
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DiICz MR-TADF emitters as potent energy transfer photocatalysts
Lea
Hämmerling
 ,
David
Hall
,
Eliott
Blin
,
Tabea
Heil
and
Eli
Zysman-Colman
*
,
David
Hall
,
Eliott
Blin
,
Tabea
Heil
and
Eli
Zysman-Colman
*
Organic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk
First published on 28th October 2025
Abstract
Photoinduced energy transfer (PEnT) reactions are a subset of photochemical reactions that involve the indirect photoactivation of substrates following an energy transfer from a photoexcited sensitizer/photocatalyst. Examples of PEnT reactions include E/Z isomerizations, [2 + 2] cycloadditions, and sigmatropic shifts. Here we introduce a family of diindolocarbazole (DiICz) multi-resonant thermally activated delayed fluorescent (MR-TADF) photocatalysts (PCs), DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, which have relatively high triplet energies (ET). We cross-compare their photocatalytic behavior and that of relevant literature reference PCs' in five distinct PEnT reactions. We demonstrate that the use of the DiICz PCs consistently leads to more rapid reaction rates and higher yields compared to the widely used 4CzIPN. DiICztBu4, DiICzMes4, and DiICztBuCz4 possess similar ET but decreasing singlet–triplet energy gaps, ΔEST, enabling for the first time a comparison of the dependency of both the reaction kinetics and the final yield on this photophysical parameter. We observed that when the reaction kinetics are fast, there is little sensitivity to quenching of the excited PC by oxygen, implying that Dexter Energy Transfer (DET) to the substrate is competitive with DET to oxygen. Importantly, this means that some of the DET reactions using these PCs can be performed in air without adversely affecting reaction yield.
Introduction
One major class of photochemical reactions consists of those that proceed via a photoinduced energy transfer (PEnT) mechanism, which typically involves an electronically excited photocatalyst (PC*) transferring its energy to a substrate (sub). The substrate in its electronically excited state (sub*) then undergoes a photochemical transformation.1–3 The majority of PEnT reactions proceed via Dexter Energy Transfer (DET) reactions. DET involves the simultaneous double electron exchange between the PC* and the sub to generate a PC in its S0 state and a sub*. This can take place from the singlet state of the 1PC* to generate 1sub* or more commonly from the triplet state of 3PC* to generate 3sub* (Fig. S1) and does not involve a change in the overall multiplicity.1 DET processes occur over shorter distances, typically less than 10 Å,4 as orbital overlap between the PC and the sub is required, which implies a collisional interaction is needed for DET to occur intermolecularly.In cases where DET occurs from the 3PC* to 3sub, it is the spectral overlap of the phosphorescence of the 3PC* and the spin-forbidden absorption of the substrate that is relevant to generate 3sub*. Notably, as the spin-forbidden absorption spectrum is of negligible intensity in most organic substrates, the low-energy onset of the low-temperature phosphorescence spectrum of the sub is used as a surrogate estimation of the energy at which spectral overlap no longer occurs, which is none other than the energy of the T1 state (ET) of the sub. It is generally accepted that DET will occur when ET(PC*) > ET(sub*), and the closer these two values are, the more likely DET is to occur.5 Given that the 3sub* has a biradical-like character, this enables a diversity of reactions such as E/Z isomerization,2,6–8 cycloadditions such as [2 + 2],9–11 sensitization of metal complexes,12–14 homolytic bond cleavages such as N2 release from benzoyl azides, scission of S–S bonds, and N–O dissociation of oxime esters to instigate the formation of carbon-centered and nitrogen-centered radicals in concert with the loss of CO2.15–17
Of the PCs typically employed in PEnT reactions, organic carbonyl-based photosensitizers have the highest ET. For instance, xanthone and acetophenone (Fig. 1) have similar ET values of 3.22 eV (74.2 kcal mol−1, 310.5 kJ mol−1) and 3.21 eV (74.0 kcal mol−1, 309.6 kJ mol−1).1,2,18 Organometallic PCs are particularly popular for PEnT reactions in part because the presence of the heavy metal center ensures an essentially quantitative triplet yield as a result of fast intersystem crossing (ISC) mediated by its large spin–orbit coupling (SOC).19,20 Two of the most commonly used organometallic PCs for PEnT reactions include [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (ET = 2.68 eV)5 for reactions requiring a relatively high ET and [Ru(bpy)3](PF6)2 (ET = 2.12 eV)21 for those that do not (Fig. 1).
 | ||
Fig. 1 Chemical structures and ET for selected literature PCs used for DET reactions and for the four TADF PCs investigated in this work. Quoted ET values for 4CzIPN, DiICztBu4, DiICzMes4, DiICztBuCz4 and DiICztBuDPA4 were estimated from ET = ES − ΔEST, with ES being the onset of the steady-state emission in DCM at room temperature and ΔEST determined in 2-MeTHF glass at 77 K. ET for fac-Ir(ppy)3 was taken from the onset of the room temperature emission in 2-MeTHF. ET for [Ru(bpy)3](PF6)2 was taken from the emission maximum at 77 K in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/EtOH glass.22ET for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was taken from the room temperature emission maximum in MeCN.23 For the organic PCs, ET is measured from the phosphorescence spectra at 77 K.5 :1 MeOH/EtOH glass.22ET for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was taken from the room temperature emission maximum in MeCN.23 For the organic PCs, ET is measured from the phosphorescence spectra at 77 K.5 | ||
Over the last decade or so, organic thermally activated delayed fluorescence (TADF) compounds have been increasingly used in the academic community as alternatives to organometallic complexes, initially as emissive materials for organic light-emitting diodes (OLEDs),24 and later, as PCs.25,26 This is because TADF compounds have accessible triplet states owing to the small energy gap (ΔEST) between their lowest excited singlet (S1) and triplet (T1) states, which allows for relatively fast ISC and reverse ISC (RISC) to take place, despite the small SOC between these states in the absence of heavy atoms.27,28 Molecules with small ΔEST are those where the donor (D) and acceptor (A) moieties are sufficiently electronically decoupled such that there is minimal orbital overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), i.e., the exchange energy is small. This spatial separation also produces an emissive excited state having charge transfer (CT) character. The most widely employed molecular design is one that adopts a strongly twisted D–A structure, exemplified in 4CzIPN and 4DPAIPN (Fig. S2).26 Owing to the distance between D and A units, the emissive S1 state possesses long-range charge transfer (LRCT) character.29 D–A TADF PCs based on carbazoyl dicyanobenzenes (CDCBs) have been extensively employed in myriad photocatalytic reactions. Examples from this family of TADF PCs cover a broad range of ET. For example, the ET values of 4DPAIPN, 4CzTPN, 2CzIPN, 4CzIPN, and 3,5-2CzBN increase from 2.30 eV30 to 2.34,31 2.72,31 2.73,32 and 3.03 eV,31 respectively. These values overlap with those of, for example, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (ET = 2.64 eV23) and benzophenone (ET = 3.00 eV33). Beyond CDCBs, other D–A TADF PEnT PCs include pDTCz-DPmS34 and DI-PF35 (Fig. S2). pDTCz-DPmS (ET = 2.93 eV) possesses a suitable ET to photocatalyze the E/Z isomerization of diisopropyl fumarate (ET = 2.7 eV),36 producing 81% of diisopropyl maleate while 4CzIPN only yields 6%, owing to its lower ET of 2.68 eV.37DI-PF (ET = 2.38 eV) yielded 66% Z-stilbene in the E/Z isomerization of E-stilbene (ET = 2.2 eV) despite its lower triplet energy, while 4CzIPN affords 87% of the desired product.35 Another class of TADF PCs contains an imidazo-phenothiazine (IPTZ) acceptor unit connected with different donor groups. The PCs ACR-IMAC, ACR-IPTZ, and SACR-IPTZ have similar triplet energies of ET = 2.76 eV,38 2.76 eV,39 and 2.77 eV,39 respectively. Their use in a range of reactions, such as the [2 + 2] cycloaddition of an indole and dimethylphenylvinylsilane, afforded near quantitative product yields.38,39 A second class of TADF compounds is the so-called multi-resonant TADF (MR-TADF) emitters. These are rigid polycyclic aromatic hydrocarbons that are typically doped with both electron-rich and electron-deficient groups. Suitable regiochemistry of these p- and n-dopants produces an alternating pattern of increasing and decreasing electron density in the excited state as compared to the ground state, leading to the necessary small exchange energy that turns on TADF. Given the short distance between D and A motifs, the emissive excited state is of short-range CT (SRCT) character.40 Compared to D–A TADF compounds, MR-TADF emitters have more intense low energy absorption bands, narrower emission spectra, smaller Stokes shifts, and their emission spectra show only minimal positive solvatochromism—all potentially attractive properties for their use as PCs.40 Our group has demonstrated the broad utility of two families of MR-TADF PCs in a range of photoinduced electron transfer (PET) and PEnT reactions.41,42 These two families contain nitrogen donor atoms and either boron or carbonyl groups as n-dopants within the polycyclic aromatic hydrocarbon framework.
We, as well as Lee and co-workers, recently showed that the acceptor moiety is not required in MR-TADF emitter design for OLEDs.43–47 Indeed, when the nitrogen atoms in diindolocarbazoles are para-disposed, there is very weak TADF as ΔEST is moderately large, resulting in slow kRISC. DiICztBu4 and DiICzMes4 have similar ET of 2.55 (in DCM)43 and 2.57 eV (in toluene),44 respectively, while their reported ΔEST values are 0.29 and 0.26 eV. Recognizing their potential as PCs, herein we investigated a series of four structurally related DiICz-based TADF PCs, DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, in DET reactions (Fig. 2); the first two have previously been reported, while DiICztBuCz4 and DiICztBuDPA4 were, coincident with this work, recently reported by Lee and co-workers.48 Across the first three compounds in this series, there is a progressive stabilization of the S1 state, while the T1 energy is largely unaffected; both S1 and T1 are stabilized in DiICztBuDPA4. Thus, the impact of ΔEST on the performance in DET is probed for the first time. While we did not observe a dependency of the efficiency of the PEnT reactions as a function of the ΔEST of the PC across five different PEnT reactions, four involving direct DET to the substrate and one nickel-sensitized dual photocatalysis reaction, we did discover that these PCs initiate fast PEnT reactions that show remarkably little sensitivity to the presence of O2, despite it being a competitive triplet quencher. In the four PEnT reactions with direct energy transfer to the substrate, DiICztBu4, DiICzMes4, and DiICztBuCz4 consistently outperform 4CzIPN both in terms of NMR yield and reaction rate.
 | ||
| Fig. 2 Chemical structures of DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4. | ||
Results and discussion
The syntheses of DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4 are detailed in the SI. The UV-vis absorption spectra of these compounds in DCM, along with that of the reference PC 4CzIPN, are shown in Fig. 3a. DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4 all exhibit low-energy absorption bands in the visible region that are more intense than that of 4CzIPN. The presence of the mesityl (Mes) groups in DiICzMes4 leads to a very slight red shift in the absorption spectrum compared to DiICztBu4, while the substitution of the diindolocarbazole with progressively stronger carbazole (Cz) and diarylamine donors results in a larger red shift of these bands (Fig. 3a and S37). A similar trend is observed in their photoluminescence (PL) spectra (Fig. 3b), where the PL maximum in DCM shifts from λPL of 440 to 442, 466, and 532 nm for DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, respectively (Fig. 3b and Table 2). Their emission is more narrowband compared to the broad PL of 4CzIPN, reflecting their more rigid structures. | ||
| Fig. 3 (a) Absorption and (b) steady-state PL spectra in DCM of DiICztBu4 (λexc = 340 nm), DiICzMes4 (λexc = 380 nm), DiICztBuCz4 (λexc = 340 nm), DiICztBuDPA4 (λexc = 400 nm), and 4CzIPN (λexc = 378 nm). | ||
The S1 and T1 energies were determined from the onsets of the steady-state PL and delayed emission (gate time: 1–9 ms) spectra in 2-MeTHF glass at 77 K, respectively (Fig. S40); the ΔEST value is the difference in energy between these two (Fig. S40 and Table 1). DiICztBu4, DiICzMes4, and DiICztBuCz4 have very similar ET of 2.58, 2.57, and 2.55 eV, respectively. As their emission profiles are similar, so too will be their spectral overlap with the sub, and thus the kDET should be comparable across these three PCs. The S1 energy, ES, however, progressively decreases from 2.91 to 2.84 and 2.77 eV (measured at 77 K in 2-MeTHF); thus, the ΔEST likewise narrows from 0.33 to 0.27 and 0.22 eV. The ES and ET for DiICztBuDPA4 are 2.54 and 2.41 eV, resulting in the smallest ΔEST of 0.13 eV. The ET values determined at cryogenic temperatures of these four derivatives are all lower than the 2.73 eV of 4CzIPN (Fig. S40e). DiICztBu4 and DiICzMes4 have previously been reported as MR-TADF emitters, and given the similar photophysical properties of DiICztBuCz4 and DiICztBuDPA4 to these two, the high molar absorptivity of the low-energy absorption band, the minimal positive PL solvatochromism, the narrowband emission and the small ΔEST, they can also be classified as MR-TADF (Table 2).
| Compound | 77 K valuesa | Room temperature ETb/eV | |||||
|---|---|---|---|---|---|---|---|
| E S/eV | E T/eV | ΔEST/eV | Toluene | EtOAc | DCM | DMF | |
| a 77 K values were measured in 2-MeTHF glass. ES was determined from the onset of the steady-state PL and ET from the onset of the gated emission acquired in a time window of 1–9 ms after excitation. ΔEST = ES − ET. λexc = 390 nm for DiICztBu4, DiICzMes4, and DiICztBuCz; λexc = 400 nm for DiICztBuDPA4 and λexc = 380 nm for 4CzIPN. b Room temperature ET values were estimated following our previously reported methodology,49 where ET(RT) = ES(RT) − ΔEST(LT), with ES(RT) being the onset of the steady-state PL in the respective solvents and ΔEST(LT) being measured in 2-MeTHF at 77 K. | |||||||
| DiICztBu4 | 2.91 | 2.58 | 0.33 | 2.60 | 2.63 | 2.60 | 2.61 |
| DiICzMes4 | 2.84 | 2.57 | 0.27 | 2.64 | 2.66 | 2.65 | 2.65 |
| DiICztBuCz4 | 2.77 | 2.55 | 0.22 | 2.58 | 2.61 | 2.58 | 2.58 |
| DiICztBuDPA4 | 2.54 | 2.41 | 0.13 | 2.44 | 2.45 | 2.40 | 2.46 |
| 4CzIPN | 2.77 | 2.73 | 0.04 | 2.67 | 2.66 | 2.56 | 2.54 |
| Compound | λ abs /nm (ε/103 M−1 cm−1) | λ PL /nm | E S /eV | E T /eV | ΔEST | τ PL /ns |
|---|---|---|---|---|---|---|
| a In DCM at 77 K, taken from the onset of steady-state PL spectrum for ES and the onset of the gated emission spectrum (1–9 ms) for ET. b In 2-MeTHF at 77 K, taken from the onset of steady-state PL spectrum for ES and the onset of the gated emission spectrum (1–9 ms) for ET. c Under degassed conditions, excited-state lifetimes in air in parentheses. d Lifetime is the average lifetime of 4CzIPN calculated as τavg = τ1 × w1 + τ2 × w2. | ||||||
| DiICztBu4 | 305 (58), 315 (67), 343 (29), 362 (54), 408 (13), 427 (16) | 440 | 2.91 | 2.58 | 0.33 | 9 (8) |
| DiICzMes4 | 300 (84), 306 (88), 315 (81), 344 (29), 364 (53), 409 (13), 430 (16) | 442 | 2.84 | 2.57 | 0.27 | 10 (8) |
| DiICztBuCz4 | 319 (152), 347 s (84), 385 s (20), 420 s (17), 444 (25) | 466 | 2.77 | 2.55 | 0.22 | 10 (8) |
| DiICztBuDPA4 | 305 (141), 343 (104), 411 (7), 485 (14) | 523 | 2.54 | 2.41 | 0.13 | 16 (13) |
| 4CzIPN | 313 (14), 325 (16), 377 (18), 450 s (7) | 544 | 2.77 | 2.73 | 0.04 | 1896 (218)d |
| fac-Ir(ppy)3 | 344 (10), 378 (13), 409 (8), 455 (3) | 520 | — | 2.58 | — | 1391 (54) |
While the low temperature (LT) measurements, at 77 K, permit a robust estimation of ΔEST, noting that in the vast majority of TADF compounds the S1 state has greater CT character than T1, this measurement will not accurately capture ES at room temperature (RT) in solution. This is because CT states are stabilized as a function of solvent polarity, and the greater the CT character, the stronger the stabilization of the state.50 As triplet states tend to show lesser CT character than singlets, the estimated ET = ES(RT) − ΔEST(LT) represents the outer bound value for what ET may be at room temperature. Thus, given the LRCT character of the S1 state of 4CzIPN, the change in ET at room temperature is more pronounced than for the four DiICz MR-TADF compounds (Table 1). This implies that in DET reactions, the spectral overlap between the PC* and the sub may change significantly, especially when the T1 state possesses some CT character. For MR-TADF compounds, this stabilization effect is small given the SRCT character of the S1 and T1 states. So, at low temperature, the ET values are 2.58, 2.57, 2.55, and 2.41 eV in 2-MeTHF for DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, respectively, while in DCM at room temperature, the ET values remain essentially the same at 2.60, 2.65, 2.58, and 2.40 eV, respectively (Table 1). This contrasts with the effect on the T1 energy of 4CzIPN (ET(LT) = 2.73 eV vs. ET(RT) = 2.56 eV; where LT is low temperature, 77 K, and RT is room temperature). This is illustrated in a comparison between the T1 energies in 2-MeTHF glass and DCM solutions, ΔET–T, which is 0.08 eV for DiICzMes4 and 0.17 eV for 4CzIPN. Thus, these four MR-TADF compounds maintain their ET in polar solvents, while for 4CzIPN, the ET is much more stabilized. The time-resolved PL decays of DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4 in DCM are shown in Fig. S40 and S41 and are listed in Table 2. There is no delayed emission, and PL lifetimes range from 9–16 ns. Given the moderately large ΔEST, it is not uncommon for MR-TADF emitters to not show delayed emission in solution as non-radiative decay competes with RISC, while in the solid state, this is largely suppressed, and delayed emission becomes apparent.
DET reactions
We began by studying intramolecular DET reactions with photocatalysts of increasing triplet energy to focus on the interaction between the PC and a single molecule. We first investigated the E/Z isomerization of an alkene, E-cinnamyl acetate, with a T1 energy similar to those of the MR-TADF emitters; notably, the structurally similar E-methyl cinnamate has a ET = 2.38 eV51 (Table S7). We expect the triplet energy of E-cinnamyl acetate, which contains a methylene group between the styrenyl moiety and the ester group, to be slightly higher compared to E-methyl cinnamate, which has a larger conjugation length. The geometric isomerization does not take place in the absence of an irradiated PC (Table 3, entries 1 and 2). After 24 h of irradiation under N2, the use of DiICztBu4, DiICzMes4, and DiICztBuCz4 as PCs yielded comparable E/Z ratios of 18/82, 19/81, and 19/81, respectively (Table 3, entries 4, 6, and 8), performing as well in this reaction as 4CzIPN (E/Z ratio of 14/86, Table 3, entry 12). Similar E/Z ratios are observed under air (E/Z = 19/81, 17/84, 20/80, and 15/85 for DiICztBu4, DiICzMes4, DiICztBuCz4, and 4CzIPN, respectively, Table 3, entries 3, 5, 7, and 11). Amazingly, the end ratio obtained for this DET reaction is unaffected despite O2 being a competitive triplet quencher. This is, however, not the case with DiICztBuDPA4, which under N2 yielded an E/Z ratio of 28/72, a lower ratio than those obtained with the other PCs, while there is effectively no isomerization observed in air (Table 3, entries 9 and 10).
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Conditions | E/Z ratio |
| a Cinnamyl acetate (0.2 mmol) in DCM (0.2 M) with 1 mol% PC loading. E/Z ratios were determined via1H NMR spectroscopy. Reported yields are the mean from at least two reactions with associated standard deviations. | |||
| 1 | None | Air | 99/1 ± 0 |
| 2 | None | N2 | 99/1 ± 0 |
| 3 | DiICztBu4 | Air | 19/81 ± 0 |
| 4 | DiICztBu4 | N2 | 18/82 ± 0 |
| 5 | DiICzMes4 | Air | 16/84 ± 1 |
| 6 | DiICzMes4 | N2 | 19/81 ± 0 |
| 7 | DiICztBuCz4 | Air | 20/80 ± 0 |
| 8 | DiICztBuCz4 | N2 | 19/81 ± 0 |
| 9 | DiICztBuDPA4 | Air | 98/2 ± 0 |
| 10 | DiICztBuDPA4 | N2 | 28/72 ± 0 |
| 11 | 4CzIPN | Air | 15/85 ± 0 |
| 12 | 4CzIPN | N2 | 14/86 ± 0 |
Fig. 4 shows the reaction progression for the E/Z isomerization of cinnamyl acetate in air at short times, while for DiICztBuDPA4, the reaction under N2 is shown. The conversion saturates after 30 min for DiICztBu4, DiICzMes4, and DiICztBuCz4, with these PCs having similar reaction rates. There is no correlation observed between ΔEST and either the E/Z ratio or reaction rate for this transformation. The absence of any substantive change in the post-reaction absorption spectra of DiICztBu4, DiICzMes4, and DiICztBuCz4 revealed that these compounds are photochemically stable to O2 under the reaction conditions (Fig. S46a–c). Given that O2 does not affect the E/Z ratio, we investigated the effect on the reaction kinetics as a function of the presence/absence of O2 by comparing the E/Z ratios after 5 min. The reaction rates slightly increase in the absence of O2, leading to slightly improved E/Z ratios of 48/52, 51/49, and 49/51 for DiICztBu4, DiICzMes4, and DiICztBuCz4, respectively, from those in air (53/47, 61/39, and 56/44, respectively); it is not clear why the change is more dramatic with DiICzMes4. Hence, O2 acts as an ineffective yet competitive quencher of the triplet excited state of these PCs, as the change in the E/Z ratio at early times is marginal. The contrasting results with DiICztBuDPA4 are discussed in the SI.
 | ||
| Fig. 4 Reaction progression for the E/Z isomerization of cinnamyl acetate in air unless otherwise noted. E/Z ratios are given in % Z isomer after x minutes (λexc = 440 nm). Cinnamyl acetate (0.2 mmol) in DCM (0.2 M) and PC (1 mol%). E/Z ratios were determined by 1H NMR spectroscopy. Reactions with DiICztBuDPA4 as the PC were performed under N2. | ||
The isomerization reaction with 4CzIPN is significantly slower than with DiICztBu4, DiICzMes4, and DiICztBuCz4, only producing a 39/61 E/Z ratio after 60 min (Fig. 4). Thus, despite similar thermodynamic driving forces (i.e., similar ET), the origin of the higher yields of the Z isomer and faster reaction kinetics could be due to either the higher molar absorptivity of these three MR-TADF compounds at the photoexcitation wavelength and/or an increased spectral overlap of the phosphorescence of the MR-TADF emitters with the spin-forbidden absorption of the sub. 4CzIPN is mostly photostable; however, a slight decrease in the absorbance of the CT band is observed in air, suggesting that some degradation is taking place if the reaction is carried out when O2 is present (Fig. S46e). Furthermore, there is a significant dependency of the kinetics of the reaction with respect to the presence/absence of O2. After 5 min, the E/Z ratios are 80/20 and 94/6 under N2 and air, respectively. These results reveal that 4CzIPN photocatalyzes this reaction much more slowly than DiICztBu4, DiICzMes4, and DiICztBuCz4 and that its triplet excited state is also more strongly quenched by O2. The excited-state lifetimes of the PCs under aerated conditions are 8, 8, 8, and 13 ns for DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, respectively. These are not affected by the presence of E-cinnamyl acetate (τPL of 8, 8, 8, and 12 ns for DiICztBu4, DiICzMes4, DiICztBuCz4, and DiICztBuDPA4, respectively, Fig. S47).
We had previously assessed MR-TADF compounds DABNA-1, tDABNA, CzBN, and DtBuCzB as photocatalysts in the E/Z isomerization of cinnamyl acetate. Of these, CzBN produced the highest conversion to the Z isomer in a ratio of 19/81 after two hours in air (in THF); the ratio remained the same after 24 hours.42 In our previous study, we did not monitor the reaction kinetics at shorter times. Thus, CzBN yields a similar E/Z ratio as the DiICz photocatalysts (19/81 for CzBN after 2 h and 18/82 for DiICztBu4 after 1 h). This is consistent with their comparable ET values (2.58 eV for CzBN and 2.58 eV for DiICztBu4, respectively, in 2-MeTHF at 77 K).42
We next investigated the E/Z isomerization of diisopropyl fumarate, which has a higher ET of 2.7 eV,31 to produce the corresponding maleate (ET = 3.1 eV)31 (Table S8). Despite the ETs of the PCs being lower than 2.7 eV (Table 2), both DiICztBu4 (ET = 2.60 in DCM) and DiICzMes4 (ET = 2.65 in DCM) photocatalyzed the reaction, producing identical E/Z ratios of 13/87 after 24 h (Table 4, entries 4 and 6). DiICztBuCz4 and 4CzIPN were less effective, producing E/Z ratios of 66/34 and 69/31, respectively (Table 4, entries 8 and 12). Surprisingly, it seems that the slightly lower ET of DiICztBuCz4 (2.58 eV in DCM) compared to DiICzMes4 (ET = 2.65 eV in DCM) and DiICztBu4 (ET = 2.60 eV in DCM) is responsible for these large changes in E/Z ratios; notably, DiICztBuCz4 is unstable under the reaction conditions (Fig. S46c). These results also clearly illustrate the effect of solvent polarity on the magnitude of the spectral overlap between the PC and sub, and the corresponding efficiency to photocatalyze the isomerization. Despite 4CzIPN having the highest ET of 2.73 eV in 2-MeTHF at 77 K, meaning that if these values were reflective of accessible triplet energies under the reaction conditions, then it should be able to photocatalyze the isomerization to a similar extent as the DiICz PCs. The triplet state of 4CzIPN, however, shows the greatest stabilization as a function of solvent polarity (ET in DCM is 2.56 eV at room temperature, Table 1). This accounts for the poorer E/Z ratio of 69/31 (Table 4, entry 12). Notably, as 4CzIPN is photostable under these reaction conditions when performed under N2, its poor performance cannot be attributed to PC degradation (Fig. S45e).
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Conditions | E/Z ratio |
| a Diisopropyl fumarate (0.2 mmol) in DCM (0.2 M) with 1 mol% PC loading. E/Z ratios were determined by 1H NMR spectroscopy. Reported yields are the mean from at least two reactions with associated standard deviations. | |||
| 1 | None | Air | 100/0 ± 0 |
| 2 | None | N2 | 100/0 ± 0 |
| 3 | DiICztBu4 | Air | 95/5 ± 1 |
| 4 | DiICztBu4 | N2 | 13/87 ± 0 |
| 5 | DiICzMes4 | Air | 92/8 ± 2 |
| 6 | DiICzMes4 | N2 | 13/87 ± 0 |
| 7 | DiICztBuCz4 | Air | 88/12 ± 0 |
| 8 | DiICztBuCz4 | N2 | 66/34 ± 4 |
| 9 | DiICztBuDPA4 | Air | 100/0 ± 0 |
| 10 | DiICztBuDPA4 | N2 | 100/0 ± 0 |
| 11 | 4CzIPN | Air | 91/9 ± 4 |
| 12 | 4CzIPN | N2 | 69/31 ± 4 |
In contrast to cinnamyl acetate (Table 3), the E/Z isomerization of diisopropyl fumarate essentially does not proceed to any appreciable extent in the presence of O2, with E/Z ratios of 96/5, 93/8, 88/12, and 91/9 for DiICztBu4, DiICzMes4, DiICztBuCz4, and 4CzIPN, respectively (Table 4, entries 3, 5, 7, and 11). DiICztBuDPA4 could not photocatalyze the reaction (Table 4, entries 9 and 10), given its ET of 2.41 eV.
To understand the divergence in the behavior of these two isomerization reactions in the presence of O2, we interrogated the reaction rates of diisopropyl fumarate under N2 (Fig. 5). If the rates are slower compared to those with cinnamyl acetate, then the lower E/Z ratios may be explained by competitive O2 quenching of the T1 state to that of the fumarate. Unlike the reaction with cinnamyl acetate, the reaction rates under N2 differ between DiICzMes4, DiICztBu4, and DiICztBuCz4, but still are all faster than 4CzIPN. The fastest conversion to the Z isomer uses DiICzMes4, yielding an E/Z ratio of 22/79 ± 3 after 60 min (13:87 after 24 h, Table 4, entry 6), while DiICztBu4 only produces an E/Z ratio of 60/41 ± 1 after 60 min, yet affords the same E/Z ratio after 24 h as DiICzMes4 (13:87 after 24 h, Table 4, entry 4). The reaction reaches its steady-state E/Z ratio after 2 h for DiICzMes4 (E/Z ratio of 12/88), while for DiICztBu4 it takes 4 h to reach the 13/87 E/Z ratio. These rates are slower than those with cinnamyl acetate, where the reaction was completed after 30 min. Given that both DiICzMes4 and DiICztBu4 are stable during the reaction when conducted under N2, photodegradation of the PC can be excluded as the origin of the slower reaction kinetics observed for DiICztBu4. As the phosphorescence of both DiICztBu4 and DiICzMes4 is the same, the spectral overlap between the phosphorescence spectra of these two PCs and the spin-forbidden absorption spectrum of the sub will therefore be similar. A plausible conclusion is that the tert-butyl groups are bulkier than mesityl groups and impede the collisional interaction more, thus adversely affecting the reaction kinetics with DiICztBu4. We observed that these two PCs do photodegrade when the reaction is carried out under air. Thus, there is a divergence in the photochemical outcome wherein the kinetics of the E/Z isomerization of cinnamyl acetate outcompetes photodegradation, while this is not the case with the E/Z isomerization of diisopropyl fumarate (Fig. S45a and b). Surprisingly, while the initial rate of isomerization using DiICztBuCz4 is comparatively as fast as using DiICzMes4, there is a significant off-cycle photodegradation both in air and N2 that effectively caps the E/Z ratio at only 66/34 ± 4 (Fig. S45c). DiICztBuDPA4 is photostable when the reaction is conducted under N2, while there are changes in the absorption spectrum when the reaction is conducted in air (Fig. S46d). Hence, the fact that the reaction does not proceed is a consequence of there being no spectral overlap between the phosphorescence of DiICztBuDPA4 and the spin-forbidden absorption of the substrate.
 | ||
| Fig. 5 Reaction progression for the E/Z isomerization of diisopropyl fumarate under N2. E/Z ratios are given in % Z isomer after x minutes irradiated at 440 nm. Diisopropyl fumarate (0.2 mmol) in DCM (0.2 M) and PC (1 mol%). E/Z ratios were determined by 1H NMR spectroscopy. | ||
4CzIPN is significantly slower and less efficient in this isomerization reaction, producing a steady-state E/Z ratio of only 69/31 ± 4 after 24 h (Table 4, entry 12); indeed, after 4 h, there is only an E/Z ratio of 95/5 ± 1 (Table S8). Under these conditions, 4CzIPN is photostable under N2; however, in air, photodegradation of 4CzIPN is observed (Fig. S46e). Thus, the poor performance of 4CzIPN can be explained by the too-small spectral overlap with the fumarate substrate.
The E/Z isomerization of diisopropyl fumarate studies demonstrate that three of these PCs can catalyze PEnT reactions with substrates having ET of 2.7 eV.31 Thus, we next explored the intramolecular [2 + 2] cycloaddition of norbornadiene (ET = 2.7 eV52) to quadricyclane (Table S9). Schmid et al. had reported a 99% yield after one hour using an anionic Ir complex having an ET of 2.99 eV (Table 5, entry 14).
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Conditions | Yield/% |
| a Norbornadiene (0.2 mmol) and PC (1 mol%) in DCM (2 mL). Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. b From ref. 52 in CD3CN, λexc = 415 nm after 1 h, norbornadiene (0.05 mmol), PC (0.3 mol%), CD3CN (0.6 mL). Reported yields are the mean from at least two reactions with associated standard deviations. | |||
| 1 | None | Air | 0 ± 0 |
| 2 | None | N2 | 0 ± 0 |
| 3 | DiICztBu4 | Air | 64 ± 4 |
| 4 | DiICztBu4 | N2, 2 h | 87 ± 1 |
| 5 | DiICztBu4 | N2 | 89 ± 2 |
| 6 | DiICzMes4 | Air | 71 ± 6 |
| 7 | DiICzMes4 | N2 | 85 ± 5 |
| 8 | DiICztBuCz4 | Air | 75 ± 2 |
| 9 | DiICztBuCz4 | N2 | 88 ± 0 |
| 10 | DiICztBuDPA4 | Air | 0 ± 0 |
| 11 | DiICztBuDPA4 | N2 | 0 ± 0 |
| 12 | 4CzIPN | Air | 0 ± 0 |
| 13 | 4CzIPN | N2 | 0 ± 0 |
| 14 | [Ir(L)2(BCF)2]− | Degassed | 99b |
The reaction does not take place in the absence of a PC (Table 5, entries 1 and 2). After 30 min reaction time using DiICztBu4, quadricyclane was obtained in an 89% yield under N2 (Table 5, entry 5). In contrast to the E/Z isomerization of fumarate, DiICzMes4 and DiICztBuCz4 perform as well as DiICztBu4, yielding 85, 88, and 89%, respectively, after 30 min (Table 5, entries 5, 7, and 9). The reaction is slightly quenched in the presence of O2, producing yields of 64, 71, and 75% after 30 min when using DiICztBu4, DiICzMes4, and DiICztBuCz4, respectively (Table 5, entries 3, 6, and 8). Given the fast reaction rates, there is thus significantly less quenching of the 3PC* by O2 than what has been observed with diisopropyl fumarate. Again, it is not surprising that DiICztBuDPA4 does not photocatalyze the reaction, given its too low ET (Table 5, entries 10 and 11). Similarly, given that 4CzIPN did not photoisomerize diisopropyl fumarate to diisopropyl maleate after 30 min to any appreciable extent, this PC did not photocatalyze the [2 + 2] cycloaddition of norbornadiene within 30 min (Table 5, entries 12 and 13). These results not only demonstrate the value of three of these MR-TADF PCs to photocatalyze a demanding PEnT reaction but also that ET as a parameter is too coarse when cross-comparing different reactions using substrates with similar ET. We had previously demonstrated that DiKTa, another MR-TADF photocatalyst (ET = 2.62 eV),41 could efficiently photoisomerize diisopropyl fumarate into diisopropyl maleate in a ratio of 10/90 in MeCN over 16 h, a similar ratio to that using DiICzMes4 (8/92); in this earlier study, the reaction progression at shorter reaction times was not analysed.41
The photostability of the PCs was tested under the reaction conditions under N2 and in air (Fig. S51). The absorption spectra of DiICztBu4, DiICzMes4, and DiICztBuCz4 show only a minimal decrease in intensity under N2, while there are greater changes with O2 present that suggest more photodegradation, which is consistent with the less efficient performance when the reaction is carried out in air (Fig. S51a–c). Interestingly, the small degree of photodegradation of the MR-TADF PCs in the [2 + 2] cycloaddition in air contrasts with the significant photodegradation in the E/Z isomerization with diisopropyl fumarate in air and the associated absence of product formation, despite both substrates having the same reported ET.
There is significant photodegradation observed for 4CzIPN under N2, and the profile is similar to that in air (Fig. S51e). The absorption band at λabs = 377 nm and the shoulder at λabs = 450 nm disappear, while a more red-shifted, less intense band at λabs = 520 nm appears. These results imply that the substrate reacts with 4CzIPN and that the [2 + 2] cycloaddition is likely a radical stepwise process as opposed to a concerted mechanism (Table 5, entry 12 and 13).
We then explored a PEnT reaction with a substrate having a higher ET, the sigmatropic shift of (S)-verbenone (ET = 3.0 eV)52 to chrysanthenone (Table S10). This rearrangement has been used in the synthesis of the natural product xishacorene B, where verbenone was directly irradiated in the first step with UV light (λexc = 365 nm), yielding 67% chrysanthenone.53 Schmid et al. showed that this transformation can be photocatalyzed using the same iridium isocyanoborato complex as was used in the [2 + 2] cycloaddition of norbornadiene,52 whereupon irradiating the solution at 415 nm produced the desired product in 80% yield after 180 min (Table 6, entry 10). With DiICztBu4, DiICzMes4, and DiICztBuCz4 as the PCs, the yields were generally low after 24 h at 29, 22, and 18%, respectively, using 1 mol% PC (Table 6, entries 2, 4, and 6); evidently, DiICztBuDPA4 cannot photocatalyze this reaction due to its too low ET (Table 6, entry 8). The yields were improved by increasing the PC loading to 5 mol%, resulting in yields of 37, 38, and 21% for DiICztBu4, DiICzMes4, and DiICztBuCz4, respectively (Table 6, entries 3, 5, and 7). Using 4CzIPN afforded only 5% product yield at 5 mol% PC loading (Table 6, entry 9).
|
|
|||
|---|---|---|---|
| Entry | PC | Loading/mol% | Yield/% |
| a (S)-verbenone (0.2 mmol) in DCM (0.1 M) with given PC loadings. Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. b From ref. 52 in CD3CN with hv = 415 nm after 3 h. Reported yields are the mean from at least two reactions with associated standard deviations. | |||
| 1 | None | — | 0 ± 0 |
| 2 | DiICztBu4 | 1 | 29 ± 3 |
| 3 | DiICztBu4 | 5 | 37 ± 1 |
| 4 | DiICzMes4 | 1 | 22 ± 2 |
| 5 | DiICzMes4 | 5 | 38 ± 6 |
| 6 | DiICztBuCz4 | 1 | 18 ± 1 |
| 7 | DiICztBuCz4 | 5 | 21 ± 2 |
| 8 | DiICztBuDPA4 | 1 | 0 ± 0 |
| 9 | 4CzIPN | 5 | 5 ± 1 |
| 10 | [Ir(L)2(BCF)2]− | 0.3 | 80b |
There is essentially no change in the absorption profiles of DiICztBu4, DiICzMes4, and DiICztBuCz4 before and after irradiation, suggesting that any photodegradation of the PCs is not the cause for the low yields in this reaction but rather the too low triplet energy (Fig. S53a–c). 4CzIPN and DiICztBuDPA4 are stable under the reaction conditions (Fig. S53d and e), and the 5% yield for 4CzIPN and the 0% yield for DiICztBuDPA4 can be attributed to each having a T1 energy that is effectively too low to enable DET with any degree of efficiency.
Thus far, we have explored four intramolecular PEnT reactions. To expand the portfolio of available reactions, we next investigated an example of a bimolecular cross-coupling reaction. This is frequently achieved by combining the photocatalytic cycle with a Ni-mediated cross-coupling reaction. In such dual-catalyzed reactions, the PC can either be involved in a PET mechanism, a so-called metallaphotoredox reaction, or in a PEnT reaction to sensitize the Ni co-catalyst by DET after it has undergone oxidative addition and transmetallation steps. This photoactivation accelerates the reductive elimination of the product.31 One such cross-coupling reaction where the PC is purported to engage in DET is an esterification involving aryl halides being cross-coupled with carboxylic acids, where Welin et al. used fac-Ir(ppy)3 as the PC (ET(fac-Ir(ppy)3) = 2.58 eV).13 In this cross-coupling reaction, an undesired side product is lactone 2 (Table 7). Several groups have employed organic PCs such as 4DPAPN or SACR-IPTZ for these reactions (Fig. S1).31,394DPAPN, after optimization of the reaction conditions, yielded 91% of 4-(trifluoromethyl)phenyl benzoate in the cross-coupling of benzoic acid with 4-bromobenzotrifluoride as substrates.31 With the same substrates but under slightly different conditions, Welin et al. reported an 86% yield using fac-Ir(ppy)3 as the PC. The reaction photocatalyzed using SACR-IPTZ yielded 99% of the coupled product between 5-bromophthalide and benzoic acid, and no protodehalogenated product was observed.39
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Conditions | Yield 1/% | Yield 2/% |
| a 5-Bromophthalide (0.188 mmol, 1.0 equiv.), benzoic acid (0.301 mmol, 1.6 equiv.), 2,2,6,6-tetramethylpiperidine (TMP) (0.375 mmol, 2.0 equiv.), Ni source (0.011 mmol, 6 mol%), 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) (0.013 mmol, 7 mol%) and PC (0.004 mmol, 2 mol%) in DCM (0.09 M). Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. | ||||
| 1 | None | Ni(COD)2 | 10 ± 1 | 11 ± 2 |
| 2 | None | NiBr2·glyme | 0 ± 0 | 0 ± 0 |
| 3 | DiICztBu4 | Ni(COD)2 | 59 ± 2 | 28 ± 0 |
| 4 | DiICztBu4 | NiBr2·glyme | 31 ± 4 | 15 ± 1 |
| 5 | DiICzMes4 | Ni(COD)2 | 65 ± 4 | 28 ± 3 |
| 6 | DiICzMes4 | NiBr2·glyme | 41 ± 0 | 19 ± 1 |
| 7 | DiICzMes4 | [Ni(dtbbpy)(OH2)4]Cl2 | 68 ± 1 | 18 ± 0 |
| 8 | DiICztBuCz4 | Ni(COD)2 | 60 ± 6 | 31 ± 2 |
| 9 | DiICztBuCz4 | NiBr2·glyme | 20 ± 1 | 20 ± 1 |
| 10 | DiICztBuDPA4 | Ni(COD)2 | 47 ± 5 | 14 ± 2 |
| 11 | DiICztBuDPA4 | NiBr2·glyme | 27 ± 3 | 11 ± 0 |
| 12 | 4CzIPN | Ni(COD)2 | 43 ± 2 | 36 ± 4 |
| 13 | 4CzIPN | NiBr2·glyme | 31 ± 0 | 19 ± 0 |
| 14 | fac-Ir(ppy)3 | Ni(COD)2 | 65 ± 1 | 15 ± 1 |
| 15 | fac-Ir(ppy)3 | NiBr2·glyme | 49 ± 5 | 16 ± 1 |
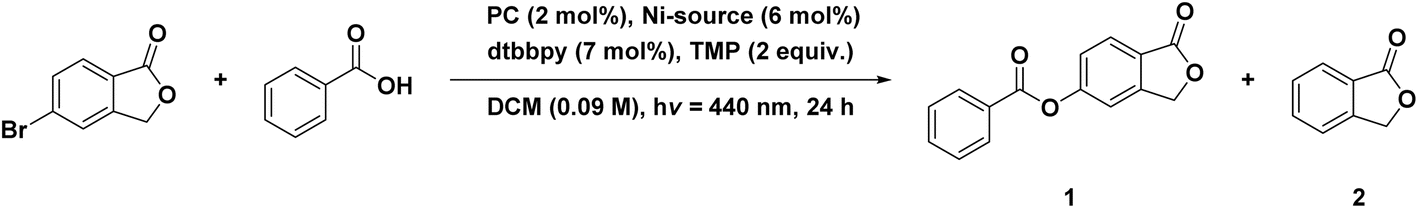
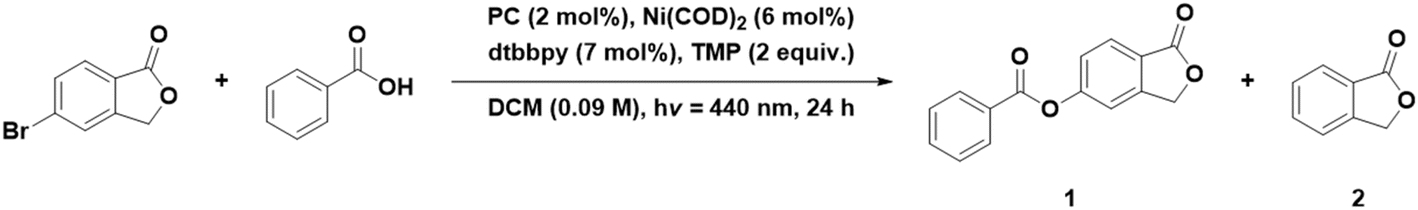
We first investigated the use of different Ni precursors in the presence of dtbbpy as an ancillary ligand under conditions similar to those reported by Hojo et al.39 We changed the solvent from DMF to DCM, as the DiICz PCs are more soluble in the latter.39 We observe a generally higher product yield in DCM when using Ni(COD)2 than with NiBr2·glyme, regardless of the choice of PC. The use of DiICzMes4 yielded 65% in combination with Ni(COD)2, while using NiBr2·glyme only results in a 41% yield (Table 7, entries 5 and 6). In both cases, side product 2 is observed in 30 and 19% yield (Table 7, entries 5 and 6). DiICztBu4, DiICzMes4, and DiICztBuCz4 perform similarly in combination with Ni(COD)2 with yields of 59, 65, and 60% of 1, respectively, while 2 formed in 28, 28, and 31% yield, respectively (Table 7, entries 3, 5, and 8). Changing the Ni source to NiBr2·glyme yielded 31/15 and 41/19% of 1 and 2 using DiICztBu4 and DiICzMes4, respectively, implying that the use of NiBr2·glyme results in more undesired protodehalogenated product for these two complexes, while with DiICztBuCz4, the reaction proceeds less readily, affording 20% of each of 1 and 2. These PCs produce higher yields of 1 in combination with Ni(COD)2 than using 4CzIPN (43%, Table 7, entry 12), and this is correlated to a greater amount of 2 forming (36%) with this latter PC. When fac-Ir(ppy)3 is employed as the PC in combination with Ni(COD)2, 65% of 1 and 15% of 2 form (Table 7, entry 14). A comparable yield is observed with DiICzMes4 (68/18% of 1 and 2, Table 7, entry 7), but the Ni source must be [Ni(dtbbpy)(OH2)4]Cl2, implying that the kinetics of the formation of the active Ni species are suboptimally aligned with the kinetics of DET with this PC. After solvent optimization with the best performing DiICz PC, DiICzMes4, the coupled product 1 was obtained in a 74% yield when using a 1:1 mixture of DMSO:toluene (Table 8, entry 4), while the control experiment with fac-Ir(ppy)3 yielded a comparable yield of 77% (Table 8, entry 6). The 1:1 DMSO:toluene solvent system had previously been shown to be optimal with SACR-IPTZ as the PC, yielding 99% of 1 (Table 8, entry 8).39 The reaction does not readily proceed in toluene, yielding only 10% product, while in DMSO an increase in the yield (64% of 1) compared to that in DCM is observed, ostensibly due to a partial suppression of the formation of 2 (Table 8, entry 1–3). Using 1:1 DMSO:toluene in combination with the preformed Ni complex [Ni(dtbbpy)(OH2)4]Cl2 and DiICzMes4 as the PC resulted in 81% yield of 1 and only 13% of 2 (Table 8, entry 7). Reducing the reaction time from 24 to 3 h resulted in a lower yield of 1 (54%) but not of 2 (13%) (Table 8, entry 5), while with fac-Ir(ppy)3 as the PC the product was obtained in 68% yield after 3 h (Table 8, entry 7). While the reaction with fac-Ir(ppy)3 is slightly faster than with DiICzMes4, effectively similar yields of 74 and 77% were obtained after 24 h using DiICzMes4 and fac-Ir(ppy)3, respectively.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | PC | Conditions | Yield 1/% | Yield 2/% |
| a 5-Bromophthalide (0.188 mmol, 1.0 equiv.), benzoic acid (0.301 mmol, 1.6 equiv.), TMP (0.375 mmol, 2.0 equiv.), Ni-source (0.011 mmol, 6 mol%), dtbbpy (0.013 mmol, 7 mol%) and PC (0.004 mmol, 2 mol%) in the respective solvent (0.09 M). Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. b Literature yield taken from ref. 39 5-bromophthalide (0.188 mmol, 1.0 equiv.), benzoic acid (0.301 mmol, 1.6 equiv.), TMP (0.375 mmol, 2.0 equiv.), Ni-source (0.011 mmol, 6 mol%), dtbbpy (0.013 mmol, 7 mol%) and PC (0.004 mmol, 2 mol%) in the respective solvent (0.09 M), λexc = 400 nm. | |||||
| 1 | DCM | DiICzMes4 | Ni(COD)2 | 60 ± 3 | 30 ± 1 |
| 2 | Toluene | DiICzMes4 | Ni(COD)2 | 10 ± 3 | — |
| 3 | DMSO | DiICzMes4 | Ni(COD)2 | 64 ± 5 | 21 ± 1 |
| 4 | DMSO:toluene 1:1 |
DiICzMes4 | Ni(COD)2 | 74 ± 2 | 15 ± 1 |
| 5 | DMSO:toluene 1:1 |
DiICzMes4 | Ni(COD)2, 3 h | 54 ± 0 | 13 ± 0 |
| 6 | DMSO:toluene 1:1 |
fac-Ir(ppy)3 | Ni(COD)2 | 77 ± 1 | 15 ± 0 |
| 7 | DMSO:toluene 1:1 |
fac-Ir(ppy)3 | Ni(COD)2, 3 h | 68 ± 2 | 16 ± 2 |
| 8 | DMSO:toluene 1:1 |
DiICzMes4 | [Ni(dtbbpy)(H2O)4]Cl2 | 81 ± 1 | 13 ± 1 |
| 9 | DMSO:toluene 1:1 |
SACR-IPTZ | Ni(COD)2 | 99 | — |
Conclusion
We have investigated four DiICz emitters as fast and efficient DET photocatalysts in five different energy transfer reactions, highlighting their ability to activate high triplet energy substrates and outperform literature reference PCs. There is scant solvent dependency of the T1 state energies of these four MR-TADF PCs, unlike D–A TADF PCs such as 4CzIPN. Surprisingly, we observed a dependency of the sensitivity of O2 quenching as a competitive triplet quencher as a function of the nature of the PEnT reaction, even when the ET of the substrate was the same. Reactions that proceed sufficiently rapidly with these PCs show little to no O2 dependency on the final yield, which is a benefit of these compounds as photocatalysts. This suggests that the quenching of the excited state by O2 is not competitive with the DET kinetics to the substrate. Excitingly, DET to substrates with triplet energies as high as 3.0 eV is feasible with DiICztBu4 and DiICzMes4, both of which catalyze the sigmatropic shift of (S)-verbenone, yielding the product in around 38% yield. We concluded that there is no connection between either the yield or the reaction rate and the ΔEST of the PC. This study reveals the particularly valuable and wide utility of DiCzMes4 as a PEnT photocatalyst for substrates possessing high triplet energies.Author contributions
E. Z.-C. conceived and managed the project and supervised the work. D. H. synthesized DiICzMes4 and DiICztBuCz4, E. B. synthesized DiICztBuDPA4 and L. H. synthesized DiICztBu4. D. H. performed the steady-state emission and absorption measurements in toluene, T. H. measured the photoluminescence quantum yields of the four photocatalysts, and the emission lifetimes of the photocatalysts in presence of cinnamyl acetate, and L. H. performed steady-state emission and absorption measurements in DCM, DMF and EtOAc, the time-resolved photoluminescence measurements in DCM, and the determination of ΔEST in 2-MeTHF. L. H. carried out photocatalysis reactions and photostability studies. L. H., D. H. and E. Z.-C. contributed to the manuscript writing and discussion.Conflicts of interest
The authors declare no competing interests.Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/22609279-98d6-4d3c-a1c1-b1f14b04b22d. Supplementary information: synthetic procedures, UV-vis absorption and photoluminescence spectra (room temperature steady-state, 77 K steady-state, and 77 K gated emission), time-resolved PL decays, photoluminescence quantum yield data, photocatalysis procedures, and photostability studies. See DOI: https://doi.org/10.1039/d5sc04014k.Acknowledgements
We thank the European Union H2020 research and innovation program for the funding under the Marie S. Curie Grant Agreement (PhotoReAct, No. 956324). We thank Geena Zoubarev for the contribution to the initial photocatalytic assessments. We thank Dr Megan Bryden, Francis Millward, and Violaine Manet for providing samples of 4CzIPN and fac-Ir(ppy)3. We thank EPSRC for financial support (EP/W522259/1, EP/W007517/1 and EP/M02105X/1).References
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Energy transfer photocatalysis: exciting modes of reactivity, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- M. Tabachnyk, B. Ehrler, S. Gelinas, M. L. Bohm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Resonant energy transfer of triplet excitons from pentacene to PbSe nanocrystals, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy transfer catalysis mediated by visible light: principles, applications, directions, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- J. B. Metternich and R. Gilmour, A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef CAS PubMed.
- B. Kweon, L. Blank, J. Soika, A. Messara, C. G. Daniliuc and R. Gilmour, Regio- and Stereo-Selective Isomerization of Borylated 1,3-Dienes Enabled by Selective Energy Transfer Catalysis, Angew. Chem., Int. Ed., 2024, 63, e202404233 CrossRef CAS PubMed.
- X. Zhang and T. Rovis, Photocatalyzed Triplet Sensitization of Oximes Using Visible Light Provides a Route to Nonclassical Beckmann Rearrangement Products, J. Am. Chem. Soc., 2021, 143, 21211–21217 CrossRef CAS PubMed.
- Z. Lu and T. P. Yoon, Visible Light Photocatalysis of [2+2] Styrene Cycloadditions by Energy Transfer, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- R. Alonso and T. Bach, A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2+2] Photocycloaddition Reactions Induced by Visible Light, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
- K. A. Rykaczewski and C. S. Schindler, Visible-Light-Enabled Paternò–Büchi Reaction via Triplet Energy Transfer for the Synthesis of Oxetanes, Org. Lett., 2020, 22, 6516–6519 CrossRef CAS PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel(II), Science, 2017, 355, 380–385 CrossRef CAS PubMed.
- S. Wang, P. Ma, S. Shaik and H. Chen, Valence-Inverted States of Nickel(II) Complexes Perform Facile C–H Bond Activation, J. Am. Chem. Soc., 2022, 144, 14607–14613 CrossRef CAS PubMed.
- E. Brachet, T. Ghosh, I. Ghosh and B. König, Visible light C–H amidation of heteroarenes with benzoyl azides, Chem. Sci., 2015, 6, 987–992 RSC.
- M. Teders, C. Henkel, L. Anhäuser, F. Strieth-Kalthoff, A. Gómez-Suárez, R. Kleinmans, A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, The energy-transfer-enabled biocompatible disulfide–ene reaction, Nat. Chem., 2018, 10, 981–988 CrossRef CAS PubMed.
- J. Majhi, R. K. Dhungana, Á. Rentería-Gómez, M. Sharique, L. Li, W. Dong, O. Gutierrez and G. A. Molander, Metal-Free Photochemical Imino-Alkylation of Alkenes with Bifunctional Oxime Esters, J. Am. Chem. Soc., 2022, 144, 15871–15878 CrossRef CAS PubMed.
- W. G. Herkstroeter, A. A. Lamola and G. S. Hammond, Mechaisms of photochemical reactions in solution. XXVIII. 1 Values of triplet excitation energies of selected sensitizers, J. Am. Chem. Soc., 1964, 86, 4537–4540 CrossRef CAS.
- A. Köhler and H. Bässler, Triplet states in organic semiconductors, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef.
- C. M. Marian, Understanding and Controlling Intersystem Crossing in Molecules, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed.
- K. Kalyanasundaram, Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Characterization of the Excited State Properties of Some New Photosensitizers of the Ruthenium (Polypyridine) Family, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- J. M. Dos Santos, D. Hall, B. Basumatary, M. Bryden, D. Chen, P. Choudhary, T. Comerford, E. Crovini, A. Danos, J. De, S. Diesing, M. Fatahi, M. Griffin, A. K. Gupta, H. Hafeez, L. Hämmerling, E. Hanover, J. Haug, T. Heil, D. Karthik, S. Kumar, O. Lee, H. Li, F. Lucas, C. F. R. Mackenzie, A. Mariko, T. Matulaitis, F. Millward, Y. Olivier, Q. Qi, I. D. W. Samuel, N. Sharma, C. Si, L. Spierling, P. Sudhakar, D. Sun, E. Tankelevičiūtė, M. Duarte Tonet, J. Wang, T. Wang, S. Wu, Y. Xu, L. Zhang and E. Zysman-Colman, The Golden Age of Thermally Activated Delayed Fluorescence Materials: Design and Exploitation, Chem. Rev., 2024, 124, 13736–14110 CrossRef CAS PubMed.
- J. Luo and J. Zhang, Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- M. A. Bryden and E. Zysman-Colman, Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- E. Zysman-Colman, Molecular designs offer fast exciton conversion, Nat. Protoc., 2020, 14, 593–594 CAS.
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. dos Santos, H. Kaji, E. Zysman-Colman, I. D. W. Samuel and C. Adachi, Exact Solution of Kinetic Analysis for Thermally Activated Delayed Fluorescence Materials, J. Phys. Chem. A, 2021, 125, 8074–8089 CrossRef CAS PubMed.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang, D. Ma, D. Zhang and L. Duan, High-efficiency and stable short-delayed fluorescence emitters with hybrid long- and short-range charge-transfer excitations, Nat. Commun., 2023, 14, 2394 CrossRef CAS PubMed.
- A. R. Meyer, M. V. Popescu, A. Sau, N. H. Damrauer, R. S. Paton and T. P. Yoon, Combined Synthetic, Spectroscopic, and Computational Insights Into a General Method for Photosensitized Alkene Aziridination, ACS Catal., 2024, 14, 12310–12317 CrossRef CAS.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, Donor-Acceptor Fluorophores for Energy-Transfer-Mediated Photocatalysis, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS PubMed.
- E T measured at 77 K in 2-MeTHF glass from the gated emission acquired in a time window of 1–9 ms after excitation.
- H. J. Timpe, K. P. Kronfeld, U. Lammel, J. P. Fouassier and D. J. Lougnot, Excited states of ketones as electron donors—ketone—iodonium salt systems as photoinitiators for radical polymerization, J. Photochem. Photobiol., A, 1990, 52, 111–122 CrossRef CAS.
- M. A. Bryden, F. Millward, T. Matulaitis, D. Chen, M. Villa, A. Fermi, S. Cetin, P. Ceroni and E. Zysman-Colman, Moving Beyond Cyanoarene Thermally Activated Delayed Fluorescence Compounds as Photocatalysts: An Assessment of the Performance of a Pyrimidyl Sulfone Photocatalyst in Comparison to 4CzIPN, J. Org. Chem., 2023, 88, 6364–6373 CrossRef CAS PubMed.
- S. Sharma and S. Sengupta, Twisted organic TADF triads based on a diindolocarbazole donor for efficient photoisomerization of stilbene and photo-arylation of heteroarenes, Org. Chem. Front., 2023, 10, 6087–6095 RSC.
- G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, Mechanisms of photochemical reactions in solution. XXII. 1 Photochemical cis-trans isomerization, J. Am. Chem. Soc., 1964, 86, 3197–3217 CrossRef CAS.
- M. Yokoyama, K. Inada, Y. Tsuchiya, H. Nakanotani and C. Adachi, Trifluoromethane modification of thermally activated delayed fluorescence molecules for high-efficiency blue organic light-emitting diodes, Chem. Commun., 2018, 54, 8261–8264 RSC.
- E. R. Sauvé, D. M. Mayder, S. Kamal, M. S. Oderinde and Z. M. Hudson, An imidazoacridine-based TADF material as an effective organic photosensitizer for visible-light-promoted [2 + 2] cycloaddition, Chem. Sci., 2022, 13, 2296–2302 RSC.
- R. Hojo, K. Bergmann, S. A. Elgadi, D. M. Mayder, M. A. Emmanuel, M. S. Oderinde and Z. M. Hudson, Imidazophenothiazine-Based Thermally Activated Delayed Fluorescence Materials with Ultra-Long-Lived Excited States for Energy Transfer Photocatalysis, J. Am. Chem. Soc., 2023, 145, 18366–18381 CrossRef CAS PubMed.
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Multiresonant Thermally Activated Delayed Fluorescence Emitters Based on Heteroatom-Doped Nanographenes: Recent Advances and Prospects for Organic Light-Emitting Diodes, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- C. Prentice, J. Morrison, A. D. Smith and E. Zysman-Colman, Multi-Resonant Thermally Activated Delayed Fluorescent (MR-TADF) Compounds as Photocatalysts, Chem.–Eur. J., 2023, 29, e202202998 CrossRef CAS PubMed.
- L. Hämmerling and E. Zysman-Colman, Building a photocatalyst library of MR-TADF compounds with tunable excited-state redox potentials, Chem Catal., 2024, 4, 101061 Search PubMed.
- V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W.-J. Son, S. O. Jeon and J. Y. Lee, Purely Spin-Vibronic Coupling Assisted Triplet to Singlet Up-Conversion for Real Deep Blue Organic Light-Emitting Diodes with Over 20% Efficiency and y Color Coordinate of 0.05, Adv. Sci., 2021, 8, 2101137 CrossRef CAS PubMed.
- D. Hall, K. Stavrou, E. Duda, A. Danos, S. Bagnich, S. Warriner, A. M. Z. Slawin, D. Beljonne, A. Köhler, A. Monkman, Y. Olivier and E. Zysman-Colman, Diindolocarbazole – achieving multiresonant thermally activated delayed fluorescence without the need for acceptor units, Mater. Horiz., 2022, 9, 1068–1080 RSC.
- H. L. Lee, S. O. Jeon, I. Kim, S. C. Kim, J. Lim, J. Kim, S. Park, J. Chwae, W.-J. Son, H. Choi and J. Y. Lee, Multiple-Resonance Extension and Spin-Vibronic-Coupling-Based Narrowband Blue Organic Fluorescence Emitters with Over 30% Quantum Efficiency, Adv. Mater., 2022, 34, 2202464 CrossRef CAS PubMed.
- V. V. Patil, J. Lim, S. M. Cho and J. Y. Lee, Highly Efficient C/N-Fused Architecture for Narrowband Deep-Blue Thermally Activated Delayed Fluorescence, Adv. Opt. Mater., 2024, 12, 2300551 CrossRef CAS.
- J. Kang, S. O. Jeon, I. Kim, H. L. Lee, J. Lim and J. Y. Lee, Color Stable Deep Blue Multi-Resonance Organic Emitters with Narrow Emission and High Efficiency, Adv. Sci., 2023, 10, 2302619 CrossRef CAS PubMed.
- J. H. Lee, T. Watanabe, L. Hartmann and T. Yasuda, Blue-to-Green Fine-Tunable Narrowband Delayed Fluorescence in Peripherally Dendritic Modified Bis-Indolocarbazoles, Angew. Chem., Int. Ed., 2025, 64, e202505191, DOI:10.1002/anie.202505191.
- M. A. Bryden, F. Millward, O. S. Lee, L. Cork, M. C. Gather, A. Steffen and E. Zysman-Colman, Lessons learnt in photocatalysis – the influence of solvent polarity and the photostability of the photocatalyst, Chem. Sci., 2024, 15, 3741–3757 RSC.
- D. K. A. Phan Huu, S. Saseendran and A. Painelli, Effective models for TADF: the role of the medium polarizability, J. Mater. Chem. C, 2022, 10, 4620–4628 RSC.
- L. Schmid, C. Kerzig, A. Prescimone and O. S. Wenger, Photostable Ruthenium(II) Isocyanoborato Luminophores and Their Use in Energy Transfer and Photoredox Catalysis, JACS Au, 2021, 1, 819–832 CrossRef CAS PubMed.
- L. Schmid, F. Glaser, R. Schaer and O. S. Wenger, High Triplet Energy Iridium(III) Isocyanoborato Complex for Photochemical Upconversion, Photoredox and Energy Transfer Catalysis, J. Am. Chem. Soc., 2022, 144, 963–976 CrossRef CAS PubMed.
- K. E. Jones, B. Park, N. A. Doering, M.-H. Baik and R. Sarpong, Rearrangements of the Chrysanthenol Core: Application to a Formal Synthesis of Xishacorene B, J. Am. Chem. Soc., 2021, 143, 20482–20490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |