
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosalient effect and reversible photochromic photoluminescence driven by cascade [2 + 2] cycloaddition reaction and water adsorption in a 0D hybrid metal halide†
Chudong
Chen‡
ab,
Ziquan
Li‡
ab,
Yonghong
Xiao
 b,
Chenghao
Ye
c,
Jianwu
Wei
a,
Ruosheng
Zeng
d,
Qi
Pang
a and
Binbin
Luo
*a
b,
Chenghao
Ye
c,
Jianwu
Wei
a,
Ruosheng
Zeng
d,
Qi
Pang
a and
Binbin
Luo
*a
aSchool of Chemistry and Chemical Engineering, Key Laboratory of Electrochemical Energy Materials, Guangxi University, Nanning 530004, P. R. China. E-mail: bbluo@gxu.edu.cn
bCollege of Chemistry and Chemical Engineering, Shantou University, Shantou 515063, P. R. China
cSchool of Power and Mechanical Engineering, Wuhan University, Wuhan 430072, P. R. China
dSchool of Physical Science and Technology, State Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, Guangxi University, Nanning 530004, P. R. China
First published on 23rd June 2025
Abstract
The ionic structure of hybrid metal halides (HMHs) is conducive to the integration of the photoactivity of organic components and mechanical properties of inorganic components. Therefore, HMHs can function as a suitable platform to advance photomechanical performance due to their great structural designability and wide compositional diversity. Herein, a notable photosalient effect was observed in 0D ionic (LH)2InCl5 (1, L = 4-styrylpyridine). Upon irradiation with 450 nm light, 1 undergoes a rare single-crystal-to-single-crystal transformation to (L2H2)[In(H2O)Cl5] (1a), along with distinct photochromic photoluminescence and various photomechanical responses, driven by the cascade photoinduced [2 + 2] cycloaddition of LH+ and water sorption by coordinatively unsaturated In3+. A detailed mechanistic investigation unraveled the photosalient effect by initiation of the cycloaddition reaction and enhancement by water sorption, which resulted in a phase transformation that is more energetically and kinetically favorable through decreasing Gibbs free energy (1.51 eV) and a kinetical energy barrier (0.29 eV). Additionally, the partially reversible transformation between LH+ monomers and dimers endows 1a with excellent photoswitch performance due to the variation in the π electron-conjugated degree for LH+. At last, by integrating powdered 1 with polymethyl methacrylate (PMMA), the 1–PMMA hybrid matrix membrane exhibited rapid and controllable responsive behaviors, including bending and curling, through controlling the direction of the concentration gradient. This work demonstrates that ionic HMHs might function as a promising platform for advanced photomechanical and photochromic materials.
Introduction
Actuating materials are thought to hold great promise in the fields of soft robotics, artificial muscles, and biomimetics because they are capable of a kinetic energy output to implement complex and rapid macroscopic movements under external stimuli including light, heat, force, and chemical reagents.1–8 Among the different stimuli, light is advantageous because it imparts a high degree of spatial and temporal control without any physical contact, as well as precise operation over properties including intensity, wavelength, and polarization.9,10Currently, the motions of photoactuators are usually triggered through photothermal (molecular sorption/desorption and phase transition) or photochemical (photoisomerization and photodimerization) processes.10 Among these strategies, [2 + 2] photocycloaddition has been widely employed to translate nanoscopic structural perturbation into macroscopic motions such as jumping, curling, bending, twisting, and disintegrating.
To trigger [2 + 2] cycloaddition in crystals, parallel olefin groups should satisfy Schmidt's topochemical criteria to undergo structural transformation.11–14 Therefore, such photoactuator materials are mostly based on molecular crystals (MCs)15–20 and coordination polymers (CPs)21–28 because of the adequate understanding of their structural design and tunability. Although a great deal of effort has been recently devoted to the development of new photocycloaddition crystals to unravel the structure–activity relationships,29–37 few effective strategies have been proposed to improve photomechanical performance due to the limited understanding of a cascade of processes including strain generation, accumulation, transmission, and release during the photomechanical response.
Different from MCs and CPs mainly assembled through non-covalent and coordinated interaction, respectively, ionic crystals (ICs) are assembled through coulombic interaction with more dense packing. Such a stacking mode usually endows them with a larger Young's modulus, implying that a more intense photomechanical response might occur than that of MCs and CPs under same variation in cell volume. Among various ICs, hybrid metal halides (HMHs) are a class of ionic semiconductors derived from halide perovskites that has recently attracted enormous attention.38–45
Because there are great advantages when structural and compositional tunability is enabled for HMHs, various strategies46–54 have been proposed to tune the structural features of HMHs, including dimensionality, layer number, and polyhedron connection modes that will affect the light-to-work energy conversion and transmission processes of interior strain in the photomechanical response. Therefore, HMHs might function as a satisfactory model to reveal the structure–activity relationships of the photomechanical response.
With this in mind, few works have successfully achieved [2 + 2] cycloaddition in HMHs. For instance, hybridizing 4-pyridineacrylic acid with BiCl3 results in photochemical reactions, including photosalient motions and photochromism after ultraviolet (UV) irradiation.55 The [2 + 2] photocycloaddition of 4-vinyl pyridine derivatives induces photochromism and photoinduced-cracking in Zn- and Pb-based HMHs.56 These two examples demonstrate the feasibility of photocycloaddition in HMHs. However, the photomechanical properties of HMHs have not been systematically explored.
In this work, 4-styrylpyridine (L) was successfully hybridized with InCl3 to form a zero-dimensional (0D) (LH)2InCl5 (denoted as 1) with coordinatively unsaturated In3+ sites. The ideally parallel stacking of LH+ dimers enables a fast and thorough photocycloaddition reaction, followed by two-step H2O sorption on In3+ sites with a trigonal bipyramidal geometry. Furthermore, the significant lattice reorganization caused by photocycloaddition and H2O sorption results in notable photomechanical motions and photochromic photoluminescence (PL) of 1.
Results and discussion
Compound 1 was prepared through a cooling crystallization method, and the synthesis details are found in the ESI.† To determine the crystal phase of the resulting sample, single crystal X-ray diffraction (SCXRD) was conducted, and the crystal structure of 1 is shown in Fig. 1a. It was observed that 1 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2 (Table S1†) and consists of discrete [InCl5]2− polyhedra and LH+ dimers with anti-parallel alignment. Different from other In-based HMHs adopting an octahedral coordination,57–60 a rare [InCl5]2− polyhedron with trigonal bipyramidal geometry is formed in 1, which is caused by the charge balance and the strong interactions between the LH+ dimer and the [InCl5]2− polyhedron.
with Z = 2 (Table S1†) and consists of discrete [InCl5]2− polyhedra and LH+ dimers with anti-parallel alignment. Different from other In-based HMHs adopting an octahedral coordination,57–60 a rare [InCl5]2− polyhedron with trigonal bipyramidal geometry is formed in 1, which is caused by the charge balance and the strong interactions between the LH+ dimer and the [InCl5]2− polyhedron.
 | ||
Fig. 1 (a) Crystal structures and phase transformation of 1, 1i, and 1a. Light source: 450 nm, 260 mW cm−2. (b) Alignments of LH+ dimers in 1. d1 is defined by the mean distance between the centroid of the phenyl and pyridyl rings. d2 is defined by the distance between the parallel C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. θ1 denotes the angle formed between two adjacent CC bonds in the top view. θ2 is defined by the angle involving the CC bond and one of the carbon atoms from the adjacent CC bond. The orange and cyan planes for θ3 are defined by the two CC bonds and their adjoining single bond. C bonds. θ1 denotes the angle formed between two adjacent CC bonds in the top view. θ2 is defined by the angle involving the CC bond and one of the carbon atoms from the adjacent CC bond. The orange and cyan planes for θ3 are defined by the two CC bonds and their adjoining single bond. | ||
As shown in Fig. 1a, the short distance between the phenyl ring and the [InCl5]2− polyhedron hinders the coordination of free Cl− or H2O with coordinatively unsaturated In3+. The strong intermolecular interactions were confirmed from the Hirshfeld surface analysis (Fig. S1†) that quantitatively determined the predominant contributions from the N–H⋯Cl (42.1%) and H⋯H (30.7%) bonding interactions. [InCl5]2− polyhedra are interconnected through coulombic and hydrogen bonding interactions with cationic LH+ (Fig. S2†). LH+ dimers exhibit two stacking modes in 1, as denoted by the cyan and orange olefins. Each stacking mode shows two types of disorder, with an occupancy of approximately 69% (solid cyan) and approximately 74% (solid orange) for the predominant alignment, respectively.
Upon irradiation of 450 nm light for 30 min in open air, a single-crystal-to-single-crystal transformation occurred from 1 to (L2H2)[In(H2O)Cl5] (1a), with the crystal data listed in Table S1.† Compared to 1, the LH+ photodimers in 1a revolve away the [InCl5]2− polyhedron upon photocycloaddition, which forces the rotation of the [InCl5]2− polyhedron and creates sufficient free space for In3+ to adsorb H2O. Once single crystals of 1 were placed in paraffin oil to slow the sorption of H2O, some 1 single crystals transferred to an intermediate phase of (L2H2)2[In(H2O)Cl5][InCl5] (1i), with complete photodimerization of LH+. Interestingly, only half of the [InCl5]2− polyhedra in 1i were coordinated with H2O molecules in paraffin oil. A further increase in the reaction time resulted in the phase transformation from 1i to 1a.
To understand the fast and thorough photocycloaddition, the alignments of the LH+ dimers in 1 are shown in Fig. 1b. Because of the similar structural change for each disordered part, only the high occupancy of disordered olefin is discussed. All LH+ dimers in 1 align in an antiparallel head-to-tail manner, with distances of 3.95 Å (mode I) and 3.79 Å (mode II) between the pyridyl and phenyl rings. Profiting from their π–π stacking interaction, the olefin bonds are perfectly aligned with an angle of θ1 = 0.0°. Although LH+ dimers slip along the lateral axis (θ2 ≠ 90) and slightly incline (θ3 ≠ 90), the distances of the olefin bonds (3.85 Å for mode I and 3.81 Å for mode II) are well satisfied with Schmidt's criteria.
Despite the crystal system and space group of 1 remaining the same after phase transformation, significant variations were observed for the crystal structure due to the thorough photocycloaddition reaction of the LH+ dimers. As shown in Fig. 2a, the average breadth of the LH+ dimers in mode I increased from 4.07 Å (1) to 6.57 (1i) and 6.2661 Å (1a), along with a reduction in the length from 11.07 Å (1) to 9.85 Å (1i) and 10.10 Å (1a). A similar structural change was also observed for LH+ dimers in mode II. In comparison with the centrosymmetric LH+ dimers in 1 and 1a, non-centrosymmetric LH+ photodimers were formed in 1i. This difference is due to the presence of [InCl5]2− and [In(H2O)Cl5]2− pairs that interact with terminal pyridinium with a different degree of force (Fig. S3†).
 | ||
| Fig. 2 (a) Geometrical variation of LH+ dimers and the [InCl5]2− polyhedron in 1, 1i, and 1a. (b) Schematic illustration of the free volume in 1i (probe radius: 1.0 Å; approx. grid spacing: 0.2 Å). (c) Lattice parameters of 1 at different irradiation times (light source: 450 nm, 260 mW cm−2). (d) Energy profile of the [2 + 2] cycloaddition reaction with constrained C–C bond length. The C15–C40 bond length was fixed during the optimization process to examine the impact of this constraint on the reaction pathway. (e) Computational study of LH+ dimers, the [InCl5]2− polyhedron, LH+ photodimers, and the [In(H2O)Cl5]2− octahedron involved in [2 + 2] cycloaddition reactions. | ||
The significant geometric variation of LH+ dimers also drives the rotation of all [InCl5]2− polyhedra, which creates sufficient free volume for the following H2O sorption. As shown in Fig. 2b, compared to the dense stacking of 1 and 1a (free volume percentage = 0%), free volume with a percentage of 5.1% is created near the [InCl5]2− and [In(H2O)Cl5]2− polyhedra in 1i. The adsorption of H2O molecules generates octahedral [In(H2O)Cl5]2− pairs with a distance of 5.72 Å and an intermolecular hydrogen bonding length of 3.25 Å. Upon phase transformation from 1i to 1a, all [InCl5]2− pairs transform to [In(H2O)Cl5]2− pairs, concurrent with a decrease in the distance of the [InCl5]2− pairs from 7.45 Å to 5.75 Å (Fig. 2a) and the reconstruction of an intermolecular hydrogen bonding network (Fig. S4†).
To further understand the process of phase transformation, the lattice parameters of a single crystal of 1 were monitored under different irradiation times. As shown in Fig. 2c, upon the photocycloaddition reaction of LH+ dimers, the a-axis length gradually decreased, along with an increase in the b-axis length within 40 s, while the c-axis length remained nearly constant and the cell volume (V) slightly increased by approximately 1.1%. Interestingly, increasing the irradiation time to 50 s resulted in a sharp increase in V to 4.3%, along with the first sudden change in a- and c-axis lengths. Additionally, a second sudden change was observed for all lattice parameters once the irradiation time was extended to 90 s.
Such an evolutionary tendency suggests a two-step phase transformation during the irradiation. Within the first 40 s, the slowly varied lattice parameters indicate that the photodimerization reaction is ongoing. Once the lattice stress sufficiently accumulates to drive the rotation of [InCl5]2−, H2O sorption will occur, accompanied by the phase transformation from 1 to 1i. When extending the irradiation time to 90 s, the H2O sorption by the other half of [InCl5]2− induces the phase transformation from 1i to 1a, resulting in a total increase in cell volume to 8.0%.
Density functional theory (DFT) calculations were also conducted to understand the cycloaddition process. Optimization of 1 was performed, using their X-ray structures as the initial structures. The C15–C40 bond length was fixed at specific values during the optimization process to examine the impact of this constraint upon the reaction pathway. As shown in Fig. 2d, this process is highly endothermic by 1.31 eV, with an electronic energy barrier of 2.09 eV. Once the distance of C15–C40 decreased to 2.0 Å, a maximum value was obtained. A further decrease in the distance to 1.6 Å will result in a local minimum value of energy, and was consistent with the experimental results of the bond length of C–C in cyclobutane, at approximately 1.57 Å (obtained at 200 K).
Fig. 2e further reveals the H2O sorption effect on the free energy variation of the phase transformation. Once H2O sorption is involved, the energy variation changes from endothermic (0.57 eV) to exothermic (−0.94 eV), with a reduction in the energy barrier of 0.29 eV. This highlights the critical role of H2O sorption in enhancing the cycloaddition reaction by synergistically modulating the thermodynamic equilibria and lowering the kinetic energy barrier.
Due to the instability of 1i, only the phase purities of powdered 1 and 1a were determined through powder X-ray diffraction (PXRD). As shown in Fig. 3a, the diffraction patterns of 1 and 1a match the simulated results, indicating the high phase purity of the as-prepared samples. To confirm the H2O sorption, the thermogravimetric analysis (TGA) of powdered 1 and 1a is shown in Fig. 3b. The decomposition temperature of 1 was approximately 193 °C, while 1a exhibited the first weight loss platform starting at approximately 150 °C with a weight loss ratio of 2.60%, corresponding well with the theoretical weight ratio (2.67%) of H2O in 1a.
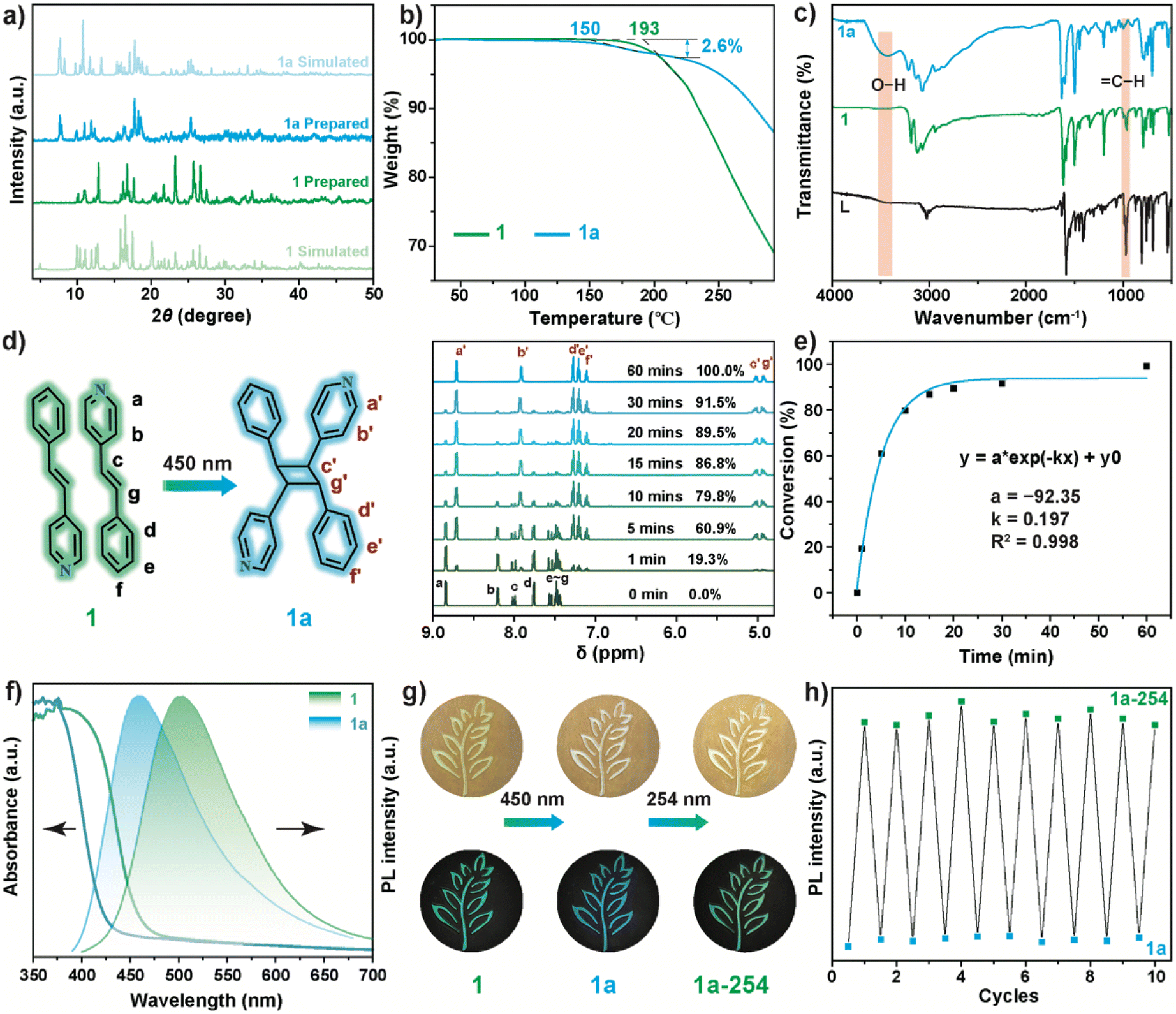 | ||
| Fig. 3 (a) PXRD patterns, (b) TGA curves, and (c) FT-IR spectroscopy of 1 and 1a. (d) 1H NMR spectra (solvent: DMSO-d6) of 1 with different irradiation times. (e) Conversion ratios for the photodimerization of 1. (f) UV-Vis diffuse reflectance and PL spectra of 1 and 1a. (g) Digital images of 1, 1a, and 1a-254 (6.1 mW cm−2). (h) Reversibility test between 1a and 1a-254; the PL intensity was monitored at 502 nm, and the irradiation time was 10 min for 254 and 450 nm. | ||
The presence of coordinated H2O was also confirmed by energy-dispersive spectroscopy (EDS) mapping. The EDS mapping (Fig. S5†) of 1a exhibits a morphology-dependent distribution for all elements including O, while this was insignificant for the O element in 1. Fourier transform infrared (FT-IR) spectroscopy (Fig. 3c) shows a notable band at 3430 cm−1 caused by the O–H stretching vibration for 1a, while this band is absent for 1. Additionally, the intensity of the C–H bending vibration peak (964 cm−1) of 1 dramatically decreased upon phase transformation, which verified the cascade [2 + 2] cycloaddition and H2O sorption reactions.
To evaluate the rate of photocycloaddition, the samples were digested in dimethyl sulfoxide-d6 (DMSO-d6) to conduct proton (1H) nuclear magnetic resonance (1H NMR) spectroscopy at various time intervals. As shown in Fig. 3d and S6,† the signals of 8.85 (Ha) and 8.21 ppm (Hb) for 1 resulted from the pyridyl ring, while the peaks of 7.76 (Hd), 7.55 (He), and 7.48 (Hf) ppm were derived from the phenyl ring, respectively. The peaks at 8.01 (Hc) and 7.44 ppm (Hg) for 1 were assigned to olefinic protons. Upon 450 nm irradiation, the intensity of these peaks gradually decreased, and a new set of peaks appeared, including two peaks at 4.93 ppm and 5.02 ppm, which were derived from cyclobutane. By integrating the peak area of Ha, the photocycloaddition proportion was determined, and is shown in Fig. 3e. The photodimerization of LH+ was nearly complete within 60 min. The fitting results indicate the first-order reaction kinetics of LH+ photocycloaddition with a rate constant of 0.197 min−1, which is higher than that in CPs.61
UV-Vis diffuse reflectance and photoluminescence (PL) spectra were also obtained to determine the influence of [2 + 2] photocycloaddition on the optical properties of the samples. The absorption onset at approximately 470 nm exhibited by 1 (Fig. 3f) was consistent with the absorption of LH·Cl (Fig. S7†). Upon illumination with 450 nm light, the absorption onset of 1a underwent a blueshift to approximately 430 nm, caused by the disruption of the π electron conjugation of LH+. Similarly, the photodimerization reaction of LH+ also greatly influenced the PL of 1. As shown in Fig. 3f, the broadband emission peaked at 502 nm and then blueshifted to 460 nm due to the photocycloaddition reaction. This photochromic PL was also observed for LH·Cl (Fig. S8†), in which the [2 + 2] photocycloaddition reaction was confirmed by the 1H NMR spectra (Fig. S9†).
These results indicate the photochromic origin resulting from the [2 + 2] cycloaddition reaction of LH+, rather than the H2O sorption by [InCl5]2− polyhedra. Consistent with the UV-Vis and PL spectra, 1 exhibited a distinct photochromic behavior from yellow-green to white under room light (Fig. 3g), along with the emission color transformation from green to blue under UV light. Upon irradiating 1a with 254 nm (1a-254) for 10 min, the color of 1a-254 reverted to a faint yellow-green, concurrent with the emissive color returning to green. This color evolution indicates the partial reversibility between LH+ photodimers and LH+ monomers, which was confirmed from the 1H NMR.
As shown in Fig. S10,† a dedimerization ratio of approximately 13.2% (8 h) was obtained. The PL spectrum of 1a-254 (Fig. S11†) also indicates the successful dedimerization of LH+ dimers. Increasing the irradiation time for 254 nm resulted in the falling of the main peak at 460 nm and the rising of the shoulder peak at 502 nm. By monitoring the PL intensity at 502 nm (Fig. 3h), excellent reversibility between 1a and 1a-254 was observed, indicating the great potential of 1a in photoswitching. However, due to the irreversible H2O sorption, 1a cannot revert back to 1, as evidenced from the PXRD patterns (Fig. S12†).
Because of the significant changes in lattice parameters caused by the cascade photocycloaddition and H2O sorption, single crystals of 1 exhibited distinct macroscopic photomechanical motions. As shown in Fig. 4a and Video S1,† once irradiated with 450 nm light, the crystal (1.22 × 0.30 × 0.15 mm3) jumps away at 0.5 s, followed by the motions of cracking, splitting, and hopping. Upon irradiation with 405 nm blue light, the crystal (1.08 × 0.72 × 0.15 mm3) shows a bursting motion at 3.0 s and thoroughly disintegrates at 25 s (Fig. 4b and Video S2†). Nevertheless, irradiating crystal 1 (0.75 × 0.35 × 0.14 mm3) with 365 nm light resulted in gradual photochromic PL with insignificant photomechanical motions (Fig. 4c and Video S3†).
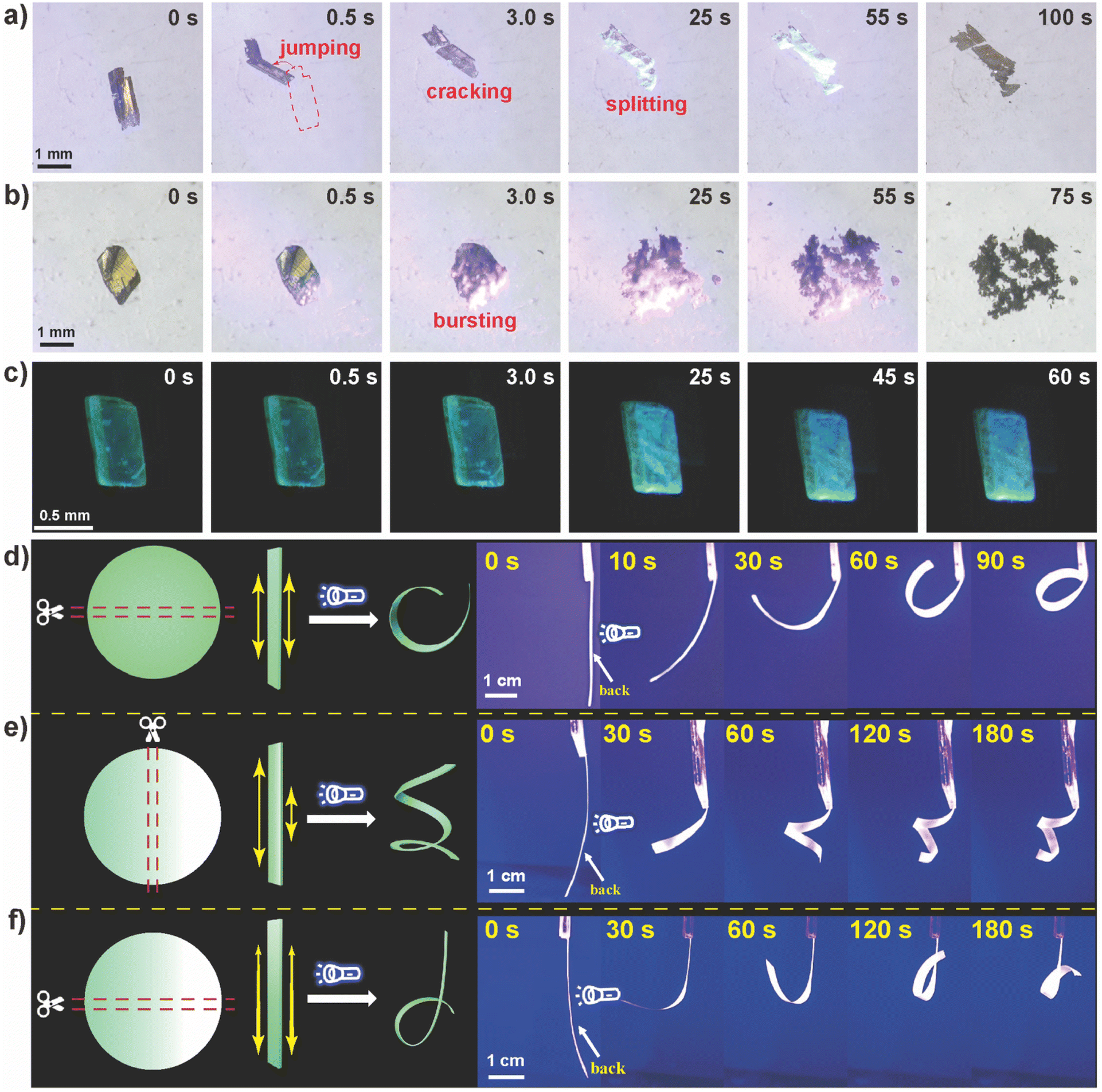 | ||
| Fig. 4 Digital images of crystal 1 under (a) 450 nm (260 mW cm−2), (b) 405 nm (260 mW cm−2), and (c) 365 nm (80 mW cm−2) irradiation with different times. Images of (d) 1–PMMA with uniform distribution, (e) 1–PMMA with a perpendicular concentration gradient, and (f) 1–PMMA with a parallel concentration gradient upon irradiation with 450 nm light. | ||
These excitation wavelength-dependent photoresponsive behaviors were attributed to the discrepancies in absorption coefficients and the power density of excited light that induces varying degrees of interior stress. To unravel the contribution of photocycloaddition and H2O sorption, crystal 1 was placed in a Schlenk tube evacuated for 2 h at 80 °C, and then filled with dry nitrogen to exclude the sorption of H2O. As shown in Fig. S13,† upon irradiating crystal 1 with 450 nm light, photomechanical behaviors such as jumping and cracking continue to be observed (Video S4†), suggesting that the photomechanical effect of 1 was initiated by the cycloaddition reaction of LH+, rather than H2O sorption. However, the photomechanical strength seems much weaker than that of 1 in open air, indicating the critical role of H2O sorption in enhancing the photosalient effect.
Although the increase in cell volume was relatively smaller than that of L-based CPs (Table S2†), the photomechanical behavior of 1 was more intense in our case. Such change was ascribed to the ionic structure of HMHs with dense stacking that exhibits relatively higher brittleness. Although an understanding of the mechanism during the photoresponsive process can be acquired through a well-defined crystal structure obtained from X-ray diffraction, it remains a great challenge to combine the motions of discrete microscale crystals into one organized macroscale system.
In an effort to produce a more predictable mechanical behavior,62 powdered 1 was mixed with polymethyl methacrylate (PMMA) to produce a hybrid matrix membrane (1-PMMA). The PXRD pattern of 1–PMMA indicated that the crystal structure of 1 is preserved in the composite membrane (Fig. S14†). The pure PMMA film did not exhibit any photomechanical behavior (Fig. S15†), indicating no photoinduced PMMA response. Upon perpendicularly exposing the backside of 1–PMMA (5.0 × 40 mm2) to the light source (Fig. 4d and Video S5†), 1–PMMA promptly bends away from the light source. The longer the irradiation time, the more severely the film bends. This was also observed at 405 and 365 nm (Fig. S16†).
To understand the kinematics and mechanism of photoinduced deformation, we quantified the bending angles (φ) as a function of exposure time (t) by recording a series of snapshot images. As shown in Fig. S17,† when exposing 1–PMMA for 10 s, the bending angle reached 50°, and a bending angle of 305° was achieved within 90 s. However, small bending angles of 182° and 112° were observed for 405 and 365 nm, respectively, within 90 s. The correlation between the bending angle and irradiation time can be described according to the following equation:63
| φ = κ(1 − exp(−t/τ)) | (1) |
Interestingly, when the front side of 1–PMMA was irradiated, the film exhibited a phototropic bending phenomenon (Fig. S18† and Video S6†) that was different from the photoresponsive behavior elicited by irradiation from the backside. To investigate this, scanning electron microscopy (SEM) and EDS mapping were conducted to reveal the powder distribution in the composite membrane. As shown in Fig. S19a,†1 is mainly distributed in the backside of the membrane due to the gravity effect, while being relatively sparse in the front side.
This was further confirmed by the EDS mapping, in which the signals of In, Cl, and N were significantly stronger on the backside. Therefore, once exposed to 450 nm illumination, 1–PMMA generated a more significant expansion on the backside (Fig. S19b†). Consequently, 1–PMMA always exhibited the bending direction away from the backside, irrespective of the irradiation direction. The κ and τ value was calculated to be 115 and 69, respectively, once the backside of 1–PMMA was irradiated (Fig. S20†). Although the irradiation direction would not alter the sensitivity of 1–PMMA toward light wavelength, the photocycloaddition ratio of LH+ on the backside was smaller due to the limitation of light penetration, and thereby, a smaller bending rate was observed for front side irradiation.
By leveraging the sedimentation of 1 during the preparation of the film, we attempted to tilt the mold at a certain angle (Fig. S21†) to prepare 1–PMMA with a concentration gradient. Interestingly, the film with a perpendicular concentration gradient (Fig. 4e and Video S7†) shows a curling responsive behavior due to the different degrees of volume expansion in both of the long sides. Once the concentration gradient was parallel to the long side of the belt film, no significant response was observed in the upper side of 1–PMMA due to the lower concentration of 1 (Fig. 4f and Video S8†), while notable bending was observed for the lower section with a higher 1 concentration.
The reversible photochromic PL of 1a encouraged us to explore its potential in anti-counterfeiting. As shown in Fig. 5a, a pine tree pattern composed of five groovy parts was filled with 1 powder. Upon 450 nm irradiation, the irradiated region exhibited a distinct photochromic PL due to the dimerization of LH+. Further irradiating at 254 nm caused a change in color from blue to green, while no change was observed for the blocked part. Thus, the pine tree pattern shows various emissive colors for different regions, which can be further erased or recovered upon 450/254 nm irradiation, greatly increasing the complexity and difficulty of decryption. Similarly, different emissive colors can be adopted for information storage.
 | ||
| Fig. 5 (a) Schematic illustration of reversible anti-counterfeiting. (b) Optical logic system for reversible information encryption and decryption. (c) A handcrafted item mimicking the blooming process of a flower. | ||
As shown in Fig. 5b, the blue- and green-emissive dots are defined to be “0” and “1”. Based on binary ASCII code, the 4 × 8 matrix pattern gives a valid code of “Stpy”, which can be successfully erased upon 450 nm irradiation. Furthermore, given the rapid photomechanical response of 1, several pieces of 1–PMMA film were tailored to petal-like shapes to mimic the blooming process of a flower. As shown in Fig. 5 and Video S9,† once the handcrafted item was irradiated with 450 nm light, the 1–PMMA films responded immediately and gradually bent away from the center of the flowerpot. By mixing 1 with PMMA, the photomechanical motions of 1 could be precisely controlled. Within 300 s, the photomechanical response was nearly completed, concurrent with the color change from yellow to white.
Conclusions
Photoactive L was first hybridized with InCl3 to form zero-dimensional 1, which exhibited notable photosalient effects under 450 nm light. The cascade mechanism involves the photoinduced [2 + 2] cycloaddition of LH+ cations and subsequent water adsorption at uncoordinated In3+ sites, driving significant lattice reorganization (8% cell volume expansion) and single-crystal-to-single-crystal transformation. This dual process amplified photomechanical responses, including jumping, splitting, and bursting. The excellent reversible photochromic PL demonstrated by 1a was due to the dimerization and dedimerization of LH+ dimers exposed to 450 and 254 nm irradiation, respectively. Additionally, an excitation wavelength-dependent photoresponse was observed with 450 nm light, enabling the most efficient photocycloaddition. By integrating 1 with PMMA, controllable bending and curling was achieved in 1–PMMA via directional concentration gradients, showcasing its potential for use in soft actuators. This work highlights HMHs as promising platforms for photoswitching and photomechanical applications and provides insights into strain manipulation through synergistic photochemical and adsorption processes.Data availability
Some data supporting this article have been included as part of the ESI.† The crystallographic data for 1, 1a, and 1i have been deposited at the CCDC under 2434257, 2434258, and 2435615. Other data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: B. Luo; data curation: C. Chen, Z. Li; formal analysis: C. Chen, Z. Li, Y. Xiao; software: C. Ye, J. Wei; writing – original draft: B. Luo, C. Chen, Z. Li; writing – review and editing: B. Luo, Q. Pang, R. Zeng; supervision: B. Luo, Q. Pang, R. Zeng; funding acquisition: B. Luo, Q. Pang.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Launch Funding of High-Level Talent Researcher of Guangxi University (ZX01080030425002), the National Natural Science Foundation of China (Grant No. 22365005), and the Open Foundation of Guangxi Key Laboratory of Processing for Nonferrous Metals and Featured Materials (2022GXYSOF14).Notes and references
- M. Lahikainen, H. Zeng and A. Priimagi, Nat. Commun., 2018, 9, 4148 CrossRef PubMed.
- C. J. Easley, F. Tong, X. Dong, R. O. Al-Kaysi and C. J. Bardeen, Chem. Sci., 2020, 11, 9852–9862 RSC.
- B. B. Rath, G. Gallo, R. E. Dinnebier and J. J. Vittal, J. Am. Chem. Soc., 2021, 143, 2088–2096 CrossRef CAS PubMed.
- K. Yadava, G. Gallo, S. Bette, C. E. Mulijanto, D. P. Karothu, I. H. Park, R. Medishetty, P. Naumov, R. E. Dinnebier and J. J. Vittal, IUCrJ, 2020, 7, 83–89 CrossRef CAS PubMed.
- J. Lin, J. Zhou, L. Li, I. Tahir, S. Wu, P. Naumov and J. Gong, Nat. Commun., 2024, 15, 3633 Search PubMed.
- J. S. Geng, L. Mei, Y. Y. Liang, L. Y. Yuan, J. P. Yu, K. Q. Hu, L. H. Yuan, W. Feng, Z. F. Chai and W. Q. Shi, Nat. Commun., 2022, 13, 2030 CrossRef CAS PubMed.
- B. B. Rath and J. J. Vittal, Acc. Chem. Res., 2022, 55, 1445–1455 CrossRef CAS PubMed.
- Q. Liu, P. Braunstein and J.-P. Lang, Acc. Mater. Res., 2025, 6, 183–194 CrossRef CAS.
- B. Zhou and D. Yan, Appl. Phys. Rev., 2021, 8, 041310 CAS.
- J. Li, X. Zhou and Z. Liu, Adv. Opt. Mater., 2020, 2000886 CrossRef CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- M. Nakagawa, S. Kusaka, A. Kiyose, T. Nakajo, H. Iguchi, M. Mizuno and R. Matsuda, J. Am. Chem. Soc., 2023, 145, 12059–12065 CrossRef CAS PubMed.
- Y. Yue, J. Dai, L. Jin, C. Liu, J. Sun, K. Ye and R. Lu, Chem. Eur J., 2023, 29, e202301525 CrossRef CAS PubMed.
- Y. Kang, X. Tang, H. Yu, Z. Cai, Z. Huang, D. Wang, J. F. Xu and X. Zhang, Chem. Sci., 2017, 8, 8357–8361 RSC.
- H. Wang, P. Chen, Z. Wu, J. Zhao, J. Sun and R. Lu, Angew. Chem., Int. Ed., 2017, 56, 9463–9467 CrossRef CAS PubMed.
- F. Tong, D. Kitagawa, I. Bushnak, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2021, 60, 2414–2423 CrossRef CAS PubMed.
- S. Dai, J. Zhong, X. Yang, C. Chen, L. Zhou, X. Liu, J. Sun, K. Ye, H. Zhang, L. Li, P. Naumov and R. Lu, Angew. Chem., Int. Ed., 2024, 63, e202320223 CrossRef CAS PubMed.
- J. Peng, C. Han, X. Zhang, J. Jia, J. Bai, Q. Zhang, Y. Wang and P. Xue, Angew. Chem., Int. Ed., 2023, 62, e202311348 CrossRef CAS PubMed.
- L. Wang, S. B. Qiao, Y. T. Chen, X. Ma, W. M. Wei, J. Zhang, L. Du and Q. H. Zhao, Chem. Sci., 2024, 15, 3971–3979 RSC.
- Y. Wei, K. Chen, S. Zhu, W. Wu, H. Zhao, X. Huang, N. Wang, L. Zhou, T. Wang, J. Wang and H. Hao, Small, 2024, 20, e2307756 CrossRef PubMed.
- M. Y. Gao, L. Liu, C. Deng, V. Bon, B. Q. Song, S. Yang, M. Schroder, S. Kaskel and M. J. Zaworotko, Angew. Chem., Int. Ed., 2024, 63, e202404084 CrossRef CAS PubMed.
- Y. J. Ma, G. Xiao, X. Fang, T. Chen and D. Yan, Angew. Chem., Int. Ed., 2023, 62, e202217054 CrossRef CAS PubMed.
- X. D. Huang, B. K. Hong, G. H. Wen, S. H. Li and L. M. Zheng, Chem. Sci., 2023, 14, 1852–1860 RSC.
- B. B. Rath and J. J. Vittal, J. Am. Chem. Soc., 2020, 142, 20117–20123 CrossRef CAS PubMed.
- I. H. Park, E. Lee, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2019, 58, 14860–14864 CrossRef CAS PubMed.
- B. B. Rath and J. J. Vittal, Chem. Mater., 2021, 33, 4621–4627 CrossRef CAS.
- S. Khan, B. Dutta, S. Naaz, A. Choudhury, P. A. Cazade, E. Kiely, S. Guerin, R. Medishetty and M. H. Mir, Commun. Chem., 2023, 6, 150 CrossRef CAS PubMed.
- C. Cao, X. R. Xue, Q. Y. Li, M. J. Zhang, B. F. Abrahams and J. P. Lang, Angew. Chem., Int. Ed., 2023, 62, e202306048 CrossRef CAS PubMed.
- I. Paul, K. A. Konieczny, R. Chavez, J. R. Shan, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2024, 146, 27988–27992 CAS.
- S. P. Yelgaonkar, G. Campillo-Alvarado and L. R. MacGillivray, J. Am. Chem. Soc., 2020, 142, 20772–20777 CrossRef CAS PubMed.
- P. Naumov, S. C. Sahoo, B. A. Zakharov and E. V. Boldyreva, Angew. Chem., Int. Ed., 2013, 52, 9990–9995 CrossRef CAS PubMed.
- N. Y. Li, B. Liu, Z. W. Zhang, H. Yao, L. L. Zhang, J. Ma, L. L. Liu and D. Liu, Inorg. Chem., 2022, 61, 18950–18956 CrossRef CAS PubMed.
- S. Bhandary, M. Belis, R. Shukla, L. Bourda, A. M. Kaczmarek and K. Van Hecke, J. Am. Chem. Soc., 2024, 146, 8659–8667 CrossRef CAS PubMed.
- Y. Zhang, T. Takeda and T. Akutagawa, J. Am. Chem. Soc., 2025, 147, 7983–7992 CrossRef CAS PubMed.
- Y. Wang, Q. Zhang, Q. Liu, B. F. Abrahams and J. P. Lang, Angew. Chem., Int. Ed., 2024, 63, e202409472 CrossRef CAS PubMed.
- Z. Y. Yang, X. Sang, D. Liu, Q. Y. Li, F. Lang, B. F. Abrahams, H. Hou, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2023, 62, e202302429 CrossRef CAS PubMed.
- X. Zhu, M. Xie, L. Gao, L. Li, P. Naumov, Q. Yu and G. Wang, Angew. Chem., Int. Ed., 2025, 64, e202416950 CrossRef CAS PubMed.
- J. Wu, X. Li, X. Lian, B. Su, J. Pang, M. D. Li, Z. Xia, J. Z. Zhang, B. Luo and X. C. Huang, ACS Nano, 2021, 15, 15354–15361 CrossRef CAS PubMed.
- M. Li and Z. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- L. Mao, J. Chen, P. Vishnoi and A. K. Cheetham, Acc. Mater. Res., 2022, 3, 439–448 CrossRef CAS.
- J. S. Yao, J. J. Wang, J. N. Yang and H. B. Yao, Acc. Chem. Res., 2021, 54, 441–451 CrossRef CAS PubMed.
- X. Liu, J. Yang, W. Chen, F. Yang, Y. Chen, X. Liang, S. Pan and W. Xiang, Nano Res., 2022, 16, 5894–5899 CrossRef.
- B. Li, Y. Wang, Y. Xu and Z. Xia, Adv. Mater., 2025, 37, e2415483 CrossRef PubMed.
- J. Li, Q. Luo, J. Wei, L. Zhou, P. Chen, B. Luo, Y. Chen, Q. Pang and J. Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202405310 CrossRef CAS PubMed.
- Y. Bai, S. Zhang, N. Luo, B. Zou and R. Zeng, Nano Res., 2024, 17, 7768–7775 CrossRef CAS.
- X. Li, A. Liu, Z. Wang, Y. Wei, Q. Lin, Y. Chen, Y. Liu and M. Hong, Nano Res., 2023, 16, 10476–10482 CrossRef CAS.
- B. Lian, J. Yao, M. Ren, B. Zou, B. Luo and R. Zeng, J. Mater. Chem. C, 2024, 12, 2944–2952 RSC.
- X. Liu, X. Li, J. Li, X. Lian, Y. Xiao, R. Zeng, S.-F. Ni, K. Xu, Y. Kuai, W.-X. Ni and B. Luo, Inorg. Chem. Front., 2023, 10, 2917–2925 RSC.
- Q. Ren, G. Zhou, Y. Mao, N. Zhang, J. Zhang and X. M. Zhang, Chem. Sci., 2024, 15, 16536–16545 RSC.
- B. Luo, Y. Guo, Y. Xiao, X. Lian, T. Tan, D. Liang, X. Li and X. Huang, J. Phys. Chem. Lett., 2019, 10, 5271–5276 CrossRef CAS PubMed.
- T. Huang, K. Li, J. Lei, Q. Niu, H. Peng and B. Zou, Nano Res., 2023, 16, 12680–12688 CrossRef CAS.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
- Z. Chen, F. Zhang, D. Yang, H. Ji, X. Chen, D. Wu, X. Li, Y. Zhang and Z. Shi, Nano Res., 2024, 14, 3068–3078 CrossRef.
- K. Zhang, Z. Qi, N. Zhang, X. Zhao, Y. Fan, L. Sun, G. Zhou, S. L. Li and X. M. Zhang, Chem. Sci., 2025, 16, 6104–6113 RSC.
- C. Xing, Z. Qi, B. Zhou, D. Yan and W. H. Fang, Angew. Chem., Int. Ed., 2024, 63, e202402634 CrossRef CAS PubMed.
- X. Liu, C. Chen, Y. Xiao, X. Li, K. Xu, X. Lian, J. Z. Zhang and B. Luo, ACS Appl. Mater. Interfaces, 2024, 16, 31313–31321 CrossRef CAS PubMed.
- Y. Xie, J. Peng, Q. Qin and B. Luo, ACS Appl. Nano Mater., 2024, 7, 1319–1326 CrossRef CAS.
- J. Li, J. Wu, Y. Xiao, L. Rao, R. Zeng, K. Xu, X.-C. Huang, J. Z. Zhang and B. Luo, Inorg. Chem. Front., 2023, 10, 7167–7175 RSC.
- D. Y. Li, H. Y. Kang, Y. H. Liu, J. Zhang, C. Y. Yue, D. Yan and X. W. Lei, Chem. Sci., 2024, 15, 953–963 RSC.
- Y. Liu, Y. Wei, Z. Luo, B. Xu, M. He, P. Hong, C. Li and Z. Quan, Chem. Sci., 2024, 15, 15480–15488 RSC.
- C. E. Mulijanto, H. S. Quah, G. K. Tan, B. Donnadieu and J. J. Vittal, IUCrJ, 2017, 4, 65–71 CrossRef CAS PubMed.
- Q. Yu, X. Yang, Y. Chen, K. Yu, J. Gao, Z. Liu, P. Cheng, Z. Zhang, B. Aguila and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 10192–10196 CrossRef CAS PubMed.
- Y. X. Shi, W. H. Zhang, B. F. Abrahams, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2019, 58, 9453–9458 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC number: 2434257 (1), CCDC number: 2434258 (1a), CCDC number: 2435615 (1i). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03344f |
| ‡ C. Chen and Z. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |