
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free vs. metal-catalyzed cyclotrimerization of didehydro[8]annulenes (COTynes): a complex pathway to non-planar PAHs – Dewar benzenes vs. benzotri[8]annulenes†
Jesús
Bello-García
 ,
Jesús A.
Varela
* and
Carlos
Saá
*
,
Jesús A.
Varela
* and
Carlos
Saá
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: carlos.saa@usc.es; jesus.varela@usc.es
First published on 8th May 2025
Abstract
The formation of non-planar PAHs from didehydro[8]annulene (COTyne) cycloadditions was investigated under both transition metal-catalyzed (Pd, Ru) and metal-free conditions. The observed reactivity depended on the planarity of the COTyne and the reaction conditions. Parent COTyne 1a dimerized into naphthocyclooctatetraene under TM-free conditions, whereas Pd(0) catalysis promoted its cyclotrimerization into benzotri[8]annulene 7. X-ray characterization and its dynamic behavior in solution was investigated. Planar dibenzoCOTyne 1b exhibited different reactivity depending on its formation method (in situ or preformed), the metal catalyst (Pd, Ru), and absence of catalysts. Under Pd(0) catalysis, cyclotrimerization yielded benzo-fused tri(dibenzo[8]annulene) 3 with moderate efficiency, regardless of how 1b was generated. The presence of K+ had no significant effect compared to tribenzoCOTyne 1c. Without metal catalysts, 1b predominantly formed the corresponding Dewar benzene derivative 2. With Ru(II) catalysts, reactivity was influenced by both the generation method of 1b and the Cp ligand. When generated in situ, 1b was an inefficient ligand for CpRu, leading to Dewar benzene formation, whereas preformed 1b produced 3 in moderate yields. The competition between Dewar benzene and benzo-fused tri(dibenzo[8]annulene) formation increased with greater steric hindrance at the Ru center (CpRu vs. Cp*Ru catalysts). Dewar benzene formation likely proceeds via a cyclobutadiene intermediate followed by cycloaddition.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) have garnered increasing attention since the discovery of planar graphene1 structures, which exhibit exceptional electronic and optical properties.2 The synthesis of planar PAHs typically requires regioselective fusion of multiple rings. Over the years, significant progress has been made in addressing this challenge.1,2c,3 Metal-catalyzed cycloadditions have proven particularly effective, as they efficiently form aromatic rings – examples include triphenylene4 and biphenylene cores,5 as well as helicenes6 and helicene-like molecules.7 More recently, non-planar (curved)8 PAHs with bowl-9 and saddle-shaped10 geometries have emerged as fascinating systems, as they can modify and improve the electronic and optical properties of their planar counterparts (Fig. 1). | ||
| Fig. 1 Benzannulated PAHs and non-planar bowl- and saddle-shaped PAHs. | ||
Although the metal-catalyzed trimerization ([2 + 2 + 2] cycloadditions) of planar arynes is well established (Scheme 1A),11 the cyclotrimerization of cycloocta-1,3,5-trien-7-ynes (COTynes or didehydro[8]annulenes) and their symmetrical benzo-fused derivatives (benzoCOTynes) into benzo-fused tri[8]annulenes via transition metal (TM) catalysis has been scarcely investigated.12 The few reported cases have produced puzzling and unexpected outcomes, as stated below.
 | ||
| Scheme 1 (A) Cyclotrimerizations of cyclic alkynes: arynes, dibenzoCOTynes and tribenzoCOTynes. (B) Transition-metal-free and metal-catalyzed cycloadditions of COTyne 1a and dibenzoCOTyne 1b. | ||
The structure of cubic graphite containing benzoannulated eight-membered rings – a hypothetical three-dimensional sp2-carbon allotrope with intriguing ion transport properties – has become one of the most attractive synthetic targets for exploring cyclotrimerizations.13 In this context, Müllen and coworkers, in their pursuit of a molecular model of cubic graphite, reported that in situ generated saddle-shaped COTyne 1d (phenanthrene-dibenzofused COTyne) could be trimerized into the (α,α,β)- or (α,α,α) stereoisomers of tri[8]annulene 4 (TDBPA) using two different transition metal catalysts, Pd(dba)2 and [CpRu(CH3CN)3]PF6 (Scheme 1A).14 More recently, our group demonstrated that in situ generated saddle-shaped tribenzoCOTyne 1c can be selectively trimerized into the non-interconvertible (α,α,β)- or (α,α,α)-benzofused tris(tribenzo[8]annulenes) 5 (TTBA) via Pd-catalyzed or K+-mediated processes, respectively (Scheme 1A). DFT calculations have been used to rationalize the selective formation of the (α,α,α)-stereoisomer, which represents a true open fragment of the proposed cubic graphite structure under K+-mediated conditions.15
In contrast, Nuckolls and coworkers previously reported the formation of Dewar benzene derivative 2 (DB) and the (α,α,β)-stereoisomer of benzofused tri(dibenzo[8]annulene) 3 (TDBA) from planar dibenzoCOTyne 1b using [CpRu(CH3CN)3]PF6 as the catalyst (Scheme 1A).16 Unfortunately, no clear mechanistic explanation was provided to account for the observed selectivity of Ru-catalyzed reactions involving both planar dibenzo- and non-planar tribenzoannulated COTynes.
From these results, it appears that both the reaction conditions and the geometry of the cyclic alkynes (COTynes), ranging from saddle-shaped to nearly planar (depending on the absence or presence of benzoannelated rings), determine the reaction outcome. To clarify these aspects and address the previously puzzling results on COTyne cyclotrimerizations, we present here the intriguing structure-dependent reactivity of COTynes, cycloocta-1,3,5-trien-7-yne (COTyne 1a) and 5,6-didehydrodibenzo[a,e][8]annulene (dibenzoCOTyne 1b), in their respective cyclotrimerizations to non-planar PAHs (Scheme 1B).
Results and discussion
COTyne 1a
Based on previously published results, it remains unclear whether the planar intermediate COTyne 1a, generated from bromoCOT 8 (ref. 17) in the presence of KOtBu, undergoes cyclotrimerization or cyclodimerization. The cyclotrimerization of COTyne 1a has been suggested based on EPR spectroscopic measurements of the tri[8]annulenylene radical. However, doubts persist due to the lack of a described synthesis procedure and spectroscopic data for the proposed cyclotrimer.18In contrast, Krebs previously reported that a similar treatment of 8 with KOtBu yielded a dimeric product of the COTyne intermediate, naphthoCOT 6, in low yield, rather than the trimeric annulene 7.19 To resolve these conflicting results and determine whether dimerization or trimerization occurs under basic conditions, we subjected bromoCOT 8 to the same basic conditions. We obtained naphthoCOT 6 in low yield (20%), with no trace of the corresponding benzotri[8]annulene 7 (Scheme 2). The molecular structure of naphthoCOT 6 was determined by X-ray diffraction analysis.20
 | ||
| Scheme 2 Transition-metal free and Pd-catalyzed cycloadditions of COTyne 1a. ORTEP drawing of naphthoCOT 6 and benzotri[8]annulene 7 showing thermal ellipsoids at the 50% probability level, H atoms have been omitted for clarity. | ||
We then explored transition metal-catalyzed processes. To our delight, treatment of bromoCOT 8 under our optimized Pd-catalyzed trimerization conditions for tribenzoCOTyne15 successfully yielded the expected (α,α,β)-benzotri[8]annulene 7 in 33–35% yield (Scheme 2). The (α,α,β) conformational structure of 7 was verified by X-ray crystallography20 and further supported by the presence of nine signals in its 1H NMR spectrum in o-C6Cl2D4 solution, indicating a symmetry plane. The signals of 7 were assigned based on 1H and 13C NMR data, as well as 2D HSQC, HMBC and COSY spectra (Fig. 2 and ESI†).
 | ||
| Fig. 2 VT-1H NMR spectra of (α,α,β)-benzotri[8]annulene 7. | ||
To investigate conformational exchange among the three degenerate conformers of (α,α,β)-benzotri[8]annulene 7, variable-temperature NMR (VT-NMR) spectroscopy was used to study the dynamics of the (α,α,β) isomer (Fig. 2). As expected, the nine signals in the 1H NMR spectrum at room temperature merged into three signals – two doublets and one singlet – when heated to 388 K. This change occurred due to rapid exchange between conformers, resulting in an averaged signal for each proton type.
The coalescence temperature (Tc) for selected protons (6,7 and 3,4) at 348 K allowed us to estimate the activation free energy (ΔG‡) for the flipping of the cyclooctatetraene ring in 7 as 17.8 kcal mol−1.21 DFT calculations yielded a theoretical ΔG‡ of 18.4 kcal mol−1 for the interconversion between degenerate (α,α,β)-7 conformers and 19.9 kcal mol−1 for the interconversion between (α,α,β) and (α,α,α) isomers. The (α,α,β) isomer was found to be 1.8 kcal mol−1 more stable than the (α,α,α) isomer.22
DibenzoCOTyne 1b
Next, we analyzed the influence of benzo-fused rings on the reactivity of COTyne. Treatment of vinyl triflate 9 (ref. 23) under similar Pd-catalyzed conditions yielded benzofused tri(dibenzo[8]annulene) 3 (TDBA) in low yield (25%, Table 1, entry 1). This result clearly indicates that the dibenzoCOTyne intermediate 1b is less reactive than the COTyne 1a in Pd-catalyzed trimerizations. Although the X-ray structure of the (α,α,β)-TDBA 3 had been previously reported, the NMR spectra were not described.16b Our results confirm the (α,α,β)-conformer, as indicated by the presence of 15 signals in the 1H NMR spectrum and 24 signals in the 13C NMR spectrum, 9 of which correspond to quaternary carbons, suggesting a plane of symmetry.22|
|
|||
|---|---|---|---|
| Entry | Starting material | Catalyst (mol%) | Product (yield)b |
| a Conditions: SM (0.2 mmol), with or w/o [M], with or w/o KOtBu in THF (0.4 M) at rt. b Isolated yield. c DCM instead of THF. d THF (0.4 M) at rt. e THF (0.4 M) at rt, in the presence of K[B(ArF5)4] (1.5 equiv.). f THF (0.4 M) at rt, in the presence of KOtBu (1.5 equiv.). | |||
| 1 | 9 | Pd2(dba)3 (5) | 3 (25%) |
| 2c | 9 | [CpRu(CH3CN)3]PF6 (10) | 10 (40%) |
| 3 | 9 | [CpRu(CH3CN)3]PF6 (10) | 2 (44%) |
| 4 | 9 | — | 2 (60%) |
| 5d | 1b | — | 2 (17%) |
| 6e | 1b | — | 2 (17%) |
| 7f | 1b | — | 11 (50%) |
| 8 | 1b | Pd2(dba)3 (5) | 3 (40%) |
| 9c | 1b | [CpRu(CH3CN)3]PF6 (10) | 3 (30%) + 2 (13%) |
| 10c | 1b | [Cp*Ru(CH3CN)3]PF6 (10) | 3 (8%) + 2 (33%) |
| 11c | 1b | Cp*RuCl(cod) (10) | 2 (52%) |

Surprisingly, when vinyl triflate 9 was reacted under basic conditions with [CpRu(CH3CN)3]PF6 as a catalyst, we obtained moderate yields of either ketone 10 (40% in DCM) or DB 2 (44% in THF) (Table 1, entries 2 and 3),24 but no TDBA 3 was detected. Interestingly, in a control experiment without a Ru catalyst, treatment of vinyl triflate 9 under basic conditions at room temperature produced DB 2 in a relatively high yield of 60% (Table 1, entry 4), establishing an alternative, transition metal-free route to this product.25
The formation of dibenzoCOTyne 1b appears to be involved in this process, as a THF solution of preformed 1b26 generated DB 2, albeit in a surprisingly low yield of 17% (Table 1, entry 5).27 The effect of K+ salts was then investigated.15 A similar outcome was observed when potassium salt K[BArF5]4 was added to a THF solution of 1b (Table 1, entry 6). However, when the reaction was conducted in the presence of KOtBu, the primary product was vinyl ether 11 (50%, Table 1, entry 7), suggesting that 1b was captured in this form.22
As expected, the Pd-catalyzed trimerization of dibenzoCOTyne 1b produced the (α,α,β)-TDBA 3 in an improved 40% yield (Table 1, entries 1 vs. 8), demonstrating that efficient cyclotrimerization requires alkyne coordination to Pd. Similar to Ru-catalyzed trimerizations, the presence of alkynes is essential for a successful cyclotrimerization.12b
In our hands, subjecting dibenzoCOTyne 1b to [CpRu(CH3CN)3]PF6 10 mol%, DCM, rt, yielded tri[8]annulene 3 as the major product (30%) and DB 2 as the minor product (13%) (Table 1, entry 9). However, using the more electron-rich and sterically hindered catalyst, [Cp*Ru(CH3CN)3]PF6, resulted in an inverted product ratio, with 3 as the minor product (8%) and DB 2 as the major product (33%) (Table 1, entry 10). Since the overall yield remained nearly unchanged, the steric hindrance at the Ru center is likely the key factor influencing this outcome.
Furthermore, employing the neutral catalyst Cp*RuCl(cod) completely inhibited the cyclotrimerization of 1b, yielding DB 2 as the sole isolated product (Table 1, entry 11).
Variable-temperature VT-1H NMR studies on (α,α,β)-TDBA 3 showed no significant variations within the examined temperature range (298–373 K, Fig. S1, ESI†), indicating no interconversion between isomers. DFT calculations further supported this, revealing very high activation free energy barriers (ΔG‡) of 44.8 kcal mol−1 for interconversion between degenerate (α,α,β) conformers and 53.0 kcal mol−1 for transformation from the (α,α,β) to the (α,α,α) isomer. Additionally, the (α,α,β) isomer was found to be 3.2 kcal mol−1 more stable than the (α,α,α) isomer.22
Transition metal-free cyclotrimerization of dibenzoCOTyne 1b: formation of Dewar benzene derivative 2 (DB)
The thermal dimerization and cyclotrimerization of linear substituted alkynes are well-documented processes, both experimentally and through DFT calculations.28 These calculations suggest that the cyclotrimerization of the cyclic alkyne dibenzoCOTyne 1b proceeds through the initial formation of a cyclobutadiene (CyBu) intermediate via a stepwise [2 + 2] diradical cycloaddition of two dibenzoCOTyne 1b units (Scheme 3a). This CyBu intermediate then undergoes another stepwise [2 + 2] diradical cycloaddition with a third dibenzoCOTyne 1b molecule, leading to the formation of the “cyclophane” DB 2 as the final cyclotrimer. Notably, DB 2 remains stable and does not undergo the typical ring-opening to the corresponding benzene, since this transformation would generate a highly strained p-[6]-cyclophane.28a,29 | ||
| Scheme 3 Plausible transition metal-free routes to DB 2: (a) from dibenzoCOTyne 1b; (b) from vinyl triflate 9 + KOtBu. | ||
Notably, entries 4 and 5 in Table 1 reveal that the yield of DB 2 is higher when dibenzoCOTyne 1b is generated in situ under basic conditions (entry 4) compared to when preformed/isolated 1b is used (entry 5). This observation prompted an investigation into an alternative pathway for the dimerization/trimerization leading to Dewar benzene formation. It is plausible that the iso-CyBu cyclobutadiene isomer could form directly from the initial vinyl triflate 9 under basic conditions (Scheme 3b). These two valence isomers can interconvert via a relatively low-energy barrier,30 and capturing this iso-CyBu isomer in a favorable [4 + 2] cycloaddition with dibenzoCOTyne 1b would directly yield DB 2.
Interestingly, unlike C sp3-annelated cyclobutadienes, no tetramerization of the CyBu or iso-CyBu cyclobutadiene intermediates was detected.31 Furthermore, cyclotrimerization to form dibenzotri[8]annulene 3 was also not observed,32 suggesting that the formation of the alternative “non-cyclophane” DB 2′ intermediate is not favored.33
Mechanistic hypotheses for Pd(0)- and Ru(II)-catalyzed cyclotrimerizations
Pd-catalyzed cyclotrimerization ([2 + 2 + 2] cycloadditions) of arynes is a well-established method for synthesizing triphenylene and its benzo-fused derivatives.11c This process involves the formation of a palladacycle intermediate (a square planar palladacyclopentadiene), followed by the insertion of a third alkyne unit and a final reductive elimination. Arynes are typically generated in situ from their corresponding o-(trimethylsilyl)triflates precursors in the presence of fluoride sources.A similar Pd-catalyzed cyclotrimerization occurs when the parent COTyne 1a and dibenzoCOTyne 1b – either preformed or generated in situ from their bromo- and triflate COT precursors 8 and 9 – are involved. Due to the non-planar structure of COTs in palladacycles, conformers must be taken into account. Additionally, the reactivity of COTynes is inversely related to the number of fused benzene rings (i.e., the more strained the COTyne, the more reactive it is).
From COTyne 1a, two possible palladacycle intermediates can form: one where both COT units are on the same face (α,α) and another where they are on opposite faces (α,β). These intermediates can interconvert through a simple flap process. Given the low steric hindrance of the unsubstituted COT ring, the insertion of a third alkyne unit should ultimately yield the thermodynamically favored (α,α,β)-tri[8]annulene 7 (Fig. 3).
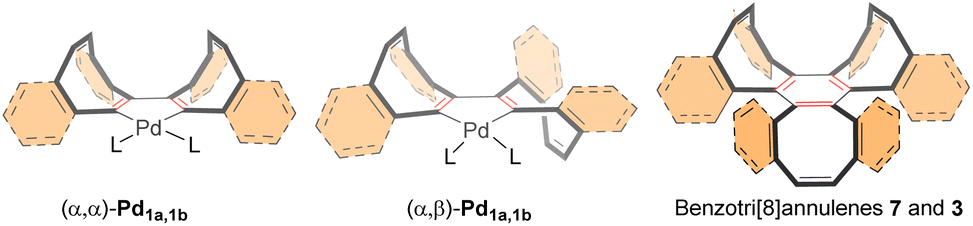 | ||
| Fig. 3 Palladacycle intermediates on the cyclotrimerization of COTyne 1a and dibenzoCOTyne 1b. | ||
A similar mechanism is expected for the formation of tri[8]annulene 3 when dibenzoCOTyne 1b (preformed or generated in situ from 9) participates in Pd-catalyzed cyclotrimerization (Fig. 3). However, in this case, significant steric hindrance from the benzofused rings could impede the COT core flap process.
According to the accepted mechanism for the Ru(II)-catalyzed alkyne cyclotrimerization,34 two possible intermediates – ruthenacyclopentadiene or ruthenacyclobiscarbene – can form, adopting either an (α,α) or (α,β) configuration. In both cases, the Cp ligand is positioned on the opposite face of the tetrahedral geometry around the Ru center (Fig. 4).35 Following coordination of the third alkyne (dibenzoCOTyne), a formal [2 + 2] cycloaddition occurs, placing the new benzene rings of the incoming dibenzoCOTyne on the opposite face of those already present in the ruthenacyclopentadiene/ruthenacyclobiscarbene intermediate. As a result, the formation of (α,α,β)-dibenzotri[8]annulene (TDBA) 3 is favored, regardless of whether the initial intermediate adopts an (α,α) or (α,β) configuration (Fig. 4).
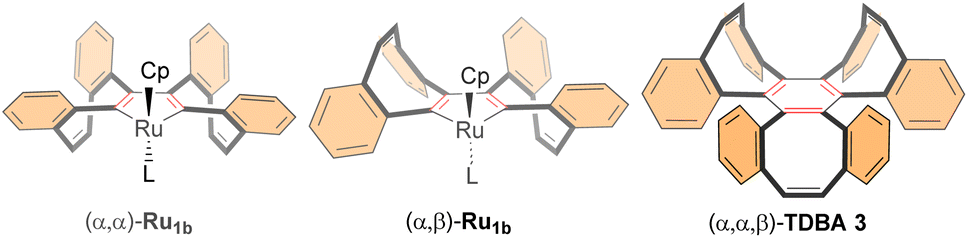 | ||
| Fig. 4 Ruthenacycle intermediates on the cyclotrimerization of dibenzoCOTyne 1b. | ||
For Ru(II)-catalyzed cyclotrimerization of preformed dibenzoCOTyne 1b (Table 1, entries 9–11), the formation of DB 2 – either predominantly (in Cp*Ru complexes) or partially – may originate from the trapped iso-CyBu cyclobutadiene isomer. This intermediate could be generated through reductive elimination from a ruthenacyclobiscarbene species.36
Conclusions
In summary, we have investigated the cycloadditions of COTynes under both catalyzed (Pd, Ru) and non-catalyzed conditions. The observed reactivity depended on the nature of the COTyne and the reaction conditions. The parent COTyne 1a led to the dimerization product, naphthocyclooctatetraene 6, under thermal conditions, whereas in the presence of Pd(0) catalysts, it underwent cyclotrimerization to form benzotri[8]annulene 7.For the planar dibenzoCOTyne 1b, its reactivity varied based on its mode of formation (in situ or preformed), the choice of metal catalyst (Pd, Ru), and the absence of catalysts. Under Pd catalysis, cyclotrimerization yielded benzo-fused tri(dibenzo[8]annulene) 3 in moderate yields, regardless of how dibenzoCOTyne 1b was generated. In contrast, under transition metal-free conditions, dibenzoCOTyne 1b consistently evolved into Dewar benzene derivative 2 (DB), although yields varied. No beneficial effect of K+ on its reactivity was observed.
Interestingly, the reactivity of dibenzoCOTyne 1b under Ru(II) catalysis depended on its formation mode and the steric properties of the Cp ligand at the Ru center. When generated in situ, 1b was unable to coordinate effectively to the CpRu catalyst, leading to DB 2 as the predominant product. However, when preformed dibenzoCOTyne 1b was used with a CpRu(II) catalyst, cyclotrimerization proceeded in moderate yields to afford benzo-fused tri(dibenzo[8]annulene) 3. The competition between DB 2 and tri[8]annulene 3 formation was more pronounced with increased steric hindrance at the Ru center (CpRu vs. Cp*Ru catalysts).
The formation of DB 2 likely proceeds through an initial cyclobutadiene intermediate, either a radical or ionic mechanism, depending on the reaction conditions, followed by cycloaddition with dibenzoCOTyne 1b.
Functionalizations and further applications of benzotribenzo-fused tri[8]annulenes are currently in progress in our lab.
Data availability
All experimental data associated with this work are provided in the ESI.† Crystallographic data for 6 and 7 have been deposited at the CCDC under 2418247 and 2427818, respectively, and can be obtained from http://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.Author contributions
J. Bello-García performed the experimental work and data analysis. J. A. Varela and C. Saá conceived the project, supervised the investigation and wrote the manuscript. All authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from MICIU (projects PID2023-151279NB-I00 and ORFEO-CINQA network RED2022-134287-T), the Xunta de Galicia (project ED431C 2022/27 and Centro singular de investigación de Galicia accreditation 2023–2027, ED431G 2023/03) and the European Union (European Regional Development Fund – ERDF). We are also grateful to the CESGA (Xunta de Galicia) for computational time. J. B.-G. thanks MICIN for a predoctoral contract.Notes and references
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- (a) Carbon-Rich Compounds: From Molecules to Materials, Wiley-VCH Verlag GmbH & Co. KGaA, M. M. Haley and R. R. Tykwinski, 2006 Search PubMed; (b) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (c) Q. Miao, Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015 CrossRef.
- (a) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef; (c) H. Ito, Y. Segawa, K. Murakami and K. Itami, J. Am. Chem. Soc., 2019, 141, 3–10 CrossRef CAS; (d) C. Tönshoff and H. F. Bettinger, Chem.–Eur. J., 2021, 27, 3193–3212 CrossRef.
- (a) D. Peña, D. Pérez and E. Guitián, Chem. Rec., 2007, 7, 326–333 CrossRef; (b) K. Murayama, Y. Sawada, K. Noguchi and K. Tanaka, J. Org. Chem., 2013, 78, 6202–6210 CrossRef CAS; (c) J.-A. García-López and M. F. Greaney, Org. Lett., 2014, 16, 2338–2341 CrossRef PubMed.
- (a) B. C. Berris, Y.-H. Lai and K. P. C. Vollhardt, J. Chem. Soc., Chem. Commun., 1982, 953–954 RSC; (b) B. C. Berris, G. H. Hovakeemian, Y. H. Lai, H. Mestdagh and K. P. C. Vollhardt, J. Am. Chem. Soc., 1985, 107, 5670–5687 CrossRef CAS; (c) R. Diercks and K. P. C. Vollhardt, Angew. Chem., Int. Ed. Engl., 1986, 25, 266–268 CrossRef; (d) J. M. Schulman and R. L. Disch, J. Am. Chem. Soc., 1996, 118, 8470–8474 CrossRef CAS; (e) C. Dengiz, Beilstein J. Org. Chem., 2023, 19, 1895–1911 CrossRef CAS PubMed.
- (a) J. Caeiro, D. Peña, A. Cobas, D. Pérez and E. Guitián, Adv. Synth. Catal., 2006, 348, 2466–2474 CrossRef CAS; (b) K. Tanaka, N. Fukawa, T. Suda and K. Noguchi, Angew. Chem., Int. Ed., 2009, 48, 5470–5473 CrossRef CAS; (c) T. Shibata, T. Uchiyama, Y. Yoshinami, S. Takayasu, K. Tsuchikama and K. Endo, Chem. Commun., 2012, 48, 1311–1313 RSC; (d) Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083 CrossRef CAS; (e) B. Heller, M. Hapke, C. Fischer, A. Andronova, I. Starý and I. G. Stará, J. Organomet. Chem., 2013, 723, 98–102 CrossRef CAS; (f) A. Jančařík, J. Rybáček, K. Cocq, J. V. Chocholoušová, J. Vacek, R. Pohl, L. Bednárová, P. Fiedler, I. Císařová, I. G. Stará and I. Starý, Angew. Chem., Int. Ed., 2013, 52, 9970–9975 CrossRef PubMed; (g) T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2017, 139, 18512–18521 CrossRef CAS; (h) A. Yubuta, T. Hosokawa, M. Gon, K. Tanaka, Y. Chujo, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2020, 142, 10025–10033 CrossRef CAS PubMed.
- (a) S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223–3227 CrossRef CAS; (b) S. Han, D. R. Anderson, A. D. Bond, H. V. Chu, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3227–3230 CrossRef CAS; (c) Y. Kimura, N. Fukawa, Y. Miyauchi, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 8480–8483 CrossRef CAS PubMed; (d) G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 18456–18459 CrossRef CAS PubMed; (e) S. H. Pun, Y. Wang, M. Chu, C. K. Chan, Y. Li, Z. Liu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 9680–9686 CrossRef CAS; (f) S. Kinoshita, R. Yamano, Y. Shibata, Y. Tanaka, K. Hanada, T. Matsumoto, K. Miyamoto, A. Muranaka, M. Uchiyama and K. Tanaka, Angew. Chem., Int. Ed., 2020, 59, 11020–11027 CrossRef CAS.
- (a) M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC; (b) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS.
- (a) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed; (b) I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaña, Chem. Commun., 2018, 54, 6705–6718 RSC; (c) Y. Duan, M. Chen, H. Hayashi, H. Yamada, X. Liu and L. Zhang, Chem. Sci., 2023, 14, 10420–10428 RSC.
- G. González Miera, S. Matsubara, H. Kono, K. Murakami and K. Itami, Chem. Sci., 2022, 13, 1848–1868 RSC.
- (a) F. Schlütter, T. Nishiuchi, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1538–1542 CrossRef PubMed; (b) J. B. Lin, T. K. Shah, A. E. Goetz, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 10447–10455 CrossRef CAS; (c) For a review, see: I. Pozo, E. Guitián, D. Pérez and D. Peña, Acc. Chem. Res., 2019, 52, 2472–2481 CrossRef CAS; (d) For [2 + 2] cyclodimerization of benzynes to biphenylenes, see: J. Lu, J. Zhang, X. Shen, D. M. Ho and R. A. Pascal, J. Am. Chem. Soc., 2002, 124, 8035–8041 CrossRef CAS; (e) B. Schuler, S. Collazos, L. Gross, G. Meyer, D. Pérez, E. Guitián and D. Peña, Angew. Chem., Int. Ed., 2014, 53, 9004–9006 CrossRef CAS; (f) S.-L. Wang, M.-L. Pan, W.-S. Su and Y.-T. Wu, Angew. Chem., Int. Ed., 2017, 56, 14694–14697 CrossRef CAS; (g) T. Ikawa, Y. Yamamoto, A. Heguri, Y. Fukumoto, T. Murakami, A. Takagi, Y. Masuda, K. Yahata, H. Aoyama, Y. Shigeta, H. Tokiwa and S. Akai, J. Am. Chem. Soc., 2021, 143, 10853–10859 CrossRef CAS PubMed; (h) D. Ehjeij, F. Rominger, U. H. F. Bunz, J. Freudenberg and K. Müllen, Angew. Chem., Int. Ed., 2024, 63, e202312040 CrossRef CAS.
- (a) H. N. C. Wong, P. J. Garratt and F. Sondheimer, J. Am. Chem. Soc., 1974, 96, 5604–5605 CrossRef CAS; (b) For metal complexes of cyclooctynes, see: M. A. Bennett and H. P. Schwemlein, Angew. Chem., Int. Ed. Engl., 1989, 28, 1296–1320 CrossRef; (c) J.-W. Han, X. Li and H. N. C. Wong, Chem. Rec., 2015, 15, 107–131 CrossRef CAS.
- (a) J. Gibson, M. Holohan and H. L. Riley, J. Chem. Soc., 1946, 456–461 RSC; (b) R. H. Baughman and C. Cui, Synth. Met., 1993, 55, 315–320 CrossRef CAS; (c) A. J. Berresheim, M. Müller and K. Müllen, Chem. Rev., 1999, 99, 1747–1786 CrossRef CAS PubMed.
- B. Ejlli, P. Nußbaum, F. Rominger, J. Freudenberg, U. H. F. Bunz and K. Müllen, Angew. Chem., Int. Ed., 2021, 60, 20220–20224 CrossRef CAS PubMed.
- J. Bello-García, J. A. Varela and C. Saá, Angew. Chem., Int. Ed., 2024, 63, e202414017 CrossRef PubMed.
- (a) M. Carnes, D. Buccella, J. Decatur, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2008, 47, 2982–2985 CrossRef CAS; (b) M. Carnes, D. Buccella, T. Siegrist, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2008, 130, 14078–14079 CrossRef CAS PubMed.
- A. K. Pati, O. El Bakouri, S. Jockusch, Z. Zhou, R. B. Altman, G. A. Fitzgerald, W. B. Asher, D. S. Terry, A. Borgia, M. D. Holsey, J. E. Batchelder, C. Abeywickrama, B. Huddle, D. Rufa, J. A. Javitch, H. Ottosson and S. C. Blanchard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24305–24315 CrossRef CAS.
- (a) M. K. Kiesewetter, R. C. Reiter and C. D. Stevenson, J. Am. Chem. Soc., 2004, 126, 8884–8885 CrossRef CAS; (b) C. D. Stevenson, Acc. Chem. Res., 2007, 40, 703–711 CrossRef CAS PubMed.
- (a) A. Krebs and D. Byrd, Justus Liebigs Ann. Chem., 1967, 707, 66–74 CrossRef CAS; (b) A. Krebs, Angew. Chem., Int. Ed. Engl., 1965, 4, 953–954 CrossRef.
- CCDC 2418247 (for 6) and 2427818 (for 7) contain the supplementary crystallographic data for this paper.
- D. Kost, E. H. Carlson and M. Raban, J. Chem. Soc. D, 1971, 656–657 RSC.
- See ESI† for more details.
- S. Chaffins, M. Brettreich and F. Wudl, Synthesis, 2002, 2002, 1191–1194 Search PubMed.
- M. J. Marsella, Acc. Chem. Res., 2002, 35, 944–951 CrossRef CAS.
- Dewar benzene derivative 2 had previously been reported when preformed dibenzoCOTyne 1b was treated with either Grubbs' second-generation carbene (traces) or [CpRu(CH3CN)3]PF6 catalysts (14% yield, DCM). See ref 16b.
- It could be isolated in 90% yield starting from triflate 9 under basic conditions. See ESI† and ref 16b for more details.
- By contrast, a solution of 1b in DCM led to a complex mixture.
- (a) Z.-K. Yao and Z.-X. Yu, J. Am. Chem. Soc., 2011, 133, 10864–10877 CrossRef CAS PubMed; (b) S. Fabig, G. Haberhauer and R. Gleiter, J. Am. Chem. Soc., 2015, 137, 1833–1843 CrossRef CAS PubMed.
- M. Dračínský, O. Castaño, M. Kotora and P. Bouř, J. Org. Chem., 2010, 75, 576–581 CrossRef.
- C. Gellini and P. R. Salvi, Symmetry, 2010, 2, 1846–1924 CrossRef CAS.
- G. Wittig and U. Mayer, Chem. Ber., 1963, 96, 342–348 CrossRef CAS.
- D. Peña, B. Iglesias, I. Quintana, D. Pérez, E. Guitián and L. Castedo, Pure Appl. Chem., 2006, 78, 451–455 CrossRef.
- G. Wittig and J. Weinlich, Chem. Ber., 1965, 98, 471–479 CrossRef CAS.
- (a) E. Rüba, R. Schmid, K. Kirchner and M. J. Calhorda, J. Organomet. Chem., 2003, 682, 204–211 CrossRef; (b) Y. Yamamoto, T. Arakawa, R. Ogawa and K. Itoh, J. Am. Chem. Soc., 2003, 125, 12143–12160 CrossRef CAS PubMed.
- Other complexes with the Cp group on the same face are likely less favorable, as they increase steric hindrance during oxidative coupling.
- (a) L. F. Veiros, G. Dazinger, K. Kirchner, M. J. Calhorda and R. Schmid, Chem.–Eur. J., 2004, 10, 5860–5870 CrossRef CAS PubMed; (b) N. V. Shvydkiy and D. S. Perekalin, Coord. Chem. Rev., 2017, 349, 156–168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2418247 and 2427818. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03035h |
| This journal is © The Royal Society of Chemistry 2025 |