
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An N-heterocyclic germylene with a versatile metal-binding pocket: insights into heterodinuclear bonding and reactivity†
Errikos
Kounalis
 a,
Rik
Sieben
a,
Léon
Witteman
a,
Martin
Lutz
b,
Marc-Etienne
Moret
*a and
Daniël L. J.
Broere
*a
a,
Rik
Sieben
a,
Léon
Witteman
a,
Martin
Lutz
b,
Marc-Etienne
Moret
*a and
Daniël L. J.
Broere
*a
aOrganic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: m.moret@uu.nl; d.l.j.broere@uu.nl
bStructural Biochemistry Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands
First published on 5th June 2025
Abstract
We report the synthesis, isolation, and characterisation of an N-heterocyclic germylene (NHGe) derived from a two-electron reduced Mg-synthon of the redox-active dippNBA ligand. This NHGe features a vacant binding pocket capable of coordinating various metals, which enables the formation of heterobimetallic Ge–Zn and Ge–Mg complexes. Electronic structure calculations reveal that the Ge–Zn interactions are weak, whilst the interactions between the metals found for the heterodinuclear Ge–Mg complex are stronger. These findings highlight how the nature of the Ge–M interactions adapts to the electron density requirements of the metal occupying the redox-active binding pocket flanking the Ge(II) centre. Notably, the Ge–Mg complex undergoes a ‘Metallo-Diels–Alder’ reaction with unsaturated C–C bonds, activating these bonds over the Ge centre and the ligand backbone – a transformation that does not proceed without Mg. This provides a compelling example of indirect cooperativity, where the Mg centre electronically stabilises a quadruply reduced ligand, upon which Ge engages in metal–ligand cooperative activation of C–C unsaturated bonds.
Introduction
Germylenes, ambiphilic Ge(II)-complexes that bear an empty p-orbital and a Ge-centred lone pair, have attracted significant attention as promising tools in main-group catalysis due to their unique electronic structure and reactivity patterns.1,2 Much like N-heterocyclic carbenes (NHCs) form a substantial subclass within the lightest of the tetrylenes, N-heterocyclic germylenes (NHGes) represent a similarly significant subclass of germylenes, distinguished by analogous bonding and electronic features.3 The ring-systems of these NHGes exhibit different degrees of unsaturation4 and ring-size, spanning from 4- to 7-membered motifs.5–10 Additionally, decoration of the N-donor with bulky substituents results in kinetic stabilisation of the reactive Ge(II) centre. The reactivity of these centres is largely due to the reduced state of Ge(II), where the Ge(II/IV) redox couple facilitates the activation of bonds like C–H and H–H via oxidative addition (Scheme 1a).11–13 This allows Ge(II) to exhibit reactivity patterns that closely parallel those of transition metals (TMs). Redox-neutral bond activation can also take place through Ge-ligand cooperativity. In these cases the bonds are activated ditopically over the Ge(II) centre and the ligand backbone, retaining the divalent state on Ge. Some examples of this type of reactivity feature ylide-like Ge-complexes bearing diketiminate (NacNac) ligands or Ge-complexes ligated by iminopyridines, allowing for the activation of a plethora of bonds (Scheme 1b).8,9,14–21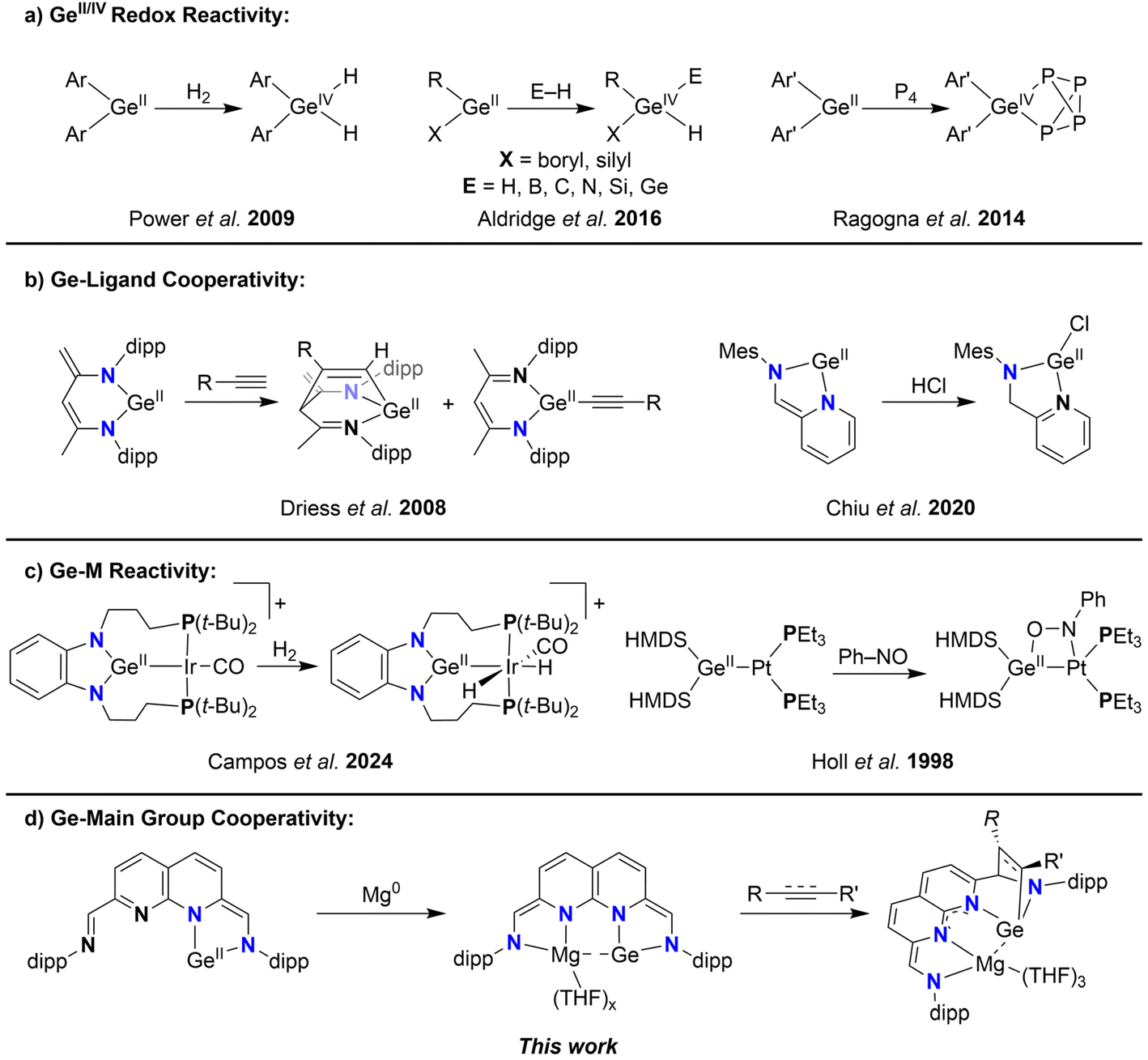 | ||
| Scheme 1 (a–c) The various types of reactivity modes described for germylenes and (d) the Ge-main group cooperativity described in this work. | ||
The ambiphilic nature of germylenes also renders them excellent ligands for transition metals, with examples spanning the whole d-block.22–24 The versatility of the potential Ge–M manifolds that can be synthesised also translates to a versatility in the loci of reactivity. The reactivity of these systems can either be localised on one of the two components of the Ge–M manifold, or be ditopic and spread over both components (Scheme 1c).25–28 In an interesting example by Cui et al., a bis-germylene was synthesised bearing a redox-active naphthyridine diimine ligand,29,30 which could act as donor of up to six electrons.31 Redox-active ligands (RALs) can act as electron reservoirs, storing electrons and eliminating the need to access energetically unfavourable oxidation states at the bound metal centre.32 This results in a flattening of the energy landscape of chemical transformations, with the metals acting as a conduit for the electrons. A compelling example is the reported four-electron reactivity of a iminopyridine–germylene complex, where the Ge(II) centre is oxidised to Ge(IV) while also acting as conduit for the electrons stored in the RAL.18 This electronic flexibility of RALs has also been leveraged in main group complexes to impart bond activation reactivity typically associated with transition metals.33 For example, a bis-Al(II) complex bound to redox-active diimine ligands can mediate the four-electron cleavage of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in azo-compounds, with the electrons required being provided by both oxidation of the Al(II) centres and the RALs.34 This promising avenue has led to the isolation of a multitude of RAL-based complexes of main group metals such as Mg,35,36 Al,37–42 Ga,43–47 Si,35 and Ge.18,31,48 In contrast to the well-reported cooperative reactivity of germylene–TM complexes, examples reporting cooperative transformations such as ditopic bond activation or synergistic redox activity have, to the best of our knowledge, not been extended to germylene-main group combinations.49
N bond in azo-compounds, with the electrons required being provided by both oxidation of the Al(II) centres and the RALs.34 This promising avenue has led to the isolation of a multitude of RAL-based complexes of main group metals such as Mg,35,36 Al,37–42 Ga,43–47 Si,35 and Ge.18,31,48 In contrast to the well-reported cooperative reactivity of germylene–TM complexes, examples reporting cooperative transformations such as ditopic bond activation or synergistic redox activity have, to the best of our knowledge, not been extended to germylene-main group combinations.49
We hypothesised that the naphthyridine diimine scaffold would enable us to integrate the previously described strategies within a single, versatile germylene scaffold. Instead of introducing two Ge(II) centres in the dinucleating naphthyridine scaffold, introduction of only one Ge(II) centre would leave a vacant redox-active pocket for a main group metal (or TM) to bind in close proximity to the germylene and potentially facilitate the envisioned cooperative reactivity (Scheme 1d). Here we report the synthesis, characterisation and electronic structure of such an NHGe. We describe the binding of Zn and Mg to the vacant site and detail the electronic structures of these heterodinuclear complexes, focussing on the interaction between the Ge(II) centre with the metal in close proximity. Lastly, we detail the activation of substrates containing C–C multiple bonds by the Ge–Mg complex, where the Mg centre plays a vital role in electronically stabilising a quadruply reduced ligand, whilst Ge engages in metal–ligand cooperative activation of C–C unsaturated bonds.
Results and discussion
Reacting the dippNBA ligand with 1 equiv. of Mg0 turnings in THF (Scheme 2) led to the slow formation of a dark red solution from which 1 was isolated as a red solid in 73% yield. Analysis of the 1H-NMR spectrum revealed full conversion of the ligand, along with several resonances consistent with the two halves of the naphthyridine motif being equivalent (see ESI Section 1.2†). The singlet at δ = 6.61 ppm, attributed to the methine linker, and the two doublets at δ = 5.70 and 4.99 ppm (3JH,H = 7.6 Hz), attributed to the naphthyridine protons, are all significantly shifted upfield, consistent with a reduced character of the naphthyridine backbone.29 A single septet for the methine protons of the dipp i-Pr groups at δ = 3.43 ppm was observed and two doublets at δ = 1.53 and 0.98 ppm (3JH,H = 6.8 Hz) were observed for the magnetically inequivalent methyl group protons of the dipp substituents. | ||
| Scheme 2 Syntheses of 1 and 2 starting from the redox-active dippNBA ligand. | ||
Single crystals suitable for analysis by X-ray diffraction were grown by layering a saturated THF solution of 1 with pentane. The dimeric nature of 1 was revealed in the solid-state structure (Fig. 1), with the two naphthyridine motifs being twisted at 67.9(10)° (dihedral angle between the two planes defined by N11–C61–N21 and N12–C62–N22, see ESI Fig. S74†) with respect to each other to accommodate a Mg centre on each side. The two-electron reduction of each ligand is evident through the bond metrics, showing a contraction of the Cmethine–Cnapy and an elongation of the Cmethine–Ndipp bonds compared to the free ligand (see ESI Table S3†). The Cmethine–Cnapy bond lengths on either side of both naphthyridine moieties are identical within error, consistent with the delocalised nature of the ligand-centred reduction, which agrees with the 1H-NMR spectrum and is also found in related two-electron reduced naphthyridine systems.29,31
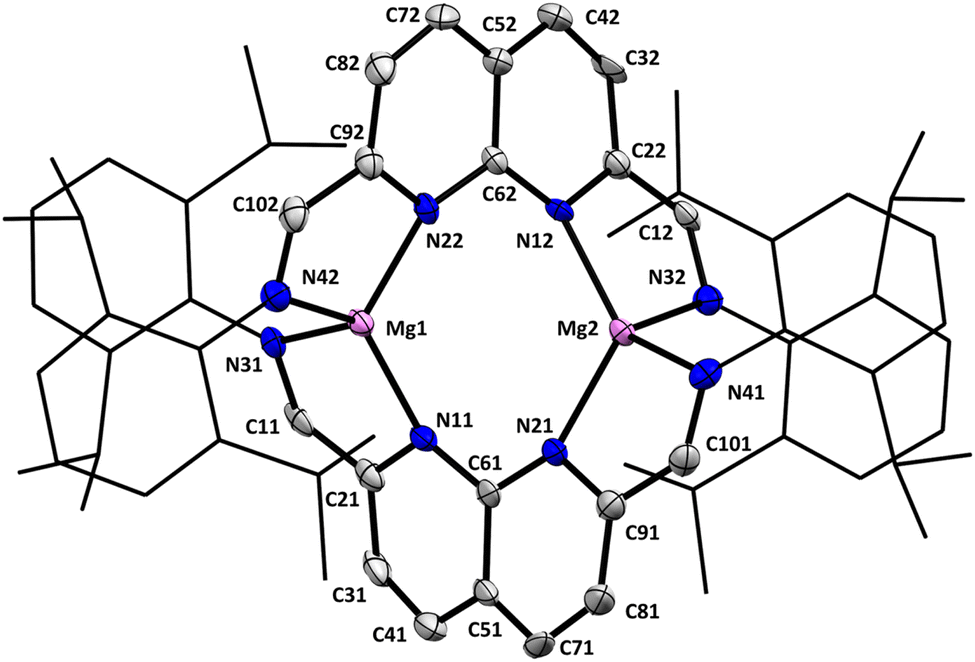 | ||
| Fig. 1 Displacement ellipsoid plot of 1 at 50% probability. Hydrogen atoms and minor disorder components are omitted for clarity. The dipp substituents are depicted as wireframe for clarity. | ||
With the reduced dippNBA-synthon 1 in hand, we set out to install the germylene in the reduced naphthyridine pocket. Treatment of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 1,4-dioxane:Et2O solution of 1 with 2 equiv. of the Ge(II) precursor GeCl2·diox (diox = 1,4-dioxane) resulted in the formation of a white precipitate, presumably polymeric [MgCl2(diox)2]∞,50 and compound 2, which was isolated as a red solid in 68% yield. 1H- and 13C{1H}-NMR analysis of 2 in C6D6 revealed a resonance count consistent with the loss of the previously observed symmetry in 1 (see ESI Section 1.3†). Two sets of naphthyridine resonances are observed, one at δ = 8.14 and 7.02 ppm (3JH,H = 7.7 Hz), and another further upfield at δ = 6.55 and 6.06 ppm (3JH,H = 9.1 Hz). Additionally, two singlets are observed at δ = 8.40 and 6.79 ppm, which we attribute to the protons attached to the pendant methine carbons. Combined, these observations are consistent with two distinct binding pockets in 2. One pocket retains its aromaticity, as evidenced by the more downfield set of naphthyridine protons and the singlet at δ = 8.40 ppm, which is consistent with the aldimine character of the methine proton. In contrast, the other binding pocket exhibits a more reduced character. This is evident from the upfield shift of the second set of naphthyridine resonances, indicating dearomatisation of the naphthyridine pocket. This dearomatisation is also reflected by the upfield shift of the resonance of the methine proton to δ = 6.79 ppm, resulting from a decrease of its aldimine character. The magnetic inequivalence observed for the dipp –CH3 resonances in 1 is absent in the non-reduced pocket of 2, where a single doublet is observed at δ = 1.18 ppm (3JH,H = 6.8 Hz). The opposite is true for the reduced pocket of 2, which is apparent through the presence of two slightly overlapping doublets at δ = 1.12 and 1.11 ppm. This magnetic inequivalence can be indicative of constrained rotation around the aniline C–N bond on the NMR timescale,51 which we ascribe to the introduction of Ge into the reduced pocket.
:1 1,4-dioxane:Et2O solution of 1 with 2 equiv. of the Ge(II) precursor GeCl2·diox (diox = 1,4-dioxane) resulted in the formation of a white precipitate, presumably polymeric [MgCl2(diox)2]∞,50 and compound 2, which was isolated as a red solid in 68% yield. 1H- and 13C{1H}-NMR analysis of 2 in C6D6 revealed a resonance count consistent with the loss of the previously observed symmetry in 1 (see ESI Section 1.3†). Two sets of naphthyridine resonances are observed, one at δ = 8.14 and 7.02 ppm (3JH,H = 7.7 Hz), and another further upfield at δ = 6.55 and 6.06 ppm (3JH,H = 9.1 Hz). Additionally, two singlets are observed at δ = 8.40 and 6.79 ppm, which we attribute to the protons attached to the pendant methine carbons. Combined, these observations are consistent with two distinct binding pockets in 2. One pocket retains its aromaticity, as evidenced by the more downfield set of naphthyridine protons and the singlet at δ = 8.40 ppm, which is consistent with the aldimine character of the methine proton. In contrast, the other binding pocket exhibits a more reduced character. This is evident from the upfield shift of the second set of naphthyridine resonances, indicating dearomatisation of the naphthyridine pocket. This dearomatisation is also reflected by the upfield shift of the resonance of the methine proton to δ = 6.79 ppm, resulting from a decrease of its aldimine character. The magnetic inequivalence observed for the dipp –CH3 resonances in 1 is absent in the non-reduced pocket of 2, where a single doublet is observed at δ = 1.18 ppm (3JH,H = 6.8 Hz). The opposite is true for the reduced pocket of 2, which is apparent through the presence of two slightly overlapping doublets at δ = 1.12 and 1.11 ppm. This magnetic inequivalence can be indicative of constrained rotation around the aniline C–N bond on the NMR timescale,51 which we ascribe to the introduction of Ge into the reduced pocket.
The high solubility of 2 in various apolar solvents precluded us from obtaining crystals suitable for analysis by X-ray diffraction. To probe whether 2 exists as a monomer or dimer (like 1) in solution, we resorted to convection-corrected Diffusion Ordered Spectroscopy (ccDOSY-NMR). This showed that 2 is monomeric in solution (see ESI Section 1.4†), similar to a recently reported structurally related NHGe reported by Cui et al.52
We subsequently explored the introduction of metals into the empty binding pocket of 2. The addition of an equimolar amount of ZnCl2 to a THF solution of 2 yielded a dark red/purple solution (Scheme 3) from which 3 was isolated in 68% yield. Analysis of the 1H-NMR spectrum (THF-d8, see ESI Section 1.5†) revealed two singlets, integrating to one proton each, at δ = 8.25 and 6.97 ppm, consistent with an aldimine and a methine proton. These chemical shifts are in agreement with one reduced binding pocket and one pocket retaining its iminopyridine character. Remarkably, the most downfield naphthyridine resonance out of the four is observed as a broad resonance lacking multiplicity rather than the expected doublet. A similar effect is observed for one of the two methine resonances of one of the dipp substituents at δ = 3.15 ppm. These observations are indicative of a dynamic change in speciation, with exchange being in the intermediate/fast regime on the timescale of the NMR experiments. In line with this interpretation, sharpening of the broad resonances in the 1H spectrum is observed upon recording the spectra at higher temperatures (see ESI Fig. S26–28†). Similarly, peak broadening and decoalescence of peaks is observed upon cooling the sample in the spectrometer in increments to 193 K (see ESI Fig. S23–25†). Interestingly, upon removal of the THF solvent, 3 changes colour to blue (see ESI Fig. S30†). This colour is observed for the solid-state, as well as in the solution-state for aromatic solvents and dioxane. Analysis of the 1H-NMR spectrum of 3 in C6D6 (see ESI Fig. S22†) revealed sharper resonances, suggesting that the observed fluxionality is likely induced by reversible THF coordination to the Zn centre, potentially followed by dissociation of the N-dipp sidearm.51
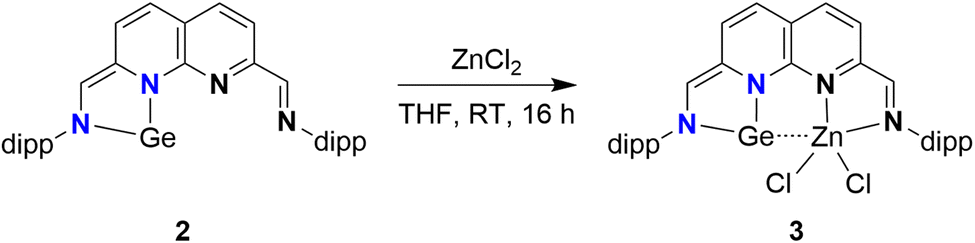 | ||
| Scheme 3 Complexation of ZnCl2 to the empty pocket of 2, yielding 3. | ||
Single crystals suitable for X-ray diffraction were grown by slow vapour diffusion of pentane into a saturated 1,4-dioxane solution of 3. The introduction of ZnCl2 to the empty pocket next to the NHGe motif was confirmed in the solid-state structure of 3 (Fig. 2). The N1–Ge–N3 bond angle of 84.15(17)° falls within the typical range for 5-membered NHGes.53 The Ge–Zn distance of 3.1110(8) Å lies between the sum of their covalent and their van der Waals radii (2.42 and 4.68 Å respectively),54 albeit appreciably larger than reported Ge–Zn distances.55 The environment around the Zn centre is best described as tetrahedral with a distortion due to the proximity of Ge. For the Ge-side of the molecule, the bond metrics are consistent with C–N single bonds (C1–N3 = 1.378(6) Å, C2–N1 = 1.410(6) Å) and the presence of a CC bond located outside of the naphthyridine ring system (C1–C2 = 1.361(7) Å). These features are in agreement with the two-electron reduction of that naphthyridine pocket. The alternating single and double C–C bonds in the respective ring of the naphthyridine motif confirm the two-electron reduction. This is in stark contrast to the other ring, where C–C bonds of more uniform length are observed (see ESI Table S4†). Additionally, the bond metrics consistent with a CN double bond (C10–N4 = 1.283(6) Å), and a C–C single bond external to the naphthyridine ring system (C9–C10 = 1.452(7) Å) further substantiate the α-diimine character of the pocket coordinating the Zn centre. The Ge⋯Cl distances (Ge⋯Cl1 = 3.527(2) Å, Ge⋯Cl2 = 3.5584(16) Å) are too long to assign any meaningful bonding interaction between these two atom pairs.56
 | ||
| Fig. 2 Displacement ellipsoid plot of 3 at 50% probability. Co-crystallised 1,4-dioxane molecules, hydrogen atoms and disordered components are omitted for clarity. The dipp substituents are depicted as wireframe for clarity. | ||
To gain insight into the electronic structure of the NHGe motif in 2 and 3, and the extent of Ge–Zn interactions in the latter, Density Functional Theory (DFT) and Natural Bond Orbitals (NBO) calculations were performed.57–62 The bond metrics of the geometry-optimised structure in the gas phase of 2 are in agreement with our proposed two-electron reduction of the naphthyridine pocket that coordinates the Ge(II) centre (for more details, see ESI Section 2.5 and Table S1†). In the gas-phase optimised geometry of 3 (see ESI Section 2.7†), the calculated Ge–Zn distance is significantly shorter than the experimental distance (2.861 Å vs. 3.1110(8) Å). This difference in the Ge–Zn distance remains even when the calculations are performed without any empirical dispersion (2.868 Å, see ESI Section 2.8†). Because distance can be an important factor when investigating the extent of electronic interactions, we ran calculations on both the freely optimised gas-phase structure of 3 (a) and a structure where the Ge–Zn distance was fixed at the distance obtained from the solid-state structure (b, see ESI Section 2.9†). It is worth mentioning that both 3a and 3b underestimate one of the Ge⋯Cl distances by approx. 0.5 Å. Analysis of the frontier Kohn Sham orbitals of 2 revealed a delocalised bonding interaction spanning the N–Ge–N motif of the NHGe in the HOMO, along with antibonding interactions between Ge and the two N-donors in the LUMO and LUMO+1 orbitals (see ESI Fig. S61†). Using NBO analysis, we further explored the electronic structure of 2, 3a, and 3b. The partial dearomatisation of the naphthyridine backbone and the presence of a CC bond located outside of the naphthyridine ring system (on the Ge side) were both observed in the Lewis structure on which the NBO calculations of 2, 3a, and 3b converged. According to the calculations, the π-bonding interaction along the N–Ge–N motif is best described as a 3-centre-4-electron (3c4e) hyperbond (see ESI Fig. S62†), which is in agreement with the reported electronic structure of related 5-membered N-heterocyclic tetrylenes.63 Structurally this can best be visualised as the superposition of the neutral germylene resonance structure (B) and the two ylide structures (A, C) depicted in Scheme 4. For 5-membered NHGe motifs like the one found in 2 and 3, an additional resonance structure has been proposed based on spectroscopic and reactivity studies: the so-called ‘chelated atom’ structure (Scheme 4, D).18,64 Based on the calculated bond metrics of the NHGe motif in both complexes, the contribution of this resonance structure – a formally Ge(0) centre stabilised by a diazabutadiene fragment – appears minor. Nonetheless, we envision that germylone-like reactivity of 2 and 3 is possible, based on the reported four-electron reactivity of a structurally similar iminopyridine–germylene complex, where the Ge centre is oxidised to Ge(IV) while also acting as conduit for the electrons stored in the redox-active ligand.18 Germanium being one of the heavier tetrels, the germylene lone pair of 2 resides in an orbital of mostly s-character (87%, 13% p-character, ESI Fig. S62c†), due to poor mixing of the s- and p-orbitals and hence lack of hybridisation.
 | ||
| Scheme 4 The described resonance structures of the NHGe motif found in complexes 2 and 3. | ||
Similarly, the Ge lone pair of 3a and 3b resides in a spherically diffuse orbital of mostly s-character (90%). Second-order perturbation analysis revealed a delocalisation energy of 15.2 kcal mol−1 from the Ge lone pair donor NBO of 3a to a Zn-based acceptor NBO of s-character (>99%). The same interaction for 3b (Fig. 3) has a delocalisation energy of 6.7 kcal mol−1, consistent with the larger Ge–Zn separation and the resulting decrease in overlap between the donor and acceptor NBOs. Both interactions are small compared to the calculated interactions found between Zn and the N-donors (32–40 kcal mol−1) and Zn and the Cl-donors (68–90 kcal mol−1). Additionally, the calculations do not reveal any additional interactions from Zn to Ge.
 | ||
| Fig. 3 Overlap of the Ge-based lone pair donor NBO (filled) with the Zn-based acceptor NBO (translucent) of 3b, where the Ge–Zn distance was kept frozen to the value obtained from the solid-state structure. 2nd order pertubation analysis revealed that the Ge–Zn interactions are weak. | ||
We subsequently investigated if the remaining binding pocket could be reduced. Treatment of a THF solution of 2 with a slight excess (2.5 equiv.) of fine mesh (325) Mg0 powder results in the formation of a dark green solution of 4, a highly reactive compound that decomposes upon complete removal of the solvent (Scheme 5). 1H-NMR analysis of 4 in THF-d8 (see ESI Section 1.6†) revealed four equally integrating naphthyridine resonances in the range of δ = 6.27–5.04 ppm. In contrast to the spectroscopic data of 2, no methine resonance with aldimine character was observed, with the two singlets attributed to the methine resonances being found at an upfield chemical shift of δ = 6.50 and 5.37 ppm. Combined, these observations are consistent with the two-electron reduction of the previously empty pocket (and formal four-electron reduction of the ligand), likely with a Mg centre being bound in a similar fashion as to a monomer of 1. The observed decomposition upon complete removal of the solvent implies that THF molecules are bound to 4. Combined with our NMR-spectroscopic observations, we propose a structure for 4 as depicted in Scheme 5.
 | ||
| Scheme 5 Reduction of red 2 with Mg0, resulting in the formation of dark green 4. | ||
Whilst the cooperative reactivity of germylene–transition metal complexes is well-documented,22,25 analogous transformations involving main-group partners have remained elusive. We therefore turned our attention to investigating whether 4 could enable (cooperative) bond activation. The addition of p-tolylacetylene to a THF solution of 4 resulted in the formation of a green solution within 5 minutes (Scheme 6, top-left). 1H-NMR analysis of the mixture in THF-d8 (see ESI Section 1.7†) revealed full conversion of 4 to a new non-symmetric species 5.
 | ||
| Scheme 6 Syntheses of adducts 5–8 out of 4, with the unsaturated C–C bonds being activated through Ge-ligand cooperativity over the Cmethine–Ge vector. | ||
The four doublets attributed to the naphthyridine protons are found relatively upfield (δ = 5.54–4.71 ppm) and the chemical shift of the two singlets attributed to the sidearm protons (δ = 5.57 and 5.07 ppm) is more consistent with assignment to an enamide character of these protons over an aldimine character. The presence of two coupling doublets at δ = 7.27 and 6.97 ppm (3JH,H = 8.1 Hz, 2H each) and a singlet at δ = 2.23 ppm (3H), are indicative of one equivalent of p-tolylacetylene being bound. We attribute the singlet at δ = 7.38 (1H) ppm to the terminal proton of the former acetylene fragment. Through a combination of 2D NMR experiments we were able to confirm the addition of one of the unsaturated C–C bonds of p-tolylacetylene over the Ge centre and the methine, resulting in a bicyclo[2,2,1]heptane motif (Scheme 6).
Storing a saturated THF/MTBE solution of 5 at −40 °C yielded single crystals suitable for analysis X-ray diffraction (Fig. 4). The solid state-structure of 5 confirms the presence of a Ge and a Mg centre bound to the dippNBA ligand. The solid-state structure also corroborates the formation of the bicyclo[2,2,1]heptane motif, consistent with our spectroscopic observations. The Ge–Mg distance of 2.9115(15) Å lies well within the sum of their van der Waals radii (4.80 Å) and outside of the sum of their covalent radii (2.62 Å).54 Additionally, the distance is significantly longer compared to other reported Ge–Mg distances.65 These observations are consistent with a dative Ge–Mg bond being present rather than a covalent bond (see below). The geometry at the Ge centre is distorted tetrahedral and the geometry around the Mg centre is distorted octahedral, with three meridionally binding THF ligands completing the coordination sphere. The C9–C10 bond length of 1.380(6) Å is consistent with a double bond, indicative of the two-electron reduction of the binding pocket coordinated to Mg(II). On the Ge side of the complex, the C1–C2 (1.511(6) Å) and C35–C36 (1.342(6) Å) bond lengths are consistent with single and double bond character, respectively. The addition of one of the unsaturated C–C bonds of the acetylene moiety over the NHGe motif can best be described as a formal [4 + 2] cycloaddition or ‘Metallo-Diels–Alder’ reaction of p-tolylacetylene with the NHGe motif from resonance structure C (Scheme 4).66
 | ||
| Fig. 4 Displacement ellipsoid plot of 5 at 50% probability. Hydrogen atoms and minor disorder components are omitted for clarity. The dipp substituents and the carbon backbones of the THF molecules are depicted as wireframe for clarity. | ||
The bond metrics of the naphthyridine motif (see also ESI Table S5†) are consistent with localised double bonds between C2–C3 (1.349(6) Å), C4–C5 (1.379(6) Å), and C7–C8 (1.368(6) Å) and provide a rationale for the relatively upfield chemical shift of the naphthyridine protons in solution. Combined with the short C6–N1 and C6–N2 distances (1.354(5) and 1.332(5) Å, respectively), the metrics point towards delocalisation of one anionic charge over the N1–C6–N2 motif.
The computationally derived bond metrics of geometry-optimised 4d (4 with 3 THF molecules bound to Mg) and 5 (see ESI Fig. S73†) further corroborated our observations from NMR-spectroscopy and the X-ray crystal structure. The alternating single/double C–C bonds observed throughout the naphthyridine backbone found for 4d are consistent with a four-electron reduced ligand featuring two bis-anionic NN pockets. The calculated bond metrics of 5 match the experimental bond metrics well (see ESI Fig. S71†) and support the description of a two-electron reduced naphthyridine-imine ligand with a delocalised anionic charge over the N1–C6–N2 motif.
To get a better understanding of the Ge–Mg interactions found for 4 and 5, electronic structure calculations were performed (for a detailed description see ESI Section 2.10†). NBO analysis indicates that the Ge–Mg interaction in 4, bound to 0–3 THF molecules (structures 4a–4d, respectively), is best described by donation from the Ge lone pair residing in a spherically diffuse donor NBO (s-character = 79.9–86.8%) to a Mg-based acceptor NBO of high s-character (>95%). These dative interactions are consistently stronger than the respective contributions from the N- and O-donors and decrease slightly upon increasing the coordination number of the Mg centre. The dative interaction is equally strong after the observed addition of p-tolylacetylene over the NHGe in 5, albeit with a small decrease of the s-character of the Ge lone pair (73.7%) and delocalisation energy. These results demonstrate that there is a strong, dative Ge–Mg interaction present in both 4 and 5. This is in contrast with our observations for Ge–Zn complex 3 and highlights the versatility of the germylene centre in 2, with the extent of Ge–M dative interactions varying as a function of the required electron density on the metal centre that occupies the redox-active binding pocket.
We subsequently set out to explore the scope of C–C unsaturated bonds we could activate with 4. Treating a THF solution of 4 with 1 equiv. of diphenylacetylene at ambient temperatures resulted in full conversion of 4 to 6 (Scheme 6, top-right). Analysis of the 1H- and 13C{1H}-NMR spectra (THF-d8, see ESI Section 1.8†) revealed similar spectra to 5. The characteristic 13C{1H} chemical shift of the bridgehead methine (δ = 85.4 ppm) and a 1H–13C HMBC cross peak between its attached proton and a carbon resonance at δ = 165.8 ppm (attributed to the alkyne carbon added to the methine carbon of the NHGe motif) confirm the addition of the alkyne triple bond over the NHGe.
To explore if CC bonds could also be activated, we treated a THF solution of 4 with styrene (Scheme 6, bottom-left). Gratifyingly, full conversion of 4 was observed to yield blue 7. Extensive 1D and 2D NMR analysis (THF-d8, see ESI Section 1.9†) showed that 7 is formed as a single diastereomer with the phenyl group pointing towards the naphthyridine motif as depicted in Scheme 6. The diastereomer with the phenyl group pointing towards the N-dipp motif was not observed, an observation we attribute to steric clashing.
Finally, to investigate the chemoselectivity of 4 towards alkenes or alkynes, we treated 4 with 2-methylbut-1-en-3-yne, a conjugated enyne (Scheme 6, bottom-right). Examination of the 1H-NMR spectrum (THF-d8, see ESI Section 1.10†) revealed full conversion of 4, yielding 8. Similar to 5, a downfield singlet resonance at δ = 7.15 ppm that shows a 1H–13C ASAP-HMQC cross-peak with a carbon resonance at δ = 152.3 ppm (vs. δ = 7.38 and 150.5 ppm found for 5), are indicative of preferential addition of the alkyne moiety to the NHGe over the alkene moiety. Consistent with this assignment, two singlet resonances in the alkene region of chemical shift are found, which are bound to the same carbon atom (δ = 110.3, confirmed through 1H–13C ASAP-HMQC NMR), and were assigned to the terminal alkene protons.
Similar “Metallo”-Diels–Alder reactions of (phenyl)acetylene with an ylide-like NHGe have been reported by the Driess group, yielding the respective bicyclo[2,2,2]octane adducts alongside the terminal C–H activation products (Scheme 1b).15 We propose that the latter is not observed for our system due to lower basicity of the methine positions of 4 compared to the diketiminate-based ylide. For the Driess system, reaction of the NHGe with diphenylacetylene does not take place, even at elevated temperatures, which was attributed to increased steric strain of the system.15 We propose that the discrepancy in observed reactivity between the two systems is influenced by two factors: the decreased steric strain around the Ge centre due to the presence of only one dipp substituent in 4 and the higher nucleophilicity of the methine carbon of the NHGe.
The reactions depicted in Scheme 6 yield bicyclo[2,2,1]heptane adducts where the unsaturated substrates are added over the Ge and methine position in one NN binding pocket, while leaving the other NN pocket containing Mg unaltered. Hence, we performed analogous reactions between 2 – containing the same germylene motif but lacking said Mg – with an equivalent of the same unsaturated substrates. However, even at prolonged reaction times or reflux conditions no conversion of 2 was observed.67 The observed discrepancy in reactivity of 2 and 4 is also reproduced computationally (see ESI Table S2†), with addition of p-tolylacetylene over the NHGe motif in 2 being endergonic by 0.7 kcal mol−1, whereas the same reaction for 4d was found to be exergonic by 25.5 kcal mol−1 and thereby suggesting that this difference is thermodynamic in nature. We hypothesise that this difference stems from the different oxidation states of the dippNBA ligand. In 2, the ligand is reduced by two electrons, with aromaticity being retained in one of the naphthyridine rings. In contrast, no aromaticity is retained in the formally quadruply reduced ligand of 4. The combined experimental and computational data do not indicate that aromaticity is gained upon cycloaddition,68 but do suggest increased delocalisation of charge over the naphthyridine N–C–N motif (see ESI Fig. S73†). We therefore propose that the majority of thermodynamic driving force originates from decreased electronic repulsion within the quadruply reduced dippNBA ligand upon cycloaddition.
The difference in reactivity between 2 and 4 highlights the crucial role of the Mg centre flanking the Ge centre, despite it not being directly involved in bond breaking or electron transfer events. We argue that the Mg centre electronically stabilises the four-electron reduced ligand sufficiently to allow for this reactive state to be accessed.69 This provides a compelling example of indirect cooperativity, where the Mg centre electronically stabilises the quadruply reduced ligand, upon which the germylene engages in metal–ligand cooperative activation of C–C unsaturated bonds.
Conclusion
In conclusion, we report the isolation, synthesis, and characterisation of NHGe 2, featuring a vacant, redox-active binding pocket that is able to bind metals in close proximity to the Ge centre. This was demonstrated by the isolation of Ge–Zn (3) and Ge–Mg (4) complexes, with the versatile redox-active binding pocket accessing different oxidation states. We envision that complex 4 presents a promising entry point into germylene–(transition) metal chemistry, through transmetalation of the Mg centre in the bis-anionic binding site with various metal precursors. Electronic structure calculations reveal that the extent of the Ge–M dative interactions can vary as a function of the required electron density on the metal centre that occupies the redox-active binding pocket, with the Ge–Zn interaction in 3 being weak, whilst strong dative Ge–Mg interactions are found for 4. The latter engages in ‘Metallo-Diels–Alder’ reactions with a multitude of substrates containing C–C unsaturated bonds, activating the C–C bond over Ge and the ligand backbone. The same reaction does not proceed prior to two-electron reduction of the vacant binding site by Mg0. This provides a well-defined example of indirect cooperativity, where the Mg centre electronically stabilises the quadruply reduced ligand, upon which the germylene engages in metal–ligand cooperative activation of C–C unsaturated bonds. Combined, these findings not only expand the understanding of germylene reactivity but also open new avenues for designing systems that leverage cooperative interactions between germylenes and proximal metals for chemical transformations.Data availability
The data supporting this article have been included as part of the ESI,† the raw data can be accessed through the Yoda repository, DOI: https://doi.org/10.24416/UU01-FJ03BR. CCDC 2427530–2427533 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
Synthesis and characterisation were done by E. K. with support from R. S. and L. W. All crystallographic measurements and the corresponding data refinement was performed by M. L. Computations were done by E. K. Project design, supervision and oversight were done by M. E. M. and D. L. J. B. Funding acquisition and administration were done by D. L. J. B. The original draft was written by E. K. and reviewing and editing was done by M. E. M. and D. L. J. B. with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Bert Klein Gebbink is kindly acknowledged for comments and suggestions on the written manuscript. This work was supported by the Netherlands Organization for Scientific Research (START-UP grant 740.018.019 to D. L. J. B.). This work also made use of the Dutch national e-infrastructure with the support of the SURF Cooperative using grant no. EINF-11244. The X-ray diffractometer has been financed by the Netherlands Organization for Scientific Research (NWO).References
- N. Mukherjee and M. Majumdar, Diverse Functionality of Molecular Germanium: Emerging Opportunities as Catalysts, J. Am. Chem. Soc., 2024, 146, 24209–24232 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Stable Heavier Carbene Analogues, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, N-Heterocyclic Carbene Analogues with Low-Valent Group 13 and Group 14 Elements: Syntheses, Structures, and Reactivities of a New Generation of Multitalented Ligands, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- W. A. Herrmann, M. Denk, J. Behm, W. Scherer, F.-R. Klingan, H. Bock, B. Solouki and M. Wagner, Stable Cyclic Germanediyls (“Cyclogermylenes”): Synthesis, Structure, Metal Complexes, and Thermolyses, Angew. Chem., Int. Ed., 1992, 31, 1485–1488 CrossRef.
- J. K. West, G. L. Fondong, B. C. Noll and L. Stahl, Heavier carbene analogues and their derivatives as κ-El, κ2-El,N, and κ2-N,N′ ligands: different reactivity patterns for acyclic and cyclic diamidogermylenes and -stannylenes, Dalton Trans., 2013, 42, 3835–3842 RSC.
- L. Kristinsdóttir, N. L. Oldroyd, R. Grabiner, A. W. Knights, A. Heilmann, A. V. Protchenko, H. Niu, E. L. Kolychev, J. Campos, J. Hicks, K. E. Christensen and S. Aldridge, Synthetic, structural and reaction chemistry of N-heterocyclic germylene and stannylene compounds featuring N-boryl substituents, Dalton Trans., 2019, 48, 11951–11960 RSC.
- I. L. Fedushkin, A. A. Skatova, V. A. Chudakova, N. M. Khvoinova, A. Y. Baurin, S. Dechert, M. Hummert and H. Schumann, Stable Germylenes Derived from 1,2-Bis(arylimino)acenaphthenes, Organometallics, 2004, 23, 3714–3718 CrossRef CAS.
- M. Driess, S. Yao, M. Brym and C. van Wüllen, A Heterofulvene-Like Germylene with a Betain Reactivity, Angew. Chem., Int. Ed., 2006, 45, 4349–4352 CrossRef CAS PubMed.
- W. Wang, S. Inoue, S. Yao and M. Driess, Reactivity of N-Heterocyclic Germylene Toward Ammonia and Water, Organometallics, 2011, 30, 6490–6494 CrossRef CAS.
- H. Arii, T. Amari, J. Kobayashi, K. Mochida and T. Kawashima, Low-Coordinate Germanium(II) Centers Within Distorted Axially Chiral Seven-Membered Chelates: Stereo- and Enantioselective Cycloadditions, Angew. Chem., Int. Ed., 2012, 51, 6738–6741 CrossRef CAS PubMed.
- Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, Reaction of Hydrogen or Ammonia with Unsaturated Germanium or Tin Molecules under Ambient Conditions: Oxidative Addition versus Arene Elimination, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, A Systematic Study of Structure and E−H Bond Activation Chemistry by Sterically Encumbered Germylene Complexes, Chem.–Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed.
- J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Reversible, Photoinduced Activation of P4 by Low-Coordinate Main Group Compounds, Chem.–Eur. J., 2014, 20, 6739–6744 CrossRef CAS PubMed.
- Y. Wu, C. Shan, Y. Sun, P. Chen, J. Ying, J. Zhu, L. Liu, L. L. Liu and Y. Zhao, Main group metal–ligand cooperation of N-heterocyclic germylene: an efficient catalyst for hydroboration of carbonyl compounds, Chem. Commun., 2016, 52, 13799–13802 RSC.
- S. Yao, C. van Wüllen and M. Drieß, Striking reactivity of ylide-like germylene toward terminal alkynes: [4 + 2] cycloaddition versus C-H bond activation, Chem. Commun., 2008, 5393–5395 RSC.
- Z. Zhao, J. Tan, T. Chen, Z. Hussain, Y. Li, Y. Wu and D. W. Stephan, Ambiphilic Behavior of Ge(II)-Pseudohalides in Inter- and Intramolecular Frustrated Lewis Pair Alkyne Addition Reactions, Inorg. Chem., 2022, 61, 18670–18677 CrossRef CAS PubMed.
- A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Dehydrogenation of LGeH by a Lewis N-Heterocyclic Carbene Borane Pair under the Formation of L′Ge and its Reactions with B(C6F5)3 and Trimethylsilyl Diazomethane: An Unprecedented Rearrangement of a Diazocompound to an Isonitrile, Inorg. Chem., 2009, 48, 7645–7649 CrossRef CAS PubMed.
- K.-H. Chen, Y.-H. Liu and C. Chiu, A Non-innocent Ligand Supported Germylene and Its Diverse Reactions, Organometallics, 2020, 39, 4645–4650 CrossRef CAS.
- A. Jana, B. Nekoueishahraki, H. W. Roesky and C. Schulzke, Stable Compounds of Composition LGe(II)R (R = OH, PhO, C6F5O, PhCO2) Prepared by Nucleophilic Addition Reactions, Organometallics, 2009, 28, 3763–3766 CrossRef CAS.
- Y. Wu, L. L. Liu, J. Su, K. Yan, J. Zhu and Y. Zhao, Synthesis of digermylene-stabilized linear tetraboronate and boroxine, Chem. Commun., 2016, 52, 1582–1585 RSC.
- Y. Wu, L. L. Liu, J. Su, K. Yan, T. Wang, J. Zhu, X. Gao, Y. Gao and Y. Zhao, Reactivity of Germylene toward Phosphorus-Containing Compounds: Nucleophilic Addition and Tautomerism, Inorg. Chem., 2015, 54, 4423–4430 CrossRef CAS PubMed.
- T. J. Hadlington, Heavier tetrylene- and tetrylyne-transition metal chemistry: it's no carbon copy, Chem. Soc. Rev., 2024, 53, 9738–9831 RSC.
- M. L. Buil, J. A. Cabeza, M. A. Esteruelas, S. Izquierdo, C. J. Laglera-Gándara, A. I. Nicasio and E. Oñate, Alternative Conceptual Approach to the Design of Bifunctional Catalysts: An Osmium Germylene System for the Dehydrogenation of Formic Acid, Inorg. Chem., 2021, 60, 16860–16870 CrossRef CAS PubMed.
- J. A. Cabeza, P. García-Álvarez, C. J. Laglera-Gándara and E. Pérez-Carreño, A Z-type PGeP pincer germylene ligand in a T-shaped palladium(0) complex, Chem. Commun., 2020, 56, 14095–14097 RSC.
- R. J. Somerville and J. Campos, Cooperativity in Transition Metal Tetrylene Complexes, Eur. J. Inorg. Chem., 2021, 2021, 3488–3498 CrossRef CAS PubMed.
- M. Fernández-Buenestado, R. J. Somerville, J. López-Serrano and J. Campos, A genuine germylene PGeP pincer ligand for formic acid dehydrogenation with iridium, Chem. Commun., 2023, 59, 8826–8829 RSC.
- K. Inomata, T. Watanabe, Y. Miyazaki and H. Tobita, Insertion of a Cationic Metallogermylene into E–H Bonds (E = H, B, Si), J. Am. Chem. Soc., 2015, 137, 11935–11937 CrossRef CAS PubMed.
- K. E. Litz, J. W. Kampf and M. M. Banaszak Holl, Activation of Arylnitroso Substrates on a Platinum−Germylene Complex Facilitating the Formation of New N−C and N−S Bonds, J. Am. Chem. Soc., 1998, 120, 7484–7492 CrossRef CAS.
- Y. Y. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Dinuclear nickel complexes in five states of oxidation using a redox-active ligand, Inorg. Chem., 2014, 53, 11770–11777 CrossRef CAS PubMed.
- Y. Y. Zhou and C. Uyeda, Catalytic reductive [4 + 1]-cycloadditions of vinylidenes and dienes, Science, 2019, 363, 857–862 CrossRef CAS PubMed.
- J. Cui, J. Weiser, F. Fantuzzi, M. Dietz, Y. Yatsenko, A. Häfner, S. Nees, I. Krummenacher, M. Zhang, K. Hammond, P. Roth, W. Lu, R. D. Dewhurst, B. Engels and H. Braunschweig, A rigid redox-active-ligand-supported bis(germylene) as a two-centre six-electron donor, Chem. Commun., 2022, 58, 13357–13360 RSC.
- E. Kounalis and D. L. J. Broere, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 4th edn, 2022, vol. 1, pp. 421–441 Search PubMed.
- G. G. Briand, Redox-active ligands – a viable route to reactive main group metal compounds, Dalton Trans., 2023, 52, 17666–17678 RSC.
- Y. Zhao, Y. Liu, L. Yang, J.-G. Yu, S. Li, B. Wu and X.-J. Yang, Mechanistic Insight into the N=N Bond-Cleavage of Azo-Compounds that was Induced by an Al–Al-bonded Compound [L2−AlII–AlIIL2−], Chem.–Eur. J., 2012, 18, 6022–6030 CrossRef CAS PubMed.
- O. T. Summerscales, T. W. Myers and L. A. Berben, Mild Reduction Route to a Redox-Active Silicon Complex: Structure and Properties of (IP2–)2Si and (IP–)2Mg(THF) (IP = α-Iminopyridine), Organometallics, 2012, 31, 3463–3465 CrossRef CAS.
- T. W. Myers, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Synthesis and characterization of bis(imino)pyridine complexes of divalent Mg and Zn, Dalton Trans., 2016, 45, 5989–5998 RSC.
- S. M. S. Subasinghe, M. R. Radzhabov and N. P. Mankad, Predictive Models for Ligand Effects on a Reactive Al-Containing Radical Intermediate from Multivariate Linear Regression Analysis, Organometallics, 2024, 43, 2854–2861 CrossRef CAS.
- S. Sinhababu, R. P. Singh, M. R. Radzhabov, J. Kumawat, D. H. Ess and N. P. Mankad, Coordination-induced O-H/N-H bond weakening by a redox non-innocent, aluminum-containing radical, Nat. Commun., 2024, 15, 1315 CrossRef CAS PubMed.
- J. Pölker, D. Schaarschmidt, J. Bernauer, M. Villa and A. Jacobi von Wangelin, BIAN-Aluminium-Catalysed Imine Hydrogenation, ChemCatChem, 2022, 14, e202200144 CrossRef PubMed.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Control of Ligand pKa Values Tunes the Electrocatalytic Dihydrogen Evolution Mechanism in a Redox-Active Aluminum(III) Complex, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- L. A. Berben, Catalysis by Aluminum(III) Complexes of Non-Innocent Ligands, Chem.–Eur. J., 2015, 21, 2734–2742 CrossRef CAS PubMed.
- L. W. T. Parsons and L. A. Berben, Expanding the Scope of Aluminum Chemistry with Noninnocent Ligands, Acc. Chem. Res., 2024, 57, 1087–1097 CrossRef CAS PubMed.
- T. M. Bass, C. R. Carr, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Syntheses of Square Planar Gallium Complexes and a Proton NMR Correlation Probing Metalloaromaticity, Inorg. Chem., 2020, 59, 13517–13523 CrossRef CAS PubMed.
- A. Arnold, T. J. Sherbow, R. I. Sayler, R. D. Britt, E. J. Thompson, M. T. Muñoz, J. C. Fettinger and L. A. Berben, Organic Electron Delocalization Modulated by Ligand Charge States in [L2 M] n– Complexes of Group 13 Ions, J. Am. Chem. Soc., 2019, 141, 15792–15803 CrossRef CAS PubMed.
- C. D. Cates, T. W. Myers and L. A. Berben, (IP)2 Ga(III) Complexes Facilitate Net Two-Electron Redox Transformations (IP = α-Iminopyridine), Inorg. Chem., 2012, 51, 11891–11897 CrossRef CAS PubMed.
- K. Kowolik, M. Shanmugam, T. W. Myers, C. D. Cates and L. A. Berben, A redox series of gallium(III) complexes: ligand-based two-electron oxidation affords a gallium–thiolate complex, Dalton Trans., 2012, 41, 7969–7976 RSC.
- T. W. Myers and L. A. Berben, A Sterically Demanding Iminopyridine Ligand Affords Redox-Active Complexes of Aluminum(III) and Gallium(III), Inorg. Chem., 2012, 51, 1480–1488 CrossRef CAS PubMed.
- A. I. Nicasio, R. J. Somerville, P. Sahagún, E. Soto, J. López-Serrano and J. Campos, Carbon–carbon bond formation and cleavage at redox active bis(pyridylimino)isoindole (BPI) germylene compounds, Dalton Trans., 2025, 54, 3039–3046 RSC.
- A transient species where intramolecular C–H activation might have been facilitated by Ge–Mg cooperativity has been proposed, but has not been spectroscopically detected, see: D. Dange, A. R. Gair, D. D. L. Jones, M. Juckel, S. Aldridge and C. Jones, Chem. Sci., 2019, 10, 3208–3216 RSC.
- R. Fischer, H. Görls, P. R. Meisinger, R. Suxdorf and M. Westerhausen, Structure–Solubility Relationship of 1,4-Dioxane Complexes of Di(hydrocarbyl)magnesium, Chem.–Eur. J., 2019, 25, 12830–12841 CrossRef CAS PubMed.
- L. Witteman, T. Evers, Z. Shu, M. Lutz, R. J. M. Klein Gebbink and M.-E. Moret, Hydrosilylation in Aryliminopyrrolide-Substituted Silanes, Chem.–Eur. J., 2016, 22, 6087–6099 CrossRef CAS PubMed.
- Y. He, C. P. Souza, J. Weiser, M. Dietz, I. Krummenacher, R. D. Dewhurst, H. Braunschweig, F. Fantuzzi and J. Cui, Single-Electron Oxidation, Chloride Abstraction, and Hydride-Induced Decomposition of a Dichloro-Bis(Germylene), Eur. J. Inorg. Chem., 2024, 27, e202400422 CrossRef CAS.
- Mean of 84.431 Å out 67 N–Ge–N angles reported for 5-membered NHGes in the CCDC.
- J. Echeverría and S. Alvarez, The borderless world of chemical bonding across the van der Waals crust and the valence region, Chem. Sci., 2023, 14, 11647–11688 RSC.
- Mean of 2.576 Å out of 114 Ge–Zn distances reported in the CCDC.
- Mean of 2.240 Å out of 2189 Ge–Cl distances reported in the CCDC.
- J. P. Foster and F. Weinhold, Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis, and F. Weinhold, NBO 7.0. Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- A. E. Reed and F. Weinhold, Natural bond orbital analysis of near-Hartree–Fock water dimer, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Electronic Structures of Main-Group Carbene Analogues, Inorg. Chem., 2007, 46, 10693–10706 CrossRef CAS PubMed.
- A. J. I. Arduengo, H. Bock, H. Chen, M. Denk, D. A. Dixon, J. C. Green, W. A. Herrmann, N. L. Jones, M. Wagner and R. West, Photoelectron Spectroscopy of a Carbene/Silylene/Germylene Series, J. Am. Chem. Soc., 1994, 116, 6641–6649 CrossRef CAS.
- Mean of 2.686 Å out of 13 Ge–Mg distances reported in the CCDC.
- M. J. Evans, M. D. Anker, A. Mouchfiq, M. Lein and J. R. Fulton, The “Metallo”-Diels–Alder Reactions: Examining the Metalloid Behavior of Germanimines, Chem.–Eur. J., 2020, 26, 2606–2609 CrossRef CAS PubMed.
- Exposing 3 to the unsaturated substrates under identical conditions results in decomposition to an intractable mixture void of the spectroscopic handles found for cycloadducts 5–9.
- Although a resonance structure with partial aromatic character of the naphthyridine backbone can be drawn, the experimental and computational bond metrics, and NMR-spectroscopic data suggest that it does not significantly contribute to the resonance hybrid.
- Attempts towards accessing the quadruply reduced ligand state through reduction of 2 or3 with 2 equiv. KC8 have thus far been unsuccessful.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2427530–2427533. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02751a |
| This journal is © The Royal Society of Chemistry 2025 |