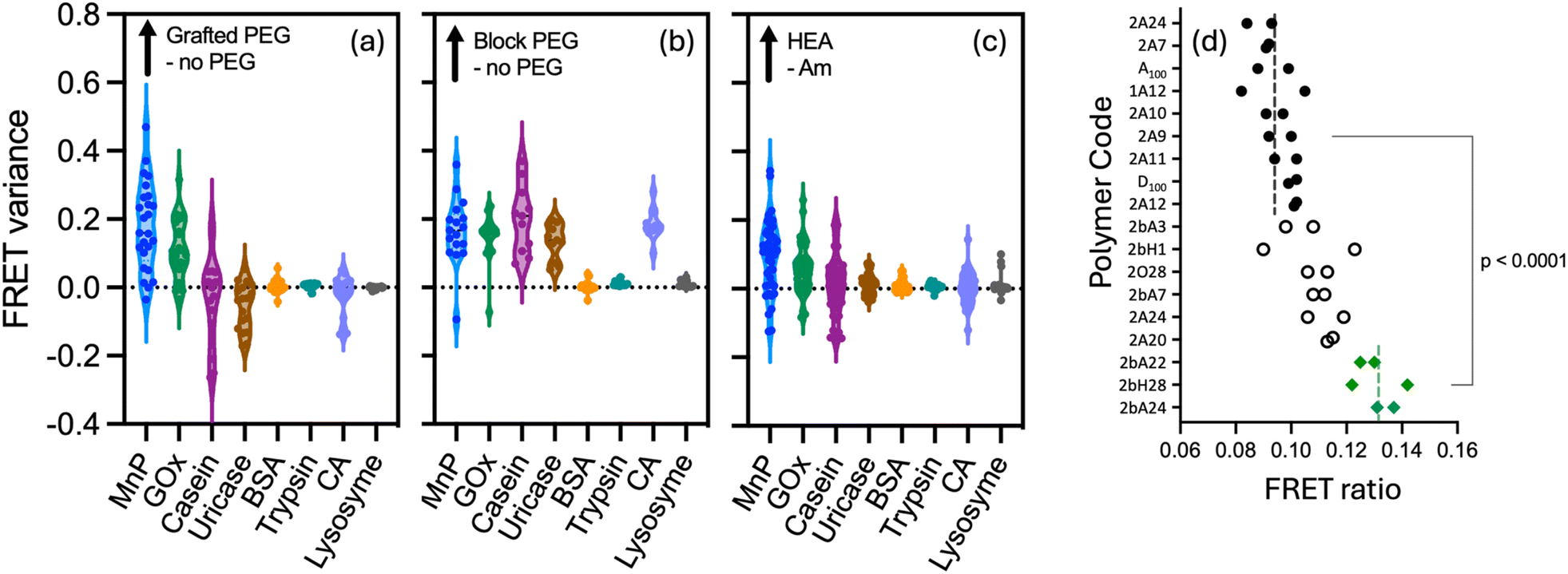 | ||
| Fig. 7 Comparison of the difference between FRET data of comparable polymers (a) +/− PEG in the side chain; (b) +/− a 2 kDa PEG block, and (c) with Hvs.A as the hydrophilic monomer across protein substrates. (d) Shows the raw FRET ratios from a screen of 18 selected polymers against TRAIL at 0.25 μM (5 μg mL−1) of the protein. | ||
We then proceeded to apply this assay to find polymer structures capable of encapsulating a therapeutically relevant protein, TNF-related apoptosis-inducing ligand (TRAIL). TRAIL is a promising chemotherapeutic protein, which our group has worked on extensively,26,40,41 but very expensive to purchase in any significant quantity. TRAIL works by clustering death receptor proteins (DR4 and DR5) which are selectively present on the surface of many cancer cells, which drives production of caspase 8 and ultimately cell death. While it is an effective drug in vitro it has shown poor performance in vivo, at least in part due to its very low circulation half-life (∼30 min in humans).42 We reasoned that encapsulation in a weak binding polymer could improve circulation half-life, without restricting its activity. TRAIL has a similar molecular weight and distribution of amino acids in its primary sequence to carbonic anhydrase (see ESI Fig. S6†). As a result, we selected 18 polymers from our library which had bound well to this protein, along with some negative controls, and screened them in duplicate against a Cy3-labelled TRAIL. As previously, analysis of the primary sequence provides only a rough starting point for determining the optimal polymer, but from this FRET screen we were able to identify three potential lead polymers with stronger binding than the others, even at only 5 μg mL−1 protein in a 40 μL sample well (see ESI† for the polymer compositions associated with each code). Fig. 7d shows each replicate, ordered by average FRET. The best leads (polymers #1–3 in this figure) show significantly elevated FRET (p < 0.0001) than the weakest 10 polymers and correspond to anionic PEG block copolymers with acrylamide (2A22 = PEG-b-P(A80-co-C20), 2A24 = PEG-b-P(A80-co-C50), and 2H28 = PEG-b-P(C100)). The pI of this ∼20 kDa TRAIL fragment is 8.89, so the fact that anionic polymers perform the best is not surprising. This data is also consistent with the finding from the protein panel screening that PEG-blocked polymers tend to work the best. We propose that these structures could be valuable leads to pursue in improving the delivery of this protein as a polyion complex.
Conclusions
The findings demonstrate the applicability and potential of HTP synthesis and screening of polymer libraries for identifying strongly or weakly binding polymers for a wide range of proteins. The sensitivity of this technique, as demonstrated in this study for GOx, enables detection at protein concentrations of 0.1 μM. While some trends can be used to direct the polymer library design towards better binding, such as the use of PEG blocks, and balancing the overall charge to complement the pI of the target protein, in many cases the strongest (and weakest) binding polymers would have been difficult to design from first principles. By screening each polymer against a panel of proteins we were able to identify moderately selective binders in most cases. The approach allowed identification of lead polymers for encapsulation of the TRAIL protein at a protein concentration of only 5 μg mL−1 in 40 μL wells.Data availability
The data supporting this article have been included as part of the ESI.† A .csv file of all FRET data is included separately.Author contributions
CB and AZM conducted the experiments and collated all of the data. The figures and the original draft were written by CB with assistance from RC. SAXS data collection and analysis was performed by CC. All authors were involved in reviewing and editing the manuscript. EJW, RC and MS were involved in supervising the experimental work and analysis. Conceptualisation by MS and RC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Australian Research Council (DP240101279, DP170101191), UNSW through an international postgraduate scholarship to AM. We are grateful to the Australian Synchrotron for providing access to the SAXS/WAXS beamline under proposals 18454 and 18455 and to Dr Nigel Kirby for his assistance at the SAXS/WAXS beamline.Notes and references
- R. Satchi, T. A. Connors and R. Duncan, Br. J. Cancer, 2001, 85, 1070–1076 CrossRef CAS PubMed.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442–1448 RSC.
- J. Chapman, A. E. Ismail and C. Z. Dinu, Catalysts, 2018, 8, 238 CrossRef.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- B. Panganiban, B. Qiao, T. Jiang, C. DelRe, M. M. Obadia, T. D. Nguyen, A. A. A. Smith, A. Hall, I. Sit, M. G. Crosby, P. B. Dennis, E. Drockenmuller, M. Olvera de la Cruz and T. Xu, Science, 2018, 359, 1239–1243 CrossRef CAS PubMed.
- R. Chapman and M. H. Stenzel, J. Am. Chem. Soc., 2019, 141, 2754–2769 CrossRef CAS PubMed.
- S. Gao, A. Holkar and S. Srivastava, Polymers, 2019, 11, 1097 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Polym. J., 2018, 50, 95–100 CrossRef CAS.
- F. Chen and M. H. Stenzel, Aust. J. Chem., 2018, 71, 768–780 CrossRef CAS.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- F. Vianello, A. Cecconello and M. Magro, Int. J. Mol. Sci., 2021, 22, 7625 CrossRef CAS PubMed.
- H. Cabral, J. Li, K. Miyata and K. Kataoka, Nat. Rev. Bioeng., 2023, 2, 214–232 CrossRef.
- T. D. Nguyen, B. Qiao and M. Olvera De La Cruz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6578–6583 CrossRef PubMed.
- M. J. Tamasi, R. A. Patel, C. H. Borca, S. Kosuri, H. Mugnier, R. Upadhya, N. S. Murthy, M. A. Webb and A. J. Gormley, Adv. Mater., 2022, 34, 2201809 CrossRef CAS PubMed.
- Z. Ruan, S. Li, A. Grigoropoulos, H. Amiri, S. L. Hilburg, H. Chen, I. Jayapurna, T. Jiang, Z. Gu, A. Alexander-Katz, C. Bustamante, H. Huang and T. Xu, Nature, 2023, 615, 251–258 CrossRef CAS PubMed.
- R. I. Dima, J. R. Banavar, M. Cieplak and A. Maritan, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4904–4907 CrossRef CAS PubMed.
- T. Jiang, A. Hall, M. Eres, Z. Hemmatian, B. Qiao, Y. Zhou, Z. Ruan, A. D. Couse, W. T. Heller, H. Huang, M. O. de la Cruz, M. Rolandi and T. Xu, Nature, 2020, 577, 216–220 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- I. Jayapurna, Z. Ruan, M. Eres, P. Jalagam, S. Jenkins and T. Xu, Biomacromolecules, 2023, 24, 652–660 CrossRef CAS PubMed.
- A. A. A. Smith, A. Hall, V. Wu and T. Xu, ACS Macro Lett., 2019, 8, 36–40 CrossRef CAS PubMed.
- C. G. Simon and S. Lin-Gibson, Adv. Mater., 2011, 23, 369–387 CrossRef CAS PubMed.
- R. Potyrailo, K. Rajan, K. Stoewe, I. Takeuchi, B. Chisholm and H. Lam, ACS Comb. Sci., 2011, 13, 579–633 CrossRef CAS PubMed.
- P. R. Judzewitsch, N. Corrigan, F. Trujillo, J. Xu, G. Moad, C. J. Hawker, E. H. H. Wong and C. Boyer, Macromolecules, 2020, 53, 631–639 CrossRef CAS.
- B. Lin, J. L. Hedrick, N. H. Park and R. M. Waymouth, J. Am. Chem. Soc., 2019, 141(22), 8921–8927 CrossRef CAS PubMed.
- Z. Li, Z. Han, M. H. Stenzel and R. Chapman, Nano Lett., 2022, 22, 2660–2666 CrossRef CAS PubMed.
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Macromolecules, 2019, 52, 3–23 CrossRef CAS.
- R. Upadhya, N. S. Murthy, C. L. Hoop, S. Kosuri, V. Nanda, J. Kohn, J. Baum and A. J. Gormley, Macromolecules, 2019, 52, 8295–8304 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- A. Z. Mustafa, B. Kent, R. Chapman and M. H. Stenzel, Polym. Chem., 2022, 13, 6108–6113 RSC.
- R. Chapman, A. J. Gormley, K. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed., 2016, 128, 4576–4579 CrossRef.
- S. Von Karstedt, A. Montinaro and H. Walczak, Nat. Rev. Cancer, 2017, 17, 352–366 CrossRef CAS PubMed.
- J. Lemke, S. von Karstedt, J. Zinngrebe and H. Walczak, Cell Death Differ., 2014, 21, 1350–1364 CrossRef CAS PubMed.
- A. Ashkenazi and V. M. Dixit, Science, 1998, 281, 1305–1308 CrossRef CAS PubMed.
- M. V. Kvach, A. V. Ustinov, I. A. Stepanova, A. D. Malakhov, M. V. Skorobogatyi, V. V. Shmanai and V. A. Korshun, Eur. J. Org Chem., 2008, 2008, 2107–2117 CrossRef.
- DANSE SasView for Small Angle Scattering Analysis, Core Shell Model, http://www.sasview.org/.
- T. Kurinomaru, K. Kuwada, S. Tomita, T. Kameda and K. Shiraki, J. Phys. Chem. B, 2017, 121, 6785–6791 CrossRef CAS PubMed.
- G. P. Smith and V. A. Petrenko, Chem. Rev., 1997, 97, 391–410 CrossRef CAS PubMed.
- Z. Han, Z. Li, M. H. Stenzel and R. Chapman, J. Am. Chem. Soc., 2024, 146, 22093–22102 CrossRef PubMed.
- Z. Han, Z. Li, R. Raveendran, S. Farazi, C. Cao, R. Chapman and M. H. Stenzel, Biomacromolecules, 2023, 24, 5046–5057 CrossRef CAS PubMed.
- R. S. Herbst, S. G. Eckhardt, R. Kurzrock, S. Ebbinghaus, P. J. O'Dwyer, M. S. Gordon, W. Novotny, M. A. Goldwasser, T. M. Tohnya, B. L. Lum, A. Ashkenazi, A. M. Jubb and D. S. Mendelson, J. Clin. Oncol., 2010, 28, 2839–2846 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc04391c |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |