
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetecting microsatellite instability in cancer via multiplexed orthogonal gap-enhanced Raman tags†
Guowei
Fu
ab,
Jin
Li
 *b,
Qian
Zhang
c,
Changjun
Lv
c,
Zhiyang
Zhang
b,
Xiaoyan
Wang
d,
Rihui
Wu
*a and
Lingxin
Chen
*be
*b,
Qian
Zhang
c,
Changjun
Lv
c,
Zhiyang
Zhang
b,
Xiaoyan
Wang
d,
Rihui
Wu
*a and
Lingxin
Chen
*be
aGuangdong Provincial Key Laboratory of Large Animal Models for Biomedicine, School of Pharmacy and Food Engineering, Wuyi University, 529020 Jiangmen, China. E-mail: wyuchemwrh@126.com
bCAS Key Laboratory of Coastal Environmental Processes and Ecological Remediation, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, 264003 Yantai, China. E-mail: jinli@yic.ac.cn; lxchen@yic.ac.cn
cDepartment of Respiratory and Critical Care Medicine, Binzhou Medical University Hospital, Binzhou Medical University, Binzhou 256603, China
dSchool of Pharmacy, Binzhou Medical University, 264003 Yantai, China
eCollege of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing 312000, China
First published on 9th May 2025
Abstract
Microsatellite instability (MSI) is a hallmark of colorectal cancer in immunotherapy, whose phenotypes mainly involve four mismatch repair (MMR) proteins (MlH1, MSH2, MSH6 and PMS2). Since these MMR proteins are highly interdependent, simultaneous detection of these proteins rather than separate detection in cancer is vital to accurately distinguish the MSI phenotypes. In this study, we fabricated four orthogonal gap-enhanced Raman tag (O-GERT) flavors with high sensitivity, superb photostability, and completely separated interference-free signal readouts. With antibody functionalization, these multicolored O-GERTs allowed one-shot detection of these four MMR proteins in cancer tissues with high specificity and spectral resolution. Based on quantitative Raman imaging, these cancer tissues were classified into microsatellite stable (MSS) or high-frequency MSI (MSI-H) subtypes. The detected MSI-H ratios for colorectal, breast and gastric cancers were 13.3%, 6.7% and 3.3%, respectively. Moreover, the correlation between the expression levels of these MMR proteins in colorectal cancer and related clinicopathologic parameters in these subtypes was established for the first time. We further demonstrated that MSI in cancer can serve as a tool for screening Lynch syndrome (a genetic disorder) and predicting potential candidates for immunotherapy by PD1/PD-L1 blockade. To the best of our knowledge, this is the first example of quantitative multiplexed Raman imaging for fast detection of MSI in cancer.
1 Introduction
Microsatellites are regions within the human genome characterized by short, repetitive DNA sequences, typically composed of 1 to 6 nucleotide repeats.1,2 Microsatellite instability (MSI), primarily caused by genetic alterations in repetitive DNA sequences due to dysfunction of the DNA mismatch repair (MMR) system, is a hallmark of many cancers, such as colorectal cancer (CRC).3–5 CRC is a malignant tumor developed in the colon or rectum, and is regarded as one of the most prevalent malignancies worldwide that poses a significant threat to human health.6,7 When the function of the MMR system in CRC is abnormal, the replication errors in microsatellites are not corrected and continuously accumulated, resulting in changes in microsatellite sequence length or base composition (Scheme 1a). MSI phenotypes are closely associated with four MMR proteins, including MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6) and postmeiotic segregation increased 2 (PMS2).8,9 Among them, MLH1 and MSH2 are the core components of the MMR system, whereas PMS2 and MSH6 form heterodimers with them (MLH1-PMS2 and MSH2-MSH6), respectively. Therefore, loss of MLH1 or MSH2 affects expression of PMS2 or MSH6, while absence of PMS2 or MSH6 generally does not affect the former two.10,11 Generally, if all four MMR proteins are concurrently expressed, the tumor is identified as microsatellite stable (MSS); otherwise, it is classified as high-frequency MSI (MSI-H) in which at least one MMR protein is lost. Since these four MMR proteins are highly interdependent, simultaneous detection of these proteins rather than separate detection in cancer is vital to accurately distinguish the MSI phenotypes (MSS or MSI-H), which plays a key role in screening Lynch syndrome12–14 as well as identifying potential beneficiaries of immunotherapy. | ||
| Scheme 1 (a) Schematic diagram of MSI in CRC, i.e. loss or gain of nucleotides from repetitive sequences. (b) Traditional IHC detection method for four MMR proteins. (c) Schematic representation of one-shot Raman imaging using multiplexed O-GERTs for MSI detection, and (d) the results were used to screen Lynch syndrome and identify potential beneficiaries of immunotherapy by programmed cell death protein 1 (PD1) blockade. | ||
Currently, immunohistochemistry (IHC) is commonly used to detect various MMR proteins for the diagnosis of MSI phenotypes in CRC (Scheme 1b). However, this semiquantitative method can only detect one target separately at a time, suffering from low throughput and poor correlation of the results.15–19 Surface-enhanced Raman scattering (SERS) spectroscopy is one of the ultrasensitive techniques down to a single-molecule detection level, benefiting from electromagnetic hotspots from plasmonic nanoparticles (NPs) that amplify the scattered signals of the attached Raman molecules.20–25 More importantly, SERS shows ultra-narrow fingerprint spectra (linewidth ca. 2 nm) with great multiplexing capability. With these merits, SERS probes have been widely utilized for various high throughput sensing and imaging applications from in vitro, ex vivo to in vivo.26–29 For example, Trau' group presented a digital nanopillar SERS platform for the simultaneous attomolar detection of four prospective cytokine biomarkers to enable dynamic tracking of immune toxicities in blood.30 Li et al. demonstrated the use of ten-color SERS nanoprobes with high spectral resolution for imaging immune checkpoint networks in breast cancer tissues to predict combination drugs.31 Noonan and co-workers reported in vivo targeted imaging of multiple vascular biomarkers using SERS NPs in a humanized mouse model, offering promise for SERS as a clinical imaging tool for cardiovascular disease in the future.32 However, to date, utility of multiplexed SERS probes for imaging MMR proteins for MSI phenotypes hasn't been reported.
In this work, we fabricated multiplexed orthogonal gap-enhanced Raman tags (O-GERTs)33 with four completely separated Raman bands in the biological silent region (1800 to 2400 cm−1) for detection of MSI in cancer tissues (Scheme 1c). By embedding orthogonal Raman reporters in interior nanogaps of ∼1 nm, these O-GERTs displayed high sensitivity, superb photostability and interference-free readouts. With antibody conjugation, these multicolored O-GERTs allowed for simultaneous quantitative detection of these four MMR proteins in various cancer tissues with ∼1 h sample-to-answer time. According to these quantitative Raman imaging, these cancer tissues were fast diagnosed as MSS or MSI-H subtypes. Moreover, for the first time, we established the correlation between the expression levels of these MMR proteins and related clinicopathologic parameters in these subtypes. These MSI phenotypic distinctions further served as a tool for screening Lynch syndrome and predicting the potential beneficiaries of immunotherapy (Scheme 1d) to facilitate personalized therapy of the subtypes.
2 Results and discussion
2.1 Design of the four multiplexed O-GERTs
O-GERTs are core–shell nanostructures that consist of orthogonal Raman reporters embedded within a gold (Au) core and shell. In this work, we employed four orthogonal reporters to prepare multicolored O-GERT flavors. The molecular structures of these orthogonal reporters are shown in Fig. 1a, including s-(4-ethynylphenyl)ethanethioate (SEE), s-(4-((trimethylsilyl)ethynyl)phenyl) ethanethioate (STE), s-(4-(phenylethynyl)phenyl)ethanethioate (SPE) and 2-mercapto-4,5,6,7-d4-benzimidazole (MDBM). These reporters bore alkyne or deuterium for signal readouts, and possessed ethanethioate or sulfhydryl groups for Au anchoring. To prepare O-GERTs, Raman reporters (SEE, STE, SPE and MDBM) were first anchored on Au cores of 20 nm by Au–S bonds, respectively (Fig. 1b). Then, Au shells of desired thickness were formed on the surface of the reporter–core conjugates by reduction of HAuCl4 to obtain multiplexed O-GERTs. These four reporter-embedded nanoparticle (NP) flavors were referred to as SEE O-GERTs, STE O-GERTs, SPE O-GERTs, and MDBM O-GERTs, respectively.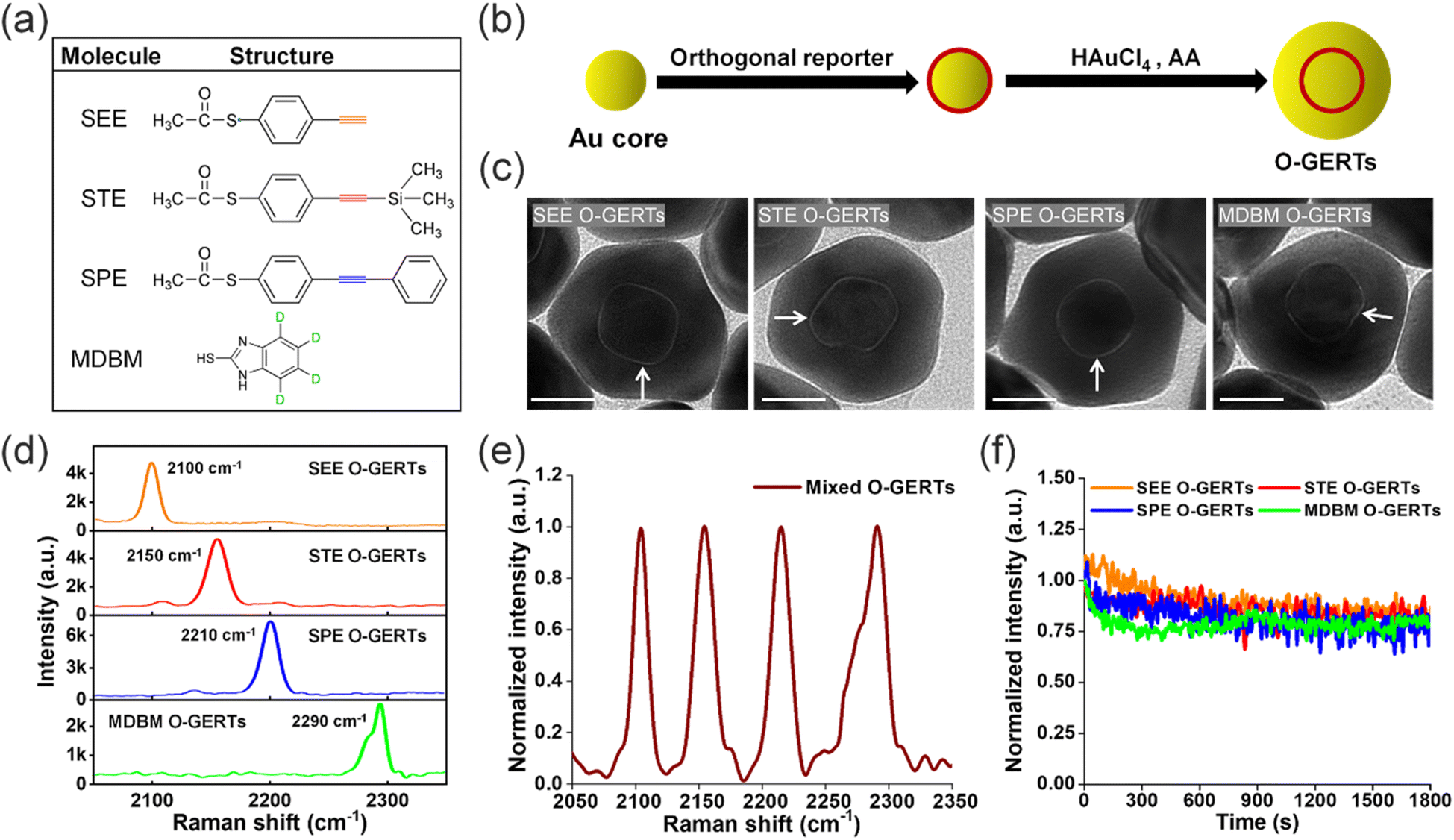 | ||
Fig. 1 (a) Structure of the orthogonal reporter molecule. (b) The synthesis process of O-GERTs. (c) TEM images of SEE O-GERTs, STE O-GERTs, SPE O-GERTs and MDBM O-GERTs. Arrows indicate internal nanogaps of O-GERTs. Scale bars = 20 nm (d) Raman spectra of the four O-GERTs at the same concentration of 0.4 nM. (e) Normalized Raman spectrum of a mixture of four O-GERTs with a molar ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3:1.0:3.0 under 785 nm excitation. Raman spectra of all O-GERTs were obtained at an exposure time of 1.0 s. (f) Evolution of normalized Raman intensities of the four O-GERTs during continuous irradiation with a 785 nm laser for 30 min. Note that a slight increase in the intensity of the SEE O-GERTs at the beginning of irradiation might have been caused by the increase in the NP density within the laser spot. :1.3:1.0:3.0 under 785 nm excitation. Raman spectra of all O-GERTs were obtained at an exposure time of 1.0 s. (f) Evolution of normalized Raman intensities of the four O-GERTs during continuous irradiation with a 785 nm laser for 30 min. Note that a slight increase in the intensity of the SEE O-GERTs at the beginning of irradiation might have been caused by the increase in the NP density within the laser spot. | ||
The core–shell structure of these O-GERTs was characterized by high-resolution transmission electron microscopy (TEM), which suggested that the average gaps between core–shell layers in these four O-GERTs were 0.9, 1.0, 1.0, and 0.9 nm, respectively (Fig. 1c and S1†). The characteristic Raman bands of four O-GERTs in the silent region under 785 nm excitation were located at 2100, 2150, 2210 and 2290 cm−1, respectively (Fig. 1d), due to stretching vibrations of the alkyne or deuterium groups. The full spectra of all O-GERTs, including the fingerprint region (600 to 2500 cm−1), were shown in Fig. S2.† The mixed Raman spectrum of these four O-GERTs with a molar ratio of 1.5:1.3:1.0:3.0 revealed that these bands in the silent region were completely separated without any spectral crosstalk (Fig. 1e), implying that these nanoprobes could realize accurately quantitative imaging of four MMR proteins. It should be noted that the non-resonant excitation of the O-GERTs with a near-infrared (NIR) laser of 785 nm (the nanoprobe absorbs at around 550 nm) not only avoids the photothermal effect, but also facilitates the subsequent detection in living mice in terms of the high penetration depth of the NIR laser.34,35 These four O-GERTs were triply diluted into a series of solutions, respectively, and were subjected to Raman imaging to determine the limits of detection (LODs) (see Methods for details). Under 785 nm excitation, the LODs of these four O-GERTs were estimated to be 0.33, 0.99, 0.11, 2.97, respectively (Fig. S3†), more than tens of times that of conventional SERS probes (80 pM) that are composed of Au NPs of 60 nm and attached crystal violet (CV) reporters on the surface (abbreviated as AuNP@CV NPs). Such high sensitivity of the O-GERTs was attributed to the combined contribution from the electromagnetic hotspots in the nanogaps and chemical charge transport effect, which improved scattered cross-sections of the Raman reporters.36
We further delved into the photostability of these O-GERTs. The O-GERTs solution samples were deposited onto a wafer and dried, followed by continuous irradiation with a 785 nm laser at a power density of 6 × 105 W cm−2 for 30 min (see Methods for details). The Raman intensities of four O-GERTs after 30 min illumination retained 74%, 80%, 78%, and 79% of their original value, respectively (Fig. 1f), demonstrating high photostability favorable for long-duration imaging. Altogether, these results indicated that the four O-GERTs were highly suitable for one-shot detection of the four MMR proteins in cancer.
2.2 O-GERTs conjugated to antibodies
Then we functionalized SEE O-GERTs, STE O-GERTs, SPE O-GERTs and MDBM O-GERTs with monoclonal antibodies against MLH1, MSH2, MSH6, and PMS2 (Table S1†), respectively. The surface modification process of these O-GERTs is shown in Fig. 2a. First, HS-PEG-COOH (MW, 2k Da) was bound on the O-GERT surface via Au–S bonds. Then the left carboxyl groups on O-GERTs were activated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS), followed by attaching these antibodies to the surface of O-GERTs via the reaction between amines and carboxyl groups.37 After the antibody conjugation, the maximum absorption peaks of the O-GERTs were slightly redshifted (Fig. 2b), for example, the characteristic absorption peak of SEE O-GERTs was shifted from 558 nm to 568 nm. Similarly, the dynamic light scattering (DLS) sizes of the four O-GERTs exhibited an increased trend (Fig. S4†). For instance, the size of SEE O-GERTs changed from 84.4 to 117.3 nm after functionalization. Using the BCA microprotein quantification kit,38 the content of the four antibodies on the four O-GERT (0.08 nM for each) surfaces was determined to be 8.01, 9.53, 8.61, and 11.08 μg mL−1, respectively (Fig. 2c). These results confirmed the successful conjugation of these antibodies to the surface of the four O-GERTs for targeted imaging of MMR proteins. In addition, the four O-GERTs are quite consistent from batch to batch in terms of size distribution as well as signal intensity, with small relative standard deviations (RSDs) (Fig. S5a and b†). We also investigated the stability of antibody-conjugated O-GERTs in two different biofluids. As shown in Fig. S5c and d,† after incubation in PBS and 10% FBS for 7 days, their hydrodynamic diameters and Raman intensities at 2100, 2150, 2210, and 2290 cm−1 remained highly stable. These results indicated that the four O-GERTs possess excellent material and signal stability suitable for biomedical applications in various environments.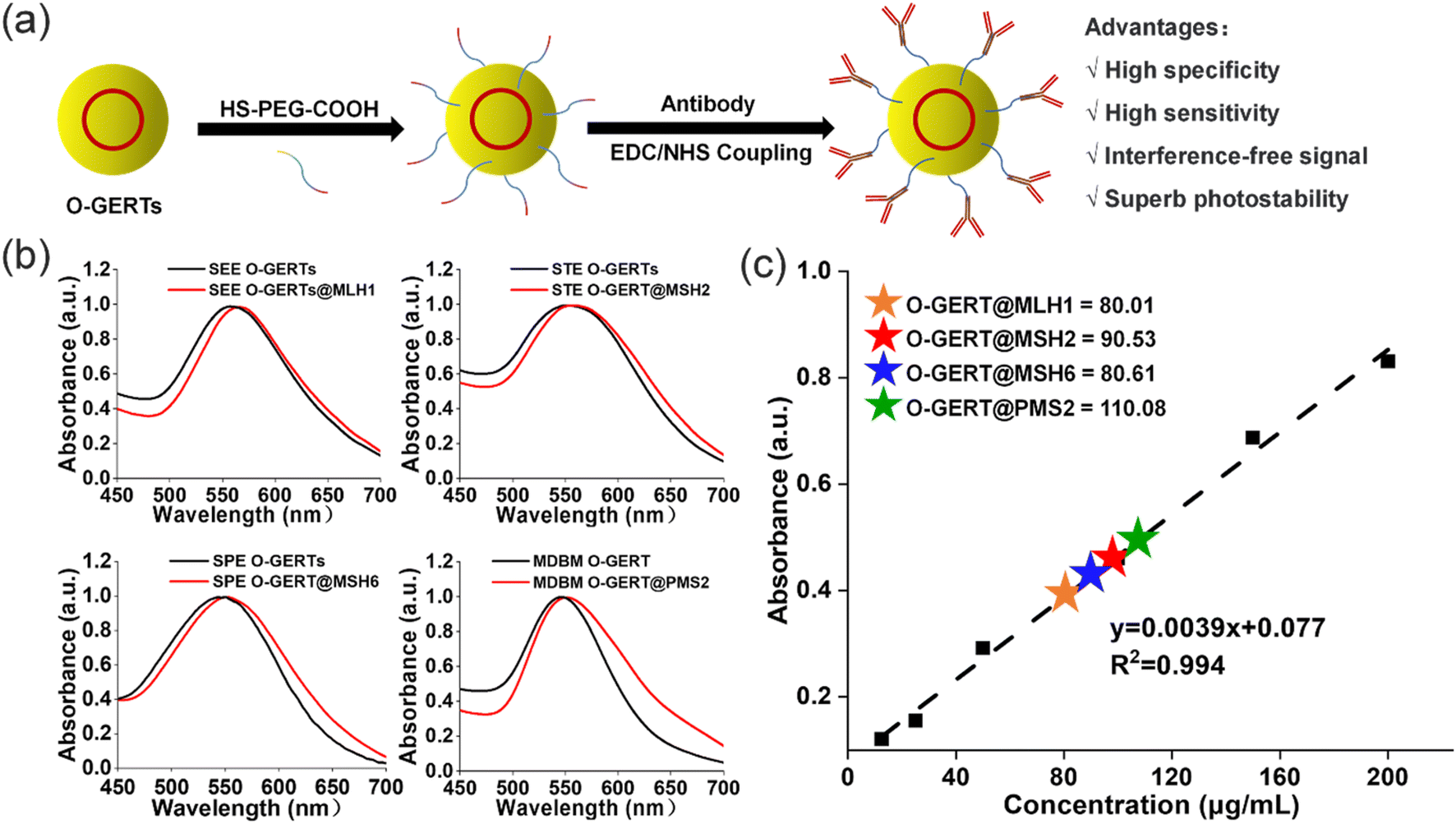 | ||
| Fig. 2 (a) Schematic diagram of the surface modification process of O-GERTs. (b) UV-vis spectra of the four O-GERTs before (black line) and after (red line) modification of the corresponding antibodies. (c) Standard curves for quantification of the antibodies on the surface of the four O-GERTs. The antibody-conjugated O-GERTs were concentrated 10-fold for analysis. | ||
To demonstrate the binding specificity of these antibody-conjugated O-GERTs in vitro, we first performed an enzyme-linked immunosorbent assay (ELISA). Antigens used in the measurements are shown in Table S2.† In this binding assay, the antigens were immobilized in the plates in the sandwich immunocomplex, while O-GERTs were used as the antibodies. With the augmentation of the amount of O-GERTs, it resulted in higher signals at 450 nm absorbance from the secondary antibody (Fig. S6a†), suggesting more O-GERTs bound to the corresponding antigens. Moreover, the antibody conjugates displayed at least 5-fold higher affinity binding to MMR proteins than the untargeted O-GERTs (Fig. S6b†). These results verified the highly specific binding capacity of antibody-conjugated O-GERTs to the MMR proteins.
2.3 Detection of MSI phenotypes of cancer by Raman imaging
To validate the feasibility of O-GERTs for MSI phenotype detection, CRC tissue samples were used for Raman imaging. We collected fresh CRC tissue samples from 30 cancer patients, and the tissue information with TNM classification is shown in Table S3.† These samples were embedded with paraffin and sectioned into thin slices of 3 μm. Four kinds of antibody-conjugated O-GERTs (SEE O-GERTs, STE O-GERTs, SPE O-GERTs and MDBM O-GERTs) mixed at a molar ratio of 1.5:1.3:1.0:3.0 were incubated with the tissue slices for 40 min (see Methods for details). Through one-shot Raman imaging, we primarily identified 4 patients as MSI-H and the others as MSS in these CRC patients (Fig. 3a). Thus, 13.3% of the MSI-H CRC tumors displayed expression loss for one or more MMR proteins.
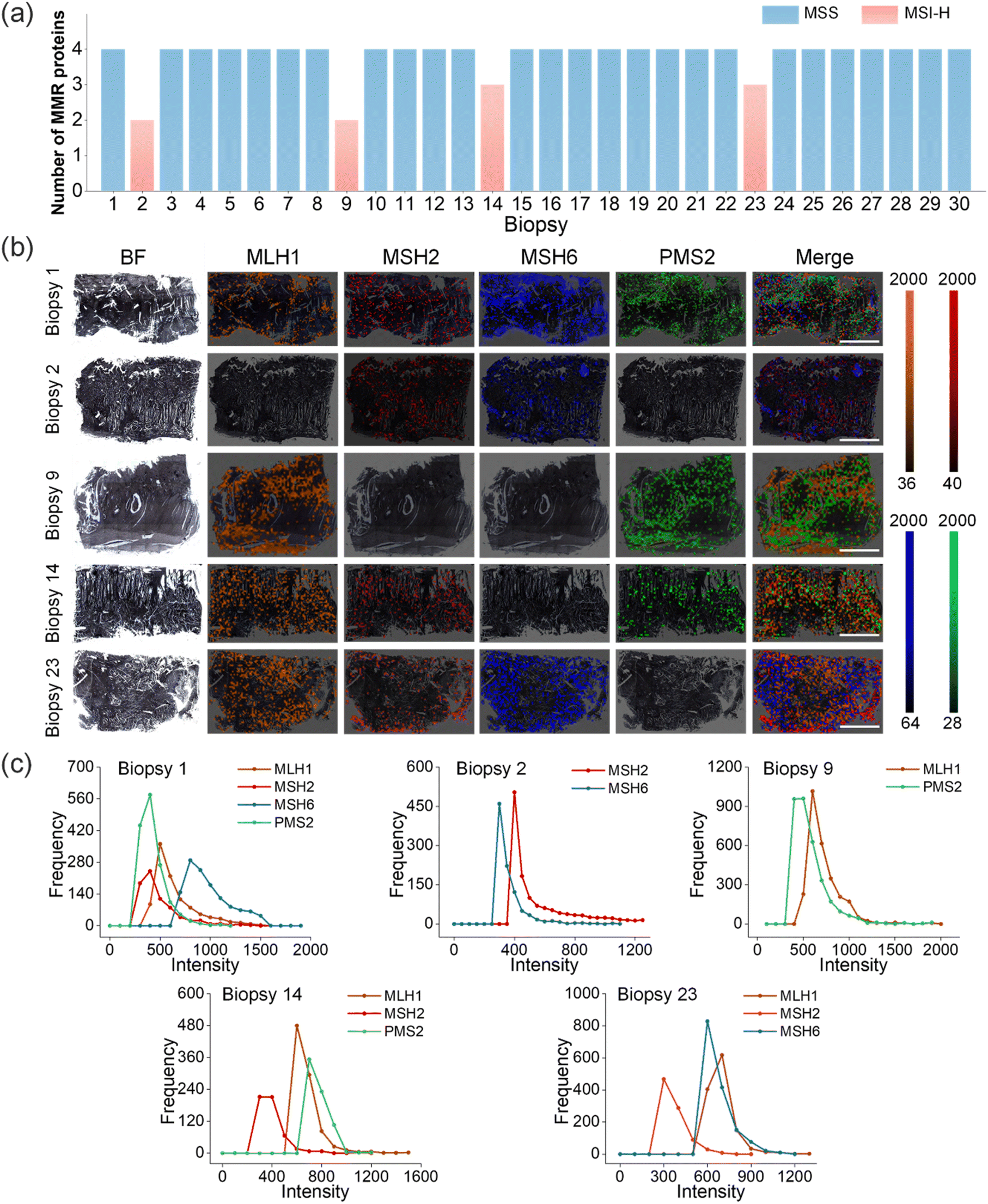 | ||
| Fig. 3 (a) Number of MMR proteins expressed in 30 CRC tissues. (b) Multiplexed Raman imaging of MMR proteins in CRC biopsies from five representative patients. Bright-field (BF) and Raman mapping images of four pseudo-colors. Scale bars = 4 mm. The orange, red, blue and green colors in the color bar correspond to the expression intensity of MLH1, MSH2, MSH6 and PMS2 proteins, respectively. (c) The expression level distribution of the four MMR proteins for each sample, plotted by the frequency and the Raman intensity of the characteristic peaks at 2100, 2150, 2210 and 2290 cm−1 for MLH1, MSH2, MSH6 and PMS2, respectively. | ||
Nonspecific binding of NPs often hampers the interpretation of imaging data.39 To account for any untargeted adsorption on CRC tissues, we added a mixture of these four O-GERTs (0.4 nM for each) to three CRC tissues. With the Raman imaging strategy, the non-specific binding patterns are shown in Fig. S7a.† The non-specific binding threshold values were calculated by the average SERS intensities (2100 cm−1 for SEE O-GERTs, 2150 cm−1 for STE O-GERTs, 2210 cm−1 for SPE O-GERTs and 2290 cm−1 for MDBM O-GERTs) in the whole non-specific binding SERS mappings of the three CRC tissues. Thus, four thresholds for untargeted SEE, STE, SPE and MDBM O-GERTs were 36, 44, 62 and 26, respectively (Fig. S7b†), which were adopted in the following Raman mappings. To confirm the specificity of the O-GERTs in CRC tissues, we used two dyes labelled O-GERTs (one for targeted conjugation and one for untargeted control) to perform the tissue imaging. This allows us to generate a map of specific-to-nonspecific binding ratios that will more accurately depict the true molecular expression profiles of the targeted protein on tissues. Ratiometric Raman images shown in Fig. S7c† demonstrated that all types of targeted NPs showed significant binding with respect to nonspecific ones. For all Raman mappings above, high specific-to-nonspecific ratios were observed (Fig. S7d†), which proved that the developed O-GERTs are capable of specifically targeting their corresponding proteins of interest. These outcomes, combined with the ELISA assay, firmly verified the binding specificity of these antibody-conjugated O-GERTs.
Following confirmation of the specificity of antibody-conjugated O-GERTs, Raman mapping on tissues using these targeted NPs was performed. The four pseudo-colors in the representative Raman mapping showed the expression of target MMR proteins, where yellow corresponded to MLH1, red to MSH2, blue to MSH6, and green to PMS2 (Fig. 3b), corresponding to Raman peaks at 2100, 2150, 2210 and 2290 cm−1, respectively. In biopsy 1, the expressions of MLH1, MSH2, MSH6, and PMS2 were all visualized, which corresponded to yellow, red, blue and green, respectively. However, the expression patterns in biopsies (2, 9, 14 and 23) were different, with loss of at least one MMR protein. For example, the expressions of MSH2 and MSH6 were observed and the expressions of MLH1 and PMS2 were lost concurrently in biopsy 2, which suggested that the loss of MLH1 resulted in the absence of PMS2. In biopsy 9, the yellow and green colors assigned to MLH1 and PMS2 were observed, and the red and blue colors corresponding to the expressions of MSH2, MSH6 were concurrently absent, implying that loss of MLH1 and MSH2 results in loss of PMS2 and MSH6. Similarly, the expressions of MLH1, MSH2 and PMS2 marked in yellow, red and green were simultaneously observed in biopsy 14, and only the expression of MSH6 marked in blue was lost, meaning that the absence of MSH6 did not cause the loss of MSH2. Likewise, in biopsy 23, yellow, red and blue colors, corresponding to MLH1, MSH2 and MSH6, were observed simultaneously, and only PMS2 expression in green was lost. Indeed, the absence of PMS2 does not lead to the absence of MLH1. The other 25 CRC Raman imaging results revealed that the four MMR proteins were all expressed (Fig. S8 and S9a†). It should be noted that a single 3-micron slice as a representative of the entire tumor for protein visualization is feasible in clinical settings,19,40 although tumors are heterogeneous. To further validate this, we imaged three slice sections of the same tumor with different depths (200 μm intervals) in three CRC tissues (Fig. S9a†). The expression levels of MMR proteins from different sites by Raman intensity statistical analysis suggested that there was no significant difference in these three slices from different tumor regions (Fig. S9b†). These data were well supported by IHC staining results (Fig. S9c and d†). These outcomes indicated that the one-shot Raman imaging could simultaneously detect these four proteins that were highly correlated with the MMR system, which laid the foundation for subsequent MSI phenotypic differentiation of cancer.
Since IHC is highly recommended as the first method for the detection of MSI by the European Society for Medical Oncology (ESMO).41 We evaluated the expression levels of the four MMR proteins in the five CRC tissues mentioned above by this method (Fig. S10†). The IHC images were quantified using ImageJ software (see Methods for details), which roughly reflected the expression levels of these MMR proteins in the five typical CRC tissues (Table S4†), consistent with Raman imaging results. It is noteworthy that IHC can only detect one biomarker at a time and requires at least 12 h to complete the whole staining process (see Methods for details), while O-GERTs realize one-shot detection of these four MMR proteins within approximately 1 h, 12-fold lower than the time taken by the IHC detection. Thus, our method not only improved the diagnostic accuracy and relevance for the co-expression correlations of these four proteins but also reduced the assay time. Previously, we had shown that O-GERTs could be used for high-speed Raman imaging down to 0.7 ms per pixel within large area scanning,22,36 due to the ultrahigh sensitivity. This meant that the imaging speed for MSI detection using these O-GERTs could have been further improved to meet clinical scanning requirements by further optimizing the sensitivity. In short, our multiplexed Raman imaging is advantageous over the IHC method, as summarized in Table S5.† Moreover, we also compared our method to multiplexed fluorescence immunohistochemistry (mIHC) that used multiple fluorescent reporters. However, these reporters suffer from exhibiting spectral overlaps when used simultaneously as well as spectral interference from autofluorescence of tissues (Fig. S11 and Table S6†). Also, comparisons of this Raman method with other methods for detecting MSI used in the clinic, such as polymerase chain reaction (PCR) and next-generation sequencing (NGS), are summarized in Table S7.†
Since the characteristic peaks of these O-GERTs located in the Raman-silent region and avoided interference from biological background, we exactly quantified the expression levels of these MMR proteins by counting the frequency of the Raman signal intensity of the bands at 2100, 2150, 2210 and 2290 cm−1 for MLH1, MSH2, MSH6 and PMS2 in the whole mapping pixels, respectively. Briefly, the frequency of each intensity interval (0–50, 50–100, 100–150, etc.) was counted and these MMR proteins distributions of the above 5 biopsies are shown in Fig. 3b. In biopsy 1, the expression levels of MSH6 were higher than those of MLH1, MSH2 and PMS2, suggesting a potentially dominant role of MSH6 in specific repair pathways. In biopsy 2, the expression levels of MSH2 were significantly higher than those of MSH6. The expression levels of MMR proteins were approximately the same in biopsy 9. In biopsy 14 and biopsy 23, MSH2 protein expression was the lowest. In short, using this method, we obtained the relative expression relations between these four MMR proteins in the biopsy samples.
Besides CRC, MSI occurs frequently in other cancer types as well, such as gastric and breast malignant tumors.42,43 To validate the versatility of O-GERTs for MSI detection, we collected clinical FFPE tissue samples from gastric and breast cancer patients (Tables S8 and S9†). Through one-shot Raman imaging using these targeted four O-GERTs, we identified 2 MSI-H subtypes from 30 patients in the gastric cancer tissues, while 1 of the 30 MSI-H subtypes for the breast cancer tissues was verified. Thus, the detected MSI-H ratios for both cancers were 6.7% and 3.3% (Fig. S12a and S13a†), respectively, in good agreement with the reported values.44,45 The representative Raman images of both tissues for the four MMR proteins are shown in Fig. S12b and S13b.† Collectively, these results suggested that multiplexed O-GERTs based Raman imaging could be used for rapid detection of MSI phenotypes across various cancer tissues.
2.4 Correlation between the expression levels of MMR proteins and clinical pathological parameters
Understanding the relationship between MSI and clinicopathology provides valuable insights for individual treatment decisions. The relative expression of MMR proteins was determined by extracting the Raman intensities of the characteristic peaks at 2100, 2150, 2210, and 2290 cm−1 in all pixels of these Raman profiles, respectively. Since different O-GERTs have different brightness at the same concentration, an adjustment of the O-GERT intensity according to their ‘brightness’ measured at the same concentration is needed when relative intensity is used to compare different markers in tissues. For relative quantification, the average SERS intensity in each Raman mapping was corrected by the brightness factors (values of 1.5, 1.3, 1.0, and 3.0 for MLH1, MSH2, MSH6, and PMS2, respectively) previously described for intensity normalization. We analyzed the correlation between the expression levels of four MMR proteins and clinicopathological parameters in the above 30 CRC patient tissues (4 patients with MSI-H and 26 patients with MSS), including clinical stage, T, N and M classification in Table S3.† The T category describes the size and extension of the primary tumor (T1: tumor diameter is ≤2 cm, and the tumor is confined to the primary site. T2: tumor diameter is between 2 and 5 cm, and the tumor is confined to the primary site. T3: tumor diameter is between 5 and 7 cm, and the tumor might significantly invade neighboring tissues or organs. T4: tumor diameter is >7 cm, and the tumor might invade the chest wall or skin). The N category describes whether the tumor has spread to nearby lymph nodes (N0: no regional lymph node metastases. N1: tumor metastasizes to ipsilateral parabronchial lymph nodes and/or ipsilateral hilar lymph nodes. N2: tumor metastasizes to ipsilateral mediastinal and/or subcarinal lymph nodes. N3: tumor metastasizes to contralateral mediastinum, contralateral hilar lymph nodes, or ipsilateral or contralateral oblique or supraclavicular lymph nodes). The M category describes whether the tumor has metastasized to distant sites (M0: no distant metastases, M1: distant metastases).Fig. 4a illustrated the relationships between the expression of MMR proteins and tumor stage. The expression level of MLH1 was significantly higher in early-stage tumors than that in advanced stage tumors. In contrast, the expression level of MSH6 was significantly lower in early-stage tumors compared to advanced stage tumors. The expression levels of MSH2 and PMS2 did not differ significantly between both tumor stages. Fig. 4b demonstrated the relationship between the expression of MMR proteins and T classification. The expression level of MLH1 was significantly higher in the T1 and T2 stages than that in the T3 and T4 stages, consistent with earlier analyses of tumor progression. Conversely, the expression level of PMS2 was significantly higher in the T4 stage than that in the T3 stage. The expression level of MSH6 was significantly higher in the T3 and T4 stages than that in the T2 stage. There was no significant difference in the expression of MSH2 across all T classifications. We also delved into the relationship between the expression of MMR proteins and N classification (Fig. 4c). It was found that the expression level of MLH1 was significantly higher in the N0 stages than that in the N1 and N3 stages, and significantly higher in the N2 stage than that in the N3 stage. Additionally, the expression level of MSH6 was significantly higher in the N0 stage compared to the N2 stage. In contrast, no significant differences were observed in the expression levels of MSH2 and PMS2 across the N stages. We finally analyzed the relationship between the expression of MMR proteins and M classification (Fig. 4d). The expression levels of MSH2 and PMS2 were higher in M0 than those in M1 stage, while MLH1 and MSH6 did not differ significantly between M0 and M1 in the expression levels.
 | ||
| Fig. 4 Box plot presenting the relative expression level of the four MMR proteins in various clinicopathologic parameters. (a) Stage (early-stage patients, n = 17; advanced stage patients, n = 13), (b) T classification (T1 patients, n = 8; T2 patients, n = 8; T3 patients, n = 8; T4 patients, n = 6), (c) N classification (N0 patients, n = 11; N1 patients, n = 7; N2 patients, n = 7; N3 patients, n = 5) and (d) M classification (M0 patients, n = 26; M1 patients, n = 4). The statistical analysis in panels (a) and (d) was determined by a two-sample Student's t-test, and in panels (b) and (c) was determined by ANOVA. Statistical significance was set at *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. denotes no significant difference, P > 0.05. | ||
From the above data, it could be seen that the expression patterns of MMR proteins from the two MSI subtypes differ greatly in clinical pathological parameters. Specifically, (1) PMS2 protein was expressed at higher levels in MSI-H patients as compared to MSS ones in the early stage. In contrast, MSH6 protein was expressed at lower levels in MSI-H patients as compared to MSS ones in the N1 stage. (2) MSI-H patients were predominantly diagnosed with early-stage tumors, characterized by small tumor diameters and tumor confinement to the primary site (T1), a low likelihood of lymph node metastasis (N0 and N1) and distant metastasis (M0; see Table S3† for details). Indeed, Robert Gryfe et al. also demonstrated that MSI-H/dMMR cases have a reduced likelihood of metastasis to lymph nodes and distant sites, with most cases being classified as stage I or II.46–48 Overall, the relationship between tumor TNM staging and MMR protein expression established in this work may help predict the progression and prognosis of MSS/MSI-H tumors for accurate treatment.49–51
2.5 MSI phenotypes for screening Lynch syndrome
Clinically, MSI phenotypes are mainly used to aid in the diagnosis of Lynch syndrome, a genetic disease that usually occurs in inherited CRC. In 2018, a pan-cancer study presented at the American Society of Clinical Oncology (ASCO) suggested that up to 50% of Lynch syndrome patients with the MSI-H subtype should receive genetic counseling and testing, regardless of tumor type or family history.52,53 Generally, the diagnosis of Lynch syndrome is not only helpful for personalized treatment of the patients, but also important for the health management of their family members.Based on the above Raman mapping results (Fig. 3b), MSI phenotypes were classified according to the expression profiles of MMR proteins to screen for Lynch syndrome. As shown in Table 1, in state 1, when all four MMR proteins were expressed simultaneously, the MSI phenotype was MSS, and the patient's disease type was diagnosed as sporadic CRC, which is an environmental rather than a genetic disease. In states 3 to 5, patients with the MSI-H phenotype had lost at least one MMR protein. For example, in state 3, both MSH2 and MSH6 proteins were lost. MSH6 and PMS2 were lost in states 4 and 5, respectively. Based on the absence of expression of the MMR proteins in the three states, these patients are tentatively diagnosed with Lynch syndrome. In state 2, when the expression of MLH1 and PMS2 was lost, it was necessary to further analyze the promoter methylation phenotype of the MLH1 gene. If the promoter of the MLH1 gene is methylated, the patient is diagnosed with sporadic CRC. Otherwise, the patient is usually diagnosed with Lynch syndrome.54,55 In short, precise phenotypic identification of MSI in cancer is useful for screening Lynch syndrome for subsequent genetic screening and treatment management.12–14
| State | MSI phenotypes | MMR proteins | Diagnostic results | |||
|---|---|---|---|---|---|---|
| MLH1 | MSH2 | MSH6 | PMS2 | |||
| 1 | MSS | + | + | + | + | Sporadic |
| 2 | MSI-H | − | + | + | − | Sporadic or Lynch syndrome |
| 3 | MSI-H | + | − | − | + | Lynch syndrome |
| 4 | MSI-H | + | + | − | + | Lynch syndrome |
| 5 | MSI-H | + | + | + | − | Lynch syndrome |
2.6 MSI phenotypes for preselection of potential immunotherapy beneficiaries
Recently, PD1/PD-L1 pathway blockade via PD1 antibody has been considered the first-line treatment for CRC patients with mismatch repair deficiency, including MSI-H status,56–58 which represented approximately 15% of tumors. We therefore examined the expression levels of PD-L1 protein in these MSS and MSI-H CRC tissues by Raman imaging with PD-L1-conjugated SPE O-GERTs (SPE O-GERT@PD-L1 NPs). First, specific binding of PD-L1-functionalized SPE O-GERTs to HCT116 cells was well validated by flow cytometry (Fig. S14†). Then, Raman imaging results showed that the expression levels of PD-L1 differed across MSI phenotypes. The expression of PD-L1 in MSI-H tissues (Fig. S15a†) was obviously higher than that in their MSS counterparts (Fig. S15b†). By further extracting the Raman intensity of characteristic peaks at 2210 cm−1 in all pixels of these Raman mappings, it was found that PD-L1 expression levels in MSI-H tissues were approximately 4 times higher than those in MSS ones (Fig. 5a). | ||
| Fig. 5 (a) The relative expression levels of PD-L1 in the four MSI-H and MSS CRC tissues (n = 4 for each group). (b) Proportions of relative expression levels of PD-L1 and four MMR proteins in MSI-H CRC tissues. (c) Heat map of the correlation of the expression levels between PD-L1 and four MMR proteins in MSI-H tissues. (d) Schematic diagram of the construction of CRC PDX mice. (e) Detection of PD-L1 protein in PDX MSI-H mice, including (1) digital photograph of PDX MSI-H mice, (2) in vivo and (3) ex vivo Raman imaging of PD-L1 protein in the tumor. (f) Detection of PD-L1 protein in PDX MSS mice, including (1) digital photograph of PDX MSS mice, (2) in vivo and (3) ex vivo ratiometric Raman imaging of PD-L1 protein in the tumor. Green and purple signals were derived from the band at 2150 and 2210 cm−1 of untargeted STE O-GERTs and PD-L1-targeted SPE O-GERTs. Scale bars = 5 mm. (g) The expression level of PD-L1 in MSI-H and MSS mice (n = 3 for each group), and values were expressed as mean ± SD. The statistical analysis in panels (a) and (g) was determined by a two-sample Student's t-test. ***P < 0.001. | ||
For these MSI-H tissues (Fig. 3b), we analyzed the relative expression levels of PD-L1 and the four MMR proteins by calculating the average Raman intensities of the corresponding mappings. As shown in Fig. 5b, in biopsy 2, the relative expression ratio of PD-L1 to MSH2 and MSH6 was 0.24:0.31:0.17. In biopsy 9, the ratio of PD-L1 to MLH1 and PMS2 was 0.24:0.47:0.73. Apparently, the relative expression levels of PD-L1 likewise showed a consistent trend in biopsy 14, where the ratio of PD-L1 to MLH1, MSH2 and PMS2 was 0.23:0.43:0.21:1.00. In biopsies 23, the ratio of PD-L1 to MLH1, MSH2 and MSH6 was 0.19:0.45:0.20:0.28. These results suggested that the relative expression of PD-L1 presented similar patterns in these MSI-H tissues. The co-expression correlation between PD-L1 and the four MMR proteins in all MSI-H tissues was further depicted in a Pearson correlation heatmap. The Pearson correlation coefficient ranges from −1 to 1, representing a negative or positive correlation, respectively. As shown in Fig. 5c, MLH1 was significantly negatively correlated with MSH2 (r = −0.71), moderately positively correlated with PMS2 (r = 0.56). MSH6 was also moderately positively correlated with MSH2 (r = 0.51), but significantly negatively correlated with PMS2 (r = −0.93) and PD-L1 (r = −0.67). PMS2 was moderately negatively correlated with MSH2 (r = −0.50) and weakly positively correlated with PD-L1 (r = 0.36). Therefore, the expression relationships among the four MMR proteins remained ambiguous and needed further study. In short, ex vivo Raman imaging corroborated that the MSI-H tumor was considered an ideal candidate for PD1/PD-L1 inhibitory immunotherapy, while the MSS tumor may not be suitable for this strategy and thus chemotherapy or targeted therapies may be preferred.
To further highlight the expression discrepancy of PD-L1 in MSI-H and MSS tumors, we further carried out in vivo Raman imaging, taking advantage of O-GERTs, which exhibit high sensitivity under a NIR laser of 785 nm that has higher penetration depth in living subjects. After confirmation of the in vitro biocompatibility of SPE O-GERT@PD-L1 NPs against three common CRC cell lines (HCT116, CT26 and MC38) (Fig. S16†), we conducted in vivo studies. In particular, we established patient-derived xenograft (PDX) mouse models (Fig. 5d) for both MSI-H and MSS subtypes by transplanting CRC tumor tissues from patients with MSI-H (biopsy 2) and MSS (biopsy 1) into mice, respectively (Fig. 5e(1) and 5f(1)). PDX mouse models have significant advantages over commonly used cell line-derived xenograft (CDX) models, because they preserve the microenvironment and heterogeneity of the original human tumor, allowing for a more realistic representation of the tumor's biological characteristics.59,60 Male NOD-PrkdcemIl2rgem mice were selected because of a high degree of immune deficiency, making them suitable for PDX models.61 After the tumors had grown to a size of around 150 mm3, we intravenously injected a mixture of untargeted STE O-GERTs and PD-L1-targeted SPE O-GERTs with the same ratios (0.4 nM and 150 μL for each) into the mice (n = 3 for each group, Fig. S17†). In vivo non-invasive Raman imaging displayed that PD-L1 expression in the MSI-H tumor (Fig. 5e(2)) was significantly higher than that in the MSS one (Fig. 5f(2)). Since both NPs, as the imaging reagents, were used in vivo under the same conditions, the results above also excluded enhanced permeability and retention (EPR) effect of NPs for nonspecific binding, and thus the targeted specificity of PD-L1-functionalized SPE O-GERTs in vivo was validated. On the other hand, ex vivo imaging of the excised tumor after 12 h postinjection of O-GERT@PD-L1 NPs showed a similar trend (Fig. 5e(3) and f(3)). The P-value for the expression level of PD-L1 in both groups is less than 0.001 (Fig. 5g), indicating that the MSI-H and MSS tumors are intrinsically different in a statistical sense. Therefore, both in vivo and ex vivo results consistently indicated that PD-L1 expression was significantly higher in MSI-H tumors than that in MSS tumors, providing a reliable basis for the use of PD1/PD-L1 blockade therapy for treating MSI-H tumors.
3 Conclusions
In summary, we have fabricated four multiplexed O-GERTs with completely separated bands in the Raman-silent region, superb sensitivity and photostability for fast detection of MSI in cancer. With targeted modification, these multicolored O-GERTs permitted one-shot imaging of four MMR proteins in various cancer tissues with high specificity and spectral resolution. Based on the Raman imaging results, these tissues were classified into MSS or MSI-H. The MSI-H ratios for CRC, breast and gastric cancers were 13.3%, 6.7% and 3.3%, respectively, well consistent with the reported values. By examining the correlation between the expression level of the four MMR proteins and their related clinicopathologic parameters, we found that CRC tissues with two MSI subtypes displayed distinct differences, which can be of great significance for individualized treatment of the patients. Moreover, we demonstrated that the MSI of cancer can be used as a screening tool for screening Lynch syndrome and identifying potential immunotherapy beneficiaries by PD1/PD-L1 blockade. We anticipate that this sample one-shot method will be a versatile nanoplatform for rapid MSI testing across multiple cancers, with the potential to complement gold-standard PCR diagnostic methodologies.4 Methods
4.1 Reagents
All chemicals were commercially obtained and used as received. Hexadecyltrimethylammonium chloride (CTAC, >98.0%), L-ascorbic acid (AA, >99.9%), and sodium hydroxide were purchased from Sigma-Aldrich. Hydrochloric acid, nitric acid, sodium borohydride (NaBH4), and tetrachloroauric acid tetrahydrate (HAuCl4·4H2O) were acquired from Sinopharm Chemical Reagent Co., Ltd. mPEG-SH (MW 2 kDa) and HS-PEG-COOH (MW 2 kDa) were purchased from Xi'an Ruixi Biotechnology Co., Ltd. HS-PEG-FITC (MW 2 kDa) was purchased from Zancheng (Tianjin) Technology Co., Ltd. L-ascorbic acid (98%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were obtained from Aladdin Biochemical Technology Co., Ltd (Shanghai, China). PBS buffer (pH 7.4) was purchased from Agilent Technologies Inc. The DAB staining kit (G1212-200TMLH1) was obtained from Wuhan Servicebio Technology Co., Ltd.4.2 Characterization
UV-vis spectra were recorded using a 220 UV-vis spectrophotometer (Thermo Fisher Scientific, Shanghai, China) with a 1 cm path-length quartz cuvette. Transmission electron microscopy (TEM) measurements were performed using a Talos F200X G2 (Thermo Fisher Scientific, Shanghai, China). Dynamic light scattering (DLS) was conducted using a Malvern Zetasizer (Nano ZS). Zeta potentials were measured using a Delsa Nano C particle analyzer (Beckman Coulter Ireland, Inc.). Raman spectra were recorded using a Renishaw inVia Raman microscope equipped with 532, 633 and 785 nm laser sources, and measured in capillaries with an average length of 10 cm with a diameter of 0.5 mm. All samples were tested at least three times. Distilled water with a resistivity of 18.0 MΩ cm was used throughout all experiments.4.3 Synthesis of O-GERTs
The preparation of O-GERTs mainly involved three steps.33 (1) Au cores with a diameter of 20 nm were prepared using a seed-mediated process. (2) Orthogonal reporters were attached to the Au cores with a monolayer. (3) The desired thickness of the Au shell was grown on the reporter conjugate by adding HAuCl4 and AA. In brief, 100 μL of 4 mM SEE solution was added to a suspension of as-synthesized Au cores (0.47 nM, 4 mL) and sonicated for 15 min in an ultrasonic bath. The reaction solution was then left to adsorb for 3 h. After washing the SEE-modified Au cores three times, the cores were re-dispersed in 1 mL of 0.05 M CTAC. Subsequently, 1 mL of the SEE-modified Au core solution was added to a mixture solution containing 20 mL of 0.05 M CTAC, 600 μL of AA (0.04 M), and 1 mL of HAuCl4 (4.86 mM). The mixture was vigorously sonicated for 10 min to produce SEE O-GERTs (0.08 nM, 2 mL). The steps for STE O-GERTs, SPE O-GERTs, and MDBM O-GERTs followed the same procedure as described above.4.4 LODs of O-GERTs
To determine the LODs of SEE O-GERTs, STE O-GERTs, SPE O-GERTs and MDBM O-GERTs, each NP was serially diluted (concentration range of 0.33–72 pM, n = 3) and placed in a 96-well plate for Raman imaging under the following conditions: power (Pex) = 150 mW, integration time = 1 s, and 5 × objective lens. All Raman images were obtained using WiRE 5.5 software (Renishaw).4.5 Photostability studies
In brief, 10 μL of O-GERTs (0.4 nM) were dropped onto a silicon wafer, dried, and subjected to continuous illumination with a 785 nm laser at a power density of 6 × 105 W cm−2 for 30 min. Raman spectra of all characteristic peaks were obtained under a 785 nm laser with an exposure time of 1.0 s per spectrum.4.6 Modification of antibodies onto O-GERTs
Surface modification of O-GERTs with antibodies was performed according to ref. 62. The antibody conjugation steps for SEE O-GERT@MLH1, STE O-GERT@MSH2, SPE O-GERT@MSH6 and MDBM O-GERT@PMS2 NPs followed similar procedures, and the first one was described as follows. Briefly, 60 μL of 10 μM HS-PEG-COOH solution was spiked dropwise with 1 mL of 0.08 nM SEE O-GERTs under vigorous stirring. After 30 min, 120 μL of freshly prepared 10 μM mPEG-SH (MW 2000 Da) was added to the solution, followed by stirring for 3 h. Then, SEE O-GERTs attached to HS-PEG-COOH were purified by three rounds of centrifugation (7200 rpm, 10 min). Next, 5 μL of 25 mM EDC and 5 μL of 25 mM NHS were added to activate the –COOH terminal groups on the surface of SEE O-GERTs at 25 °C. Through three rounds of centrifugation (7200 rpm, 10 min), excess EDC and NHS were removed from the O-GERTs. Finally, the activated carboxyl groups on the surface of SEE O-GERTs were reacted with MLH1 (0.69 mg mL−1, 5 μL) for 2 h at 25 °C. The reaction mixture was subjected to three rounds of centrifugation (7200 rpm, 10 min) to remove excess antibodies, and the obtained SEE O-GERT@MLH1 was then redispersed in PBS and stored at 4 °C.4.7 Quantification of antibodies on the surface of O-GERTs
The amounts of antibodies bound to the surface of O-GERTs were estimated using the BCA protein assay kit with bovine serum albumin (BSA) as a reference. The mass concentrations of these antibodies on O-GERTs were determined from the corresponding standard curves.4.8 ELISA binding assay
The ELISA binding assay was performed following the reference with slight changes.37 The experimental procedure of four antibody-conjugated O-GERTs for ELISA was similar, and SEE O-GERT@MLH1 NPs were described. First, 100 μL of the target antigen solution (1 μg mL−1) was added to each well of a 96-well plate and incubated overnight at 4 °C. Then, the plate was washed three times with PBS buffer to remove unbound antigen, followed by adding 200 μL of blocking buffer to each well. After incubation for 1 h, the plate was washed three times to minimize non-specific binding. Second, 100 μL of SEE O-GERT@MLH1 NPs at varying concentrations (0 to 8 μg mL−1) were added to each well and incubated for 2 h at room temperature, followed by washing three times to remove unbound NPs. Next, a goat anti-rabbit IgG F(ab)2/HRP secondary antibody was added. After incubation for 2 h, 100 μL of 3,3′,5,5′-tetramethylbenzidine was added to each well, and the plate was incubated for 30 min at room temperature. The reaction was then stopped by adding 50–100 μL of 0.2 M sulfuric acid solution to each well. Finally, the absorbance of the above solution at 450 nm was measured using a microplate reader.4.9 SERS mapping of cancer tissues
The human tumor samples were provided by Binzhou Medical University Hospital, Binzhou Medical University. The approval number was [2023] LW-84. For Raman imaging, cancer tissues were cut into 3 μm thick sections and stained with a mixture of SEE O-GERT@MLH1, STE O-GERT@MSH2, SPE O-GERT@MSH6 and MDBM O-GERT@PMS2 NPs (0.4 nM for each). Note that the 4 NP solutions completely covered the tissues and are in excess to ensure sufficient antibody-protein conjugation, and thus the molar ratio of the 4 NP batches was not crucial. After incubation for 40 min, the stained biopsies were thoroughly washed with PBS to remove non-specifically adsorbed O-GERTs. Subsequently, Raman mapping on tissues was performed using a streamline high-speed acquisition mode under the following measurement conditions: excitation wavelength (λex) = 785 nm, incident power (Pex) ≈ 30 mW and acquisition time = 0.5 s.4.10 IHC
Briefly, clinical CRC tissue sections were deparaffinized with xylene, hydrated with ethanol and treated with 3% H2O2 for 10 min. After rinsing three times with PBS, the sections were subjected to antigen retrieval by microwave heating (200 watts, 30 min) in citrate buffer (pH 5.5), followed by washing with PBS. The sections were then incubated with BSA solution (pH 5.5, 2 mM) for 30 min to block non-specific adsorption and washed with PBS. Next, these sections were incubated with each diluted primary antibody (MLH1, MSH2, MSH6, and PMS2, respectively) and placed in the refrigerator at 4 °C overnight. On the next day, HRP (horseradish peroxidase) labeled secondary antibody was added dropwise to the sections and incubated for 30 min at 37 °C, followed by rinsing three times with PBS to remove unbound secondary antibodies. Then DAB (3,3′-diaminobenzidine) color working solution was added dropwise and incubated for 5–10 min at room temperature. The color development reaction was observed under a microscope until a desirable brown or dark brown precipitate appeared. After the reaction was terminated, the sections were sequentially dehydrated with a gradient of 60–100% alcohol and sealed with neutral gum for microscopic observation.4.11 Quantification of IHC and Raman images in CRC tissues
Quantification of IHC results followed the methods in ref. 63. Briefly, we first extracted the average gray values using ImageJ. The values measured using ImageJ are then displayed in the graph Fig. S9d† as the reciprocal staining intensity (RSI), where RSI = 255 – mean gray value. For Raman images, the relative expression of these MMR proteins was quantified based on the frequency of the Raman signal intensity of bands at 2100, 2150, 2210 and 2290 cm−1, respectively.64 Briefly, the backgrounds in the Raman image were first deducted using WiRE 5.5 software (Renishaw). Then the frequency of the intensity interval (0–50, 50–100, 100–150, etc.) of the characteristic Raman bands at 2100, 2150, 2210 and 2290 cm−1 was counted, and these MMR protein distributions of tissues were plotted as Gaussian curves.4.12 Cell culture and cytotoxicity
MC38 and CT26 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin and maintained at 37 °C in a humidified atmosphere containing 5% CO2. The cytotoxicity of SPE O-GERTs@PD-L1 NPs was assessed using the CCK-8 assay. CT26 and MC38 cells (6 × 103 cells per well) were seeded in 96-well plates and incubated for 24 h at 37 °C. Subsequently, different concentrations of the SPE O-GERT@PD-L1 NPs (0, 0.1, 0.2, 0.5, 1.0, 1.5, and 2.0 nM) were added. After an additional 24 or 48 h of incubation, the wells were washed twice with PBS. CCK-8 solution was then added to each well and the cells were incubated for another 2 h. Cell viability was determined by measuring the optical density (OD) at 450 nm using a microplate reader.4.13 Flow cytometry
The binding specificity of O-GERT@PD-L1 NPs to PD-L1 proteins on the cell surface was assessed using flow cytometry. HCT116 cells were used as PD-L1-positive controls, and CT26 cells were used as negative controls. O-GERT@PD-L1 NPs were first fluorescently labelled by attaching HS-PEG-FITC to the NP surface. The samples were prepared by aliquoting 2 × 105 cells (100 μL) and adding 15 μL of 0.8 nM O-GERT@PD-L1 NPs to achieve a final NP concentration of 120 pM. The NP-cell mixtures were incubated for 30 min at 4 °C, washed once in 2 mL PBS + 1% BSA, and resuspended in 300 μL PBS + 1% BSA for analysis. For epitope blocking experiments, prior to adding NPs, HCT116 cells were suspended in anti-PD-L1 ranging from 1 ng mL−1 to 100 μg mL−1 for 30 min on ice, washed once with 2 mL PBS + 1% BSA and added 15 μL of 0.8 nM O-GERT@PD-L1NPs to achieve a final NP concentration of 120 pM. The mixture of cells with O-GERT@PD-L1 NPs was washed once with 2 mL PBS + 1% BSA, and then resuspended in 300 μL PBS + 1% BSA. Flow cytometry analysis was performed on a BD FACSAria™ flow cytometer.4.14 PDX models
Male NOD-PrkdcemIl2rgem mice (6 to 8 weeks) were obtained from Hangzhou Ziyuan Experimental Ltd (Hangzhou, China). All animal experiments in this work were approved by the Ethics Committee for Animal Experiments of Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences (No. KJ-LL-015). PDX models were established following the references with slight changes.65,66 Fresh tumor tissues from CRC patients were obtained from Binzhou Medical University. Upon receipt of the fresh tumor tissues, they were stored at 4 °C and commenced modeling within 2 h. After excision of the necrotic tumor tissue, the remaining tissue was sliced into multiple pieces with a size of 2 × 2 × 2 mm3 each. Subsequently, each tumor piece was immersed in a mixture of matrix gel and cellular culture medium (DMEM/F-12), followed by subcutaneous implantation into the dorsum of the mice. The tumor formation was monitored daily after post-inoculation. When the tumors reached 150–200 mm3, the tumor tissue was excised and transplanted into the next generation. The above procedure was repeated to obtain the third generation of the PDX mouse model for subsequent experiments.4.15 Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0. One-way ANOVA and Student's t-test were utilized for statistical analyses. A value of P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. In the box plot, the line inside each box represents the median, and the lower and upper edges of the box represent the first and third quartiles, respectively. Lower whisker and upper whisker represent the minimum and maximum values of the data, respectively (Fig. 4).Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences” and approved by the Animal Ethics Committee of “Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences” (No. KJ-LL-015).Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
G. F.: investigation and writing – original draft. J. L.: conceptualization, writing – review & editing and funding acquisition. Q. Z. and C. L.: resources. Z. Z. and X. W.: software and formal analysis. R. W.: methodology. L. C.: supervision and project administration.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 82102108, 22376216, 42476186 and 22006162).References
- E. Vilar and S. B. Gruber, Nat. Rev. Clin. Oncol., 2010, 7, 62–153 CrossRef.
- F. Gelsomino, M. Barbolini, A. Spallanzani, G. Pugliese and S. Cascinu, Cancer Treat. Rev., 2016, 51, 19–26 CrossRef CAS.
- J. C. Toh, P. H. Bokey, L. Chan, C. Spring, K. J. Dent and O. F. Dent, Br. J. Surg., 2017, 104, 1250–1259 CrossRef CAS.
- R. Collaborative, Br. J. Surg., 2022, 109, 632–636 CrossRef.
- E. A. H. Kheirelseid, N. Miller, K. H. Chang, C. Curran, E. Hennessey, M. Sheehan and M. J. Kerin, J. Gastrointest. Oncol, 2013, 4, 397–408 CAS.
- C. Eng, A. A. Jácome, M. Agarwal, M. H. Hayat, M. X. Byndloss, A. N. Holowatyj, C. Bailey and C. H. Lieu, Lancet Oncol., 2022, 23, e116–e128 CrossRef.
- D. Sun, Y. Chen and J. Y. Fang, Natl. Sci. Rev., 2019, 6, 1138–1148 CrossRef CAS.
- W. Chen and W. L. Frankel, Mod. Pathol., 2019, 32, 1–15 CrossRef CAS.
- D. C. Chung and A. K. Rustgi, Ann. Intern. Med., 2003, 138, 560–570 CrossRef CAS PubMed.
- S. I. Tou, E. R. Drye, P. B. Boulos and S. J. Hollingsworth, Br. J. Cancer, 2004, 90, 2006–2012 CrossRef CAS.
- M. J. Hall, J. N. Bodor, J. Xiu, R. Feldman, A. Grothey, R. M. Goldberg, W. M. Worrilow, J. J. Hwang, E. S. Kim, H. J. Lenz, D. Raghavan, A. F. Shields, J. Marshall, W. M. Korn and M. E. Salem, J. Clin. Oncol., 2019, 37, 505 Search PubMed.
- M. B. Yurgelun and F. Kastrinos, J. Clin. Oncol., 2019, 37, 263–265 CrossRef.
- J. Walkowska, T. Kallemose, G. Jönsson, M. Jönsson, O. Andersen, M. H. Andersen, I. M. Svane, A. Langkilde, M. Nilbert and C. Therkildsen, OncoImmunology, 2019, 8, 1515612 CrossRef.
- H. Dust, E. Stoffel, E. Koeppe and S. Bolde, Am. J. Gastroenterol., 2018, 113, S154 CrossRef.
- K. Li, H. Luo, L. Huang, H. Luo and X. Zhu, Cancer Cell Int., 2020, 20, 16 CrossRef PubMed.
- M. L. Nádorvári, I. Kenessey, A. Kiss, T. Barbai, J. Kulka, E. Rásó and J. Tímár, J. Transl. Med., 2024, 22, 150 CrossRef.
- P. Vikas, H. Messersmith, C. Compton, L. Sholl, R. R. Broaddus, A. Davis, M. Estevez-Diz, R. Garje, P. A. Konstantinopoulos, A. Leiser, A. M. Mills, B. Norquist, M. J. Overman, D. Sohal, R. C. Turkington and T. Johnson, J. Clin. Oncol., 2023, 41, 1943–1948 CrossRef CAS PubMed.
- X. Li, G. Liu and W. Wu, Exp. Hematol. Oncol., 2021, 10, 37 CrossRef CAS.
- M. Angelo, S. C. Bendall, R. Finck, M. B. Hale, C. Hitzman, A. D. Borowsky, R. M. Levenson, J. B. Lowe, S. D. Liu, S. Zhao, Y. Natkunam and G. P. Nolan, Nat. Med., 2014, 20, 436–442 CrossRef CAS.
- J. Li, F. G. Liu and J. Ye, J. Phys. Chem. Lett., 2022, 13, 6496–6502 CrossRef CAS.
- Z. Wang, S. Zong, L. Wu, D. Zhu and Y. Cui, Chem. Rev., 2017, 117, 7910–7963 CrossRef CAS PubMed.
- J. Li, C. He, H. J. Qu, F. Shen and J. Ye, J. Mater. Chem. C, 2022, 10, 7273–7282 RSC.
- S. J. Liao, J. Cao, W. Zhu, W. Li, J. M. Hu and A. G. Shen, Trends Anal. Chem., 2023, 169, 117357 CrossRef CAS.
- J. Li, H. Q. Liu, P. F. Rong, W. Zhou, X. H. Gao and D. B. Liu, Nanoscale, 2018, 10, 8292–8297 RSC.
- C. Zhu, J. Li, X. Chen, L. Fu, Z. Zhang, Y. Wang, X. Wang and L. Chen, Adv. Funct. Mater., 2025, 25, 2417341 CrossRef.
- C. Dong, X. Fang, J. Xiong, J. Zhang, H. Gan, C. Song and L. Wang, ACS Nano, 2022, 16, 14055–14065 CrossRef CAS PubMed.
- J. Li, B. G. Deng and J. Ye, Biomaterials, 2023, 300, 122211 CrossRef CAS PubMed.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- L. Song and J. Li, Small, 2025, 21, e2407787 CrossRef PubMed.
- J. Li, A. Wuethrich, A. A. I. Sina, H. H. Cheng, Y. Wang, A. Behren, P. N. Mainwaring and M. Trau, Nat. Commun., 2021, 12, 1087 CrossRef CAS.
- J. Li, F. Liu, X. Bi and J. Ye, Biomaterials, 2023, 302, 122327 CrossRef CAS.
- J. Noonan, S. M. Asiala, G. Grassia, N. MacRitchie, K. Gracie, J. Carson, M. Moores, M. Girolami, A. C. Bradshaw, T. J. Guzik, G. R. Meehan, H. E. Scales, J. M. Brewer, I. B. McInnes, N. Sattar, K. Faulds, P. Garside, D. Graham and P. Maffia, Theranostics, 2018, 8, 6195–6209 CrossRef CAS PubMed.
- J. Li, F. G. Liu, C. He, F. Shen and J. Ye, Nanophotonics, 2022, 11, 1549–1560 CrossRef CAS.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci., 2009, 106, 13511–13516 CrossRef CAS.
- C. Qi, L. Shen, J. Li, X. Sun, L. Song, J. Chen, Y. Wu, J. Choo and L. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 67333–67343 CrossRef CAS.
- Y. Zhang, Y. Gu, J. He, B. D. Thackray and J. Ye, Nat. Commun., 2019, 10, 3905 CrossRef.
- Y. C. Ou, X. Wen, C. A. Johnson, D. Shae, O. D. Ayala, J. A. Webb, E. C. Lin, R. C. DeLapp, K. L. Boyd, A. Richmond, A. Mahadevan-Jansen, M. Rafat, J. T. Wilson, J. M. Balko, M. N. Tantawy, A. E. Vilgelm and R. Bardhan, ACS Nano, 2019, 14, 651–663 CrossRef.
- W. Lian, D. Tu, P. Hu, X. Song, Z. Gong, T. Chen, J. Song, Z. Chen and X. Chen, Nano Today, 2020, 35, 100943 CrossRef CAS.
- R. M. Davis, B. Kiss, D. R. Trivedi, T. J. Metzner, J. C. Liao and S. S. Gambhir, ACS Nano, 2018, 12, 9669–9679 CrossRef CAS PubMed.
- C. Giesen, H. A. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS.
- Z. Diao, Y. Han, Y. Chen, R. Zhang and J. Li, Crit. Rev. Oncol. Hemat., 2021, 157, 103171 CrossRef.
- S. A. Martin, C. J. Lord and A. Ashworth, Clin. Cancer Res., 2010, 16, 5107–5113 CrossRef CAS PubMed.
- S. S. Joshi and B. D. Badgwell, Ca-Cancer J. Clin., 2021, 71, 264–279 CrossRef.
- J. Zheng, K. Du, G. Ren, Z. Xie, R. Li, G. Zheng, F. Feng, W. Duan, D. Huang and H. Zhang, J. Clin. Oncol., 2022, 40, 16018 CrossRef.
- E. Sajjadi, K. Venetis, R. Piciotti, M. Invernizzi, E. Guerini-Rocco, S. Haricharan and N. Fusco, Cancer Cell Int., 2021, 21, 266 CrossRef CAS PubMed.
- R. Gryfe, H. Kim, E. T. Hsieh, M. D. Aronson, E. J. Holowaty, S. B. Bull, M. Redston and S. Gallinger, N. Engl. J. Med., 2000, 342, 69–77 CrossRef CAS PubMed.
- N. N. Yassen, D. M. Abouelfadl, N. F. Abbas, A. S. A. Soliman and M. E. Shabana, Bull. Natl. Res. Cent., 2023, 47, 155 CrossRef.
- H. Elrefaey, D. Mohamed, M. A. Azim, S. E. Shorbagy and D. Helal, J. Adv. Med. Med. Res., 2023, 35, 21–42 CrossRef.
- S. Popat, R. Hubner and R. S. Houlston, J. Clin. Oncol., 2005, 23, 6009–6018 CrossRef.
- H. T. Lynch and A. de la Chapelle, N. Engl. J. Med., 2003, 348, 919–932 CrossRef CAS PubMed.
- R. Gryfe, H. Kim, E. T. Hsieh, M. D. Aronson, E. J. Holowaty, S. B. Bull, M. Redston and S. Gallinger, N. Engl. J. Med., 2000, 342, 69–77 CrossRef CAS.
- P. Huang, Y. Deng, J. Zhang, M. Huang, J. Huang, J. Liang, M. Huang, J. Zhao, C. Gao, W. Xie and S. Cai, J. Clin. Oncol., 2019, 37, 1574 Search PubMed.
- A. Mehta, G. Gupta and J. Curr, Oncol, 2018, 1, 55–60 Search PubMed.
- M. Esteller, N. Engl. J. Med., 2008, 358, 1148–1159 CrossRef CAS.
- C. R. Boland and A. Goel, Gastroenterology, 2010, 138, 2073–2087 CrossRef CAS PubMed.
- T. André, K. K. Shiu, T. W. Kim, B. V. Jensen, L. H. Jensen, C. Punt, D. Smith, R. Garcia-Carbonero, M. Benavides, P. Gibbs, C. de la Fouchardiere, F. Rivera, E. Elez, J. Bendell, D. T. Le, T. Yoshino, E. Van Cutsem, P. Yang, M. Z. H. Farooqui, P. Marinello and L. A. Diaz Jr, N. Engl. J. Med., 2020, 383, 2207–2218 CrossRef.
- Q. Jiao, Y. Ren, A. N. Ariston Gabrie, Q. Wang, Y. Wang, L. Du, X. Liu, C. Wang and Y. S. Wang, Biomed. Pharmacother., 2020, 123, 109745 CrossRef CAS.
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, N. Engl. J. Med., 2012, 366, 2443–2454 CrossRef CAS.
- Y. Liu, W. Wu, C. Cai, H. Zhang, H. Shen and Y. Han, Signal Transduct. Target Ther., 2023, 8, 160 CrossRef.
- H. Suto, Y. Funakoshi, Y. Nagatani, Y. Imamura, M. Toyoda, N. Kiyota, H. Matsumoto, S. Tanaka, R. Takai, H. Hasegawa, K. Yamashita, T. Matsuda, Y. Kakeji and H. Minami, J. Cancer Res. Ther., 2021, 17, 1358–1369 CrossRef CAS.
- S. Okada, K. Vaeteewoottacharn and R. Kariya, Cells, 2019, 8, 889 CrossRef CAS.
- Z. Cheng, N. Choi, R. Wang, S. Lee, K. C. Moon, S. Y. Yoon, L. Chen and J. Choo, ACS Nano, 2017, 11, 4926–4933 CrossRef CAS PubMed.
- K. Cizkova, T. Foltynkova, M. Gachechiladze and Z. Tauber, Acta Histochem. Cytochem., 2021, 54, 21–29 CrossRef CAS PubMed.
- S. C. Tsao, J. Wang, Y. Wang, A. Behren, J. Cebon and M. Trau, Nat. Commun., 2018, 9, 1482 CrossRef.
- K. J. Lee, E. J. Ko, Y. Y. Park, S. S. Park, E. J. Ju, J. Park, S. H. Shin, Y. A. Suh, S. M. Hong, I. J. Park, K. p. Kim, J. J. Hwang, S. J. Jang, J. S. Lee, S. Y. Song, S. Y. Jeong and E. K. Choi, Biomaterials, 2020, 255, 120151 CrossRef CAS.
- J. Jung, H. S. Seol and S. Chang, Cancer Res. Treat., 2018, 50, 1–10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01912e |
| This journal is © The Royal Society of Chemistry 2025 |