
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoding the influence of monomer structures on the electrical double layer of alkaline fuel cells†
Xiao-Hui
Yang
ab,
Lin
Zhuang
c and
Jun
Cheng
 *abd
*abd
aState Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China
bNational Engineering Research Center of High-end Electronic Chemicals (Reconstructed), Xiamen University, Xiamen, 361005, China
cCollege of Chemistry and Molecular Sciences, Hubei Key Lab of Electrochemical Power Sources, Wuhan University, Wuhan, 430072, China
dInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen, China. E-mail: chengjun@xmu.edu.cn
First published on 24th June 2025
Abstract
Alkaline polymer electrolytes (APEs) are extensively used in fuel cells and electrolysers due to their safety and low-cost advantages. Recently, scientists have explored their potential as electrochemical reaction catalysts, allowing APEs to function as ion transport channels and actively participate in catalytic reactions, exhibiting bifunctional characteristics. However, the key challenge in designing such materials lies in ensuring that the catalytic groups embedded within the polymer backbone can interact effectively with the electrode surface or reaction sites. Failure to do so would render them unable to participate in the electrocatalytic reactions occurring at the interface. This necessitates a deep understanding of the electric double-layer (EDL) structure at the electrode/APE interface. Despite numerous studies on improving their stability, a general understanding of the interfacial EDL structure is still lacking. To address this gap, we adopted state-of-the-art simulation approaches, combining constant potential and finite field methods, to investigate the EDL structures of two APEs (QAPS and QAPPT) at different electrode potentials. Our research findings indicate that, although these two APEs contain the same cationic groups, they exhibit distinctly different EDL structures on negatively charged surfaces. Specifically, when the electrode was negatively charged, the phenylene unit on the backbone of QAPPT was perpendicular to the surface, while the backbone of QAPS was displaced away from the surface. This study underscores the importance of interfacial double layers in influencing the performance of polymer electrolytes, aiding in understanding the polarisation behaviour on charged surfaces of polymer electrolytes and guiding how to modulate functional groups on these APEs to achieve catalytic effects. Furthermore, this work provides an effective method for studying the EDL structure of electrode/APE interfaces, which can be widely applied to research on various complex interfaces.
Introduction
State-of-the-art fuel cell technologies often employ cation exchange membranes (CEMs), with Nafion membranes as a typical example,1 showing great energy conversion efficiency advantages.2 Alternatively, anion-exchange membranes (AEMs), which transport free hydroxide ions, offer cost advantages by enabling the use of non-noble metal catalysts and reducing catalyst poisoning in alkaline environments.3–7 Recent developments in AEM technology, such as improved chemical stability and ion conductivity, have enabled densities comparable to those achieved with CEMs.8–12 These advances suggest promising opportunities for AEMs, particularly in applications that require cost efficiency and reduced environmental impact.13 However, challenges such as maintaining stability under varying humidity and high operating temperatures require further investigation.14–17From an electrochemical perspective, polymers are among the most intricate electrolytes, as their properties depend on the molecular structure and type of monomers used.18,19 Over the past decade, researchers have focused on improving their macroscopic properties, such as conductivity, stability, and water uptake,20–23 which are essential for their application in fuel cells and electrolysers. These macroscopic properties are closely linked to the microstructural organisation of the polymer and the interfacial processes at the EDL, a key interfacial structure where electrochemical reactions occur.24 A deep understanding of the EDL structure, which governs ion distribution and local electrostatic interactions near the electrode surface, is crucial for enabling catalytic effects through structural modifications.4,25,26 Further investigation into EDL tailoring could facilitate the design of advanced polymer electrolytes for sustainable energy applications.24
The well-known Gouy-Chapman-Stern (GCS) model often fails to describe the complexity of the electrode/polyelectrolyte interface due to its inability to account for the unique molecular properties of polyelectrolytes.27 Specifically, the model overlooks the single-ion conduction behaviour, arising from immobilised counterions, and the diverse structural characteristics of monomers.28 At the core of this challenge is the structure of the EDL, which reflects the dynamic response of monomers to variations in electrode potential, including changes in charge distribution and molecular orientation.29,30 The molecular flexibility of the monomers, influenced by their intrinsic structural features such as bond rotational freedom, plays a crucial role in shaping the EDL structure.9 Consequently, polyelectrolytes with different flexibilities exhibit distinct ion distributions and interfacial phenomena, leading to a wide range of electrochemical behaviours on electrode surfaces.24,31
Understanding how differences in monomer composition and backbone rigidity influence the interfacial EDL structure is a central research question that offers valuable insights into polymer behaviour under varying electrode potentials.32 The monomer composition, such as the functional group polarity and charge density, directly impacts ion distributions, while backbone rigidity determines molecular flexibility and alignment, influencing the dynamic formation of the EDL.2,33,34 Accessing the EDL structure at the electrode/polyelectrolyte interface, however, presents significant challenges due to the nanoscopic scale of the system and the complexity of interfacial interactions. Current approaches, including electrochemical characterisation,35–37 molecular dynamics simulations,38–42 and adsorption energy calculations,43,44 have provided valuable insights but remain limited in capturing the dynamic, atomic-scale interactions inherent to such systems. Thus, methodologies capable of achieving atomic-level resolution while balancing computational cost are highly desirable.45–47
Among advanced methodologies, ab initio molecular dynamics (AIMD) stands out for its ability to explicitly resolve interfacial atomic structures at the quantum chemistry level. AIMD simulations have demonstrated exceptional accuracy in reproducing the EDL structure and water polarisation, providing critical insights into the dynamic behaviour of electrode interfaces.48–53 However, AIMD simulations require high computational cost, making them impractical for simulating large-scale electrode/polyelectrolyte systems. As a cost-effective alternative, classical molecular dynamics (CMD) simulations enable the investigation of larger systems while incorporating critical interfacial structures.42,47,54–57 By implementing enhancements such as polarisable electrode models and finite field methods,58,59 CMD simulations can capture electrode polarisation effects and control electrode potentials. This approach allows CMD to reveal atomic-scale changes in the EDL structure as a function of the electrode potential, providing accessible but detailed insights into interfacial behaviour at a fraction of the computational cost of AIMD.27,60
This work used CMD simulations with a polarisable electrode model and the finite field method to study APE interface structures. We chose graphite as the electrode, which is commonly used as the supporting electrode in fuel cells. Two commonly used alkaline polymers, quaternary ammonia polysulfone (QAPS) and quaternary ammonia poly(N-methyl-piperidine-co-p-terphenyl) (QAPPT), were selected together with OH− anions and water to form electrolytes (Fig. 1a). The choice of these two polymers is due to their representative nature and distinctive structural characteristics. QAPS features a main chain with several functional groups, allowing flexibility in its molecular structure. In contrast, the monomer of QAPPT incorporates a terphenyl structure, which imparts rigidity. When the electrode surface was negatively charged, a distinct interfacial structure emerged at the graphite/QAPPT interface, with the phenylene unit oriented perpendicular to the surface, while this interfacial structure was absent at the graphite/QAPS interface. This observation suggests that the rigidity and monomer composition of polymer backbones play a critical role in determining the interfacial EDL structures. We believe that these results provide new insights into the influence of the polymer structure on electrochemical interfaces, highlighting potential design strategies for optimising polymer electrolytes in energy applications.
 | ||
| Fig. 1 (a) Molecular structures of QAPS and QAPPT. (b) A snapshot of the graphite/QAPS model constructed in this work and corresponding elements in the model are also included. | ||
Computational details
The systems modelled in this study represent full electrochemical cells, as illustrated in Fig. 1a. Each cell comprises three layers of graphite electrodes and is filled with either QAPS or QAPPT polymers serving as the electrolyte. In our simulations, each QAPS electrolyte contains 6 polymer chains, with each chain composed of 30 monomers. Similarly, the QAPPT electrolyte consists of 5 polymer chains, each containing 30 monomers. To maintain consistency with experimental conditions, the hydration number (λ), defined as the ratio of water molecules to OH− anions, was set to λ = 20 based on data from ref. 61. Consequently, each QAPS electrolyte includes 3600 water molecules and 180 OH− anions, while the QAPPT electrolyte consists of 3000 water molecules and 150 OH− anions.We conducted simulations using the LAMMPS package,62 covering a range of cell voltages from 0 V to 6 V. To precisely control the cell voltages within the CMD simulations, we employed the constant potential method.58,63 This method utilises the Siepmann–Sprik potential model64 to describe the charge of each electrode atom using a Gaussian charge distribution. The magnitude of this distribution can be adjusted according to a variational procedure to maintain a constant electrostatic potential during the simulation, allowing the electrode charges to fluctuate in response to local charge variations of electrolyte ions to keep the potential constant.65 This method later was implemented as a plug-in code named USER-CONP2 by Tee and co-workers,66 further streamlining the management of voltage control within the LAMMPS package. Each simulation trajectory consisted of an initial pre-equilibrium period lasting more than 100 ns, followed by a production time of about 50 ns. The equilibrium was validated through two key properties: bulk densities and interfacial structures. To expedite the convergence of these properties, we employed techniques such as the annealing approach to optimise the initial structure67 and the multi-timescale rESPA method to speed up the simulation.68 Additionally, we employed the “piston approach” to implement the NPT ensemble within 2D periodic boundary conditions. Ultimately, our ensembles successfully replicated the expected bulk densities, and as long as the electrode potential remained fixed, the EDL structures proved independent of initial configurations.
Results and discussion
We initially modelled a graphite/QAPS system. Typically, only the cell potential is considered in a full electrochemical cell simulation. When examining the potential change of a single electrode, we adopted the conventional approach that uses the electrostatic potential of the electrolyte bulk phase as a reference. Given that polymers constitute a more heterogeneous electrolyte (Fig. 1b) compared to aqueous solutions, we constructed a large unit cell along the surface normal direction (18 nm) in the simulation to ensure better convergence of the electrostatic potential in the middle of the electrolyte. This area in the middle of the electrolyte was justified as the bulk region of the full electrochemical cell because the density deviation of this area at different electrode potentials was less than 2% (at the same time, the single component deviation was less than 5%); see the ESI† for details. By aligning the electrostatic potentials of the bulk region with different cell potentials, we plotted the potential profile of graphite/QAPS in Fig. 2a, and the thin black line in the centre represents the bulk region. | ||
| Fig. 2 (a) Potential profile of the graphite/QAPS system with respect to the potential of zero charge. (b) Schematic representation and density profiles of the interfacial QAPS structure, the corresponding water density profile is also included for comparison. | ||
The potential profile of graphite/QAPS (Fig. 2a) with respect to the potential of zero charge (PZC) allows us to investigate the interfacial double-layer structure at various potentials. Based on our electrochemical intuition and previous studies,60 when the electrode is positively charged, OH− anions tend to approach the electrode surface, while quaternary ammonium cations are repulsed from the electrode surface together with the backbone. Therefore, when the potential is higher than PZC, the EDL structure of the graphite/APE interface should be similar to the double-layer interface in a high-pH aqueous solution. In contrast, when the surface is negatively charged, quaternary ammonium cations tend to approach the electrode surface, while OH− anions move away from the surface. However, quaternary ammonium cations are fixed on the polymer chain, and OH− anions are already distant from the surface, so determining the EDL structure of graphite/APEs is challenging.
To study the EDL structure when the electrode surface is negatively charged, we analysed the density distribution of polymer and water molecules at negative potentials in the graphite/QAPS electrolyte system, as shown in Fig. 2b. Even if the electrode potential decreases from PZC to −2.6 V (vs. PZC), the density of QAPS does not change significantly. This is unexpected, as electrostatic attractions fail to pull the cations to the surface. One possible explanation is that distant cations bonded on the backbone cannot move freely and that the electrostatic interaction between the cations and the electrode is not strong enough to overcome the reorganisation energy of the polymer structure. Furthermore, when the electrode potential becomes more negative (−3.4 V), the density of QAPS near the surface appears to decrease and accumulate in regions ca. 8 Å away from the surface. At the same time, the density of water increases significantly near the surface. We speculate that the EDL changes at a very low electrode potential, and thus the backbone of QAPS is pushed away from the surface and occupied by water molecules. However, we still need the full picture at the atomic level to understand this sudden decrease in polymer density when the electrode potential is E = −3.4 V. To this end, we compared the EDL structures at different electrode potentials, as shown in Fig. 3.
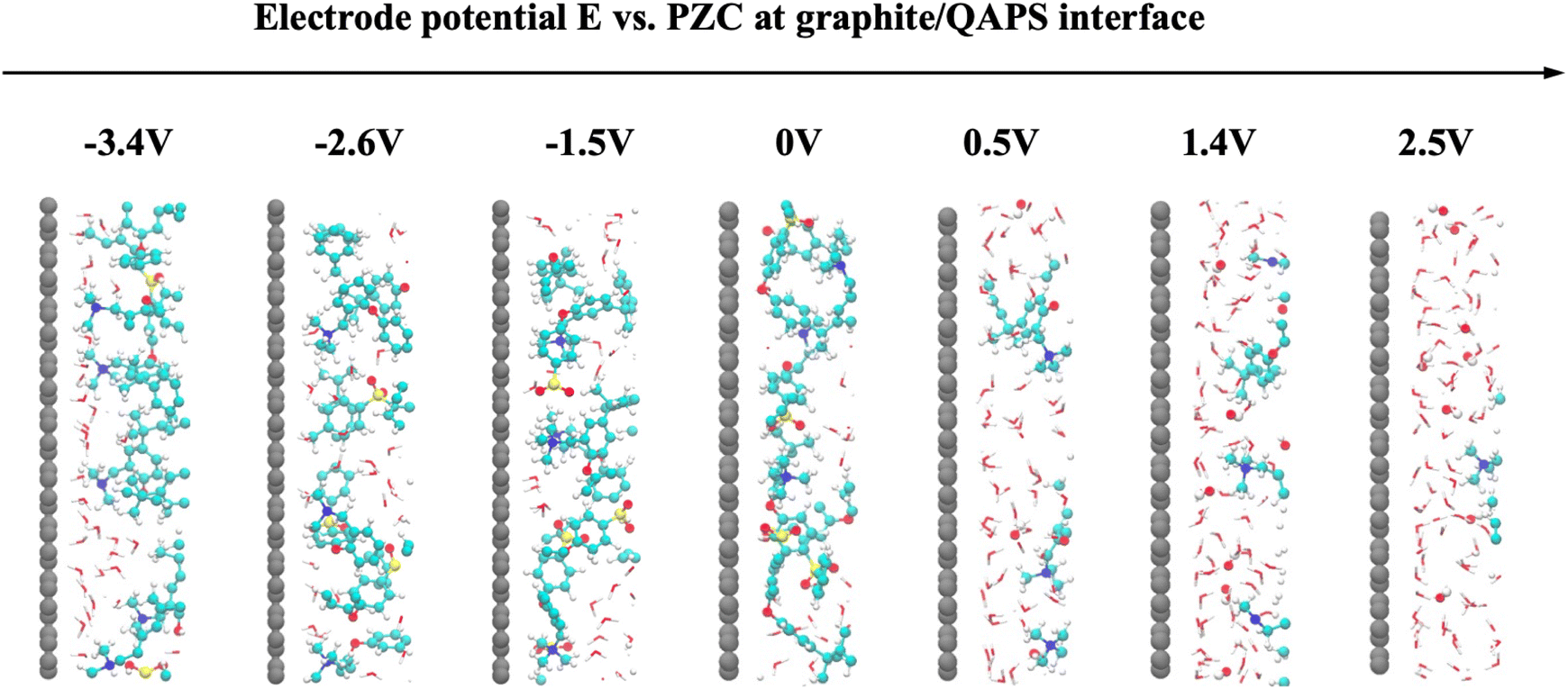 | ||
| Fig. 3 Enlarged snapshots of graphite/QAPS interfacial structures (within ca. 1.5 nm of the surface) at different electrode potentials. For a clear view of the polymer structures, only molecules within 1 nm thickness were included. | ||
The interfacial structure aligns with our electrochemical understanding of the graphite/QAPS interface when the electrode potential (E) is positive. In Fig. 3, no polymer is observed near the surface, which is consistent with the typical EDL structure described by the GCS model for the graphite/water solution interface. Conversely, when E < 0, quaternary ammonium cations accumulate near the surface, pulling the backbone structure of the polymer towards the surface. Interestingly, when the electrode potential decreases from −2.6 V to −3.4 V, more quaternary ammonium cations accumulate on the surface, and at the same time, the main chain structure is pushed away from the surface, consistent with the observed changes in QAPS density profiles in Fig. 2b. Therefore, with the help of atomic views in simulation, we determined the evident density decrease of the EDL at −3.4 V, which in turn facilitates the approach of as many cations as possible to the surface.
To validate whether the EDL structures in Fig. 2b and 3 apply to all polyelectrolyte systems, simulation of the graphite/QAPPT interface was also conducted. In this model, we expand the surface area from approximately 9 nm2 to 25 nm2 to sample a more extensive EDL structure. Similarly, we plotted the potential distribution profile for graphite/QAPPT (Fig. 4a), with the bulk region represented by the thin black line in the central region. In contrast to the graphite/QAPS interface, the polymer backbones of QAPPT remain near the surface at different electrode potentials, and the polymer density profile exhibits three distinct peaks in Fig. 4b. Interestingly, these three sharp peaks perfectly match the location of the carbon atoms when the phenylene unit is perpendicular to the surface. Furthermore, as the potential decreases from −0.7 V to −3.5 V, there is no gradual increase in density at the three peaks. From a statistical point of view, this means that the EDL structure undergoes little change when the potential drops from −0.7 V to −3.5 V. Therefore, even if the surface charge density is low (i.e., E = −0.7 V), the phenylene unit has transitioned to the perpendicular adsorbed style.
 | ||
| Fig. 4 (a) Potential profile of the graphite/QAPPT system with respect to the potential of zero charge. (b) Schematic representation and density profiles of the interfacial QAPPT structure, the corresponding water density profile is also included for comparison. | ||
We further studied the atomic view of the graphite/QAPPT interface structure at different electrode potentials (Fig. 5). When the electrode is positively charged, quaternary ammonium cations are repelled from the surface, resulting in an EDL structure at the graphite/QAPPT interface resembling the GCS model. At negative potentials, quaternary ammonium cations migrate toward the surface. Rather than being displaced in the graphite/QAPS system, the QAPPT backbone forms a regular pattern consisting of three layers of carbon atoms. This observation is consistent with the distribution in Fig. 4b, and the phenylene unit of the QAPPT maintains a perpendicular orientation relative to the electrode surface when E < 0.
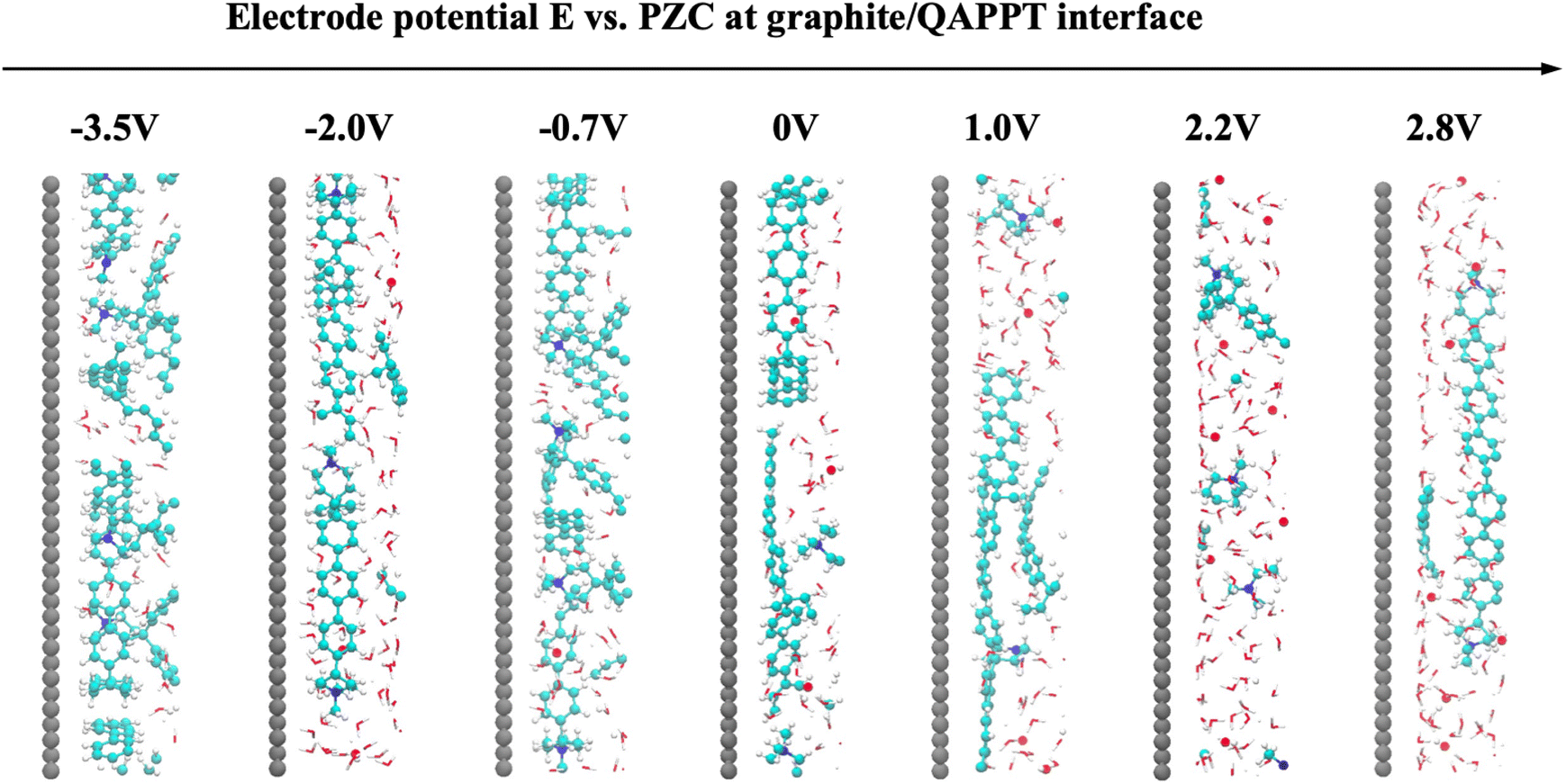 | ||
| Fig. 5 Enlarged snapshots of graphite/QAPPT interfacial structures (within ca. 1.5 nm of the surface) at different electrode potentials. For a clear view of the polymer structures, only molecules within 1 nm thickness were included. | ||
QAPS and QAPPT have shown different EDL structures at graphite surfaces, particularly when the electrode was negatively charged. We speculate that these different characteristics of EDL structures originate from their flexibility. Unlike the QAPS polymer, the C–C sigma bonds between the phenylene unit in the triphenyl group are difficult to rotate or bend, increasing the rigidity of the QAPPT backbone. Thus, we closely examined the structure of a single polymer chain at a cell voltage of 4 V. As shown in Fig. 6a and b, QAPS exhibits a high degree of rotational freedom, leading to increased flexibility. In contrast, the terphenyl structure in QAPPT restrains the degree of freedom, resulting in a rigid backbone structure. Though both monomers contain phenylene units, their flexibility differs significantly. Further analysis of the single polymer chain within a 1 nm range from the surface is presented in Fig. 6c and d. The flexible QAPS polymer chain can bend and rotate, allowing the fixed quaternary ammonium cations to approach the electrode surface readily. In contrast, because of the rigidity of QAPPT, quaternary ammonium cations are constrained to approach the surface alongside the triphenyl and pyridine rings. Only in this way can the quaternary ammonium cations accumulate close to the surface as the potential becomes more negative.
 | ||
| Fig. 6 (a) and (b) Snapshots of a single polymer chain in graphite/QAPS (a) and graphite/QAPPT (b) models when the cell voltage is 4 V. (c) and (d) The corresponding top and side views of the single polymer in (a) and (b) at the negatively charged interface. Note that the rest of the electrolyte species have been removed from snapshots for a clear view of polymer backbones. | ||
The findings in Fig. 6 provide valuable insight into the microscopic structural characteristics of APEs when the electrode is negatively charged. A subsequent question that arises is what electrode potential is required to achieve the vertical adsorption of the phenylene unit at the graphite/QAPPT interface. As shown in Fig. 5, at E = 0, some phenylene units were adsorbed parallel to the surface, and this structure decreases significantly at E = −0.7 V. To further investigate the adsorption structure of phenylene units at the surface under negative potential, we applied a cell voltage of 1.6 V to a PZC model. As shown in Fig. 7, the adsorption structure of the phenylene unit changed from parallel to vertical to the surface with the potential decrease from PZC to −0.7 V. Note that the capacitance is often underestimated in CMD simulation. According to our previous graphite/water interface model results,60 a surface charge density of −5.6 μC cm−2 (−0.7 V vs. PZC in CMD simulations) corresponds to an electrode potential of approximately −0.48 V vs. PZC in AIMD simulations. Therefore, a small voltage is applied to the electrodes, and an evident structural transition of triphenyl at the interface occurs. Please note that our force field in this work has properly accounted for the π–π interactions between the phenylene and the graphite surface.69 Therefore, we infer that once the electrode is negatively charged, the QAPPT polymer at the interface is adsorbed perpendicular to the surface.
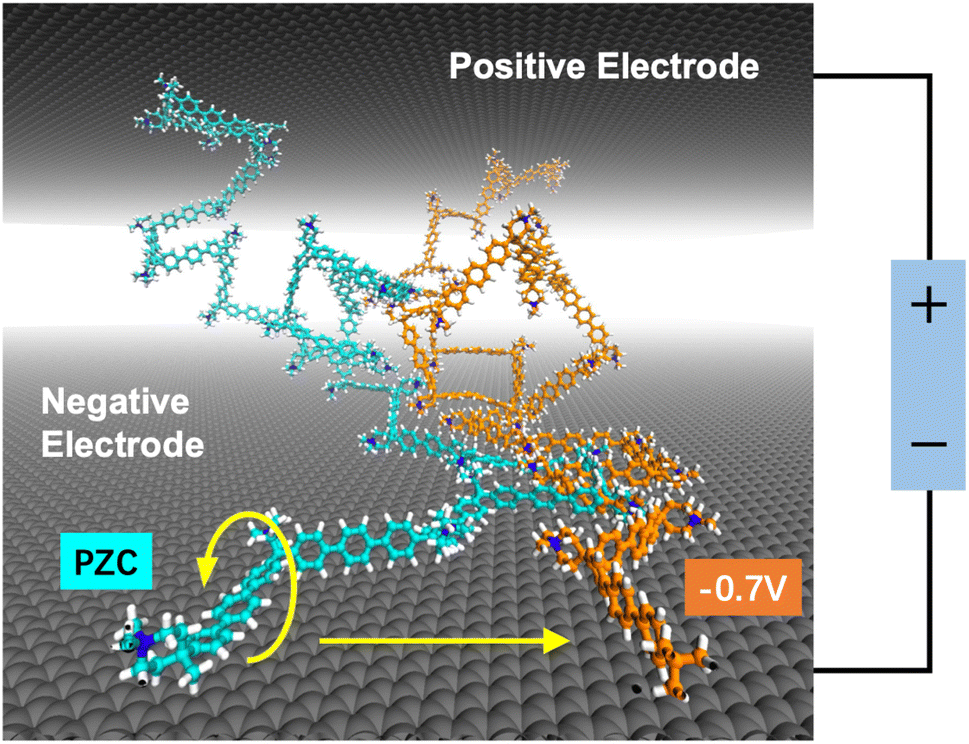 | ||
| Fig. 7 Snapshots of two polymer chains in the graphite/QAPPT model. The different colours represent the different electrode potentials they were experiencing. The structure in cyan is the polymer chain at PZC, while the structure in orange represents the polymer chain at an electrode potential of −0.7 V vs. PZC. | ||
From an atomic-level interface perspective, the rigid structure and perpendicular adsorption of QAPPT are inferred to create more space for water transport channels compared to QAPS. This configuration potentially exposes more active sites on the catalyst, leading to superior fuel cell performance compared to QAPS.3,10,70 In Fig. 8, we compared the water density of different APEs at the same electrode potential, and the result shows a higher water density in the QAPPT than in the QAPS, indicating that more water channels could be formed.
 | ||
| Fig. 8 Comparison of water densities in the EDL region when electrode potential is about −2.7 V vs. PZC. | ||
Our findings show that at charged electrode surfaces, the monomer structure can not only determine the flexibility of APEs, and thus contribute to the construction of stable gas/water channels, but also significantly affect the EDL structure. In Fig. 6d, although a comparable number of QAPPT monomers are adsorbed on the surface, they occupy a relatively smaller surface area compared to QAPS. Consequently, more active sites on the electrode surface remain exposed, providing greater opportunities to incorporate active groups through structural modifications of the monomer, thus improving catalytic performance in fuel cells or electrolysers.33,71
In realistic membrane electrode assembly (MEA) devices, various electrodes are often used, such as Cu for the CO2RR and Pt/Ru for the HOR. Please note that although the applied potential in our simulations exceeds the practical operating window of alkaline fuel cells, the simulation framework does not explicitly represent electrons and cannot capture faradaic processes such as hydrogen evolution or carbon corrosion. This provides mechanistic insights without inducing faradaic reactions, which are beyond the scope of classical MD. In the operational voltage range of the electrodes, the corresponding electrode could have a high charge density, significantly affecting the configuration and properties of the EDL. As presented in Fig. 2, the QAPS backbone moves away from the surface. This separation may hinder the functional groups from effectively interacting with the reactants, resulting in negligible catalytic activity. In contrast, the QAPPT backbone remains adhered to the surface among all negative potentials, as shown in Fig. 4. Thus, the QAPPT backbone can be functionalised with catalytic groups, as demonstrated by Zhuang and co-workers.11 Additionally, restricting the movement of the QAPPT backbone, making the polymer chain more rigid as a whole, can form more stable transport channels.61,72
Conclusions
In summary, this study utilised CMD simulations with a polarisable electrode model and the finite field method to investigate interfacial structures in graphite/polyelectrolyte systems. Focussing on two polymer electrolytes, QAPS and QAPPT, we analysed their behaviour at varying electrode potentials. The results demonstrate that differences in structural rigidity lead to distinct EDL structures for different polymer electrolytes. These findings suggest that previous studies may have overlooked the significant impact of monomer structures on EDL configurations, which could play a critical role in electrocatalysts. In addition to their great importance in the formation of table gas/water channels, the catalytic properties of bifunctional polymers are inherently linked to the monomer structure. Only in this way can the catalytic functional group approach the surface and contribute to the reaction, instead of being displaced away from the surface (e.g., the electrode/QAPS interface). This work underscores the importance of an atomic-level understanding and highlights how the simulation method can reveal the EDL structures of novel alkaline polymer electrolytes, aiding in the fundamental understanding of promising bifunctional monomers prior to experimental validation.Data availability
The data supporting this article have been included as part of the ESI.† (Given the large size of the output file, we have included the full input file within the ESI.† This enables the rapid generation of corresponding results whenever required).Author contributions
Conceptualization: X.-H. Y. and J. C.; investigation: X.-H. Y.; supervision: J. C. and L. Z.; writing – review & editing: X.-H. Y., J. C. and L. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grants No. 21991151, 21991150, 22402167, 22021001, 22225302, and 92161113), the Fundamental Research Funds for the Central Universities (20720220009 and 20720230090), the Laboratory of AI for Electrochemistry (AI4EC) and IKKEM (Grant No. RD2023100101 and RD2022070501).References
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed
.
- Y. Su, Y. Cheng, Z. Li, Y. Cui, C. Yang, Z. Zhong, Y. Song, G. Wang and L. Zhuang, J. Energy Chem., 2024, 88, 543–551 CrossRef CAS
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed
- J. Han, Y. Zhang, X. Zheng, Y. Lu, W. Li, X. Zhou, Z. Ren, Y. Liu, M. Hu, L. Xiao and L. Zhuang, ChemSusChem, 2024, 17, e202300985 CrossRef CAS PubMed
- Y. Xie, G. Wang, H. Tang, G. Wang, L. Xiao and L. Zhuang, J. Membr. Sci., 2024, 709, 123057 CrossRef CAS
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC
- J. Han, J. Pan, C. Chen, L. Wei, Y. Wang, Q. Pan, N. Zhao, B. Xie, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2019, 11, 469–477 CrossRef CAS PubMed
- Y. Gao, Y. Yang, R. Schimmenti, E. Murray, H. Peng, Y. Wang, C. Ge, W. Jiang, G. Wang, F. J. DiSalvo, D. A. Muller, M. Mavrikakis, L. Xiao, H. D. Abruña and L. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119883119 CrossRef CAS PubMed
- W. Li, Z. Yin, Z. Gao, G. Wang, Z. Li, F. Wei, X. Wei, H. Peng, X. Hu, L. Xiao, J. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS
- B. Huang, C. Lei, X. Sun, L. Luo, G. Wang, L. Zhuang and L. Xiao, Int. J. Hydrogen Energy, 2023, 48, 35453–35462 CrossRef CAS
- X. Tian, R. Ren, F. Wei, J. Pei, Z. Zhuang, L. Zhuang and W. Sheng, Nat. Commun., 2024, 15, 76 CrossRef CAS PubMed
- J. Ma, D. Meng, Y. Zhang, H. Ma, Z. Ren, J. Zhang, L. Xiao, L. Zhuang, L. Li and L. Chen, J. Power Sources, 2023, 587, 233687 CrossRef CAS
- Q. Li, M. Hu, C. Ge, Y. Yang, L. Xiao, L. Zhuang and H. D. Abruña, Chem. Sci., 2023, 14, 10429–10434 RSC
- T. Ning, Y. Hu, R. Ren, Z. Yi, G. Wang, L. Xiao, J. Lu and L. Zhuang, J. Membr. Sci., 2023, 685, 121900 CrossRef CAS
- Chinese Society of Electrochemistry, J. Electrochem., 2024, 30, 2024121 Search PubMed
- H. Lim, I. Jeong, J. Choi, G. Shin, J. Kim, T.-H. Kim and T. Park, Appl. Surf. Sci., 2023, 610, 155601 CrossRef CAS
- Y. Zhou, H. Yu, F. Xie, Y. Zhao, X. Sun, D. Yao, G. Jiang, J. Geng and Z. Shao, Int. J. Hydrogen Energy, 2023, 48, 5266–5275 CrossRef CAS
- S. Chen, H. Wang, J. Zhang, S. Lu and Y. Xiang, J. Membr. Sci., 2020, 605, 118105 CrossRef CAS
- J. Pan, H. Zhu, H. Cao, B. Wang, J. Zhao, Z. Sun and F. Yan, J. Membr. Sci., 2021, 620, 118794 CrossRef CAS
- S. Willdorf-Cohen, A. Zhegur-Khais, J. Ponce-Gonzaález, S. Bsoul-Haj, J. R. Varcoe, C. E. Diesendruck and D. R. Dekel, ACS Appl. Energy Mater., 2023, 6, 1085–1092 CrossRef CAS PubMed
- B. Huang, X. Wang, W. Li, W. Tian, L. Luo, X. Sun, G. Wang, L. Zhuang and L. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202304230 CrossRef CAS PubMed
- F. Dorchies, A. Serva, D. Crevel, J. D. Freitas, N. Kostopoulos, M. Robert, O. Sel, M. Salanne and A. Grimaud, J. Am. Chem. Soc., 2022, 144, 22734–22746 CrossRef CAS PubMed
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H.-Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abrun a, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed
- K. Goloviznina, E. Bendadesse, O. Sel, J.-M. Tarascon and M. Salanne, ACS Appl. Mater. Interfaces, 2023, 15, 59380–59388 CrossRef CAS PubMed
- Y.-X. Yang, X.-H. Yang, M.-L. Huang, L.-W. Wu, Z. Liu, J. Cheng and Y.-F. Huang, J. Phys. Chem. Lett., 2024, 15, 701–706 CrossRef CAS PubMed
- Z. Mi, T. Wang, L. Xiao, G. Wang and L. Zhuang, J. Am. Chem. Soc., 2024, 146, 17377–17383 CrossRef CAS PubMed
- J.-B. Le, A. Chen, Y. Kuang and J. Cheng, Natl. Sci. Rev., 2023, 10, nwad105 CrossRef CAS PubMed
- Y.-H. Wang, X. Jin, M. Xue, M.-F. Cao, F. Xu, G.-X. Lin, J.-B. Le, W.-M. Yang, Z.-L. Yang, Y. Cao, Y. Zhou, W. Cai, Z. Zhang, J. Cheng, W. Guo and J.-F. Li, Joule, 2023, 7, 1652–1662 CrossRef CAS
- L. Xue, Z. Gao, T. Ning, W. Li, J. Li, J. Yin, L. Xiao, G. Wang and L. Zhuang, Angew. Chem., Int. Ed., 2023, 62, e202309519 CrossRef CAS PubMed
- C.-X. Liu, Z.-P. Zou, M.-X. Hu, Y. Ding, Y. Gu, S. Liu, W.-J. Nan, Y.-C. Ma, Z.-B. Chen, D.-P. Zhan, Q.-G. Zhang, L. Zhuang, J.-W. Yan and B.-W. Mao, J. Electrochem., 2024, 30, 2303151 Search PubMed
- Y. Li, S. Intikhab, A. Malkani, B. Xu and J. Snyder, ACS Catal., 2020, 10, 7691–7698 CrossRef CAS
- Z. Gao, L. Xue, X. Hu, J. Yin, L. Xiao, G. Wang, J. Lu and L. Zhuang, Electrochim. Acta, 2023, 458, 142509 CrossRef CAS
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Adv. Energy Mater., 2023, 13, 2203488 CrossRef CAS
- Y. Ding, C.-X. Liu, Y.-C. Ma, M.-X. Hu, S. Liu, Y. Gu, L. Zhuang, Q.-G. Zhang, B.-W. Mao and J.-W. Yan, ACS Appl. Mater. Interfaces, 2023, 15, 31057–31066 CrossRef CAS PubMed
- G. Wang, G. Wang, L. Zhuang and L. Xiao, Electrochim. Acta, 2024, 510, 145348 CrossRef
- C. Chen, Y.-L. S. Tse, G. E. Lindberg, C. Knight and G. A. Voth, J. Am. Chem. Soc., 2016, 138, 991–1000 CrossRef CAS PubMed
- R. Jinnouchi, K. Kudo, K. Kodama, N. Kitano, T. Suzuki, S. Minami, K. Shinozaki, N. Hasegawa and A. Shinohara, Nat. Commun., 2021, 12, 4956 CrossRef CAS PubMed
- H.-J. Chen, M.-H. Tang and S.-L. Chen, J. Electrochem., 2023, 29, 2207061 Search PubMed
- S. Ntim and M. Sulpizi, J. Phys. Chem. B, 2024, 128, 1936–1942 CrossRef CAS PubMed
- S. Bi, L. Knijff, X. Lian, A. v. Hees, C. Zhang and M. Salanne, ACS Nano, 2024, 18, 19931–19949 Search PubMed
- M. Ünlü, D. Abbott, N. Ramaswamy, X. Ren, S. Mukerjee and P. A. Kohl, J. Electrochem. Soc., 2011, 158, B1423–B1431 Search PubMed
- I. T. McCrum, M. A. Hickner and M. J. Janik, J. Electrochem. Soc., 2018, 165, F114–F121 Search PubMed
- N. Ramaswamy and S. Mukerjee, Chem. Rev., 2019, 119, 11945–11979 CrossRef CAS PubMed
- T. E. Gartner and A. Jayaraman, Macromolecules, 2019, 52, 755–786 CrossRef CAS
- X. Yang, Y. Zhuang, J. Zhu, J. Le and J. Cheng, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 12, e1559 Search PubMed
- J.-L. Li, Y.-F. Li and Z.-P. Liu, J. Electrochem., 2022, 28, 2108511 Search PubMed
- M. Karibayev, S. Kalybekkyzy, Y. Wang and A. Mentbayeva, Molecules, 2022, 27, 3574 Search PubMed
- Z. Zhu and S. J. Paddison, Front. Chem., 2022, 10, 981508 CrossRef CAS PubMed
- A. Chen, J.-B. Le, Y. Kuang and J. Cheng, J. Chem. Phys., 2022, 157, 094702 Search PubMed
- P. Vogel, N. Moöller, M. N. Qaisrani, P. Bista, S. A. L. Weber, H.-J. Butt, B. Liebchen, M. Sulpizi and T. Palberg, J. Am. Chem. Soc., 2022, 144, 21080–21087 Search PubMed
- Y. Sun, C.-R. Wu, F. Wang, R.-H. Bi, Y.-B. Zhuang, S. Liu, M.-S. Chen, K. H.-L. Zhang, J.-W. Yan, B.-W. Mao, Z.-Q. Tian and J. Cheng, Chem. Sci., 2024, 15, 12264–12269 RSC
- C. Bacon, A. Serva, C. Merlet, P. Simon and M. Salanne, Electrochim. Acta, 2023, 455, 142380 Search PubMed
- S. Ntim and M. Sulpizi, Phys. Chem. Chem. Phys., 2023, 25, 22619–22625 RSC
- C. Zhang, J. Cheng, Y. Chen, M. K. Y. Chan, Q. Cai, R. P. Carvalho, C. F. N. Marchiori, D. Brandell, C. M. Araujo, M. Chen, X. Ji, G. Feng, K. Goloviznina, A. Serva, M. Salanne, T. Mandai, T. Hosaka, M. Alhanash, P. Johansson, Y.-Z. Qiu, H. Xiao, M. Eikerling, R. Jinnouchi, M. M. Melander, G. Kastlunger, A. Bouzid, A. Pasquarello, S.-J. Shin, M. M. Kim, H. Kim, K. Schwarz and R. Sundararaman, J. Phys. Energy, 2023, 5, 041501 Search PubMed
- W. Pu and Z. Sun, Phys. Chem. Chem. Phys., 2025, 27, 4634–4642 RSC
- T. Dufils, G. Jeanmairet, B. Rotenberg, M. Sprik and M. Salanne, Phys. Rev. Lett., 2019, 123, 195501 CrossRef CAS PubMed
- P. Lemaire, A. Serva, M. Salanne, G. Rousse, H. Perrot, O. Sel and J.-M. Tarascon, ACS Appl. Mater. Interfaces, 2022, 14, 20835–20847 CrossRef CAS PubMed
- X.-Y. Li, X.-F. Jin, X.-H. Yang, X. Wang, J.-B. Le and J. Cheng, J. Chem. Phys., 2023, 158, 084701 CrossRef CAS PubMed
- J. Pan, C. Chen, Y. Li, L. Wang, L. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 7, 354–360 RSC
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. i. t. Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 Search PubMed
- L. Scalfi, T. Dufils, K. G. Reeves, B. Rotenberg and M. Salanne, J. Chem. Phys., 2020, 153, 174704 CrossRef CAS PubMed
- J. I. Siepmann and M. Sprik, J. Chem. Phys., 1995, 102, 511–524 CrossRef CAS
- S. K. Reed, O. J. Lanning and P. A. Madden, J. Chem. Phys., 2007, 126, 084704 CrossRef PubMed
- S. R. Tee and D. J. Searles, J. Chem. Phys., 2022, 156, 184101 CrossRef CAS PubMed
- B. V. Merinov and W. A. Goddard, J. Membr. Sci., 2013, 431, 79–85 Search PubMed
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef CAS
- J. Björk, F. Hanke, C.-A. Palma, P. Samori, M. Cecchini and M. Persson, J. Phys. Chem. Lett., 2010, 1, 3407–3412 CrossRef
- Q. Li, H. Peng, Y. Wang, L. Xiao, J. Lu and L. Zhuang, Angew. Chem., Int. Ed., 2019, 58, 1442–1446 Search PubMed
- K. Zhao, M. Luo, Y. Zhang, X. Chang and B. Xu, Nat. Catal., 2025, 1–12 CAS
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2014, 8, 177–181 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Force field parameters, simulation details, and density profiles of graphene/QAPS and graphene/QAPPT full cell simulations at different electrode potentials. See DOI: https://doi.org/10.1039/d5sc00492f |
| This journal is © The Royal Society of Chemistry 2025 |