
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective chemoenzymatic phytate transformations provide access to diverse inositol phosphate derivatives†
Georg Markus
Häner
 a,
Guizhen
Liu
a,
Esther
Lange
b,
Nikolaus
Jork
a,
Klaus
Ditrich
c,
Ralf
Greiner
d,
Gabriel
Schaaf
b and
Henning J.
Jessen
*ae
a,
Guizhen
Liu
a,
Esther
Lange
b,
Nikolaus
Jork
a,
Klaus
Ditrich
c,
Ralf
Greiner
d,
Gabriel
Schaaf
b and
Henning J.
Jessen
*ae
aInstitute of Organic Chemistry, University of Freiburg, Germany. E-mail: henning.jessen@oc.uni-freiburg.de
bInstitute of Crop Science and Resource Conversation, Department of Plant Nutrition, University of Bonn, Germany
cWhite Biotechnology Research – Biocatalysis BASF SE, A030, Carl-Bosch-Strasse 38, 67056 Ludwigshafen am Rhein, Germany
dDepartment of Food Technology and Bioprocess Engineering, Max Rubner-Institut, Federal Research Institute of Nutrition and Food, Haid-und-Neu-Straße 9, Karlsruhe 76131, Germany
eCIBSS – Centre for Integrative Biological Signaling Studies, Freiburg, Germany
First published on 23rd June 2025
Abstract
Phosphorylated myo-inositols (InsPs) are essential cytoplasmic signaling molecules, while their lipidated analogs (PtdInsPs) play a crucial role in membrane signaling. Stereoselective synthesis of these compounds has been achieved through various methods, predominantly using the meso compound myo-inositol as a starting material. However, phytate (InsP6), also a meso compound, is the most abundant inositol derivative in plants – far more prevalent than myo-inositol itself. Despite its abundance, phytate has been rarely used in synthetic strategies for accessing a variety of chiral inositol phosphates and their derivatives through selective dephosphorylations on a preparative scale. Here, we report gram-scale (stereo)selective dephosphorylations of phytate using phytases and demonstrate the application of these products in generating modified InsPs through a transient phosphitylation approach. Notably, the bacterial effector XopH efficiently desymmetrizes meso-phytate to yield enantiomerically pure 1-OH-InsP5. This transformation renders the 1-position accessible for further modifications, which, in biological systems, is where glycerolphosphate diesters are attached. By using selective dephosphorylations with phytases in concert with chemoselective telescoping reaction sequences, this approach greatly advances the stereoselective synthesis of inositol phosphates and their derivatives, such as glycerophosphoinositols, from abundant InsP6.
Introduction
Westheimer famously stated “phosphate esters and anhydrides dominate the living world”, but the less-cited second half of his statement adds that they “are seldom used as intermediates by organic chemists”.1 This is still true to date. Here, we address this disparity in the context of myo-inositol phosphate chemistry. Myo-inositol (hereafter inositol) phosphates (InsPx), inositol-pyrophosphates (PP-InsPx) and their membrane bound brethren (phosphatidyl inositol phosphates, PtdInsPx) are a family of highly phosphorylated second messenger molecules, influencing an enormous variety of biological processes (e.g. phosphate homeostasis or membrane trafficking events).2–6 Recent analytical advances propelled the field forward, (re)discovering previously understudied isomers (e.g. different PP-InsP's)7, and metabolites (e.g. 2-InsP1 or 1,2,3-InsP3)8,9 in biological samples via MS10–12- and NMR13 based methods.To elucidate the identity of the signaling molecules and understand their precise biological functions, the synthesis of defined isomers and analogs is crucial.2,3,6,14 Typically, desymmetrization of the parent meso compound myo-inositol is considered as a reliable entry point.15,16 This meso trick is often performed with the help of chiral auxiliaries, allowing the synthesis of e.g. InsP7 (ref. 17–20) and PtdInsP21–24 isomers relying on asymmetric phosphorylations.20,23,25 Desymmetrizations with peptide catalysts,26–31 for selective phosphorylations, or the use of lipases,21,32,33 for selective esterifications, were established as alternatives to chiral auxiliaries. The use of chemoenzymatic approaches, including dioxygenases, for the stereoselective synthesis of inositol derivatives from distant precursors like benzene, has been comprehensively reviewed.34 In summary, the desymmetrization of myo-inositol has been established as a corner stone of diverse synthetic approaches towards InsP and PtdInsP derivatives.
In contrast, the complementary approach – desymmetrization of the fully phosphorylated meso compound InsP6via selective dephosphorylations as a starting point for synthesis – is almost completely absent from literature. This is not understandable from a supply perspective: InsP6 is the major phosphate storage molecule in plants and is available on a large scale. In fact, it is a far more abundant starting material compared to inositol itself. However, chemistry needs to be developed further to transform the highly charged polyphosphorylated intermediates that would arise from selective phytate dephosphorylations, which we identify as one major obstacle in this approach, in line with the initial statement by Westheimer.
In the 90's, selective phytate dephosphorylation with baker's yeast leading mainly to 1,2,6-InsP3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35,36 was reported (Scheme 1). The synthetic utility of the resulting chiral trisphosphate was proven in the generation of diverse therapeutically and synthetically relevant derivatives via oxidation,37O-acylation,38 carbamoylation,35P-alkylation39 and ultimately acidic phosphate cleavage.35,36 In 1999 selective dephosphorylation of 1,4,5,6-InsP4 (or enantiomeric 3,4,5,6-InsP4) with InsP5/InsP4-phosphohydrolase to 1,5,6-InsP3 (or 3,4,5-InsP3 respectively) was shown, highlighting other synthetic applications of selective enzymatic dephosphorylations. However, the required starting materials had to be synthesized in enantiomerically pure form starting from hydroquinone.40
35,36 was reported (Scheme 1). The synthetic utility of the resulting chiral trisphosphate was proven in the generation of diverse therapeutically and synthetically relevant derivatives via oxidation,37O-acylation,38 carbamoylation,35P-alkylation39 and ultimately acidic phosphate cleavage.35,36 In 1999 selective dephosphorylation of 1,4,5,6-InsP4 (or enantiomeric 3,4,5,6-InsP4) with InsP5/InsP4-phosphohydrolase to 1,5,6-InsP3 (or 3,4,5-InsP3 respectively) was shown, highlighting other synthetic applications of selective enzymatic dephosphorylations. However, the required starting materials had to be synthesized in enantiomerically pure form starting from hydroquinone.40
 | ||
| Scheme 1 Desymmetrization of phytate via selective dephosphorylation using phytases. Previously, 1,2,6-InsP3 was obtained by baker's yeast digest.35,36 Here, selective dephosphorylation with expressed XopH on a preparative scale was established. The resulting chiral 1-OH-InsP5 was transformed into the corresponding chiral 1-InsP1 chemoenzymatically with the phytase Natuphos, thus inverting the phosphorylation pattern. | ||
Here, we present a dephosphorylation approach to enzymatically desymmetrize abundant meso-phytate and use the single product as an entry point to access phosphorylated inositol derivatives (see Scheme 1 for an overview). We rely on the recently discovered unique phytase activity of XopH, a bacterial effector protein found in Xanthomonas euvesicatoria. XopH dephosphorylates InsP6 exclusively at the 1-position, resulting in enantiopure chiral 1-hydroxy-inositol-pentakisphosphate (1-OH-InsP5).41 Notably, the 1-position holds particular significance in InsP chemistry, serving as the attachment point for the glycerolphosphate diester in PtdInsPs.
Additionally, we demonstrate that commercial Natuphos, originating from Aspergillus ficuum, has potential in InsP chemistry, although no desymmetrization is observed in this case, as the sole product is the meso compound 2-InsP1, in accordance with observations in naturally occurring phytases of varying provenance.42–48 This commercial phytase was previously compared to the follow-up product Natuphos E for the synthesis of InsP3–5via selective dephosphorylations.49
The products of the enzymatic dephosphorylations are then transformed into other phosphorylated inositols using a transient phosphitylation approach, that relies on the lability of mixed P(III)–P(V) anhydrides in contrast to the higher stability of P(III) triesters. Natuphos can then be used to remove unwanted phosphates and thus invert the phosphorylating pattern of the starting material (Scheme 1).
Results and discussion
1-OH-InsP5 has previously been obtained by selective dephosphorylation using the bacterial effector XopH on a small scale in vitro to assign enzyme specificity.41 We have now scaled this reaction to access 1-OH-InsP5 as starting material for further chemical transformations (Scheme 2). | ||
| Scheme 2 Synthesis of 1-OH-InsP52via selective dephosphorylation of InsP6 with XopH. | ||
During scale-up it was observed that larger amounts of phytate in the reaction led to some precipitation. To account for possible XopH precipitation, the reaction time was extended accordingly and complete consumption of InsP6 was observed after 8 h.
The crude enzymatic digest of InsP6 was purified by strong anion exchange (SAX) chromatography on a Q-sepharose column using an NH4HCO3 (1 M, pH = 8.0) gradient as eluent. Next, to ensure solubility in organic solvents for further transformations, 1-OH-InsP5 was converted to its tetrabutylammonium (TBA) salt.
One of our goals was to install a phosphate diester in the 1-position of 1-OH InsP5 as this would enable direct access to PtdInsPs and analogues, which was previously difficult to achieve. It is known that phosphates react rapidly with other P-derived electrophiles. For example, unprotected nucleoside phosphates (with free OH groups) can be phosphorylated with P-amidites.50 Also P-imidazolides51–57 and diamidophosphates58 can be used to construct P-anhydrides even in water as solvent with high chemoselectivity. This suggests that a chemical modification of phosphorylated inositols, such as 1-OH-InsP5, will first take place at the phosphate esters, before the alcohol will react.
This has significant implications regarding reaction design. Here, we focused on the use of P-amidites, with the expectation that initially P(III)–P(V) anhydrides would form, followed by a reaction of the OH group forming a P(III) triester. A strategy was therefore required to selectively cleave the mixed anhydrides again after transiently blocking the P(V) esters, while preserving the P(III) triester – an approach that would have to be based on transient phosphitylation. To identify mild conditions that enable the cleavage of a P(III)–P(V) anhydride, adenosine diphosphate (ADP) was used as a model compound (see ESI Fig. 1†). Screening revealed that pyridinium hydrobromide (in the following Pyr × HBr) rapidly cleaves the mixed anhydride. This approach was then applied to 1-OH-InsP5, enabling selective modification at the 1-position while ensuring transient P(III)–P(V) anhydrides were cleaved in the presence of Pyr × HBr.
1-OH-InsP5 TBA salt 2 was reacted with an excess of bis-fluorenylmethyl-P-amidite 1 (see ESI Fig. 2,† fluorenylmethyl = Fm) and was then analyzed by 31P-NMR spectroscopy. Due to the number of phosphates in the starting material 2, several P(III)–P(V)-anhydride signals were observed, as indicated by the characteristic chemical shifts in 31P-NMR (125 to 130 ppm).59 Addition of pyridinium hydrobromide led to complete disappearance of the P(III)–P(V)-anhydride signals, indicating cleavage. However, subsequent oxidation using mCPBA led to a complex mixture. To understand the failure of the desired synthesis, the crude material was analyzed by capillary electrophoresis mass spectrometry (CE-MS).7 We found a variety of InsP6 derivatives where adjacent phosphate groups had undergone condensation reactions to cyclic pyrophosphate derivatives. Moreover, the products were bearing either one or two Fm groups (Scheme 3a). Optimization of the reaction conditions did neither lead to reduced cyclizations nor reduced Fm cleavage. As an alternative, we reasoned that a controlled cyclization should furnish a more defined cyclic pyrophosphate mixture, and a controlled hydrolysis of those intermediates could then result in a defined product.
 | ||
| Scheme 3 Synthesis of InsP6 derivatives modified solely at the 1-position. CE-qTOF-MS (background electrolyte (in the following BGE): NH4OAc 35 mM pH = 9.7, CE voltage: 30 kV, CE current: 23 μA, injection: 100 mbar, 15 s (30 nL)) analysis of the transient phosphitylation of 1-OH-InsP5 using different P-amidites. The depicted structures are color coded by identical mass. (a) Using P-amidite 1 a complex mixture was obtained. (b) Reduction of components was achieved via controlled cyclization using cyclization prone P-amidite 9. The depicted cyclic pyrophosphates are just examples of possible structures. (c) The obtained cyclic intermediates were transformed into a single product in situ via subsequent acid treatment. (d) Using an optimized transient phosphitylation work-flow 1-Fm-InsP68 and 1-DEACM-InsP611 were synthesized in one telescoping reaction sequence starting from 1-OH-InsP5. | ||
P-amidite 9, containing a phenyl moiety as a good leaving group instead of Fm, was designed and synthesized to promote the cyclization reactions (Scheme 3b). Interestingly, no P(III)–P(V) anhydrides were detectable by 31P-NMR after global phosphitylation (see ESI Fig. 3†). However, addition of Pyr × HBr was necessary to assure quantitative decomposition of putatively in situ formed P(III)–P(V) anhydrides. The reaction outcome was analyzed by CE-MS. Under these new conditions a much more defined mixture of only mono- and bis-cyclic InsP6 derivatives bearing exactly one Fm-group were obtained after oxidation. While the number of possible products decreased drastically, it was still not possible to identify the formed isomers. This assignment would also not be necessary, if it were possible to hydrolyze the cyclic pyrophosphates. One would expect the mixture to converge into a single product: InsP6 with an Fm protecting group located at the phosphate in the 1-position. Cyclic pyrophosphate hydrolysis was therefore studied next using inositoltrispyrophosphate (ITPP) as a model compound (see ESI Tables 4, 5 and Fig. 4†).60,61 Reaction conditions were identified that led to complete hydrolysis of the anhydrides (either HBr at 80 °C or ZnCl2 and HCl at 37 °C). After optimization, the anhydrides in the crude mixture of 1-Fm-InsP6 derivatives obtained above were hydrolysed and the reaction progress was monitored by 31P-NMR (see ESI Fig. 5†). Upon disappearance of all pyrophosphate signals, the reaction mixture was diluted with water, neutralized with NH4HCO3 (pH = 8.0, 1 M) and purified by SAX on a Q-sepharose column using a NaClO4 (1 M) gradient as eluent.
CE-MS analysis of the material before purification revealed formation of 1-Fm-InsP68 as main product (Scheme 3c), while InsP6 and InsP5 were the sole side products and readily removable by SAX as shown by CE-MS analysis after purification. The position of the Fm group was initially verified via 2D-NMR spectroscopy (see ESI Fig. 7 and 8†). Overall, in the whole telescoping sequence, 1-Fm-InsP68 was synthesized in 45% yield directly from 1-OH-InsP5 (Scheme 3d). To demonstrate further utility of this method, we used the sequence to introduce a 7-(diethylamino)-4-(hydroxymethyl)-coumarin (in the following DEACM) moiety at the phosphate in the 1-position. Thus, photocaged 1-DEACM-InsP611 was synthesized, simply by exchanging the cyclization prone P-amidite (Scheme 3d).
We next studied the dephosphorylation of the Fm modified InsP68 using the promiscuous commercial phytase Natuphos as a very mild and potentially selective alternative to acidic hydrolysis.35,36 Its action on InsP6 led to quantitative dephosphorylation to 2-InsP1 within 10 min (see ESI Table 6†).49 Importantly, this is also an interesting starting material for the introduction of further modifications. Isomer identity was assigned via NMR in accordance with literature.62
Phytases initiate the stepwise dephosphorylation of phytate with a high stereo- and regioselectivity.63 2-InsP1 with the phosphate in axial position is the endpoint of many naturally occurring phytases,42–48 but phytases stopping at different InsP derivatives (e.g. InsP5, InsP3) are known.42,63–66 However, unlike XopH these enzymes often deliver mixtures of myo-inositol phosphate intermediates. The dephosphorylation of InsP6 to 2-InsP1 by Natuphos produces 5 equivalents of phosphate (Pi), which were removed by precipitation with Ba(OAc)248 or CaCl2. The product was then obtained by precipitation from EtOH. Following this approach, 1 g of phytate was digested to 2-InsP1 in 86% yield (Scheme 4).48 Next, the digest of chiral 1-Fm-InsP68 by Natuphos was monitored using 31P-NMR (see ESI Fig. 10†) and a slower reaction compared to unprotected phytate was observed (>24 h for complete conversion). However, a complete digestion of the phosphate monoesters to deliver phosphate diester 1-Fm-InsP113 was achieved after 2 days of incubation. The product 13 was purified by reversed-phase medium pressure liquid chromatography (RP-MPLC) to remove excess Pi. Subsequent basic deprotection led to 1-InsP114 in 58% over two steps, starting from enantiomerically pure 1-Fm-InsP68.
 | ||
| Scheme 4 Dephosphorylation of InsP6 derivatives with Natuphos (10500 U mL−1, 3150 U was used for the digest of phytate, 2100 U was used for the digest of 8) in NH4OAc (50 mM, pH 6.3) leads to defined InsP1 isomers. 1,2-InsP2 was obtained as dephosphorylation product of 1-Fm-InsP68 using the 6-Phytase from E. coli (7500 U mL−1, 1 U phytase was used) in HEPES-buffer (50 mM HEPES, 10 mM NaCl, 5% glycerol, 2 mM DTT, 0.5 mM, MgCl2, pH = 4.0; at 28 °C for 45 min) and subsequent basic deprotection. | ||
With 1-InsP1 in hand, enantiomer identity of the product was further corroborated using optical rotation and comparison to an original standard.20,26,67 This additionally affirms our assignment, that the 1-position in 1-OH-InsP5 is modified in our transient phosphitylation approach. Depending on the reaction time (<48 h), we observed transient accumulation of an InsP2 species resulting from incomplete dephosphorylation. Based on the selectivity of Natuphos described above, we tentatively assigned the isomer as 1-Fm-1,2-InsP2. Indeed, basic deprotection of the Fm group with piperidine after purification led to formation of enantiopure 1,2-InsP2 validated by CE-MS experiments and spiking with defined InsP2 isomers (see ESI Fig. 11†).9 In summary, the synthesis of enantiopure 1-InsP1 from InsP6 using a chemoenzymatic approach is possible in 3 steps.
Next, we screened a small panel of phytases to potentially obtain other phosphorylated inositol isomers or a higher accumulation of defined intermediates. As starting materials, either InsP6, 1-DEACM-InsP611 or 1-Fm-InsP68 were studied. The resulting InsP mixtures were analyzed by CE-MS (see Scheme 5 and ESI Fig. 13†). All tested phytases hydrolyzed InsP6 to 2-InsP1, as confirmed by CE-MS spiking experiments with [18O]-2-InsP1 (see ESI Fig. 14†). 1-DEACM-InsP611 was hydrolyzed by all tested phytases to a mixture of 1-DEACM-InsP1 and 1-DEACM-InsP2, which is not useful from a preparative perspective. However, 1-Fm-InsP68 was predominantly (>80%) dephosphorylated under comparable conditions to 1-Fm-1,2-InsP2. Again, isomer assignment was achieved after basic deprotection and subsequent CE-MS spiking experiments (Scheme 5 and ESI Fig. 12†).9 Decomposition of [13C]-labeled InsP6 (by incubation at 100 °C) led to a mixture of all InsP2 isomers, which was used in the following for spiking experiments, to verify the initial assignment (see Scheme 5).9
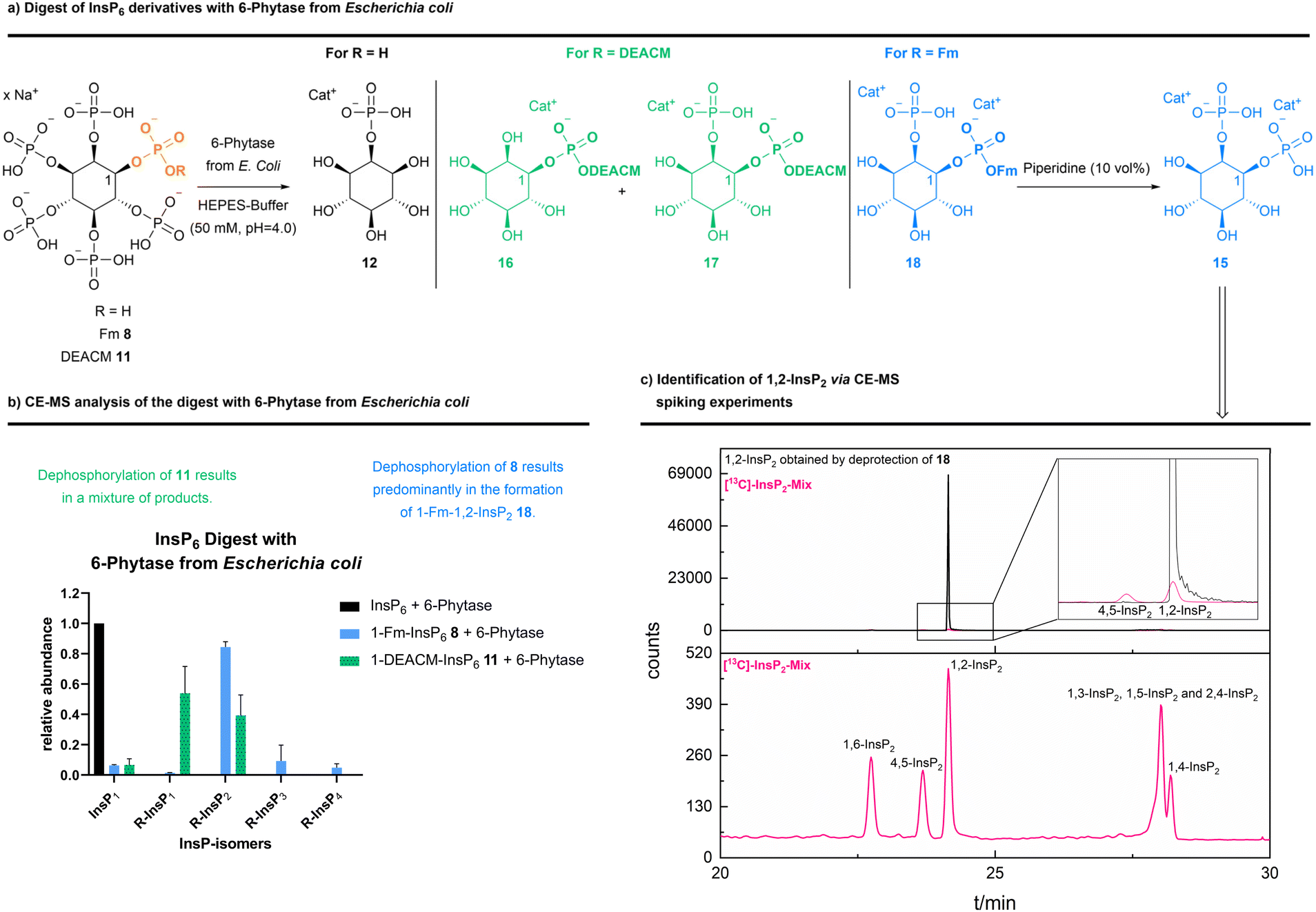 | ||
| Scheme 5 (a) Different InsP6 (15 mM) were dephosphorylated with 6-phytase from Escherichia coli (7500 U mL−1 at pH = 5.0, 1 U phytase was used) (used buffer: 50 mM HEPES, 10 mM NaCl, 5% glycerol, 2 mM DTT, 0.5 mM, MgCl2, pH = 4.0; at 28 °C for 45 min). CE-qTOF-MS (BGE: NH4OAc 35 mM pH = 9.7, CE voltage: 30 kV, CE current: 23 μA, injection: 100 mbar, 15 s (30 nL)). (b) Analysis revealed different InsPs as product mixtures, depending on the used InsP6 derivative. 1-Fm-InsP68 was relatively cleanly dephosphorylated to an InsP2 derivative. (c) The formed 1,2-InsP2 was identified via CE-QQQ-MS (BGE: NH4OAc 35 mM pH = 9.7, CE voltage: 30 kV, CE current: 23 μA, injection: 100 mbar, 10 s (20 nL)) spiking experiments9 using a [13C]-InsP2 mix (obtained via decomposition of [13C]-InsP6 at 100 °C), after basic deprotection (piperidine (10 vol%)) of 1-Fm-InsP218. | ||
Glycerophosphoinositols (hereafter referred to as GroPIns) are produced in vivo by phospholipase A2 cleavage of the glycerol-phosphate-diester in PtdInsP's. These metabolites are active as cellular signals.68–70 Previous syntheses of GroPIns were for example achieved via saponification of PtdIns.71
We envisioned synthetic access to GroPIns and non-natural derivatives using the unprotected InsP1 isomers obtained in this study. Minnard recently demonstrated the catalytic ring-opening of epoxides with phosphate diester nucleophiles using Co(salen) complexes.72 Herein, we extend this approach to InsP1 TBA salts as phosphate monoester nucleophiles. We started our exploration of suitable reaction conditions (see ESI Table 7 and Fig. 15†) with 2-InsP112 since it is readily available from InsP6. (S)-glycidol was used as electrophile. Jacobsen's Co(III) salen catalyst73 was added to promote ring opening of the epoxide (Scheme 6).
 | ||
| Scheme 6 Synthesis of 1-GroPIns 21 and the non-natural 2-GroPIns derivative 20via Co(III) catalyzed epoxide ring opening of S-(−)-glycidol with InsP1 isomers. | ||
The reactions did not go to completion. No improvements were observed neither for longer reaction times nor for additional epoxide additions, and the crude reaction mixture was directly purified via SAX after an overnight reaction. This allowed isolation of the desired GroPIns derivatives and re-isolation of unreacted InsP1 starting material. The optimized reaction conditions gave access to 2-GroPIns 20 in 29% isolated yield from unprotected 2-InsP112. Natural 1-GroPIns 21 was then obtained under analogous conditions in 25% yield. These isolated yields are comparatively low, however, this is compensated by re-isolation of the starting material (up to 65%). Of note, the required enantiopure 1-InsP1 is now available on larger scale using the XopH dephosphorylation approach described in this paper.
Conclusion
The stereoselective synthesis of inositol phosphates and their derivatives is usually based on desymmetrization of myo-inositol. However, dephosphorylation of abundant meso InsP6 as a complementary approach has rarely been studied and was abandoned despite promising studies in the 1990s.35,36 Here, we demonstrate the synthetic potential of selective enzymatic dephosphorylations as a tool to access defined InsPx isomers and their transformation into otherwise difficult to access derivatives.After enzymatic desymmetrization using XopH, 1-OH-InsP5 is converted into phosphate diesters attached selectively to the 1-position using a transient phosphitylation approach with a cyclization prone P-amidite. Acidic ring opening of the ensuing cyclic pyrophosphate esters achieves convergence of complex mixtures into a single product.
This methodology unlocks new synthetic pathways, allowing hydroxyl group phosphorylation even in the presence of unprotected phosphates. The obtained InsP6 derivatives were subjected to dephosphorylation by different phytases. With this approach, it is possible to obtain enantiopure 1-InsP1, 1,2-InsP2 and other derivatives. To further demonstrate the versatility of the obtained building blocks, other metabolites (2-InsP1,8 1-GroPIns68,70 and 2-GroPIns) were synthesized using Co(III) catalyzed glycidol epoxide openings.
Selective dephosphorylation of InsP6 can be considered as an alternative desymmetrization route compared to classical approaches starting from inositol. One can imagine an even wider scope of selective dephosphorylations with appropriate phytases that have the potential to transform the way of how we generally think about synthesizing InsPs. Our paper provides a new entry point into the world of phosphorylated metabolites using phosphates as key strategic components that are still too “seldom used as intermediates by organic chemists”.1
Data availability
Experimental procedures, characterization data and data supporting this article have been included as part of the electronic (ESI).†Author contributions
G. M. H. and H. J. designed the molecules. G. M. H. synthesized and analyzed the compounds. G. L. helped with compound characterization by CE-MS. G. M. H. drafted the initial manuscript and prepared the figures. K. D. and R. G. provided expertise regarding phytases and provided different phytases for experiments. E. L. and G. S. provided XopH digests and enzymes and helped with scaling-up and the phyatese screening. N. J. helped with purification of molecules and isomer assignment. H. J. conceived the project. All authors provided input on the final version of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr S. Braukmüller and C. Warth from the Analytical Service Team of the University of Freiburg for NMR and HRMS measurements, respectively. We thank Dr Thomas Haas for initial experiments with XopH digests. This research was supported by the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy (CIBSS-EXC-2189-Project ID 390939984, to Henning J. Jessen and SCHA 1274/5-1 to G. Schaaf). HJJ acknowledges support from the VW Foundation (VW Momentum Grant 98604).Notes and references
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- S. Kim, R. Bhandari, C. A. Brearley and A. Saiardi, Trends Biochem. Sci., 2024, 49, 969–985 CrossRef CAS PubMed.
- M. Nguyen Trung, D. Furkert and D. Fiedler, Curr. Opin. Chem. Biol., 2022, 70, 102177 CrossRef CAS.
- Y. Posor, W. Jang and V. Haucke, Nat. Rev. Mol. Cell Biol., 2022, 23, 797–816 CrossRef CAS PubMed.
- E. Riemer, N. J. Pullagurla, R. Yadav, P. Rana, H. J. Jessen, M. Kamleitner, G. Schaaf and D. Laha, Front. Plant Sci., 2022, 13, 1–18 Search PubMed.
- M. D. Best, H. Zhang and G. D. Prestwich, Nat. Prod. Rep., 2010, 27, 1403 RSC.
- D. Qiu, M. S. Wilson, V. B. Eisenbeis, R. K. Harmel, E. Riemer, T. M. Haas, C. Wittwer, N. Jork, C. Gu, S. B. Shears, G. Schaaf, B. Kammerer, D. Fiedler, A. Saiardi and H. J. Jessen, Nat. Commun., 2020, 11, 6035 CrossRef CAS PubMed.
- M. Nguyen Trung, S. Kieninger, Z. Fandi, D. Qiu, G. Liu, N. K. Mehendale, A. Saiardi, H. Jessen, B. Keller and D. Fiedler, ACS Cent. Sci., 2022, 8, 1683–1694 CrossRef CAS.
- G. Liu, E. Riemer, R. Schneider, D. Cabuzu, O. Bonny, C. A. Wagner, D. Qiu, A. Saiardi, A. Strauss, T. Lahaye, G. Schaaf, T. Knoll, J. P. Jessen and H. J. Jessen, RSC Chem. Biol., 2023, 4, 300–309 RSC.
- J. J. Carroll, C. Sprigg, G. Chilvers, I. Delso, M. Barker, F. Cox, D. Johnson and C. A. Brearley, Methods Ecol. Evol., 2024, 15, 530–543 CrossRef.
- M. Ito, N. Fujii, S. Kohara, S. Hori, M. Tanaka, C. Wittwer, K. Kikuchi, T. Iijima, Y. Kakimoto, K. Hirabayashi, D. Kurotaki, H. J. Jessen, A. Saiardi and E. Nagata, J. Biol. Chem., 2023, 299, 102928 CrossRef CAS PubMed.
- X. Liu, P. W. Villalta and S. J. Sturla, Rapid Commun. Mass Spectrom., 2009, 23, 705–712 CrossRef CAS.
- R. K. Harmel, R. Puschmann, M. Nguyen Trung, A. Saiardi, P. Schmieder and D. Fiedler, Chem. Sci., 2019, 10, 5267–5274 RSC.
- M. P. Thomas, S. J. Mills and B. V. L. Potter, Angew. Chem., Int. Ed., 2016, 55, 1614–1650 CrossRef CAS PubMed.
- K. S. Bruzik and M. D. Tsai, J. Am. Chem. Soc., 1992, 114, 6361–6374 CrossRef CAS.
- R. J. Kubiak and K. S. Bruzik, J. Org. Chem., 2003, 68, 960–968 CrossRef CAS.
- M. L. Shipton, F. A. Jamion, S. Wheeler, A. M. Riley, F. Plasser, B. V. L. Potter and S. J. Butler, Chem. Sci., 2023, 14, 4979–4985 RSC.
- I. Pavlovic, D. T. Thakor and H. J. Jessen, Org. Biomol. Chem., 2016, 14, 5559–5562 RSC.
- M. Wu, L. S. Chong, S. Capolicchio, H. J. Jessen, A. C. Resnick and D. Fiedler, Angew. Chem., Int. Ed., 2014, 53, 7192–7197 CrossRef CAS.
- S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912–6916 CrossRef CAS PubMed.
- A. M. Joffrin, A. M. Saunders, D. Barneda, V. Flemington, A. L. Thompson, H. J. Sanganee and S. J. Conway, Chem. Sci., 2021, 12, 2549–2557 RSC.
- T. Aiba, S. Suehara, S. Choy, Y. Maekawa, H. Lotter, T. Murai, S. Inuki, K. Fukase and Y. Fujimoto, Chem.–A Eur. J., 2017, 23, 8304–8308 CrossRef CAS PubMed.
- T. Aiba, M. Sato, D. Umegaki, T. Iwasaki, N. Kambe, K. Fukase and Y. Fujimoto, Org. Biomol. Chem., 2016, 14, 6672–6675 RSC.
- M. Bru, S. P. Kotkar, N. Kar and M. Köhn, Chem. Sci., 2012, 3, 1893 RSC.
- E. Durantie, S. Huwiler, J.-C. Leroux and B. Castagner, Org. Lett., 2016, 18, 3162–3165 CrossRef CAS.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 11653–11656 CrossRef CAS PubMed.
- B. R. Sculimbrene, Y. Xu and S. J. Miller, J. Am. Chem. Soc., 2004, 126, 13182–13183 CrossRef CAS PubMed.
- K. J. Kayser-Bricker, P. A. Jordan and S. J. Miller, Tetrahedron, 2008, 64, 7015–7020 CrossRef CAS.
- C. M. Longo, Y. Wei, M. F. Roberts and S. J. Miller, Angew. Chem., Int. Ed., 2009, 48, 4158–4161 CrossRef CAS PubMed.
- P. A. Jordan, K. J. Kayser-Bricker and S. J. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20620–20624 CrossRef CAS PubMed.
- B. D. Chandler, A. L. Burkhardt, K. Foley, C. Cullis, D. Driscoll, N. Roy D'Amore and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 412–418 CrossRef CAS PubMed.
- M. G. Vasconcelos, R. H. C. Briggs, L. C. S. Aguiar, D. M. G. Freire and A. B. C. Simas, Carbohydr. Res., 2014, 386, 7–11 CrossRef CAS.
- M. F. P. Ribeiro, K. C. Pais, B. S. M. de Jesus, R. Fernandez-Lafuente, D. M. G. Freire, E. A. Manoel and A. B. C. Simas, European J. Org. Chem., 2018, 2018, 386–391 CrossRef CAS.
- J. Duchek, D. R. Adams and T. Hudlicky, Chem. Rev., 2011, 111, 4223–4258 CrossRef CAS.
- C. Blum, S. Karlsson, G. Schlewer, B. Spiess and N. Rehnberg, Tetrahedron Lett., 1995, 36, 7239–7242 CrossRef CAS.
- C. Blum, N. Rehnberg, B. Spiess and G. Schlewer, Carbohydr. Res., 1997, 302, 163–168 CrossRef CAS.
- M. Malmberg and N. Rehnberg, Synlett, 1996, 1996, 361–362 CrossRef.
- M. Malmberg and N. Rehnberg, J. Carbohydr. Chem., 1996, 15, 459–464 CrossRef CAS.
- A. Lindahl, M. Malmberg and N. Rehnberg, J. Carbohydr. Chem., 1996, 15, 549–554 CrossRef CAS.
- S. Adelt, O. Plettenburg, R. Stricker, G. Reiser, H.-J. Altenbach and G. Vogel, J. Med. Chem., 1999, 42, 1262–1273 CrossRef CAS.
- D. Blüher, D. Laha, S. Thieme, A. Hofer, L. Eschen-Lippold, A. Masch, G. Balcke, I. Pavlovic, O. Nagel, A. Schonsky, R. Hinkelmann, J. Wörner, N. Parvin, R. Greiner, S. Weber, A. Tissier, M. Schutkowski, J. Lee, H. Jessen, G. Schaaf and U. Bonas, Nat. Commun., 2017, 8, 2159 CrossRef PubMed.
- U. Konietzny and R. Greiner, Int. J. Food Sci. Technol., 2002, 37, 791–812 CrossRef CAS.
- R. Greiner, N. Carlsson and M. L. Alminger, J. Biotechnol., 2000, 84, 53–62 CrossRef CAS PubMed.
- M. Wyss, R. Brugger, A. Kronenberger, R. Rémy, R. Fimbel, G. Oesterhelt, M. Lehmann and A. P. G. M. van Loon, Appl. Environ. Microbiol., 1999, 65, 367–373 CrossRef CAS.
- T. Hayakawa, K. Suzuki, H. Miura, T. Ohno and I. Igaue, Agric. Biol. Chem., 1990, 54, 279–286 CAS.
- I. Maiti, Phytochemistry, 1974, 13, 1047–1051 CrossRef CAS.
- P. E. Lim and M. E. Tate, Biochim. Biophys. Acta, Enzymol., 1973, 302, 316–328 CrossRef CAS.
- D. Cosgrove, Aust. J. Biol. Sci., 1970, 23, 1207 CrossRef CAS PubMed.
- R. Greiner, Prep. Biochem. Biotechnol., 2021, 51, 985–989 CrossRef CAS.
- G. S. Cremosnik, A. Hofer and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 286–289 CrossRef CAS.
- K. Qian, B. Hanf, C. Cummins and D. Fiedler, Angew. Chem., Int. Ed., 2025, 64, e202419147 CrossRef CAS.
- N. W. Brown, S. K. Schlomach, A. M. Marmelstein and D. Fiedler, ChemBioChem, 2023, 24, e202200407 CrossRef CAS PubMed.
- A. M. Marmelstein, J. A. M. Morgan, M. Penkert, D. T. Rogerson, J. W. Chin, E. Krause and D. Fiedler, Chem. Sci., 2018, 9, 5929–5936 RSC.
- L. Yates, A. Marmelstein and D. Fiedler, Synlett, 2014, 25, 2239–2245 CrossRef CAS.
- M. Strenkowska, P. Wanat, M. Ziemniak, J. Jemielity and J. Kowalska, Org. Lett., 2012, 14, 4782–4785 CrossRef CAS PubMed.
- S. Golojuch, M. Kopcial, D. Strzelecka, R. Kasprzyk, N. Baran, P. J. Sikorski, J. Kowalska and J. Jemielity, Bioorg. Med. Chem., 2020, 28, 115523 CrossRef CAS.
- B. A. Wojtczak, M. Bednarczyk, P. J. Sikorski, A. Wojtczak, P. Surynt, J. Kowalska and J. Jemielity, J. Org. Chem., 2023, 88, 6827–6846 CrossRef CAS PubMed.
- H. Lin, L. J. Leman and R. Krishnamurthy, Chem. Sci., 2022, 13, 13741–13747 RSC.
- H. J. Jessen, T. Dürr-Mayer, T. M. Haas, A. Ripp and C. C. Cummins, Acc. Chem. Res., 2021, 54, 4036–4050 CrossRef CAS PubMed.
- L. F. Johnson and M. E. Tate, Can. J. Chem., 1969, 47, 63–73 CrossRef CAS.
- K. C. Fylaktakidou, J.-M. Lehn, R. Greferath and C. Nicolau, Bioorg. Med. Chem. Lett., 2005, 15, 1605–1608 CrossRef CAS PubMed.
- D. C. Billington, R. Baker, J. J. Kulagowski, I. M. Mawer, J. P. Vacca, S. J. DeSolms and J. R. Huff, J. Chem. Soc., Perkin Trans. 1, 1989, 1423 RSC.
- D. Menezes-Blackburn, R. Greiner and U. Konietzny, in Enzymes in Farm Animal Nutrition, CABI, 2022, pp. 103–123 Search PubMed.
- A. Hara, S. Ebina, A. Kondo and T. Funaguma, Agric. Biol. Chem., 1985, 49, 3539–3544 CrossRef CAS.
- L. Barrientos, J. J. Scott and P. Murthy, Plant Physiol., 1994, 106, 1489–1495 CrossRef CAS.
- J. Kerovuo, J. Rouvinen and F. Hatzack, Biochem. J., 2000, 352, 623 CrossRef CAS PubMed.
- D. C. Billington, R. Baker, J. J. Kulagowski and I. M. Mawer, J. Chem. Soc. Chem. Commun., 1987, 314 RSC.
- D. Corda, P. Zizza, A. Varone, B. M. Filippi and S. Mariggiò, Cell. Mol. Life Sci., 2009, 66, 3449–3467 CrossRef CAS.
- D. Corda, P. Zizza, A. Varone, K. S. Bruzik and S. Mariggiò, Biochem. Soc. Trans., 2012, 40, 101–107 CrossRef CAS PubMed.
- L. Patrussi, S. Mariggiò, D. Corda and C. T. Baldari, Front. Immunol., 2013, 4, 1–6 CAS.
- K. S. Bruzik, Z. Guan, S. Riddle and M.-D. Tsai, J. Am. Chem. Soc., 1996, 118, 7679–7688 CrossRef CAS.
- R. L. H. Andringa, M. Jonker and A. J. Minnaard, Org. Biomol. Chem., 2022, 20, 2200–2204 RSC.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02844b |
| This journal is © The Royal Society of Chemistry 2025 |