
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerfluoroarene–arene interaction cocrystal of perfluorocarbazoles toward IEF-enhanced photocatalysis†
Wenbo
Hu
 ,
Heng
Li
and
Bingxin
Yuan
*
,
Heng
Li
and
Bingxin
Yuan
*
College of Chemistry, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: bxyuan@zzu.edu.cn
First published on 21st June 2025
Abstract
The photocatalytic generation of highly reactive oxygen species (ROS) such as H2O2 and ˙OH, using molecular oxygen without sacrificial reagents or metal catalysts, is a significant challenge. To address this, we introduce a novel approach by adding a second perfluoroarene layer to arene–perfluoroarene cocrystals, which enhances the dipoles between the π-hole and π-donor layers, thereby increasing the internal electric field (IEF) and improving charge separation. We successfully synthesized a series of perfluorocarbazole derivatives through a Pd-catalyzed C–Br/C–F amination reaction. Three cocrystals, Pe–FPC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), Pe–FMC (1:2), and TP–FPC (1:1) were synthesized, with Pe–FPC (1:2) exhibiting exceptional photocatalytic activity. Pe–FPC (1:2) effectively degrades RhB, MB, and Eosin B in a short time period under air, demonstrating excellent stability over seven cycles. It also catalyzes the oxidation of aromatic and aliphatic aldehydes to carboxylic acids, outperforming small-molecule catalysts in aqueous-phase reactions. Mechanistic studies show that h+, ˙O2−, ˙OH, 1O2, and H2O2 synergistically contribute to the photoinduced oxidation process. The Pe–FPC (1:2) cocrystal also shows a high H2O2 production yield (2640 μmol h−1 g−1), with enhanced surface potential and a lower photoluminescence lifetime, confirming the improved IEF and charge separation. This work validates our hypothesis that introducing a second perfluoroarene layer enhances photocatalytic efficiency, opening new avenues for the design of high-performance photocatalysts for energy and environmental applications.
:2), Pe–FMC (1:2), and TP–FPC (1:1) were synthesized, with Pe–FPC (1:2) exhibiting exceptional photocatalytic activity. Pe–FPC (1:2) effectively degrades RhB, MB, and Eosin B in a short time period under air, demonstrating excellent stability over seven cycles. It also catalyzes the oxidation of aromatic and aliphatic aldehydes to carboxylic acids, outperforming small-molecule catalysts in aqueous-phase reactions. Mechanistic studies show that h+, ˙O2−, ˙OH, 1O2, and H2O2 synergistically contribute to the photoinduced oxidation process. The Pe–FPC (1:2) cocrystal also shows a high H2O2 production yield (2640 μmol h−1 g−1), with enhanced surface potential and a lower photoluminescence lifetime, confirming the improved IEF and charge separation. This work validates our hypothesis that introducing a second perfluoroarene layer enhances photocatalytic efficiency, opening new avenues for the design of high-performance photocatalysts for energy and environmental applications.
Introduction
The worsening climate crisis and the rapid depletion of natural resources have driven the pursuit of sustainable technologies.1,2 Among these, photocatalysis has gained significant attention as an effective approach for utilizing solar energy to produce fuels and chemicals. In particular, photocatalytic technology stands out as a highly promising method for water pollutant degradation and energy storage, as it efficiently harnesses molecular oxygen, sunlight, and other renewable resources.3–6 Central to photocatalytic oxidation processes is the generation of reactive oxygen species (ROS), particularly hydroxyl radicals (˙OH), which exhibit unparalleled oxidative power for decomposing organic pollutants and enabling energy-carrying chemical transformations.7–9 However, producing ˙OH selectively and efficiently remains challenging. The direct oxidation of water to ˙OH by photogenerated holes is thermodynamically unfavorable because of the high electrode potential of the H2O/˙OH system (2.38 V vs. NHE).10,11 To overcome this limitation, researchers have focused on alternative pathways, such as the in situ photodecomposition of hydrogen peroxide (H2O2).8,12,13 For practical applications, photocatalysts must simultaneously achieve two goals: high H2O2 production yields and efficient decomposition into ˙OH, while maintaining robust charge separation and electron transfer kinetics.A fundamental challenge in photocatalysis lies in the ultrafast timescales of critical processes.14,15 Electron–hole (e−–h+) pairs form within femtoseconds of photon absorption but recombine within picoseconds unless effectively separated and transported to catalytic sites.16–18 Maximizing photocatalytic efficiency requires effective charge separation and suppression of charge recombination (Fig. 1a). Traditional strategies to suppress recombination—such as heterojunction design, phase engineering, and elemental doping—have shown limited success.6,17,19–21 These methods often suffer from interfacial energy losses and insufficient driving forces for charge migration.
 | ||
| Fig. 1 (a) Schematic representation of the photocatalytic oxidation and reduction half-reactions under illumination and recombination of electrons (e−) and holes (h+). VB: valence band, CB: conduction band. (b) Comparative illustrations of assemblies featuring intermolecular dipole (previous work) and boosted photocatalytic ROS formation via IEF enhancement, achieved by enlarged dipole (as presented in this study). (c) Cocrystallization schemes of Pe–FPC, Pe–FMC and TP–FPC. | ||
Recent advances highlight internal electric field (IEF) engineering as a transformative solution. Built-in electric fields can spatially separate charges and direct their flow both within the bulk and at the surface of photocatalysts, significantly enhancing photocatalytic efficiency.22–25 In organic photocatalysts, IEF strength can be amplified through molecular dipole engineering. This involves introducing substituents with contrasting electronegativities to manipulate electron density distributions.23,26 The choice of substituents, especially those with varying electronegativities, has a profound impact on the electronic distribution and molecular dipole moment of organic photocatalysts. By strategically selecting appropriate substituents based on the electronic structure of the modified photocatalyst, both the molecular dipole and IEF can be significantly strengthened.26–28 For example, Zhu et al. developed carboxyl-functionalized perylene diimide (PDI) nanofibers that leverage dipole-induced IEFs.28 The combination of terminal carboxyl moieties and the electron-deficient perylene core contributed to the formation of an internal electric field, which in turn facilitated charge carrier migration and separation. This supramolecular system achieved metal-free photocatalytic water oxidation, demonstrating the potential of tailored molecular design.
Organic cocrystals provide a versatile platform for optimizing photocatalytic properties. These materials are assembled through noncovalent interactions—such as π–π stacking, hydrogen bonding, and halogen bonding—enabling precise control over molecular organization.29–33 These multi-component systems enable the creation of well-ordered molecular architectures with customizable physicochemical properties that are often unattainable in single-component materials. Notably, cocrystals have demonstrated significant promise in the development of advanced photocatalysts, exhibiting superior attributes such as enhanced electrical conductivity, ferroelectric behavior, and improved light absorption.34–39 By carefully selecting molecular components, researchers can precisely tailor cocrystal properties, offering a strategic pathway toward next-generation photocatalytic materials.
A key noncovalent interaction leveraged in these cocrystals is the arene–perfluoroarene (A–P) interaction, which arises from electrostatic attraction between fluorinated and non-fluorinated aromatic molecules.40,41 Many perfluoroarenes, such as octafluoronaphthalene (OFN), readily form stable 1:1 molecular complexes with arenes, adopting a nearly parallel stacking arrangement in the solid state.42–44 The electronegative nature of perfluoroarenes, featuring an electrophilic π-hole, alters the π-electron distribution of arenes, inducing polarization and generating a strong intermolecular dipole moment within A–P pairs.45–48 This increased dipole interaction strengthens the internal electric field (IEF), thereby facilitating the separation and transport of photogenerated charge carriers in spatially confined regions. For instance, Xiong and colleagues incorporated OFN into 1:1 cocrystals with an anthracene derivative, where OFN prevents electron exchange between fluorophores, enhancing emission efficiency.49 It also acts as an electrophilic center (π-hole), altering π-electron distribution in arenes and creating a strong dipole moment between AP pairs. This resulted in a photocatalytic hydrogen evolution reaction (HER) rate of 2.45 mmol g−1 h−1, 15.2 times higher than the self-assembled anthracene derivative monomers.50 Wang and coworkers adopted a similar approach, preparing 1:1 cocrystals of OFN with 9,10-dimethylanthracene (DMA) and perylene (Pe), which enhanced the internal electric field, improving charge separation and photocatalytic degradation efficiency.34
While A–P interactions have been shown to effectively enhance internal electric fields (IEF), current A–P systems face two critical limitations. First, the predominance of 1:1 A–P configurations in current studies limits the maximal IEF intensity achievable through dipole stacking. Second, the limited availability of stable perfluoroarene building blocks hinders the exploration of higher-order cocrystals. For instance, 1:2 A–P configurations—which could theoretically intensify IEFs through multi-layer dipole alignment—remain largely unexplored (Fig. 1b). This gap contrasts sharply with inorganic photocatalysts like bismuth semiconductors, where layered dipolar structures naturally create strong IEFs.51–55 Addressing these limitations, we aim to develop a novel 1:2 arene–perfluoroarene cocrystal system engineered. By introducing an additional perfluoroarene layer into conventional A–P (1:1) assemblies, we amplify the IEF through synergistic dipole–dipole interactions, achieving unprecedented photocatalytic performance. However, the available selection of perfluoroarenes is still limited, with 1:1 arene–perfluoroarene complexes being the most common. The rarer 1:2 configurations are largely unexplored.56–58 To overcome this limitation, we synthesized a novel series of perfluorocarbazole (PFC) derivatives that not only expand the perfluoroarene library but also facilitate the formation of both 1:2 and 1:1 arene–perfluoroarene cocrystals (Fig. 1c). This design amplifies the IEF through synergistic dipole–dipole interactions, mimicking the layered dipole alignment in inorganic semiconductors. Among them, Pe–FPC (1:2) achieves efficient H2O2 and ˙OH production. This results in exceptional performance in the photodegradation of dye pollutants and the photo-oxidation of aldehydes to carboxylic acids. Our findings highlight that the introduction of an additional perfluoroarene layer by forming 1:2 arene–perfluoroarene cocrystal is an effective strategy to enhance internal electric fields, leading to an excellent H2O2 production rate of 2640 μmol g−1 h−1 without any sacrificial agents or metal catalyst under air atmosphere. Additionally, Pe–FPC exhibited over 90% degradation efficiency of Rhodamine B (RhB) within 15 minutes under visible light. The catalyst also demonstrated excellent stability, maintaining its catalytic activity after seven degradation cycles without any noticeable decline. This stability is likely attributed to the presence of three types of weak supramolecular interactions—C–H⋯F, C–H⋯N, and C–H⋯π—within the cocrystal, which reinforce its robustness during the catalytic process. Furthermore, the AP cocrystal catalyzed the aerobic oxidation of both aromatic and aliphatic aldehydes into carboxylic acids in aqueous media under mild conditions.
Results and discussion
Optimization substrate scope
Initially, we focused on synthesizing perfluorocarbazoles, as carbazole derivatives are well-known for their promising optoelectronic properties, including high quantum yields, excellent charge carrier mobility, and exceptional thermal and photochemical stability. We began by attempting a cascaded coupling reaction between 2-bromo-2′,3,3′,4,4′,5,5′,6,6′-nonafluoro-1,1′-biphenyl (1) and aniline (2a). The first trial, employing Pd(OAc)2 as the catalyst and XPhos as the ligand in toluene at 110 °C, did not result in any product formation (Table 1, entry 1). Subsequent ligand screening identified RuPhos as an effective ligand, yielding the target product 3a in 23% yield (entries 2–4). Further optimization of the catalytic system revealed that Pd(dba)2, combined with XPhos, improved the yield to 60% (entry 5). Additionally, replacing tBuOK with tBuONa as the base led to a further increase in yield to 75% (entry 6). Control experiments, conducted without the Pd catalyst or phosphine ligands, produced only trace amounts of 3a, highlighting the critical role of the Pd catalyst and phosphine ligands in driving the reaction (entries 7 and 8). In the absence of a base, no product was formed, thus establishing that tBuONa is essential for driving this reaction (entry 9, see the ESI† for details).|
|
||||
|---|---|---|---|---|
| Entry | [Pd] | Ligand | Base | Yieldc (3a, %) |
| a 1 (0.05 mmol), 2a (0.06 mmol), [Pd] (5 mol%), ligand (5 mol%), base (3 equiv.), 1 atm N2, toluene (1 mL), 110 °C, 12 h. b Base (2 equiv.). c Isolated yields. | ||||
| 1 | Pd(OAc)2 | XPhos | t BuONab | 0 |
| 2 | Pd(OAc)2 | t BuXPhos | t BuONa | 0 |
| 3 | Pd(OAc)2 | DCPF | t BuONa | Trace |
| 4 | Pd(OAc)2 | RuPhos | t BuONa | 23 |
| 5 | Pd(dba)2 | XPhos | t BuOK | 60 |
| 6 | Pd(dba)2 | XPhos | t BuONa | 75 |
| 7 | — | XPhos | t BuONa | Trace |
| 8 | Pd(dba)2 | — | t BuONa | Trace |
| 9 | Pd(dba)2 | XPhos | — | 0 |

The substrate scope of perfluorocarbazoles was systematically investigated under the optimized reaction conditions (Table 2). Arylamines bearing both electron-donating groups, such as methoxy and amino, and electron-withdrawing substituents, such as chloro and bromo, reacted with 1 to afford the corresponding perfluorinated carbazoles 3b (FMC)-3e in moderate yields (53–62%). Notably, 3-aminopyridine and pentafluoroaniline were also well-tolerated, giving products 3f and 3g in yields of 59% and 83%, respectively. Of particular interest is the preservation of the bromo group in product 3e under palladium-catalyzed conditions, providing a functional handle for subsequent derivatization. The absolute structures of 3f and 3g were unequivocally determined by X-ray crystallography (see ESI† for details). As shown in the crystal structure (Fig. S1†), C–H⋯F, F⋯F, and π–π interactions were observed in the polyfluorinated carbazole derivative 3f (FPC), with C–H⋯F and F⋯F distances of 2.64 Å and 2.94 Å, respectively, and an interplanar distance of 3.37 Å between the perfluorocarbazole rings.

Cocrystal preparation and structure
With the synthesized perfluorocarbazoles in hand, we proceeded to fabricate AP cocrystals by selecting Pe (perylene) or triphenylene (TP) as π-donors, and FPC or FMC as π-hole acceptors. Single-crystal X-ray diffraction (XRD) analysis revealed that the Pe–FPC and Pe–FMC cocrystals exhibit a 1:2 donor–acceptor (D–A) stoichiometry. Pe–FPC crystallizes in the triclinic crystal system and belongs to the ![[P with combining macron]](https://www.rsc.org/images/entities/char_0050_0304.gif) 1 space group. The crystal packing is illustrated in Fig. 2a and d and S2.† In the Pe–FPC cocrystal, one Pe molecule binds to two FPC molecules, forming a sandwich-type AP complex via face-to-face stacking along the [100] direction. The mean interplanar distances are 3.57 Å between Pe and FPC with 10.7° fold angle, 3.36 Å between FPC–FPC, and 10.51 Å between Pe–Pe. The interactions between FPC and the perylene donor are characterized by aromatic-perfluoroaromatic (AP) interactions, distinct from charge transfer (CT) interactions. Specifically, weak C–H⋯π interactions are observed between FPC and the perylene molecule at a distance of 2.56 Å, while C–H⋯F interactions are present with the shortest distance of 2.90 Å. Furthermore, C–H⋯N interactions between FPC and perylene are detected at distances of 2.80 Å, 2.98 Å, and 3.17 Å, respectively. These weak but multiple non-covalent interactions within the cocrystal effectively stabilize the AP complex. The Pe–FMC cocrystal, on the other hand, crystallizes in the orthorhombic crystal system and belongs to the Pbca space group (Fig. 2b and e and S3†). Here, two FMC molecules sandwich a single Pe molecule, with perfluorocarbazole rings and perylene arranged in a face-to-face stacking configuration. The fold angle of perfluorocarbazole rings and perylene is 2.7°, with a face-to-face separation of 3.44 Å, indicative of π–π stacking interactions. Alteration of the N-substituted aryl group in the perfluorocarbazole led to a change in molecular stacking from 1:2 to 1:1, resembling the conventional AP cocrystals formed by OFN. In the TP–FPC (1:1) cocrystal, TP and FPC molecules alternate to form a column-like mixed stack (Fig. 2c and f and S4†). The extension of these non-covalent interactions (in Pe–FPC, Pe–FMC, and TP–FPC) within a 2D plane facilitates the formation of three distinct 2D superstructure networks.59
1 space group. The crystal packing is illustrated in Fig. 2a and d and S2.† In the Pe–FPC cocrystal, one Pe molecule binds to two FPC molecules, forming a sandwich-type AP complex via face-to-face stacking along the [100] direction. The mean interplanar distances are 3.57 Å between Pe and FPC with 10.7° fold angle, 3.36 Å between FPC–FPC, and 10.51 Å between Pe–Pe. The interactions between FPC and the perylene donor are characterized by aromatic-perfluoroaromatic (AP) interactions, distinct from charge transfer (CT) interactions. Specifically, weak C–H⋯π interactions are observed between FPC and the perylene molecule at a distance of 2.56 Å, while C–H⋯F interactions are present with the shortest distance of 2.90 Å. Furthermore, C–H⋯N interactions between FPC and perylene are detected at distances of 2.80 Å, 2.98 Å, and 3.17 Å, respectively. These weak but multiple non-covalent interactions within the cocrystal effectively stabilize the AP complex. The Pe–FMC cocrystal, on the other hand, crystallizes in the orthorhombic crystal system and belongs to the Pbca space group (Fig. 2b and e and S3†). Here, two FMC molecules sandwich a single Pe molecule, with perfluorocarbazole rings and perylene arranged in a face-to-face stacking configuration. The fold angle of perfluorocarbazole rings and perylene is 2.7°, with a face-to-face separation of 3.44 Å, indicative of π–π stacking interactions. Alteration of the N-substituted aryl group in the perfluorocarbazole led to a change in molecular stacking from 1:2 to 1:1, resembling the conventional AP cocrystals formed by OFN. In the TP–FPC (1:1) cocrystal, TP and FPC molecules alternate to form a column-like mixed stack (Fig. 2c and f and S4†). The extension of these non-covalent interactions (in Pe–FPC, Pe–FMC, and TP–FPC) within a 2D plane facilitates the formation of three distinct 2D superstructure networks.59
 | ||
| Fig. 2 Superstructures of Pe–FPC, Pe–FMC, and TP–FPC. Schematic representation of the components and stoichiometric ratios of Pe–FPC (a), Pe–FMC (b), and TP–FPC (c). Stacking modes of Pe–FPC (d), Pe–FMC (e), and TP–FPC (f) co-crystals in a 2D plane. For clarity, hydrogen atoms are omitted. Different colors denote symmetry equivalence. The dashed lines indicate π⋯π distances (in Å) and dihedral angles. | ||
The three cocrystals were subsequently prepared in larger quantities using liquid-assisted grinding (Fig. 3a–c).60 Powder X-ray diffraction (PXRD) analysis was conducted on the ground samples to confirm the composition of the complexes. As shown in Fig. S5,† the diffraction pattern of the Pe–FPC powder exhibited new peaks distinct from those of the individual monomers, and these peaks coincided with those predicted by the CIF simulation for the Pe–FPC single crystal. This indicates that a novel crystal structure was formed upon grinding the two monomers. Using the same approach as for Pe–FPC, the successful synthesis of Pe–FMC and TP–FPC cocrystals was also confirmed through PXRD analysis (Fig. S6 and S7†).
 | ||
| Fig. 3 (a) Diagram of molecular cocrystals (Pe–FPC (a), Pe–FMC (b), and TP–FPC (c)). Optical photographs of Pe–FPC (d), Pe–FMC(e), and TP–FPC (f). Photographs of Pe–FPC (g), Pe–FMC (h), and TP–FPC (i) under the illumination of 365 nm UV light. Optical and fluorescent images of micro-nano crystals: optical (j) and fluorescent images (m) of Pe–FPC, optical (k) and fluorescent images (n) of Pe–FMC, optical (l) and fluorescent images (o) of TP–FPC. | ||
Further investigation of the non-covalent interaction forces beyond chemical bonding to reveal the property–structure relationship were conducted. At the macroscopic level, under sunlight, Pe–FPC and Pe–FMC were observed as yellow powders, while TP–FPC appeared white (depicted in Fig. 3d–f). Upon exposure to 365 nm UV light, both Pe–FPC and Pe–FMC emitted greenish-yellow fluorescence, while TP–FPC showed weak fluorescence (Fig. 3g–i). The optical micrographs revealed that the micro-nano cocrystals predominantly exhibited a 1D rod-like morphology, with the green and yellow fluorescence typical of these compounds (Fig. 3j–o). SEM analysis of Pe–FPC (Fig. S8†) indicated that the cocrystal particles aggregated into irregular clusters, with sizes ranging from 0.2 to 0.6 μm, while Pe–FMC clusters measured between 0.5 and 1.5 μm (Fig. S9†).
AP interaction of cocrystals
Fig. 4 illustrates the absorption and fluorescence spectra for the Pe–FPC, Pe–FMC, and TP–FPC cocrystals. The absorption spectra of the cocrystals closely resemble the combined absorption profiles of the donor and acceptor components, with no significant redshift observed. The absorption of perylene spans from 240 to 550 nm, while FPC absorbs from 240 to 400 nm (Fig. 4a). In contrast to perylene's emission, Pe–FPC exhibits a blue-shifted emission peak centered at 523 nm (Fig. 4d). The fluorescence emission spectrum further supports the presence of AP interactions while no CT interaction was observed. Pe–FMC shows a similar trend in its absorption and emission, with varying degrees of blue shift (Fig. 4b and e). The blue shift observed in the fluorescence spectra, induced by the AP interactions, can be attributed to the periodic incorporation of the electron acceptors (FPC or FMC) between the donor molecules through π–π interactions, creating molecular barriers. These barriers, formed by FPC acting as a “diluent,” effectively increase the spatial separation between donors, significantly minimizing the redshift typically induced by exciton delocalization.43,61,62 The fluorescence spectra of solid-state TP–FPC exhibited an even more pronounced blue shift compared to the monomeric emission due to AP interactions, including a distinct charge-transfer (CT) absorption band around 400 nm, which results from THF-assisted, loosely packed and non-parallel stacking (Fig. 4c and f).63 This feature is primarily due to electron transfer from TP to FPC. The relatively low Stokes shifts for Pe–FPC and Pe–FMC suggest the absence of ground-state CT interactions, indirectly confirming the presence of AP interactions within these cocrystals. Notably, the photoluminescence quantum efficiency (PLQE) of Pe–FPC was determined to be 9.31%, which exceeds the PLQE of the pure Pe powder (4.6%). This enhancement is rationalized by the periodic insertion of FPC molecules in a 1:2 molecular ratio, which prevents intermolecular interactions and electron transfer between the arene–perfluoroarene components.48
 | ||
| Fig. 4 UV-vis absorption spectra of Pe–FPC (a), Pe–FMC (b), and TP–FPC (c). Fluorescence emission spectra of Pe-FPC (d), Pe–FMC (e), and TP–FPC (f). | ||
Infrared (IR) spectroscopy analysis revealed that the spectrum of Pe–FPC is a superposition of the individual spectra of Pe and FPC, confirming successful cocrystallization (Fig. S10a†). The electron-rich environment of Pe induces a weakening of the C–F bond, as evidenced by a shift in the stretching vibration from 1281 cm−1 to 1277 cm−1. The Ar–H stretching vibration of Pe remains unchanged at 3050 cm−1. Similarly, in the Pe–FMC cocrystal, the C–F stretching vibration is also shifted from 1253 cm−1 to 1250 cm−1, indicating bond weakening (Fig. S10b†). In the case of TP–FPC, the Ar–H stretching vibration undergoes a shift from 3023 cm−1 to 3043 cm−1, attributable to the π⋯π stacking interactions between the donor and acceptor units. Furthermore, the C–F stretching vibration shifts from 1355 cm−1 to 1351 cm−1, indicating a weakening of the C–F bond due to the electron-rich environment surrounding TP (Fig. S10c†).
Photocatalytic degradation performance
The photocatalytic activity of the as-prepared AP cocrystals was evaluated for molecular oxygen activation to produce ROS in pure water under UV-visible irradiation, without the need for a sacrificial agent or metal catalyst. In the beginning, Rhodamine B (RhB) was selected as a model water pollutant to assess the photodegradation efficiency of the photocatalysts. The photocatalytic degradation experiments were conducted under a 10 W blue LED (425 nm) light source, where Pe–FPC demonstrated excellent photodegradation performance. As shown in Fig. 5a, after 30 minutes dark treatment, Pe–FPC achieved >95% degradation of RhB within 15 minutes under visible light irradiation, while under similar conditions, the Pe and FPC monomers degraded only 41% and 18% of RhB, respectively. The corresponding reaction rate constants (kap) for FPC, Pe, and Pe–FPC were 0.0183 min−1, 0.0416 min−1, and 0.175 min−1, respectively, indicating that the photocatalytic rate of Pe–FPC is 9.56 and 4.21 times higher than that of FPC and Pe, respectively (Fig. 5b). Moreover, after dark treatment for 30 minutes, Pe–FMC exhibited 95% degradation of RhB within 25 minutes (Fig. 5c). In contrast, Pe and FMC degraded only 64% and 19% of RhB, respectively, under identical conditions (425 nm blue LED). The corresponding kap values for FMC, Pe, and Pe–FMC were 0.0088 min−1, 0.0416 min−1, and 0.113 min−1, respectively, demonstrating that the photocatalytic rate of Pe–FMC is 15.1 times higher than that of FMC and 3.20 times higher than that of Pe (Fig. 5d). However, under the same conditions, TP–FPC degraded only 32% of RhB, a rate that is 14.5 times and 9.3 times lower than that of Pe–FPC and Pe–FMC, respectively (Fig. 5e and f). Recent photocatalytic materials for the photodegradation of RhB are summarized in Table S2.† Among these, our Pe–FPC (1:2) cocrystal demonstrates exceptional photocatalytic degradation activity. These results confirm that the incorporation of an additional perfluoroarene to enhance the internal electric field (IEF) effectively improves photocatalytic efficiency. Thus, the strategy of increasing the molecular barrier thickness is shown to be a successful approach for enhancing photocatalytic performance.
 | ||
| Fig. 5 Photocatalytic degradation of RhB. The degradation curves (a) and pseudo-first-order kinetics fitting curves. (b) For Pe, FPC, and Pe–FPC with time under 425 nm blue LED (10 W) light illumination. The degradation curves (c) and pseudo-first-order kinetics fitting curves (d) for Pe, FMC, and Pe–FMC with time under 425 nm blue LED (10 W) light illumination. The degradation curves (e) and pseudo-first-order kinetics fitting curves (f) for TP, FPC, and TP–FPC with time under 425 nm blue LED (10 W) light illumination. (g) Cycling degradation performance of Pe-FPC. (h) PXRD of the cocrystal before and after using as photocatalyst for pollutant degradation. | ||
Photocatalysts often face challenges such as rapid deactivation, poor reusability, difficult recovery, and cleaning issues, which hinder their widespread application and increase costs. To assess the long-term stability and reusability of the Pe–FPC cocrystal, we conducted a series of cycling experiments (Fig. 5g). Remarkably, the Pe–FPC catalyst demonstrated excellent chemical stability, with no significant loss of photocatalytic activity after seven consecutive cycles. PXRD analysis of the recovered cocrystal confirmed that its diffraction pattern remained essentially unchanged after multiple cycles, indicating that the cocrystal structure was preserved and did not undergo degradation during the repeated catalytic processes (Fig. 5h). This enhanced stability can be attributed to the formation of a robust supramolecular network within the cocrystal, held together by three types of weak non-covalent interactions: C–H⋯F, C–H⋯N, and C–H⋯π. These interactions contribute to the structural integrity of the cocrystal, effectively preventing disruption during cycling and ensuring sustained catalytic activity over extended use.
To assess the versatility of the Pe–FPC cocrystal for the degradation of various water pollutants, its photocatalytic performance was also evaluated for the removal of Methylene Blue (MB) and Eosin B. The results indicated that the cocrystal photocatalyst exhibited comparable efficiency in degrading these additional dye pollutants under visible light irradiation. Kinetic analysis, as shown in Fig. S12 of the ESI,† reveals that the Pe–FPC photocatalyst facilitated the degradation of MB and Eosin B at rates similar to those observed for Rhodamine B. These findings underscore the applicability of the cocrystal strategy, demonstrating its ability to enhance the photocatalytic degradation of a range of dye pollutants.
Radical oxygen species detection
The most common active species in photocatalytic degradation of organic pollutants are photogenerated holes (h+), hydroxyl radicals (˙OH), singlet oxygen (1O2), and hydrogen peroxide (H2O2). Preliminary evidence for the types of radicals generated during the photocatalytic process has been obtained through radical scavenging experiments. Ascorbic acid (AA), tocopherol, triethanolamine (TEOA), and tBuOH (TBA) were employed as scavengers for superoxide radicals (˙O2−), singlet oxygen (1O2), photogenerated holes (h+) and ˙OH, respectively, with a scavenger concentration of 10 mM, as commonly described in the literature.64 The addition of triethanolamine (TEOA) resulted in significant inhibition of the degradation process, indicating that h+ species play a crucial role in the photocatalytic reactions. The inclusion of ascorbic acid (AA) notably suppressed the degradation of pollutants, pointing to the involvement of superoxide radicals (˙O2−) in the reaction system. In contrast, the addition of tocopherol and TBA led to moderate inhibition of photocatalysis, indicating that singlet oxygen (1O2) and ˙OH are generated during the illumination of the cocrystal (Fig. 6a). Control experiments conducted under a nitrogen atmosphere confirmed the essential role of O2 in the generation of ˙O2− and 1O2 during photocatalytic degradation (Fig. 6b). To further identify the active species, electron spin resonance (ESR) spectroscopy was employed. Using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a trapping agent, ESR spectra revealed the presence of ˙OH signals under light irradiation in the Pe–FPC system, which is consistent with the results from the radical scavenging experiments (Fig. 6c). Meanwhile, significant amounts of H2O2 were detected during the photodegradation process. The concentrations of H2O2 and ˙OH in the Pe–FPC system increased over time, further confirming their involvement in the degradation of RhB (Fig. 6d and e). The H2O2 production rate of Pe–FPC in deionized water under an air atmosphere was measured at 2640 μmol h−1 g−1, which is comparable to the performance of several previously reported photocatalysts for H2O2 production, as summarized in Table S3.† The synergistic effects of h+, ˙O2−, ˙OH, 1O2, and H2O2 thus effectively contribute to the degradation of challenging water pollutants, demonstrating the robust photocatalytic performance of the Pe–FPC system.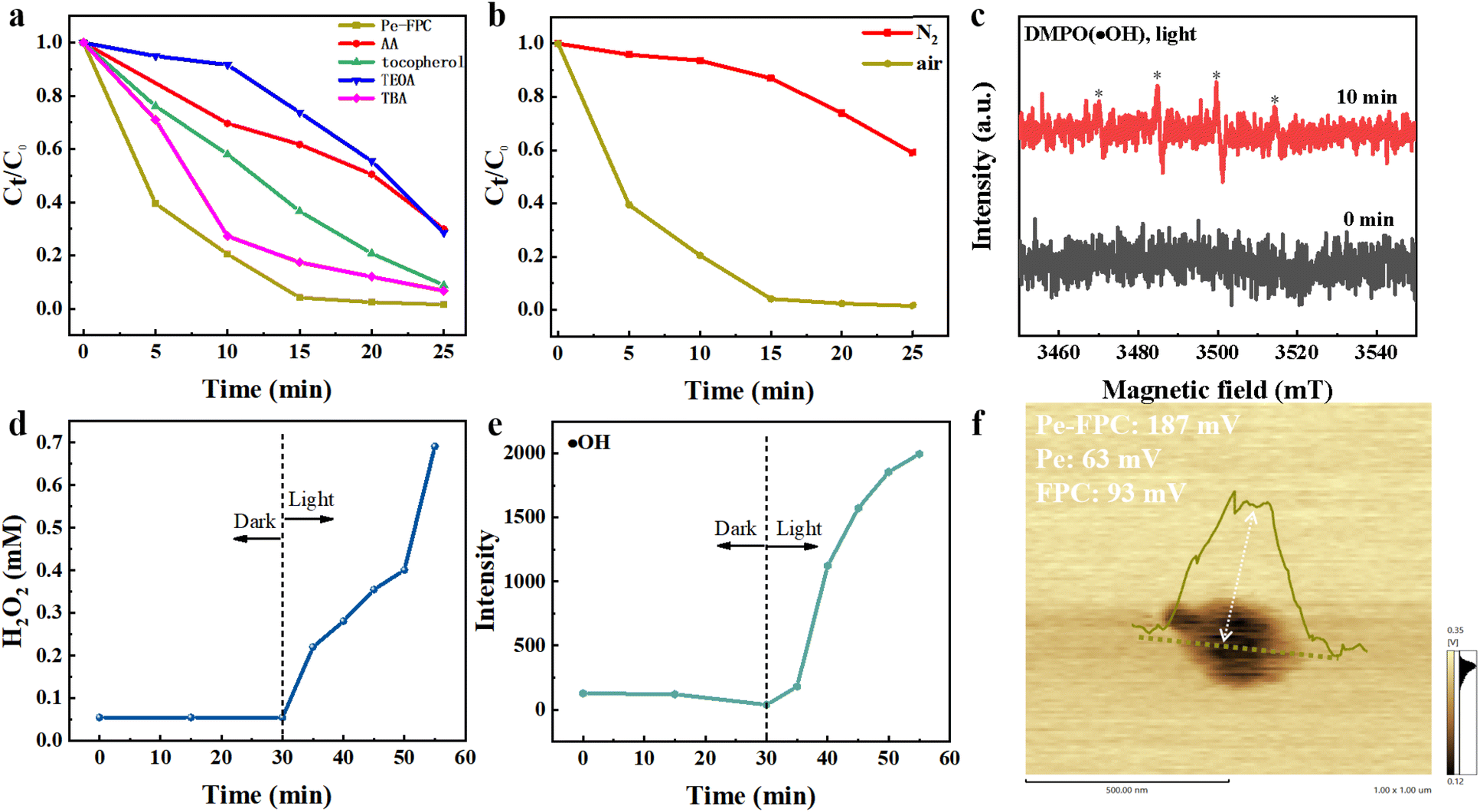 | ||
| Fig. 6 Photocatalytic degradation reaction mechanism. (a) Quenching experiments of different free radical capture agents (10 mM) on the degradation of RhB by Pe–FPC. (b) Degradation curves of Pe–FPC under different gas conditions. (c) EPR spectra of ˙OH (DMPO) under illuminations of 15 min and 0 min. (d) Performance of Pe–FPC in the photocatalytic H2O2 production under visible light irradiation in water and air atmosphere by potassium iodide method. (e) Determination of ˙OH in photocatalytic degradation reaction of Pe–FPC. (f) Surface potentials of Pe–FPC detected with KPFM. | ||
Carrier separation and transport
To investigate the charge separation and transport mechanisms following photon absorption, Atomic Force Microscopy (AFM) was employed to probe the internal electric field (IEF) within the system. For this purpose, we applied the method utilized by Zhu et al., which has been successfully used to quantitatively measure IEF intensity in organic supramolecular systems.34 The IEF is a critical kinetic factor that influences the separation of photogenerated charge carriers, with its strength directly correlating to the surface potential. Surface potential measurements were conducted on both the Pe–FPC cocrystal and its individual components using AFM coupled with a Kelvin probe force microscopy (KPFM) technique, as shown in Fig. 6f. The surface potentials of the individual components, Pe (63 mV) and FPC (93 mV), were significantly lower than that of the Pe–FPC cocrystal (187 mV). Notably, the surface potential of Pe–FPC is approximately three times higher than that of Pe, indicating a significantly enhanced IEF within the cocrystal. This result suggests that the cocrystal structure promotes more efficient charge separation compared to the individual components. For comparison, in a previous study by Zhu et al., the surface potential of a 1:1 Pe-OFN cocrystal was approximately twice as high as that of Pe.34 In our case, the surface potential of the Pe–FPC (1:2) cocrystal is three times as high as that of Pe. This substantial difference underscores the effectiveness of our design, which involves increasing the perfluoroarene layer (acting as a π-hole layer) from a single to a double layer by forming a 1:2 arene–perfluoroarene cocrystal. The additional perfluoroarene layer increases the dipole interactions between the ordered π-donor and π-hole layers, thereby creating a more pronounced IEF in the Pe–FPC system compared to a 1:1 arene–perfluoroarene cocrystal. This enhanced IEF in the Pe–FPC (1:2) cocrystal facilitates the efficient separation of photogenerated excitons into free carriers (electrons and holes), further improving photocatalytic efficiency. This finding is corroborated by time-resolved fluorescence spectroscopy, which revealed a significantly lower photoluminescence (PL) lifetime for Pe–FPC (4.69 ns) compared to Pe (33.77 ns) (Fig. S16†). The much shorter PL lifetime in the cocrystal suggests that photogenerated excitons in Pe–FPC are more effectively separated into charge carriers, promoting higher photocatalytic efficiency.65,66 In the electrochemical impedance spectroscopy, a smaller curvature in the Nyquist plot for Pe–FPC suggested a reduced charge transport resistance relative to the individual cocrystal monomers (Fig. S19†). In conclusion, our results validate the hypothesis that increasing the number of molecular barrier layers to enhance the IEF is a successful strategy. The introduction of a second perfluoroarene layer in the Pe–FPC (1:2) cocrystal effectively promotes charge separation, leading to enhanced photocatalytic efficiency. This strategy opens new avenues for designing high-performance photocatalysts for energy and environmental applications.
Photocatalytic oxidation performance
To further assess the practical application potential of the Pe–FPC cocrystal in organic synthesis under aqueous conditions, its photocatalytic activity was evaluated in the oxidation reaction of 5-hydroxymethylfurfural (HMF). HMF is regarded as a top biomass-derived building block for producing valuable products, with 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) being one of the key products of HMF oxidation. HMFCA is an important biomass-based furan compound with widespread applications, notably as a renewable monomer in the synthesis of polyesters. Initially, HMF was used as the substrate, with the Pe–FPC cocrystal serving as the photocatalyst under an oxygen atmosphere in water. After 6 hours, HMFCA was obtained with a 74% GC yield (Table 3, entry 1). Prolonging the reaction time to 12 hours resulted in a yield of 92% (entry 2). The effect of the gaseous environment was then studied using both air and nitrogen atmospheres. Under air, the reaction achieved an 85% yield after 24 hours, whereas, under nitrogen, no conversion of the substrate was observed, highlighting the critical role of oxygen in the reaction (entries 3, 4). When no light or catalyst was applied, the reaction barely proceeded (entries 5, 6). Using the monomers, perylene or FPC, as catalysts resulted in only 40% and trace yields, respectively (entries 7, 8), demonstrating that the cocrystal catalyst is far more effective than the monomers alone. Replacing molecular oxygen with hydrogen peroxide (H2O2) as the oxidant resulted in lower conversion and a decrease in product yield (entry 9), and extending the reaction time to 24 hours decreased the yield further to 60%, accompanied by the formation of over-oxidized products (entry 10). Increasing the reaction temperature to 80 °C or introducing bases (Na2CO3, K2CO3) also led to the formation of over-oxidized products (entries 11–13).|
|
|||
|---|---|---|---|
| Entry | Variations | Conv. (%) | Y (%) |
| a HMF (0.2 mmol), Pe–FPC (4.1 mg), O2 (1 atm), H2O (1 mL), 425 nm blue LEDs, 10 W, rt, 6 h. The conversion and yield were determined by GC. | |||
| 1 | — | 95 | 74 |
| 2 | 12 h | 98 | 92 |
| 3 | Air, 24 h | 95 | 90 |
| 4 | N2, 12 h | 5 | Trace |
| 5 | No light, 12 h | 10 | Trace |
| 6 | No cocrystal, 12 h | 10 | Trace |
| 7 | Pe (4.1 mg), 12 h | 50 | 40 |
| 8 | FPC (4.1 mg), 12 h | 10 | Trace |
| 9 | H2O2 (3 equiv.) | 60 | 60 |
| 10 | 24 h | 99 | 60 |
| 11 | 80 °C | 99 | 35 |
| 12 | Na2CO3 | 99 | 20 |
| 13 | K2CO3 | 99 | 20 |

Subsequently, the substrate scope of the aldehyde-to-carboxylic acid oxidation catalyzed by Pe–FPC was explored. As shown in Table 4, various benzaldehyde derivatives, including para-methyl, meta-methyl, ortho-methyl, para-isopropyl, and 3,4-dimethyl benzaldehydes, exhibited good reactivity, producing the corresponding carboxylic acids in yields ranging from 84% to 96% (6a–6f). Electron-donating groups, such as para-methoxy (6g), 3,4-dimethoxy (6h), and meta-trifluoromethoxy (6i) substituted benzaldehydes also showed good reactivity, yielding the target products in the range of 66–89%. Halogen-substituted benzaldehydes, including para-fluoro, para-chloro, para-bromo, and ortho-bromo derivatives (6j–6m), afforded the corresponding products in yields of 60–85%. Electron-withdrawing groups, such as meta-trifluoromethyl (6n) and para-nitro (6o) benzaldehydes, produced their respective oxidation products with yields of 87% and 93%. Heterocyclic compounds such as HMF (6p) were also well-suited for the reaction, resulting in a high yield of 92%. However, quinoline (6q) and pyridine (6r) substrates did not hinder the reactivity, producing the corresponding products in yields of 92% and 93%, respectively. Thiophene (6s) also gave a good yield of 86%. Furthermore, aliphatic aldehydes such as pivaldehyde (6t), heptaldehyde (6u), and decaldehyde (6v) were successfully oxidized to their corresponding carboxylic acids with good to high isolated yields ranging from 65% to 88%. These results demonstrate the broad substrate scope and high reactivity of the Pe–FPC cocrystal in catalyzing the oxidation of aldehydes to carboxylic acids, highlighting its promising potential for applications in biomass conversion and organic synthesis.

The active species involved in the photocatalytic oxidation reactions were further analyzed using Electron Paramagnetic Resonance (EPR) spectroscopy. As shown in Fig. 7a, after 10 minutes of illumination, a distinct resonance peak corresponding to the hydroxyl radical (˙OH) was observed. Based on previously reported photodegradation mechanisms involving H2O2,11 we hypothesize that superoxide radicals (˙O2−) and singlet oxygen (1O2) are generated in the reaction system but persist only briefly before rapidly converting into ˙OH and H2O2. Furthermore, the observed decrease in the TEMPO radical signal under illumination provides additional evidence for the presence of photogenerated holes (h+), despite some variations due to restricted motion (Fig. 7b).67 Based on mechanistic investigations, we propose a reaction mechanism (Fig. 7c). Under irradiation, molecular oxygen (O2) is reduced by the cocrystal catalyst to generate ˙O2−, which can subsequently undergo a two-step single-electron reduction pathway to form H2O2. The H2O2 then decomposes efficiently to yield ˙OH, which, along with ˙O2−, 1O2, and H2O2, contributes to the overall photocatalytic degradation of pollutants in water. During the oxidation reaction, benzaldehyde interacts with ˙O2− to generate benzoyl radicals and the corresponding hydroperoxyl radical (˙HO2). The benzoyl radicals further react with ˙OH, ultimately leading to the formation of the carboxylic acid product. These results highlight the crucial role of reactive oxygen species (ROS) in facilitating the photocatalytic oxidation process and confirm the effectiveness of the Pe–FPC cocrystal catalyst in promoting oxidation reactions under visible-light irradiation.
 | ||
| Fig. 7 Photocatalytic aerobic oxidation reaction mechanism. (a) EPR spectra of ˙OH (DMPO) under illuminations of 10 min and 0 min in O2 atmosphere. (b) EPR spectra of h+ (TEMPO) under dark and light conditions. (c) Diagram of the proposed photocatalytic process. | ||
Conclusions
We successfully designed and synthesized a series of novel perfluorocarbazole derivatives via a Pd-catalyzed cascade C–Br/C–F amination reaction. These compounds expand the perfluoroarene library, offering versatile building blocks for constructing arene–perfluoroarene cocrystals. Three cocrystals—Pe–FPC (1:2), Pe–FMC (1:2), and TP–FPC (1:1)—were fabricated, with their structures confirmed by single-crystal X-ray diffraction. Notably, the 1:2 arene–perfluoroarene cocrystal is a rare achievement in this field. The cocrystals were synthesized in larger quantities using liquid-assisted grinding. The Pe–FPC (1:2) cocrystal demonstrated exceptional photocatalytic activity, activating molecular oxygen to generate reactive oxygen species (ROS) in pure water under visible light irradiation, without the need for a sacrificial agent or metal catalyst. It effectively degraded RhB, MB, and Eosin B, maintaining stability over seven cycles. The cocrystal's stability is attributed to its intermolecular interactions, such as C–H⋯π, C–H⋯F, and C–H⋯N. Additionally, this study marks the first use of cocrystals in organic transformations, where Pe–FPC catalyzed the oxidation of various aromatic and aliphatic aldehydes to carboxylic acids, outperforming traditional small-molecule aqueous-phase catalysts. Mechanistic studies reveal that the synergistic effects of h+, ˙O2−, ˙OH, 1O2, and H2O2 are crucial for the photoinduced oxidation process. The Pe–FPC (1:2) cocrystal exhibited a high H2O2 production yield of 2640 μmol h−1 g−1 in deionized water under air, further supporting its photocatalytic performance. Surface potential measurements showed that Pe–FPC's surface potential is approximately three times higher than that of Pe, indicating a significantly enhanced internal electric field (IEF), which promotes the efficient separation of photogenerated excitons into free carriers. This was corroborated by time-resolved fluorescence spectroscopy, which revealed a much shorter photoluminescence (PL) lifetime for Pe–FPC (4.69 ns) compared to Pe (33.77 ns), signifying more efficient charge separation and higher photocatalytic efficiency. These findings confirm that increasing the number of perfluoroarene layers to enhance the IEF is an effective strategy for improving photocatalytic performance. The introduction of a second perfluoroarene layer in Pe–FPC (1:2) successfully promotes charge separation, opening new avenues for designing high-performance photocatalysts for energy and environmental applications.
Data availability
All relevant data are within the manuscript and its additional files.Author contributions
B. Yuan supervised the project. B. Yuan and W. Hu designed the experiments. W. Hu performed and analyzed the experiments. W. Hu, H. Li and B. Yuan prepared this manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Key-Area Research and Development Program of Guangdong Province (No. 2020B010188003) and Natural Science Foundation of Henan Province (No. 222300420528). The authors are also grateful to the supports from Center of Advanced Analysis & Gene Sequencing of Zhengzhou University.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- K. Kümmerer, Angew. Chem., Int. Ed., 2017, 56, 16420–16421 CrossRef.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, W. Liu, Y. Lou, Y. Zhang, Y. Dong, J. Xu, C. Pan and Y. Zhu, Appl. Catal., B, 2022, 300, 120762 CrossRef CAS.
- L. Yang, Z. Chen, Q. Cao, H. Liao, J. Gao, L. Zhang, W. Wei, H. Li and J. Lu, Adv. Mater., 2024, 36, 2306758 CrossRef CAS PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- P. Mahbub, A. Smallridge, A. Irtassam and T. Yeager, Chem. Eng. J., 2022, 427, 131762 CrossRef CAS.
- N. Li, Y. Ma and W. Sun, Molecules, 2024, 29, 3995 CrossRef CAS PubMed.
- Q. Zhang and Y. Luo, High Power Laser Sci. Eng., 2016, 4, e22 CrossRef.
- A. Asghar, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2015, 87, 826–838 CrossRef CAS.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- X. Sun, R. Wang and D. Su, Chin. J. Catal., 2013, 34, 508–523 CrossRef CAS.
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234 RSC.
- Y. Wang, J. Hu, T. Ge, F. Chen, Y. Lu, R. Chen, H. Zhang, B. Ye, S. Wang, Y. Zhang, T. Ma and H. Huang, Adv. Mater., 2023, 35, 2302538 CrossRef CAS PubMed.
- Y. Guo, W. Shi and Y. Zhu, EcoMat, 2019, 1, e12007 CrossRef.
- Z. Chen, D. Yao, C. Chu and S. Mao, Chem. Eng. J., 2023, 451, 138489 CrossRef CAS.
- S. Huang, X. Yang, L. Zhou, J. Lei, L. Wang, Y. Liu and J. Zhang, J. Environ. Sci., 2024, 50, 2917–2969 CAS.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, Adv. Mater., 2019, 31, 1806626 CrossRef PubMed.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 4965–4968 CrossRef CAS PubMed.
- J. Wang, W. Shi, D. Liu, Z. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS.
- L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Adv. Mater., 2019, 31, 1902328 CrossRef PubMed.
- Akhtaruzzaman, S. Khan, B. Dutta, T. S. Kannan, G. Kumar Kole and M. Hedayetullah Mir, Coord. Chem. Rev., 2023, 483, 215095 CrossRef CAS.
- L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243–19256 CrossRef CAS PubMed.
- E. Stoler and J. Warner, Molecules, 2015, 20, 14833–14848 CrossRef CAS PubMed.
- X. Wang, Z. Wang, X. Wang, F. Kang, Q. Gu and Q. Zhang, Angew. Chem., Int. Ed., 2024, e202416181 CAS.
- L. Wang, J. Deng, M. Jiang, C. Zhen, F. Li, S. Li, S. Bai, X. Zhang and W. Zhu, J. Mater. Chem. A, 2023, 11, 11235–11244 RSC.
- T. Xue, C. Ma, L. Liu, C. Xiao, S.-F. Ni and R. Zeng, Nat. Commun., 2024, 15, 1455 CrossRef CAS PubMed.
- L. Wang, J. Deng, S. Bai, Y. Wu and W. Zhu, Small, 2024, 2406352 CrossRef CAS PubMed.
- K. Liu, L. Wang, S. Li, H. Liu, D. Zhang, M. Jiang, W. Chen, F. Jiao, X. Zhang and W. Hu, Adv. Funct. Mater., 2023, 33, 2306871 CrossRef CAS.
- R. A. Wiscons, N. R. Goud, J. T. Damron and A. J. Matzger, Angew. Chem., Int. Ed., 2018, 57, 9044–9047 CrossRef CAS PubMed.
- S. Li, Z. Zhao, T. Ma, P. Pachfule and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202112298 CrossRef CAS PubMed.
- S. Zhang, A. Chen, Y. An and Q. Li, Matter, 2024, 7, 3317–3350 CrossRef CAS.
- W. Wang, W. X. Wu, Y. Zhang and W. J. Jin, Chem. Phys. Rev., 2024, 5, 031303 CrossRef CAS.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9696–9711 CrossRef CAS PubMed.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef PubMed.
- Q. Zhou, X. Zhang, L. Ning, Y. Song, Y. Wang, J. Feng, C. Sun, J. Li, Q. Gong, Q. Zhang and Y. Huang, Small Methods, 2025, 9, 2401439 CrossRef CAS PubMed.
- H. Zhang, J. Han, X. Jin and P. Duan, Angew. Chem., Int. Ed., 2021, 60, 4575–4580 CrossRef CAS PubMed.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS PubMed.
- H. Yin, Y. Ma, H. Lin, S. Chen, Z. Zhou, S. Zhuo and X. Wang, Chin. J. Chem., 2022, 40, 1149–1155 CrossRef CAS.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef PubMed.
- H. Ye, G. Liu, S. Liu, D. Casanova, X. Ye, X. Tao, Q. Zhang and Q. Xiong, Angew. Chem., Int. Ed., 2018, 57, 1928–1932 CrossRef CAS PubMed.
- Z. Gao, J. Sun, L. Shi, W. Yuan, H. Yan and W. Tian, Angew. Chem., Int. Ed., 2025, e202423174 CAS.
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC.
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed.
- Y. Su, L. Zhang, W. Wang and D. Shao, ACS Sustain. Chem. Eng., 2018, 6, 8704–8710 CrossRef CAS.
- J. Li, G. Zhan, Y. Yu and L. Zhang, Nat. Commun., 2016, 7, 11480 CrossRef CAS PubMed.
- J. Li, L. Zhang, Y. Li and Y. Yu, Nanoscale, 2014, 6, 167–171 RSC.
- Q. J. Shen, H. Q. Wei, W. S. Zou, H. L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 1010–1015 RSC.
- Z. Wang, F. Dötz, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 1247–1250 CrossRef CAS PubMed.
- A. S. Batsanov, J. A. K. Howard, D. Albesa-Jové, J. C. Collings, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault and T. B. Marder, Cryst. Growth Des., 2012, 12, 2794–2802 CrossRef CAS.
- B. Li, L. Liu, Y. Wang, K. Liu, Z. Zheng, S. Sun, Y. Hu, L. Li and C. Li, Nat. Commun., 2024, 15, 2535 CrossRef CAS PubMed.
- Y. Huang, Q. Gong, J. Ge, P. Tang, F. Yu, L. Xiao, Z. Wang, H. Sun, J. Yu, D.-S. Li, Q. Xiong and Q. Zhang, ACS Nano, 2020, 14, 15962–15972 CrossRef CAS PubMed.
- H. Ye, G. Liu, S. Liu, D. Casanova, X. Ye, X. Tao, Q. Zhang and Q. Xiong, Angew. Chem., Int. Ed., 2018, 57, 1928–1932 CrossRef CAS PubMed.
- Z. Gao, J. Sun, L. Shi, W. Yuan, H. Yan and W. Tian, Angew. Chem., Int. Ed., 2025, e202423174 CAS.
- Z. Wang, F. Yu, W. Chen, J. Wang, J. Liu, C. Yao, J. Zhao, H. Dong, W. Hu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17580–17586 CrossRef CAS PubMed.
- Q. Shen, L. Wei, R. Bibi, K. Wang, D. Hao, J. Zhou and N. Li, J. Hazard. Mater., 2021, 413, 125376 CrossRef CAS PubMed.
- Y. Li, L. Yang, H. He, L. Sun, H. Wang, X. Fang, Y. Zhao, D. Zheng, Y. Qi, Z. Li and W. Deng, Nat. Commun., 2022, 13, 1355 CrossRef CAS PubMed.
- L. Yang, M. Lv, Y. Song, K. Yin, X. Wang, X. Cheng, K. Cao, S. Li, C. Wang, Y. Yao, W. Luo and Z. Zou, Appl. Catal., B, 2020, 279, 119341 CrossRef CAS.
- N. Byra, S. Krukowski, J. Sadlo and W. Kolodziejski, Materials, 2022, 15, 2043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and synthesis, useful information, 1H NMR spectra, 13C NMR spectra, MS data, characterization details. CCDC 2425781, 2425783, 2425788, 2425789 and 2425790. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02837j |
| This journal is © The Royal Society of Chemistry 2025 |