
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electroreductive room-temperature C–H activations with RuCl3·nH2O precatalyst via cathodic ruthenium(III/II) manifold†
Takuya
Michiyuki
 a,
Tristan
von Münchow
a,
Zhipeng
Lin
a,
Binbin
Yuan
a,
João C. A.
Oliveira
a and
Lutz
Ackermann
*ab
a,
Tristan
von Münchow
a,
Zhipeng
Lin
a,
Binbin
Yuan
a,
João C. A.
Oliveira
a and
Lutz
Ackermann
*ab
aWöhler Research Institute for Sustainable Chemistry, Georg-August-Universität Göttingen, Tammannstraße 2, 37077, Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
bDZHK (German Centre for Cardiovascular Research), Potsdamer Straße 58, 10875 Berlin, Germany
First published on 27th June 2025
Abstract
We, herein, disclose a strategy to directly utilize user-friendly RuCl3·nH2O for ortho- as well as meta-C–H functionalizations at low temperatures. The key to success was the in situ formation of the active ruthenium catalyst through cathodic electron transfer, setting the stage for C–H activations under exceedingly mild reaction conditions. The robustness of our electrocatalysis process was highlighted by the late-stage diversification of compounds of relevance to chemical, agrochemical, and pharmaceutical industries, as well as simple amines as terminal reductants for the electroreduction. Detailed mechanistic studies by, among others, spectroelectrochemical analysis provided strong evidence for a cathodic reduction manifold.
Introduction
Ruthenium-catalyzed C–H activation chemistry has evolved into an indispensable chemical toolbox for selectively functionalizing arene scaffolds.1–10 For instance, site-selectivity can be precisely modulated11–17 without exploiting elaborate auxiliaries, providing instant access to ortho-18–27 or meta-decorated frameworks.28–41 Our group indeed discovered that the addition of phosphine ligands can alter the site-selectivity in C–H benzylations from ortho- to unusual meta-selectivities.14 Precise selectivity control can also be achieved depending on the nature of the electrophilic substrates.13,16 Among such transformations, C–H arylations hold a significant potential for the assembly of valuable biaryl scaffolds,6,9,10 with carboxylate-assisted ruthenium(II/IV)-catalyzed C–H activations representing the most robust and efficient strategy.23 In recent years, substantial efforts have been made to render reaction conditions milder. Notable advancements include the use of light irradiation, as disclosed by Ackermann and Greaney,42–45 as well as the employment of arene ligand-free ruthenium precatalysts.46,47Despite the considerable progress,48–52 the majority of existing approaches heavily depend on ruthenium(II) precatalysts, which require lengthy organometallic syntheses by experts with glovebox techniques. This reliance jeopardizes the resource efficiency and user-friendly nature of the overall processes. Importantly, when tracing back the synthesis routes, one can realize that ruthenium(II) complexes, such as [RuCl2(p-cymene)]2, [Ru(OAc)2(p-cymene)], arene ligand-free complexes Ru1 and Ru2, originate from RuCl3·nH2O (Scheme 1A).46,47,53 Therefore, the direct employment of RuCl3·nH2O as the catalyst offers a more sustainable and user-friendly strategy for executing C–H activation. In 2007, our group first disclosed the use of arene ligand-free RuCl3·nH2O for ortho-C–H arylation.24 Subsequent studies also demonstrated that RuCl3·nH2O can be utilized for ortho-54–62 as well as meta-C–H functionalizations.63–65 Nevertheless, rather harsh conditions with reaction temperatures from 100–140 °C are generally required, representing a considerable limitation.
 | ||
| Scheme 1 (A) Ruthenium(II) complexes prepared from RuCl3·nH2O. (B) This study. Het: heterocycle. | ||
To directly unlock the inherent reactivity of ruthenium catalysis using RuCl3·nH2O, we hypothesized whether, upon the electroreduction of a ruthenium(III) species, an active ruthenium(II) intermediate could be formed,54,57,59 setting the stage for economically-attractive, yet user-friendly C–H activation. As a result of our studies, we, herein, disclose the direct use of RuCl3·nH2O for C–H activations at mild reaction temperatures, enabling ortho-C–H arylations and meta-C–H alkylations across a wide array of substrates, including challenging pharmaceutical, agrochemical, and naturally-occurring molecules (Scheme 1B). Thus, we harness electricity as a sustainable reductant, eliminating the need for potentially hazardous chemical redox agents.66–69 A notable feature of our findings is that both a sacrificial anode or a simple amine could be employed as the terminal oxidant. Detailed spectroelectrochemical studies uncovered the key reduction of the ruthenium(III) precatalyst.
Results and discussion
Reaction optimization
To probe our original hypothesis, we performed the C–H arylation under electroreductive conditions using transformable oxazoline 1a and aryl bromide 2a as the model substrates in the presence of RuCl3·nH2O as the bench-stable and commercially available precatalyst (Table 1). We thus employed NaOAc for a base-assisted internal electrophilic substitution (BIES) C–H activation manifold7,10 with K2CO3 as the stoichiometric base. We probed dipolar DMA as the solvent to ensure conductivity, along with a zinc plate as the sacrificial anode. Gratifyingly, we indeed obtained the desired arylation product 3 in 85% yield after electrolysis using a non-precious zinc plate anode and a nickel foam cathode (j = 2.5 mA cm−2, 1.8 F mol−1, entries 1 and 2). We were delighted to observe that the reaction proceeded at 35 °C, significantly lower than the temperatures required in previous studies using RuCl3·nH2O.24,54–62N-Methyl-2-pyrrolidone (NMP) as the reaction medium led to a lower yield (entry 3). Other leaving groups of aryl halides, such as chloro and iodo groups, proved viable, with the bromo group being superior in the ruthenium electrocatalysis (entry 4). Thus far, chloroarenes gave inferior results, likely due to their higher bond dissociation energies (BDE).24,62 A control experiment without electricity highlighted the indispensable role of the electroreduction (entry 5). Likewise, no product formation or a diminished yield was observed in the absence of either RuCl3·nH2O or NaOAc (entries 6 and 7). Particularly, the crucial presence of NaOAc suggests that carboxylate-assisted C–H activation is the operative working mode.23 We also tested other bench-stable ruthenium(II) complexes, such as [RuCl2(p-cymene)]2 or [Ru(OAc)2(PPh3)2], albeit with limited success (entry 8).|
|
||
|---|---|---|
| Entry | Deviations from the above conditions | 3 (%)b |
| a Reaction conditions: 1a (0.50 mmol), 2a (1.5 equiv.), RuCl3·nH2O (10 mol%), NaOAc (30 mol%), K2CO3 (2.0 equiv.), DMA (3.0 mL), 24 h, 35 °C, CCE at 2.0 mA, Zn anode, Ni cathode, undivided cell. b Determined by 1H-NMR analysis using dibromomethane as the internal standard. c Isolated yield. d Catalyst loading: 5.0 mol%. CCE: constant current electrolysis, DMA: N,N-dimethylacetamide, N.D.: not detected. | ||
| 1 | None | 90 (85)c |
| 2 | Room temperature | 37 |
| 3 | NMP instead of DMA | 78 |
| 4 | Aryl iodide or aryl chloride instead of 2a | 60/73 |
| 5 | No electricity but with electrodes | N.D. |
| 6 | No RuCl3·nH2O | N.D. |
| 7 | No NaOAc | 23 |
| 8 | [RuCl2(p-cymene)]2d or [Ru(OAc)2(PPh3)2] instead of RuCl3·nH2O and no electricity | N.D. |

Electrocatalysis robustness
With the optimal conditions in hand, we next explored the viable (hetero)aryl bromide scope for the electro-enabled C–H arylation (Fig. 1A). Various para-substituted aryl bromides were well tolerated, regardless of the electronic nature of their substituents (3–10). We were particularly pleased to find that valuable electrophilic functional groups, such as ester (3), ketone (4), hydroxy (9), and chloro (10) groups, were fully tolerated by the electrocatalysis, without any significant signs of cross-electrophile couplings (CEC)70–80 induced by the electricity. Furthermore, meta- and more challenging ortho-substituted aryl bromides gave the desired arylation products likewise (11–17). To our delight, the ruthena-electrocatalysis proved compatible with hetaryl bromides, such as dibenzofuran, 1,3-benzodioxoles, and NH-free indoles (18–22). | ||
| Fig. 1 (Hetero)aryl bromide and heterocycle scope. aReaction conditions: 1 (0.50 mmol), 2 (1.5–3.0 equiv.), RuCl3·nH2O (10 mol%), NaOAc (30 mol%), K2CO3 (2.0 equiv.), DMA (3.0 mL), 24 h, 35 °C, CCE at 2.0 mA, Zn anode, Ni cathode, undivided cell. Ar: 4-(CO2Me)C6H4 for products 23–27. | ||
Next, we turned our attention to evaluating heterocycle-attached arenes (Fig. 1B). The benzene-substituted oxazoline yielded the difunctionalization product 23 in excellent yield. The electrocatalysis proved tolerant of the fluoro-attached aryl oxazoline (24). Notably, the chloro group on the para-position of the aromatic ring remained intact (25), demonstrating the high electrocatalysis chemo-selectivity and its potential for further diversification. We were pleased to selectively obtain arylation products using pyridine and pyrazole-derived substrates (26 and 27).
We have confirmed, thus far, that our electrochemical C–H arylation efficiently proceeded under mild conditions to furnish various arylation products. Hence, we next explored the challenging late-stage diversification of complex molecules (Fig. 2). Thus, we employed commercially available pharmaceutical and agrochemical compounds bearing chloro leaving groups. For instance, fenofibrate and chlorpropham were successfully incorporated into the aryl oxazoline scaffolds, even when sensitive ester and carbamate functional groups were present (28 and 29). The drug haloperidol with a free OH-hydroxyl group was fully tolerated by the ruthenium electrocatalysis (30). To our delight, ezetimibe and δ-tocopherol furnished the target arylation products 31 and 32. Furthermore, glyburide was also tolerated, leading to the arylation product bearing a sulfonylurea fragment, one of the important building blocks in medicinal chemistry (33).81 We were also pleased that the azapeptide drug, atazanavir, underwent selective functionalization on the phenylpyridine scaffold (34).
 | ||
| Fig. 2 Electro-late-stage diversification. General reaction conditions: 1 (0.50 mmol), 2 (1.5–3.0 equiv.), RuCl3·nH2O (10 mol%), NaOAc (30 mol%), K2CO3 (2.0 equiv.), DMA (3.0 mL), 24 h, 35 °C, CCE at 2.0 mA, Zn anode, Ni cathode, undivided cell. a Reaction temperature: 50 °C. bRuCl3·nH2O: 20 mol%, NaOAc: 60 mol%, K2CO3: 3.0 equiv., 70 °C. Ar: m-xylyl for product 34. Bn: benzyl. | ||
To replace the zinc anode, we evaluated N,N-diisopropylethylamine (DIPEA) as a terminal reductant with a stable anode material.82 Upon experimentation,83 the desired products 3, 15, and 24 were selectively obtained without a sacrificial anode (Fig. 3). Detailed mass-spectrometry analysis confirmed the Hünig base (DIPEA) serving as the terminal reductant.
 | ||
| Fig. 3 Sacrificial anode-free electrochemical ruthenium-catalyzed ortho-C–H arylation. Reaction conditions: 1 (1.2 equiv.), 2 (0.50 mmol), RuCl3·nH2O (10 mol%), NaOAc (30 mol%), K2CO3 (2.0 equiv.), DIPEA (2.0 equiv.), DMA (3.0 mL), 24 h, 35 °C, CCE at 2.0 mA, platinum anode, GF cathode, undivided cell. | ||
The ruthena-electrocatalysis was not limited to ortho-C–H arylations. Indeed, it also proved to be applicable to challenging meta-C–H alkylations, when employing secondary and tertiary alkyl (pseudo)halides (Fig. 4). By the judicious choice of additive and solvent, we accomplished the meta-C–H alkylations of arenes, thus giving the desired products 36 and 37. Inspired by our previous findings,84 we also tested the pyridinium salt derived from aspartic acid and successfully isolated the corresponding meta-secondary alkylation product 38.
 | ||
| Fig. 4 Electrochemical ruthenium-catalyzed meta-C–H alkylation. Reaction conditions: 1 (0.50 mmol), 35 (2.0–3.0 equiv.), RuCl3·nH2O (10 mol%), (PhO)2PO2H (30 mol%), K2CO3 (2.0 equiv.), nBu4NPF6 (50 mM), THF (3.0 mL), 50 °C, CCE at 1.0 mA, 4 h, and 13 h at 50 °C, Zn anode, Ni cathode, undivided cell. THF: tetrahydrofuran. | ||
Mechanistic studies
Having validated the robustness of the new ruthenium electrocatalysis, we turned our attention to elucidating the modus operandi. First, we performed a control experiment wherein 10 mol% of electrons were supplied within the initial 40 minutes, and the current was switched off thereafter (Fig. 5A). Here, the arylation product 3 was obtained in 59% yield. This outcome suggests that electricity is primarily required to induce the formation of the key monocyclometalated ruthenium(II) intermediate (Fig. 5B, left). The involvement of such a species was verified by the experiment using a well-defined monocyclometalated complex Ru3, in which we obtained the desired arylation product 24 in 79% yield, further corroborating the electrochemical initiation being operative (Fig. 5B, right).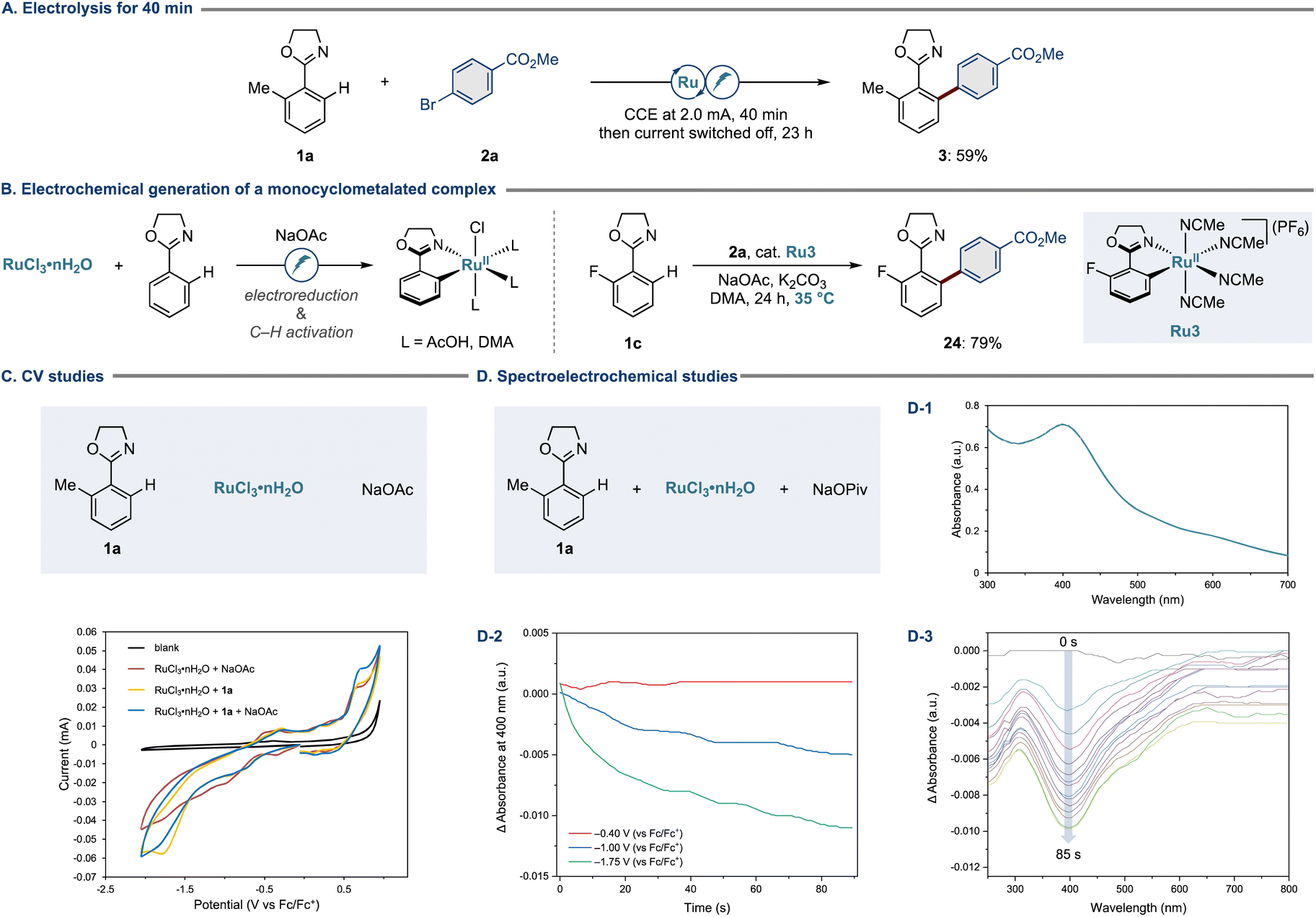 | ||
| Fig. 5 (A) Electrolysis for 40 min. (B) Electrochemical generation of a monocyclometalated complex. (C) CV studies. (D-1) UV/vis absorption spectrum of the DMA solution containing substrate 1a (0.13 mM), RuCl3·nH2O (0.13 mM), and NaOPiv (0.13 mM). (D-2) Spectroelectrochemical analysis of the DMA solution containing substrate 1a (0.2 mM), RuCl3·nH2O (0.2 mM), NaOPiv (0.2 mM), and nBu4NPF6 (0.1 M) at 35 °C under N2. Measurements were performed at −0.40, −1.00, or −1.75 V. (D-3) Spectroelectrochemical analysis performed at −1.75 V (vs. Fc/Fc+) over 85 s. | ||
To rationalize the electrochemical features of our electrocatalysis, we conducted detailed cyclic voltammetry (CV) studies using RuCl3·nH2O, substrate 1a, and NaOAc (Fig. 5C). Interestingly, the presence of substrate 1a resulted in a pronounced irreversible reduction event at −1.7 V, implying coordination of arene 1a to ruthenium. In all cases, oxidation events were observed at 0.7 V. To gain more insights, we selected specific voltage values from the cyclic voltammograms and sought to observe the potential-dependent consumption of the ruthenium(III) precatalyst by means of spectroelectrochemistry. Thus, we conducted potentiostatic analysis mode and focused on the absorption of the ruthenium(III) precatalyst at 400 nm (Fig. 5D-1). As depicted in Fig. 5D-2, whereas there was no significant change in the absorbance at 400 nm during the measurements at −0.40 and −1.00 V, spectroelectrochemical analysis at −1.75 V allowed us to detect a notable decrease in absorbance, which was also visible from the spectra with entire wavelengths (Fig. 5D-3). Overall, these observations are indicative of an electrochemical reduction of the ruthenium(III) precatalyst to afford a catalytically relevant ruthenium(II) complex. Also, electrochemical analysis by a rotating disk electrode was supportive of a ruthenium(III)/ruthenium(II) scenario.83,85,86
DFT calculations
To rationalize the modus operandi of the electrocatalysis, detailed density functional theory (DFT) calculations were performed for the key oxidative addition step, considering both mono- and biscycloruthenated species Ru-I and Ru-IV, respectively, at the PBE0-D4/def2-TZVPP-SMD(DMA)//TPSS-D3(BJ)/def2-SVP level of theory (Fig. 6). The biscycloruthenated complex Ru-IV, generated via two C–H ruthenations, exhibits an energy barrier of 20.2 kcal mol−1 for the oxidative addition with aryl bromide 2a, leading to the ruthenium(IV)-aryl species Ru-VI. Notably, the oxidative addition pathway involving the monocycloruthenated species is energetically disfavored with a considerably higher activation barrier of 25.5 kcal mol−1. These results are consistent with earlier computational studies by Ackermann19 and very recent findings by Macgregor87 for the oxidative addition occurring on the biscyclometalated ruthenium(II) intermediates. | ||
| Fig. 6 Computed relative Gibbs free energies on the oxidative addition step for both (a) monocycloruthenated and (b) biscycloruthenated species at the PBE0-D4/def2-TZVPP-SMD(DMA)//TPSS-D3(BJ)/def2-SVP level of theory. | ||
Proposed mechanism
On the basis of our experimental and computational findings, we propose a catalytic cycle for the electro-induced C–H arylations as depicted in Fig. 7. The reaction commences upon cathodic ruthenium(III) reduction, along with carboxylate-assisted C–H activation to form complex Ru-I. Next, biscyclometalated intermediate Ru-IV is formed and undergoes oxidative addition with aryl bromide 2. Reductive elimination from the thus formed ruthenium complex Ru-IV releases the arylation product and regenerates the complex Ru-I.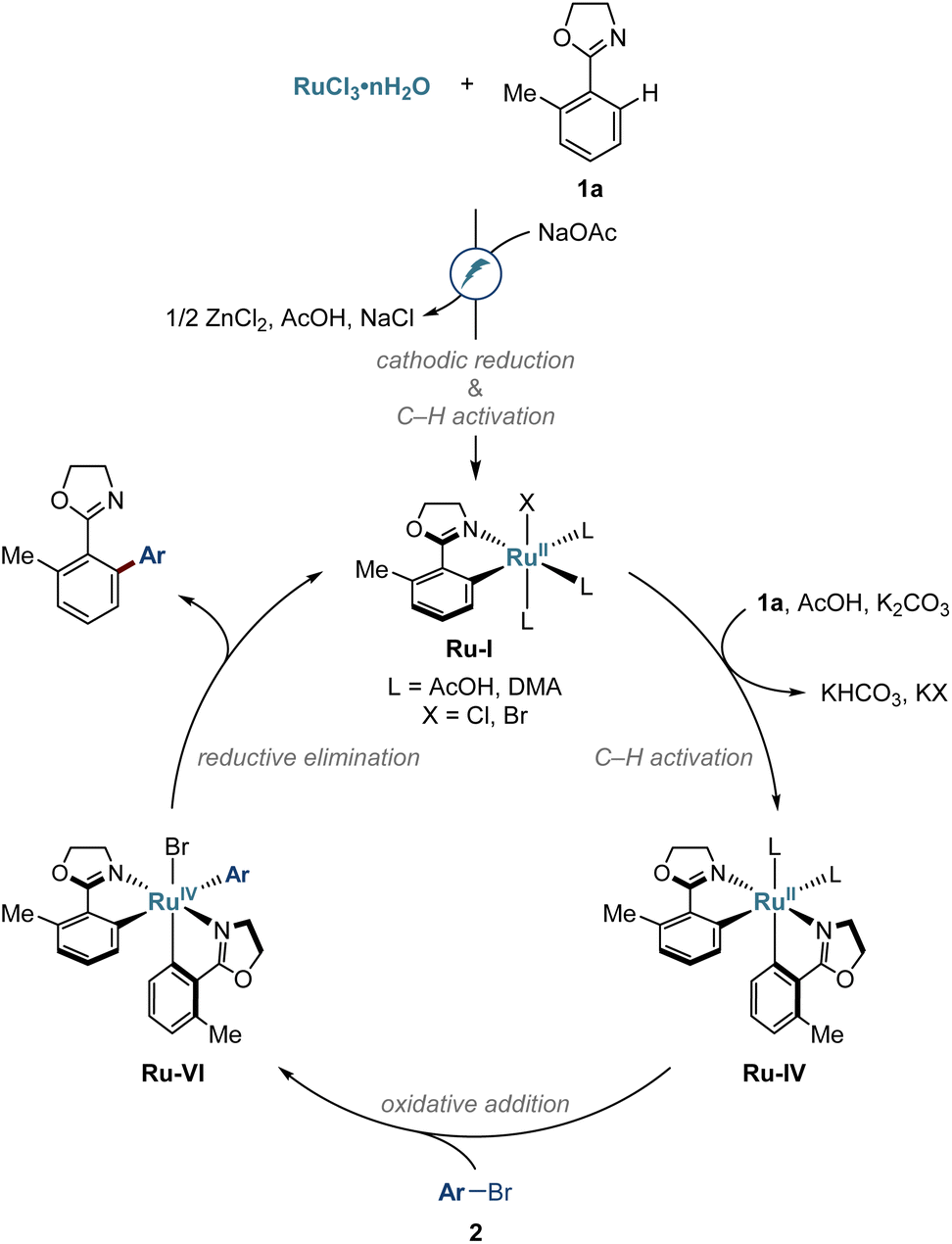 | ||
| Fig. 7 Proposed mechanism. | ||
Conclusions
In conclusion, we have realized a low-temperature ruthenium-catalyzed C–H activation directly employing commercially-available RuCl3·nH2O as a resource-economical precatalyst. The reaction provided selective access to ortho- or meta-functionalized products from a wide array of substrates, including structurally-complex drug molecules and natural products. Detailed electroanalysis provided strong support for a ruthenium(III)/ruthenium(II) manifold enabled by cathodic reduction. A sacrificial anode could be avoided. Overall, our electrocatalysis strategy offers a user-friendly and general strategy to replace the established ruthenium(II) precatalysts in the field of C–H activation chemistry to enable catalysis under exceedingly mild conditions.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization, L. A.; methodology, T. M.; investigation, T. M.; spectroelectrochemical analysis, T. v. M.; rotating disk electrode experiments, T. M. and Z. L.; DFT calculations, B. Y. and J. C. A. O.; writing – original draft, all authors; writing – review & editing, all authors; funding acquisition, L. A.; resources, L. A.; supervision, L. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the DZHK, the ERC Advanced Grant No. 101021358, the DFG (SPP2363, Gottfried Wilhelm Leibniz award to L. A.), the European Union's Horizon 2020 research and innovation program (Marie Skłodowska-Curie Grant Agreement No. 860762 to T. M.), the FCI (Kekulé-Fellowship no. 110091 to T. v. M.), and the CSC (scholarship to Z. L. and B. Y.). We thank Dr M. John (University of Göttingen) for assistance with NMR analysis and Dr H. Frauendorf (University of Göttingen) for HRMS.Notes and references
- J. H. Docherty, T. M. Lister, G. McArthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- H. Liang and J. Wang, Enantioselective C−H Bond Functionalization Involving Arene Ruthenium(II) Catalysis, Chem.–Eur. J., 2023, 29, e202202461 CrossRef CAS PubMed.
- K. Korvorapun, R. C. Samanta, T. Rogge and L. Ackermann, Remote C–H Functionalizations by Ruthenium Catalysis, Synthesis, 2021, 53, 2911–2946 CrossRef CAS.
- G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Recent Advances in Ruthenium(II)-Catalyzed C−H Bond Activation and Alkyne Annulation Reactions, Adv. Synth. Catal., 2019, 361, 654–672 CrossRef CAS.
- J. A. Leitch and C. G. Frost, Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- G.-F. Zha, H.-L. Qin and E. A. B. Kantchev, Ruthenium-catalyzed direct arylations with aryl chlorides, RSC Adv., 2016, 6, 30875–30885 RSC.
- L. Ackermann, Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C–H/Het–H Bond Functionalizations, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Weakly Coordinating Directing Groups for Ruthenium(II)- Catalyzed C–H Activation, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- L. Ackermann, Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- X.-Y. Gou, J. C. A. Oliveira, S. Chen, S. L. Homölle, S. Trienes, T. von Münchow, B.-S. Zhang and L. Ackermann, Ruthenaelectro-catalyzed C–H phosphorylation: ortho to para position-selectivity switch, Chem. Sci., 2025, 16, 824–833 RSC.
- X. Chen, H. C. Gülen, J. Wu, Z.-J. Zhang, X. Hong and L. Ackermann, Close-Shell Reductive Elimination versus Open-Shell Radical Coupling for Site-Selective Ruthenium-Catalyzed C−H Activations by Computation and Experiments, Angew. Chem., Int. Ed., 2023, 62, e202302021 CrossRef CAS PubMed.
- K. Korvorapun, M. Moselage, J. Struwe, T. Rogge, A. M. Messinis and L. Ackermann, Regiodivergent C−H and Decarboxylative C−C Alkylation by Ruthenium Catalysis: ortho versus meta Position-Selectivity, Angew. Chem., Int. Ed., 2020, 59, 18795–18803 CrossRef CAS PubMed.
- K. Korvorapun, R. Kuniyil and L. Ackermann, Late-Stage Diversification by Selectivity Switch in meta-C–H Activation: Evidence for Singlet Stabilization, ACS Catal., 2020, 10, 435–440 CrossRef CAS.
- X.-G. Wang, Y. Li, L.-L. Zhang, B.-S. Zhang, Q. Wang, J.-W. Ma and Y.-M. Liang, Ruthenium(II)-catalyzed selective C–H difluoroalkylation of aniline derivatives with pyrimidyl auxiliaries, Chem. Commun., 2018, 54, 9541–9544 RSC.
- K. Korvorapun, N. Kaplaneris, T. Rogge, S. Warratz, A. C. Stückl and L. Ackermann, Sequential meta-/ortho-C–H Functionalizations by One-Pot Ruthenium(II/III) Catalysis, ACS Catal., 2018, 8, 886–892 CrossRef CAS.
- W. Liu and L. Ackermann, Ortho- and Para-Selective Ruthenium-Catalyzed C(sp2)–H Oxygenations of Phenol Derivatives, Org. Lett., 2013, 15, 3484–3486 CrossRef CAS PubMed.
- H. Simon, A. Zangarelli, T. Bauch and L. Ackermann, Ruthenium(II)-Catalyzed Late-Stage Incorporation of N-Aryl Triazoles and Tetrazoles with Sulfonium Salts via C−H Activation, Angew. Chem., Int. Ed., 2024, 63, e202402060 CrossRef CAS PubMed.
- T. Rogge and L. Ackermann, Arene-Free Ruthenium(II/IV)-Catalyzed Bifurcated Arylation for Oxidative C−H/C−H Functionalizations, Angew. Chem., Int. Ed., 2019, 58, 15640–15645 CrossRef CAS PubMed.
- M. Schinkel, I. Marek and L. Ackermann, Carboxylate-Assisted Ruthenium(II)-Catalyzed Hydroarylations of Unactivated Alkenes through C–H Cleavage, Angew. Chem., Int. Ed., 2013, 52, 3977–3980 CrossRef CAS PubMed.
- E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Autocatalysis for C–H Bond Activation by Ruthenium(II) Complexes in Catalytic Arylation of Functional Arenes, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef CAS PubMed.
- L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Ruthenium-Catalyzed Regioselective Direct Alkylation of Arenes with Unactivated Alkyl Halides through C–H Bond Cleavage, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente and A. Althammer, Assisted Ruthenium-Catalyzed C−H Bond Activation: Carboxylic Acids as Cocatalysts for Generally Applicable Direct Arylations in Apolar Solvents, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS PubMed.
- L. Ackermann, A. Althammer and R. Born, [RuCl3(H2O)n]-Catalyzed Direct Arylations with Bromides as Electrophiles, Synlett, 2007, 2833–2836 CrossRef CAS.
- L. Ackermann, Phosphine Oxides as Preligands in Ruthenium-Catalyzed Arylations via C−H Bond Functionalization Using Aryl Chlorides, Org. Lett., 2005, 7, 3123–3125 CrossRef CAS PubMed.
- S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Ruthenium Complex-Catalyzed Direct Ortho Arylation and Alkenylation of 2-Arylpyridines with Organic Halides, Org. Lett., 2001, 3, 2579–2581 CrossRef CAS PubMed.
- S. G. Ouellet, A. Roy, C. Molinaro, R. Angelaud, J.-F. Marcoux, P. D. O'Shea and I. W. Davies, Preparative Scale Synthesis of the Biaryl Core of Anacetrapib via a Ruthenium-Catalyzed Direct Arylation Reaction: Unexpected Effect of Solvent Impurity on the Arylation Reaction, J. Org. Chem., 2011, 76, 1436–1439 CrossRef CAS PubMed.
- S. Chen, Z. Xu, B. Yuan, X.-Y. Gou and L. Ackermann, Difunctionalization of bicyclo[1.1.0]butanes enabled by merging C−C cleavage and ruthenium-catalysed remote C−H activation, Nat. Synth., 2025, 4, 655–663 CrossRef CAS PubMed.
- X. Luo, P. Hou, J. Shen, Y. Kuang, F. Sun, H. Jiang, L. J. Gooßen and L. Huang, Ligand-enabled ruthenium-catalyzed meta-C−H alkylation of (hetero)aromatic carboxylic acids, Nat. Commun., 2024, 15, 5552 CrossRef CAS PubMed.
- J. Wu, N. Kaplaneris, J. Pöhlmann, T. Michiyuki, B. Yuan and L. Ackermann, Remote C−H Glycosylation by Ruthenium(II) Catalysis: Modular Assembly of meta-C-Aryl Glycosides, Angew. Chem., Int. Ed., 2022, 61, e202208620 CrossRef CAS PubMed.
- X.-Y. Gou, Y. Li, W.-Y. Shi, Y.-Y. Luan, Y.-N. Ding, Y. An, Y.-C. Huang, B.-S. Zhang, X.-Y. Liu and Y.-M. Liang, Ruthenium-Catalyzed Stereo- and Site-Selective ortho- and meta-C−H Glycosylation and Mechanistic Studies, Angew. Chem., Int. Ed., 2022, 61, e202205656 CrossRef CAS PubMed.
- K. Jing, Z.-Y. Li and G.-W. Wang, Direct Decarboxylative Meta-Selective Acylation of Arenes via an Ortho-Ruthenation Strategy, ACS Catal., 2018, 8, 11875–11881 CrossRef CAS.
- G. M. Reddy, N. S. Rao and H. Maheswaran, Highly meta-selective halogenation of 2-phenylpyridine with a ruthenium(I) catalyst, Org. Chem. Front., 2018, 5, 1118–1123 RSC.
- Z. Fan, H. Lu, Z. Cheng and A. Zhang, Ligand-promoted ruthenium-catalyzed meta C–H chlorination of arenes using N-chloro-2,10-camphorsultam, Chem. Commun., 2018, 54, 6008–6011 RSC.
- S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, meta-C−H Bromination on Purine Bases by Heterogeneous Ruthenium Catalysis, Angew. Chem., Int. Ed., 2017, 56, 1557–1560 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, Meta-Selective CAr–H Nitration of Arenes through a Ru3(CO)12-Catalyzed Ortho-Metalation Strategy, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef CAS PubMed.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, N-Acyl Amino Acid Ligands for Ruthenium(II)-Catalyzed meta-C–H tert-Alkylation with Removable Auxiliaries, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Catalytic meta-selective C–H functionalization to construct quaternary carbon centres, Chem. Commun., 2015, 51, 12807–12810 RSC.
- N. Hofmann and L. Ackermann, meta-Selective C–H Bond Alkylation with Secondary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, Ruthenium-Catalyzed Meta Sulfonation of 2-Phenylpyridines, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- L. Ackermann, N. Hofmann and R. Vicente, Carboxylate-Assisted Ruthenium-Catalyzed Direct Alkylations of Ketimines, Org. Lett., 2011, 13, 1875–1877 CrossRef CAS PubMed.
- A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Ortho C–H arylation of arenes at room temperature using visible light ruthenium C–H activation, Chem. Sci., 2020, 11, 4439–4443 RSC.
- K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C−H Arylations at Ambient Temperature, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed.
- A. Sagadevan and M. F. Greaney, meta-Selective C−H Activation of Arenes at Room Temperature Using Visible Light: Dual-Function Ruthenium Catalysis, Angew. Chem., Int. Ed., 2019, 58, 9826–9830 CrossRef CAS PubMed.
- P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Visible-Light-Enabled Ruthenium-Catalyzed meta-C−H Alkylation at Room Temperature, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS PubMed.
- G. McArthur, J. H. Docherty, M. D. Hareram, M. Simonetti, I. J. Vitorica-Yrezabal, J. J. Douglas and I. Larrosa, An air- and moisture-stable ruthenium precatalyst for diverse reactivity, Nat. Chem., 2024, 16, 1141–1150 CrossRef CAS PubMed.
- M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals, Nat. Chem., 2018, 10, 724–731 CrossRef CAS PubMed.
- A. Dey, R. Kancherla, K. Pal, N. Kloszewski and M. Rueping, Rapid and scalable ruthenium catalyzed meta-C–H alkylation enabled by resonant acoustic mixing, Commun. Chem., 2024, 7, 295 CrossRef CAS PubMed.
- S. Trienes, S. Golling, M. H. Gieuw, M. Di Matteo and L. Ackermann, Visible light-induced ruthenium(II)-catalyzed hydroarylation of unactivated olefins, Chem. Sci., 2024, 15, 19037–19043 RSC.
- P. Hauk, S. Trienes, F. Gallou, L. Ackermann and J. Wencel-Delord, Next-generation functional surfactant for mild C−H arylation under micellar conditions, Chem Catal., 2024, 4, 101146 CrossRef CAS.
- X. Lv, Y. Cheng, Y. Zong, Q. Wang, G. An, J. Wang and G. Li, Visible-Light-Mediated Ruthenium-Catalyzed para-Selective Alkylation of Unprotected Anilines, ACS Catal., 2023, 13, 7310–7321 CrossRef CAS.
- Y. Wang, S. Chen, X. Chen, A. Zangarelli and L. Ackermann, Photo-Induced Ruthenium-Catalyzed Double Remote C(sp2)−H/C(sp3)−H Functionalizations by Radical Relay, Angew. Chem., Int. Ed., 2022, 61, e202205562 CrossRef CAS PubMed.
- M. A. Bennett, T.-N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, 16. (η6-Hexamethylbenzene)Ruthenium Complexes, Inorg. Synth., 1982, 21, 74–78 CrossRef CAS.
- L. Jian, H.-Y. He, J. Huang, Q.-H. Wu, M.-L. Yuan, H.-Y. Fu, X.-L. Zheng, H. Chen and R.-X. Li, Combination of RuCl3·xH2O with PEG – a simple and recyclable catalytic system for direct arylation of heteroarenes via C–H bond activation, RSC Adv., 2017, 7, 23515–23522 RSC.
- Y.-L. Zou, Z.-Y. Wang, Y.-M. Feng, Y.-G. Li and E. A. B. Kantchev, Solvent and Base in One: Tetra-n-butylammonium Acetate as a Multi-Purpose Ionic Liquid Medium for Ru-Catalyzed Directed Mono- and Di-o-C–H Arylation Reactions, Eur. J. Org Chem., 2017, 2017, 6274–6282 CrossRef CAS.
- B. Zhao, RuCl3-Catalyzed Regioselective Diarylation with Aryl Tosylates via C-H Activation, Synth. Commun., 2013, 43, 2110–2118 CrossRef CAS.
- L. A. Adrio, J. Gimeno and C. Vicent, One-pot direct C–H arylation of arenes in water catalysed by RuCl3·nH2O–NaOAc in the presence of Zn, Chem. Commun., 2013, 49, 8320–8322 RSC.
- M. Seki, Highly Efficient Catalytic System for C–H Activation: A Practical Approach to Angiotensin II Receptor Blockers, ACS Catal., 2011, 1, 607–610 CrossRef CAS.
- N. Luo and Z. Yu, RuCl3·x H2O-Catalyzed Direct Arylation of Arenes with Aryl Chlorides in the Presence of Triphenylphosphine, Chem.–Eur. J., 2010, 16, 787–791 CrossRef CAS PubMed.
- L. Ackermann and R. Vicente, Catalytic Direct Arylations in Polyethylene Glycol (PEG): Recyclable Palladium(0) Catalyst for C−H Bond Cleavages in the Presence of Air, Org. Lett., 2009, 11, 4922–4925 CrossRef CAS PubMed.
- K. Cheng, Y. Zhang, J. Zhao and C. Xie, Peroxide-Promoted Regioselective Arylation of 2-Phenylpyridines and Related Substrates with Aryl Iodides, Synlett, 2008, 1325–1330 CAS.
- L. Ackermann, A. Althammer and R. Born, [RuCl3(H2O)n]-catalyzed direct arylations, Tetrahedron, 2008, 64, 6115–6124 CrossRef CAS.
- S. Yang, B. Yan, L. Zhong, C. Jia, D. Yao, C. Yang, K. Sun and G. Li, AIBN for Ru-catalyzed meta-CAr–H alkylation, Org. Chem. Front., 2020, 7, 2474–2479 RSC.
- C. Jia, S. Wang, X. Lv, G. Li, L. Zhong, L. Zou and X. Cui, Ruthenium-Catalyzed meta-CAr–H Bond Difluoroalkylation of 2-Phenoxypyridines, Eur. J. Org Chem., 2020, 2020, 1992–1995 CrossRef CAS.
- G. Li, D. Li, J. Zhang, D.-Q. Shi and Y. Zhao, Ligand-Enabled Regioselectivity in the Oxidative Cross-coupling of Arenes with Toluenes and Cycloalkanes Using Ruthenium Catalysts: Tuning the Site-Selectivity from the ortho to meta Positions, ACS Catal., 2017, 7, 4138–4143 CrossRef CAS.
- X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS.
- M. C. Leech and K. Lam, A practical guide to electrosynthesis, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, Organic Electrochemistry: Molecular Syntheses with Potential, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, Organic electrosynthesis: from academia to industry, React. Chem. Eng., 2020, 5, 977–990 RSC.
- L. E. Ehehalt, O. M. Beleh, I. C. Priest, J. M. Mouat, A. K. Olszewski, B. N. Ahern, A. R. Cruz, B. K. Chi, A. J. Castro, K. Kang, J. Wang and D. J. Weix, Cross-Electrophile Coupling: Principles, Methods, and Applications in Synthesis, Chem. Rev., 2024, 124, 13397–13569 CrossRef CAS PubMed.
- M. C. Franke and D. J. Weix, Recent Advances in Electrochemical, Ni-Catalyzed C−C Bond Formation, Isr. J. Chem., 2024, 64, e202300089 CrossRef.
- Y. Liu, P. Li, Y. Wang and Y. Qiu, Electroreductive Cross-Electrophile Coupling (eXEC) Reactions, Angew. Chem., Int. Ed., 2023, 62, e202306679 CrossRef CAS PubMed.
- E. Richmond and J. Moran, Recent Advances in Nickel Catalysis Enabled by Stoichiometric Metallic Reducing Agents, Synthesis, 2018, 50, 499–513 CrossRef CAS.
- D. J. Weix, Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- J. Gu, X. Wang, W. Xue and H. Gong, Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations, Org. Chem. Front., 2015, 2, 1411–1421 RSC.
- D. A. Everson and D. J. Weix, Cross-Electrophile Coupling: Principles of Reactivity and Selectivity, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed.
- C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. J. von Wangelin, Reductive Cross-Coupling Reactions between Two Electrophiles, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent advances in homogeneous nickel catalysis, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- T. Moragas, A. Correa and R. Martin, Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds, Chem.–Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed.
- M. Durandetti and J. Périchon, Nickel-Catalyzed Electrochemical Coupling of Aryl, Heteroaryl or Vinyl Halides with Activated Alkyl Chlorides: Synthetic and Stereochemical Aspects, Synthesis, 2004, 3079–3083 CrossRef CAS.
- T. De Ventura and V. Zanirato, Recent Advances in the Synthesis of Sulfonylureas, Eur. J. Org Chem., 2021, 2021, 1201–1214 CrossRef CAS.
- J. Wu, R. Purushothaman, F. Kallert, S. L. Homölle and L. Ackermann, Electrochemical Glycosylation via Halogen-Atom-Transfer for C-Glycoside Assembly, ACS Catal., 2024, 14, 11532–11544 CrossRef CAS PubMed.
- For detailed information, see the ESI.†.
- W. Wei, H. Yu, A. Zangarelli and L. Ackermann, Deaminative meta-C–H alkylation by ruthenium(II) catalysis, Chem. Sci., 2021, 12, 8073–8078 RSC.
- S. I. Etkind, D. A. Vander Griend and T. M. Swager, Electroactive Anion Receptor with High Affinity for Arsenate, J. Org. Chem., 2020, 85, 10050–10061 CrossRef CAS PubMed.
- C. Amatore, M. Azzabi, P. Calas, A. Jutand, C. Lefrou and Y. Rollin, Absolute determination of electron consumption in transient or steady state electrochemical techniques, J. Electroanal. Chem., 1990, 288, 45–63 CrossRef CAS.
- P. Domingo-Legarda, S. E. Neale, A. Carpentier, C. L. McMullin, M. Findlay, I. Larrosa and S. A. Macgregor, The Mechanism of Ruthenium-Catalyzed Directed C–H Arylation of Arenes: The Key Role of Bis-Cyclometalated Intermediates, Angew. Chem., Int. Ed., 2025, 64, e202506707 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02780b |
| This journal is © The Royal Society of Chemistry 2025 |