DOI:
10.1039/D5SC01320H
(Edge Article)
Chem. Sci., 2025,
16, 12623-12634
Novel lanthanide(III)/gallium(III) metallacrowns with appended visible-absorbing organic sensitizers for molecular near-infrared imaging of living cells†
Received
19th February 2025
, Accepted 14th May 2025
First published on 12th June 2025
Abstract
Optical imaging of biological samples in the near-infrared (NIR) domain is of major interest for non-invasive experiments due to reduced light scattering, absorption and autofluorescence. Taking advantage of the maleimide moieties in the [Ln2Ga8(shi)8(mip)4]2− metallacrowns (MCs; shi3− = salicylhydroximate; mip2− = 5-maleimido-isophthalate; Ln = NdIII, GdIII, ErIII, YbIII and YIII), we appended visible-absorbing thiol-coumarin (C) sensitizers through a thiol-Michael addition click reaction, affording the functionalized [Ln2Ga8(shi)8(C-mip)4]2− MCs. By using this approach, the sensitization of NIR-emitting NdIII, ErIII and YbIII was achieved upon excitation in the visible range (λexc = 420 nm) through the appended coumarins. NIR epifluorescence microscopy of living HeLa cells incubated with the NdIII derivative confirmed unambiguous detection of the signal arising from the 4F3/2 → 4I11/2 electronic transition in the range of 1050–1080 nm in the cells. These results demonstrate that the [Ln2Ga8(shi)8(mip)4]2− MC can be used as a versatile molecule for tuning the chemical and photophysical properties of the [Ln2Ga8] MC family.
Introduction
Optical imaging of biological samples in the near-infrared (NIR) I and II windows has become a tool of increasing importance for biological research and medical diagnostics.1–4 Detection in the NIR I and II windows allows for the removal of unwanted contributions from the native fluorescence of biological materials (autofluorescence), simplifying experiments and preventing any ambiguity in the interpretation of results. The key requirement for such detection is the availability of reporters emitting in the NIR I and II windows. However, today, compounds with such emission properties suitable for biological imaging remain scarce, even more so lanthanide (LnIII) based ones. Trivalent LnIII are a class of metal ions that possess unique photophysical properties to address the needs of optical biological imaging in the NIR I and II spectral domains. Due to their unique electronic configurations, they exhibit sharp emission bands that cover a large range of the visible and NIR spectra. In this respect, they are of great interest for various practical applications such as biological NIR imaging,5–13 solar energy conversion14–16 or telecommunications.17,18 As most of the f–f transitions are forbidden by the Laporte rule, LnIII display low molar absorption coefficients, making their direct sensitization inefficient. This limitation can be overcome by using a highly absorbing organic chromophore that can sensitize the LnIII through the so called “antenna effect”. In this process, the chromophoric moiety (the antenna) absorbs as much light as possible, transferring the resulting energy to the accepting levels of the luminescent LnIII metal ion.
Metallacrowns (MCs)19 are a class of metal complexes that have structural similarities to crown ethers. Instead of the repeating C–C–O motif, various metals are incorporated into the repeating M–N–O unit to form x-MCs-y of different sizes, where x and y refer to the number of atoms forming the MC ring and the number of metals (and ligands), respectively (Scheme 1).
 |
| | Scheme 1 Left: representation of the 12-MC-4 MC core (M–N–O coloured: N: blue; O: red; MC bonds: orange) formed by four shi3− ring ligands and four GaIII atoms, with the LnIII occupying the central cavity. Top right: salicylhydroximate (shi3−) ring ligand. | |
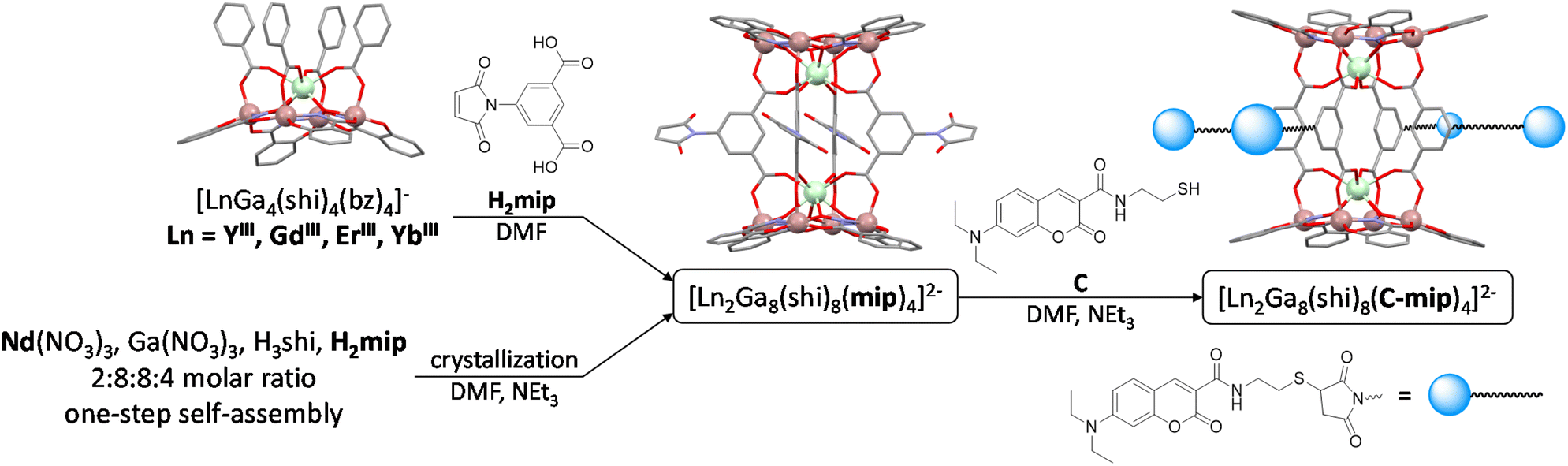
The large range of metals that can be incorporated in these MCs enable the preparation of a wide variety of materials that have applications as luminescent probes for optical imaging,12,20,21 nanothermometers,22,23 white light generation,24,25 single molecule magnets (SMMs),26–30 magneto refrigerants,31–33 or contrast agents for magnetic resonance imaging (MRI).34–36 Previously, we took advantage of the properties of [YbZn16pyz16]3+ MCs (pyz2− = pyrazine hydroximate) to create combined cell fixation and NIR counter staining agents20 or use them for the specific NIR imaging of necrotic cells.12 The uniquely rigid structure37 of [LnZn16L16]3+ MCs provides outstanding LnIII luminescence quantum yields and high stability in biological media. However in both aforementioned examples of NIR imaging, MCs were not taken up by living cells.12 We have demonstrated that the behavior of [LnZn16L16]3+ MCs can be modulated by using a mixed-ligand approach.38 In another attempt of NIR imaging applications using MCs, living HeLa cells were incubated with [Yb2Ga8(shi)8(ip)4]2− MCs (shi3− = salicylhydroximate; ip2− = isophthalate). A NIR signal arising from YbIII was detected on the surface of HeLa cells due to non-specific interaction but MC uptake was not confirmed.39 A current limitation of NIR-emitting LnIII-based MCs for imaging applications is the high energy of their excitation wavelengths which implies the use of light that can be detrimental to biological samples. The excitation wavelengths of MCs are defined by the nature of the chromophoric building blocks that they are made of. By expanding the aromatic system of the ligand, the excitation wavelength can be red-shifted at the cost of reduced solubility.23,40,41 As an alternative, non-linear two-photon absorption (2PA) spectroscopy can be used to circumvent the need for lower energy excitation by the simultaneous absorption of two photons of half the energy required by the one-photon absorption (1PA) process.9,10,13,42 In our work, we focus on 1PA as it uses lower power excitation sources, and more versatile and easy to use equipment than 2PA. 1PA relies on chemical modifications of the ligands to gain access to lower-energy excitations.10,13,21,43–45 As the functionalization of the [LnZn16L16]3+ MC scaffold is highly challenging, we focused our efforts on the [Ln2Ga8]2− MC and recently reported its functionalization with four additional RuII complexes.21 In these (RuII)4–[Ln2Ga8] MCs, the appended RuII complexes display a metal-to-ligand charge transfer (MLCT) band located in the visible range that can be used for the sensitization of NIR-emitting LnIII. However, biological imaging with these (RuII)4–[Ln2Ga8] MCs might be limited because of the toxicity induced by the generation of reactive oxygen species due to the efficient population of the 3MLCT electronic states of the RuII complexes.
In this work, we present a new strategy for the functionalization of the [Ln2Ga8] MC scaffold using a conjugated organic sensitizer, a coumarin chromophore, that is not part of the MC building blocks, but is attached to it, allowing the sensitization of NIR-emitting LnIII. As the basis of our design, we chose MCs where GaIII is used in the M–N–O repeating unit along with salicylhydroximate ligands to coordinate LnIII (Scheme 1) leading to complexes with high NIR luminescence intensity, such as the [LnGa4(shi)4(bz)4]pym MCs (hereafter [LnGa4]; bz− = benzoate; pym+ = pyridinium)46 and their corresponding [Ln2Ga8(shi)8(ip)4](NH4)2 MC dimers (hereafter [Ln2Ga8]).39 More specifically, in the [Ln2Ga8] MC dimers, the 5 position of the isophthalate bridging ligands can be used for the functionalization of these MCs. The 5-maleimido-isophthalic acid bridging ligand (Scheme 2, H2mip) was previously designed for this purpose.47 It allows for a convenient functionalization of these MCs via a base mediated thiol-Michael addition click reaction.21,47–49 In [LnGa4] or [Ln2Ga8] MCs, LnIII are typically sensitized through an excitation in the absorption band of the GaIII/shi scaffold in the UV region (λmax = 320 nm), a wavelength that can result in significant damage to living cells. We have chosen to use coumarins as an additional LnIII sensitizer in our effort to (i) add an excitation band of the complex at lower energy on the basis of their absorption wavelength and (ii) to promote the biocompatibility of the MC through the coumarins.50,51 Coumarins and carbostyrils have been previously reported to sensitize LnIII emission in the visible range52–56 and in the NIR range.57 However, the wavelength corresponding to the maximum of the absorption band of these complexes usually lies at around λmax = 310–340 nm. In this work, we took into account that the addition of a dimethyl amino group in the 7-position of the coumarin scaffold results in the shift of the maximum absorption to ≈420 nm.58 This wavelength is sufficiently differentiated from the UV band of the MC scaffold (λmax = 320 nm) to probe the sensitization of the NIR emitting LnIII through the 7-diethylamino-coumarin (Scheme 2, C) in this new generation of MCs. In addition, our goal was not only to analyze the NIR luminescence properties of these functionalized [Ln2Ga8] MCs but also to study their behavior and performance as imaging agents in proof-of-concept experiments on living HeLa cells.
 |
| | Scheme 2 Preparation of the [Ln2Ga8(shi)8(mip)4]2− MC precursors and their functionalization with C through a thiol-Michael addition click reaction. | |
[Ln2Ga8(shi)8(mip)4]2− MCs were prepared either by reacting [LnGa4] MCs with the H2mip bridging ligand (Ln = YIII, GdIII, ErIII and YbIII) or in a one-step self-assembly reaction (Ln = NdIII). They were further reacted with the thiol-coumarin antenna C through a base mediated thiol-Michael addition click reaction, affording the coumarin-functionalized [Ln2Ga8(shi)8(C-mip)4]2− MCs (Scheme 2). The synthesized MCs were characterized by ESI-MS, elemental analysis, and UV-vis and IR spectroscopy. 1H NMR characterization of the MCs was performed using the diamagnetic YIII analogues. X-ray single crystal analysis was performed on the [Nd2Ga8(shi)8(mip)4]2− MC. The LnIII-centered luminescence properties of these MCs were recorded for the NdIII, ErIII and YbIII analogues. The GdIII analogue was used for the determination of the singlet (S1) and triplet (T1) state energy levels of C in these MCs. HeLa cell imaging experiments (NIR epifluorescence microscopy and visible confocal microscopy) and cytotoxicity assays were performed to evaluate the potential of [Ln2Ga8(shi)8(C-mip)4]2− MCs for biological imaging.
Experimental section
Organic synthesis
All reagents and chemicals were purchased from commercial sources and used without further purification unless otherwise stated. Silica (215–400 mesh) was used for flash column chromatography. 5-Maleimido-isophthalic acid H2mip,47 7-(diethylamino)-3-coumarin carboxylic acid 3 (ref. 58–61) and S-tritylcysteamine linker 6 (ref. 62) were synthesized according to or by modifying existing procedures (see Section S1, ESI† for detailed procedures). 1H and 13C NMR spectra of synthesized molecules are reported in Fig. S1–S8, ESI.†
Preparation of [Ln2Ga8(shi)8(mip)4]pym2 MCs
[Ln2Ga8(shi)8(mip)4]pym2 MCs were prepared as previously reported.47 1.0 × 10−1 mmol of [LnGa4(shi)4(bz)4]pym MCs (1.0 eq., Ln = YIII, GdIII, ErIII, YbIII) and 55.8 mg of 5-maleimido-isophthalic acid (H2mip, 2.10 × 10−1 mmol, 2.1 eq.) were dissolved in 2 mL of dimethylformamide. Precipitation occurred within 10 min and the stirring was continued overnight. The solution was evaporated to dryness and the solid was dispersed in 5 mL of methanol. The solid was filtered using a Büchner funnel, thoroughly washed with methanol, and dried in air to yield [Ln2Ga8(shi)8(mip)4]pym2 MCs as beige solids.
[Y2Ga8(shi)8(mip)4]pym2.
The synthetic yield was 66% based on [YGa4(shi)4(bz)4]pym. ESI-MS (MeOH/DMSO 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, soft-negative mode, m/z): [Y2Ga8(shi)8(mip)4 + 4MeOH]2−: 1550.76 (calc.), 1550.74 (exp.). Elemental analysis of [Y2Ga8(shi)8(mip)4]pym2·9.3H2O: calc.: % C 41.48, % H 2.52, % N 5.94; found: % C 41.53, % H 2.57, % N 5.93.
:10, soft-negative mode, m/z): [Y2Ga8(shi)8(mip)4 + 4MeOH]2−: 1550.76 (calc.), 1550.74 (exp.). Elemental analysis of [Y2Ga8(shi)8(mip)4]pym2·9.3H2O: calc.: % C 41.48, % H 2.52, % N 5.94; found: % C 41.53, % H 2.57, % N 5.93.
[Gd2Ga8(shi)8(mip)4]pym2.
The synthetic yield was 67% based on [GdGa4(shi)4(bz)4]pym. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Gd2Ga8(shi)8(mip)4]2−: 1554.73 (calc.), 1554.75 (exp.). Elemental analysis of [Gd2Ga8(shi)8(mip)4]pym2·8.5H2O: calc.: % C 40.00, % H 2.38, % N 5.73; found: % C 40.11, % H 2.50, % N 5.70.
[Er2Ga8(shi)8(mip)4]pym2.
The synthetic yield was 76% based on [ErGa4(shi)4(bz)4]pym. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Er2Ga8(shi)8(mip)4]2−: 1564.73 (calc.), 1564.76 (exp.). Elemental analysis of [Er2Ga8(shi)8(mip)4]pym2·12.6H2O: calc.: % C 38.93, % H 2.56, % N 5.58; found: % C 38.94, % H 2.32, % N 5.34.
[Yb2Ga8(shi)8(mip)4]pym2.
The synthetic yield was 86% based on [YbGa4(shi)4(bz)4]pym. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Yb2Ga8(shi)8(mip)4]2−: 1570.74 (calc.), 1570.76 (exp.). Elemental analysis of [Yb2Ga8(shi)8(mip)4]pym2·12.3H2O: calc.: % C 38.86, % H 2.53, % N 5.57; found: % C 38.82, % H 2.36, % N 5.39.
Preparation of [Nd2Ga8(shi)8(mip)4](HNEt3)2 MC by slow evaporation of DMF
275.6 mg of salicylhydroxamic acid (H3shi, 1.8 mmol, 8.0 eq.), 235.1 mg of 5-maleimido-isophthalic acid (H2mip, 0.9 mmol, 4.0 eq.), 460.3 mg of Ga(NO3)3 hydrate (1.8 mmol, 8.0 eq.) and 197.3 mg of Nd(NO3)3·6H2O (0.45 mmol, 2.0 eq.) were dissolved in 18 mL of dimethylformamide and 1097 μL of triethylamine (7.88 mmol, 35.0 eq.) were added to the solution. The solution was stirred for 30 min, filtered, and set for crystallization by slow evaporation of the solvent. Small needles grew after a few weeks and were carefully collected using a shortened/cut Pasteur pipette, and filtered and dried in air to yield 38.0 mg of [Nd2Ga8(shi)8(mip)4](HNEt3)2 MC as a light-yellow solid (1.16 × 10−2 mmol, 5% based on Nd(NO3)3·6H2O). ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Nd2Ga8(shi)8(mip)4]2−: 1541.71 (calc.), 1541.68 (exp.). Elemental analysis of [Nd2Ga8(shi)8(mip)4](HNEt3)2·10.4H2O: calc.: % C 40.09, % H 3.04, % N 5.64; found: % C 40.11, % H 3.21, % N 5.77.
Preparation of [Nd2Ga8(shi)8(mip)4](HNEt3)2 MC by diffusion of tBuOMe
92.0 mg of salicylhydroxamic acid (H3shi, 6.01 × 10−1 mmol, 8.0 eq.), 78.4 mg of 5-maleimido-isophthalic acid (H2mip, 3.00 × 10−1 mmol, 4.0 eq.), 153.6 mg of Ga(NO3)3 hydrate (6.01 × 10−1 mmol, 8.0 eq.) and 65.6 mg of Nd(NO3)3·6H2O (0.15 mmol, 2.0 eq.) were dissolved in 12 mL of dimethylformamide. 370 μL of triethylamine (2.65 mmol, 35.0 eq.) were added to the solution which was stirred for 30 min, filtered, and set for crystallization by slow diffusion of tert-butyl methyl ether (tBuOMe). Cubic crystals grew within two weeks and were collected using a shortened/cut Pasteur pipette, and filtered and dried in air to yield 102.8 mg of [Nd2Ga8(shi)8(mip)4](HNEt3)2 MC as a light-yellow solid (3.13 × 10−2 mmol, 41% based on Nd(NO3)3·6H2O). ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Nd2Ga8(shi)8(mip)4]2−: 1541.71 (calc.), 1541.71 (exp.). Elemental analysis of [Nd2Ga8(shi)8(mip)4](HNEt3)2·3.4H2O·4.9DMF: calc.: % C 42.34, % H 3.40, % N 7.14; found: % C 42.38, % H 3.57, % N 7.01.
Preparation of [Ln2Ga8(shi)8(C-mip)4](HNEt3)2 MCs
7.5 × 10−3 mmol of [Ln2Ga8(shi)8(mip)4]X2 MC (1.0 eq., X = pyridinium for Ln = YIII, GdIII, ErIII and YbIII; X = HNEt3+ for NdIII) and 4.5 × 10−2 mmol of C (6.0 eq.) were suspended in 2.0 mL of dimethylformamide. 21 μL of triethylamine (0.15 mmol, 20.0 eq.) were added to the suspension which quickly dissolved. The stirring was continued for 30 min and the solution was evaporated to dryness. The solid was suspended in 20 mL of methanol, filtered using a Büchner funnel, thoroughly washed with CH2Cl2 (3 × 30 mL) and methanol (2 × 30 mL) and dried in air to yield the corresponding [Ln2Ga8(shi)8(C-mip)4](HNEt3)2 MCs as yellow solids.
[Y2Ga8(shi)8(C-mip)4](HNEt3)2.
The synthetic yield was 54% based on [Y2Ga8(shi)8(mip)4]pym2. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Y2Ga8(shi)8(C-mip)4]2−: 2126.95 (calc.), 2126.91 (exp.). Elemental analysis of [Y2Ga8(shi)8(C-mip)4](HNEt3)2·12.3H2O: calc.: % C 46.19, % H 4.06, % N 6.58; found: % C 45.94, % H 3.80, % N 6.46.
[Gd2Ga8(shi)8(C-mip)4](HNEt3)2.
The synthetic yield was 70% based on [Gd2Ga8(shi)8(mip)4]pym2. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Gd2Ga8(shi)8(C-mip)4]2−: 2195.97 (calc.), 2195.93 (exp.). Elemental analysis of [Gd2Ga8(shi)8(C-mip)4](HNEt3)2·12.7H2O: calc.: % C 44.81, % H 3.96, % N 6.39; found: % C 44.67, % H 3.81, % N 6.28.
[Nd2Ga8(shi)8(C-mip)4](HNEt3)2.
The synthetic yield was 61% based on [Nd2Ga8(shi)8(mip)4](HNEt3)2. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Nd2Ga8(shi)8(C-mip)4]2−: 2181.95 (calc.), 2181.92 (exp.). Elemental analysis of [Nd2Ga8(shi)8(C-mip)4](HNEt3)2·16.9H2O: calc.: % C 44.36, % H 4.09, % N 6.32; found: % C 43.98, % H 3.70, % N 6.16.
[Er2Ga8(shi)8(C-mip)4](HNEt3)2.
The synthetic yield was 74% based on [Er2Ga8(shi)8(mip)4]pym2. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Er2Ga8(shi)8(C-mip)4]2−: 2205.98 (calc.), 2205.94 (exp.). Elemental analysis of [Er2Ga8(shi)8(C-mip)4](HNEt3)2·12.6H2O: calc.: % C 44.64, % H 3.94, % N 6.36; found: % C 44.40, % H 3.69, % N 6.24.
[Yb2Ga8(shi)8(C-mip)4](HNEt3)2.
The synthetic yield was 81% based on [Yb2Ga8(shi)8(mip)4]pym2. ESI-MS (MeOH/DMSO 90:10, soft-negative mode, m/z): [Y2Ga8(shi)8(C-mip)4]2−: 2211.48 (calc.), 2211.44 (exp.). Elemental analysis of [Yb2Ga8(shi)8(C-mip)4](HNEt3)2·11.4H2O: calc.: % C 44.74, % H 3.90, % N 6.38; found: % C 44.52, % H 3.68, % N 6.27.
Analytical measurements
1H and 13C NMR spectra were recorded on a Bruker Avance Neo 500 (500 MHz) or a Varian MR400 (400 MHz) spectrometer. Chemical shifts are given in ppm with respect to TMS. ESI-MS spectra were recorded on an Agilent 6230 TOF HPLC-MS spectrometer in negative or positive modes using the following ionization and cone potentials: 240 V and 65 V for organic molecules and 350 V and 50 V for MCs. Stock solutions of MCs at 1 mg mL−1 in DMSO were prepared and further diluted to 80–200 μg mL−1 in MeOH/DMSO 90:10. Recorded data were processed using the Agilent MassHunter software. Elemental analyses were performed by Atlantic Microlab. UV-vis spectra were recorded in DMSO on a Varian Cary 100 Bio UV-visible spectrophotometer using 1 cm Hellma QS or QX quartz glass cuvettes. IR spectra were collected in the solid state on a Thermo-Nicolet IS-50 spectrometer (ATR-FTIR). Recorded data were processed using Thermo Scientific OMNIC Specta software.
Photophysical properties
Photophysical measurements were performed on powder samples or freshly prepared solutions of MCs in DMSO placed in 2.4 mm i.d. quartz capillaries or Suprasil® cells. Emission and excitation spectra were measured on a modified Horiba-Jobin-Yvon Fluorolog 3 spectrofluorimeter equipped with visible and NIR photomultiplier tubes (PMTs: R13456 and H10330-75 from Hamamatsu) upon excitation with a continuous xenon lamp. Phosphorescence spectra of GdIII MCs were measured at 77 K in time-resolved mode. All spectra were corrected for the instrumental functions. Luminescence lifetimes were determined under excitation at 355 nm provided by a Nd:YAG laser (YG 980; Quantel). The signals in the NIR range were detected with a Hamamatsu H10330-75 PMT connected to an iHR320 monochromator (Horiba Scientific). The output signals from the detectors were fed into a 500 MHz bandpass digital oscilloscope (TDS 754C; Tektronix). Luminescence lifetimes are averages of at least three independent measurements. Quantum yields were determined with a modified Fluorolog 3 spectrofluorimeter based on the absolute method using an integration sphere (Model G8, GMP SA, Renens, Switzerland). Each sample was measured several times. The experimental error for the determination of quantum yields is estimated as 10%.
NIR luminescence imaging
Images of quartz capillaries containing MC DMSO solutions (CMC = 3.04 μM; A = 0.5 for a 1 cm optical path length at λabs = 421.4 nm) were recorded using a custom-designed NIR-II Kaer Labs imaging system that includes a ZEPHIR 1.7× camera (Photon etc., Montréal, Quebec, Canada). The sample was excited using a Nikon C-HGFI Intensilight source combined with a 417/60 nm bandpass (BP) filter. Emission signals were recorded using an 800 nm longpass (LP) filter along with additional BP filters depending on the nature of the monitored LnIII: YbIII – BP996/70 nm; NdIII – BP1065/30 nm and BP1365/130 nm; ErIII – BP1530/30 nm. The recorded images were treated using the Fiji distribution of ImageJ/ImageJ2.63
Single crystal X-ray crystallography
Single-crystal X-ray crystallographic data for [Nd2Ga8(shi)8(mip)4]2− were collected at 150 K (Oxford Cryosystems low temperature device) on a Bruker Quest diffractometer with a fixed chi angle, a Mo Kα wavelength (λ = 0.71073 Å) sealed fine focus X-ray tube, a single crystal curved graphite incident beam monochromator and a Photon II area detector. Data were collected, reflections were indexed and processed, and the files scaled and corrected for absorption using APEX4 (ref. 64) and SADABS.65 The space groups were assigned using XPREP within the SHELXTL suite of programs66,67 and solved by direct methods using ShelXT68 and refined by full matrix least squares against F2 with all reflections using Shelxl2018 (ref. 69 and 70) using the graphical interface Shelxle.71 The unit cell metrically fits a double orthorhombic F-centered cell but the structure realizes only monoclinic C-centered symmetry. No twinning by the higher metric symmetry was observed. Attempts have been made to refine the structure in C2 and P1 space groups (without inversion centers and mirror planes) under inclusion of possible twin operations. 1:1 disorder, however, persisted and no better fit to the data was observed. Thus, the highest possible symmetry, C2/m, was used (see Section S3, ESI† for additional details). The deposition number CCDC 2412013 contains the supplementary crystallographic data.
HeLa cell culture
The HeLa (human cervical adenocarcinoma) cell line from ATCC was used for viability assays and cell imaging. Cells were cultured in Dulbecco's modified Eagle's medium (Gibco; 4.5 g per L glucose, no pyruvate, GlutaMAX) supplemented with 10% fetal bovine serum (FBS; Gibco), and 1% of streptomycin and penicillin antibiotics (Gibco). HeLa cells were routinely seeded at densities of 10′000–20′000 cells cm−2, reaching 80% confluency at the end of an experiment. Cells were cultured at 5% CO2, 37 °C and 95% humidity.
Viability assay
HeLa cells were seeded in 96-well black wall clear bottom plates at a density of 1.5 × 104 cells per well and cultured at 37 °C in a 5% humidified CO2 atmosphere until the next day. After 24 h of attachment, cells were incubated with [Ln2Ga8(shi)8(C-mip)4]2− MC at different concentrations for 24 h (4 μL of MC in DMSO stock solutions at concentrations of 0.001–5 mg mL−1 were added to the cells in Opti-MEM (200 μL), giving final MC concentrations of 0.2–100 μg mL−1, or 0.04–8.75 μM). DMSO was used as a control (2% in Opti-MEM). After 24 h, the cells were incubated with alamarBlue (10% v/v; DL1025-Invitrogen) for 1 h at 37 °C in a 5% humidified CO2 atmosphere. The fluorescence intensity of the alamarBlue was recorded on a CLARIOstar® Plus (BMG Labtech, λexc = BP545/20 nm, λem = BP600/40 nm).
NIR HeLa cell imaging
HeLa cells were seeded in an eight-well 1.5 Borosilicate glass Lab-Tek™ II (Nunc) at a density of 2 × 104 cells per well and cultured at 37 °C in a 5% humidified CO2 atmosphere. After 24 h, the cell culture medium was removed and the cells were washed twice with Opti-MEM at 298 K. They were then incubated with [Nd2Ga8(shi)8(C-mip)4]2− MC at 40 μg mL−1 (8.75 μM) for 3 h (4 μL of a 2 mg per mL MC stock solution in DMSO were added to the cells in Opti-MEM (200 μL), giving a final MC concentration of 40 μg mL−1). DMSO was used as a control (2% in Opti-MEM). The cells were washed twice with Opti-MEM prior to epifluorescence and confocal microscopy experiments. Brightfield and epifluorescence microscopy images were acquired with an inverted Nikon Eclipse Ti microscope equipped with an EMCCD Evolve 512 (Roper Scientific) and ZEPHIR 1.7× (Photon etc., Montréal, Quebec, Canada) photometric cameras. The samples were excited using a Nikon C-HGFI Intensilight source combined with BP414/46 nm or BP417/60 nm filters. C-centered emission in the visible range was collected using a BP482/35 nm filter while the NdIII signal in the NIR II was selected with a BP1065/30 nm filter. Images were obtained with Nikon Plan Apoλ 20× and Nikon Apo TIRF 60× objectives. Laser confocal scanning microscopy was performed on an inverted Nikon Eclipse Ti microscope equipped with a Nikon C2+ confocal system. The samples were excited with a 405 nm laser while the C-centered fluorescence was collected with a spectral detector in the range of 475–600 nm. Images were acquired with Nikon Plan Apoλ 20× and/or Nikon Apo TIRF 60× objectives. The recorded images were treated using the Fiji distribution of ImageJ/ImageJ2.63 Brightness and contrast were adjusted automatically (brightfield images) or manually (fluorescence and NIR images). The merging of brightfield and NIR images was performed using the “Landmark Correspondences” plugin in Fiji (ImageJ/ImageJ2).63
Results and discussion
Organic synthesis
The Knœvenagel condensation of Meldrum's acid 2 with 4-(diethylamino)-2-hydroxybenzaldehyde 1 in refluxing ethanol, followed by a recrystallization from boiling ethanol yielded the pure coumarin 3-carboxylic acid 3 (Scheme S1, ESI†). Its activation as an N-hydroxy succinimide ester60 and further coupling to the S-tritylcysteamine linker 6 (ref. 62) ultimately provided the thiol-coumarin antenna C after deprotection of the trityl group.
Design of MCs
The use of the H2mip bridging ligand that contains a maleimido group offers several advantages. Only one MC needs to be prepared and can subsequently be functionalized with various thiol-containing compounds of interest. Additionally, H2mip is a small bridging ligand and reduces the risk of steric hindrance and solubility issues that come with bigger chromophore-containing bridges that render MC preparation more challenging.
Preparation of MCs
The [LnGa4(shi)4(bz)4]pym (Ln = YIII, GdIII, ErIII and YbIII) MCs were prepared by modifying a procedure that we published previously46 (Section S2.1, ESI†). The corresponding [Ln2Ga8(shi)8(mip)4]pym2 MC dimers were then obtained by substituting the benzoates in the [LnGa4] precursors with the H2mip bridging ligands.47 As [NdGa4] could not be isolated, which can be explained by the larger size of NdIII,46 [Nd2Ga8(shi)8(mip)4](HNEt3)2 was prepared in a one-step self-assembly reaction using a 2:8:8:4 molar ratio of Nd(NO3)3, Ga(NO3)3, H3shi and H2mip in DMF in the presence of NEt3 as a base. Crystals of [Nd2Ga8(shi)8(mip)4](HNEt3)2 were obtained either by slow evaporation of DMF or by diffusion of tBuOMe.
Functionalization of MCs
[Ln2Ga8(shi)8(C-mip)4](HNEt3)2 MCs were prepared by base-mediated thiol-Michael addition click reactions of C onto the maleimido groups of the corresponding [Ln2Ga8(shi)8(mip)4]2− MCs. Because the strength and nature of the base governs the amount of thiolate present in solution,72 NEt3 was used as its pKa (10.75 in water)73 is slightly above the pKa of the cysteamine moiety in C (10.53 in water).73 As a result, the functionalization of the MCs reaches completion within minutes, as confirmed by ESI-MS. A slight excess of C was used (6 eq. per MC, or 1.5 eq. per maleimido substituent, Section S2.2, ESI†) to ensure a complete functionalization of the [Ln2Ga8(shi)8(mip)4]2− MCs. An excess of NEt3 (20 eq. per MC) was used to ensure the complete substitution of the pyridinium counter-cations in the [Ln2Ga8(shi)8(mip)4]pym2 MC precursors (Fig. S10, ESI;† Ln = YIII, GdIII, ErIII and YbIII), yielding the coumarin-functionalized triethylammonium derivatives [Ln2Ga8(shi)8(C-mip)4](HNEt3)2 MCs (Fig. 2). Because the [Nd2Ga8(shi)8(mip)4]2− MC precursor is already prepared in a one-step process as the HNEt3+ derivative, the pyridinium substitution does not apply in this case.
Molecular structure of [Nd2Ga8(shi)8(mip)4](HNEt3)2 in the solid state
X-ray quality crystalline beige needles were grown by slow evaporation of a concentrated DMF solution containing Nd(NO3)3, Ga(NO3)3, H3shi and H2mip in a 2:8:8:4 molar ratio with 35.0 eq. of NEt3. The analysis of the structure obtained by single crystal X-ray diffraction with the SHAPE software74,75 reveals that, as expected for the [LnGa4] and [Ln2Ga8] MC families, the NdIII metal ion adopts a square antiprism coordination geometry (Table S2, ESI†). The HNEt3+ counter-cations could not be located within the resolved section of the molecular structure and are likely residing within the unresolved section related to disordered elements of the structure. Their presence was evidenced by 1H NMR measurements on the isolated [Nd2Ga8(shi)8(mip)4](HNEt3)2 MC (Fig. S27, ESI†). This molecular structure obtained by X-ray diffraction (Fig. 1) also highlights the location of the four maleimide moieties pointing outwards, giving ample space for further functionalization of the MC.
 |
| | Fig. 1 Structure of [Nd2Ga8(shi)8(mip)4]2− obtained from single crystal X-ray diffraction (deposition number CCDC 2412013). Left: side view; right: top-down view. Color code: NdIII: cyan; GaIII: rose; O: red; N: light blue; C: grey. Hydrogen atoms and solvent molecules are omitted for clarity. While the above picture is a good general representation of the MC, it does not accurately reflect the crystal structure content, which is more complex due to the presence of symmetry elements (Fig. S25, ESI†). | |
MC characterization
MCs were characterized by ESI-MS (Fig. S12–S21, ESI†), elemental analysis, and UV-vis (Fig. S22 and S23, ESI†) and ATR-FTIR spectroscopy (Fig. S24, ESI†). The diamagnetic YIII analogues were prepared and characterized by 1D and 2D 1H NMR experiments (Fig. S9–S11, ESI†). The results fully support the formation of the expected MCs. The spectra of both [Y2Ga8(shi)8(mip)4]2− and [Y2Ga8(shi)8(C-mip)4]2− MCs show the typically observed39,47 splitting of the e and f protons (Fig. 2, S10 and S11, ESI†). The cause of this splitting is likely due to an inequivalence of the two [LnGa4] subunits in the [Ln2Ga8] MCs, resulting in a lower symmetry of the MC in solution compared to the one observed in the solid state on single crystals.76 In [Y2Ga8(shi)8(C-mip)4]2−, the signals of the two geminal protons g are split as they become chemically non-equivalent after the connection of C to the maleimide groups using the thiol-Michael addition click reaction.
 |
| | Fig. 2
1H NMR spectrum of [Y2Ga8(shi)8(C-mip)4](HNEt3)2 in DMSO-d6 at 298 K. | |
UV-vis absorption of the MCs
The absorption spectra of [Ln2Ga8(shi)8(mip)4]2− MCs (Ln = YIII, GdIII, NdIII, ErIII and YbIII) contain two broad absorption bands located at high energy39 with λmax ≈ 268 and 318 nm. These signals are assigned to the electronic structure of the MC-based scaffold (Fig. 3, black and red full lines). Following the functionalization of [Ln2Ga8(shi)8(mip)4]2− MCs with the coumarin derivative C, the [Ln2Ga8(shi)8(C-mip)4]2− MCs display in addition to the band in the UV region (λmax = 314.5–320.5 nm, ε = 4.28 × 104 to 6.50 × 104 M−1 cm−1) a broad band in the visible range with high molar extinction coefficients (λmax = 421.0–421.5 nm, ε = 1.57 × 105 to 1.65 × 105 M−1 cm−1). The extinction coefficients are in line with the value corresponding to four C moieties (Fig. 3, black dotted line), confirming the complete functionalization of all maleimide sites of the MC. The presence of this additional band should result in the possibility to sensitize NIR-emitting LnIII in the [Ln2Ga8(shi)8(C-mip)4]2− MCs with an excitation wavelength shifted by 104 nm towards lower energy which corresponds to a decrease of ≈7700 cm−1. In addition, the molar extinction coefficients of all the MCs formed with different LnIII also increase by a factor of ≈3. This is highly desirable as the overall brightness of the MC results from the combination of the absorption (through the molar extinction coefficient) and the quantum yield of the NIR-emitting MC.77
 |
| | Fig. 3 UV-vis absorption spectra of [Gd2Ga8(shi)8(mip)4]2− (black line, CMC = 1.5 × 10−5 M), [Gd2Ga8(shi)8(C-mip)4]2− (red line, CMC = 2.5 × 10−6 M) and the thiol-coumarin antenna C multiplied four times (black dotted line) in DMSO at 298 K. The UV-vis spectra of YIII, NdIII, ErIII and YbIII MCs are presented in the ESI (Fig. S22 and S23†). | |
Photophysical properties
LnIII-centered luminescence properties of MCs were studied both in the solid state and in DMSO solution (Fig. 4 and S28–S30, ESI;†Tables 1 and S3, ESI†). Upon excitation into the C-centered electronic band at λmax = 420 nm, the characteristic emission signal of YbIII centered at 991 nm, arising from the 2F5/2 → 2F7/2 electronic transition, is observed (Fig. 4, S29 and S30, ESI†), as well as three independent sharp NIR signals for the MC formed with NdIII (centered at 884 nm due to the 4F3/2 → 4I9/2 electronic transition; 1063 nm due to the 4F3/2 → 4I11/2 electronic transition and 1338 nm due to the 4F3/2 → 4I13/2 electronic transition) and one signal for the ErIII analogue (centered at 1524 nm due to the 4I13/2 → 4I15/2 electronic transition). Moreover, upon monitoring the emission of the main transition of NdIII and ErIII, the excitation spectra display a broad band matching with the envelope of the corresponding absorption spectra of C in [Ln2Ga8(shi)8(C-mip)4]2− MCs (Fig. S29, ESI†). In the excitation spectrum of YbIII, this band is less apparent (Fig. S29 and S30, ESI†). These results confirm that the sensitization of NdIII, ErIII and YbIII in [Ln2Ga8(shi)8(C-mip)4]2− is occurring through the electronic states of the C sensitizer. The NdIII MC was chosen as the most promising candidate for living cell imaging due to its ability to emit the largest amount of NIR photons as it displays the highest quantum yield in the NIR range within the studied series of MCs (Table 1). Therefore, we evaluated its photophysical properties in aqueous media at the same concentration used for cell imaging and proved that they are preserved under these conditions (H2O/DMSO 100:2, CMC = 8.75 μM, Table 1 and Fig. S31, ESI†).
 |
| | Fig. 4 Corrected and normalized emission spectra of [Ln2Ga8(shi)8(C-mip)4]2− MCs upon excitation into the C-centered electronic band at λexc = 420 nm (50 μM in DMSO, 298 K). | |
Table 1 LnIII-centered quantum yields values (QLLn) and observed luminescence lifetimes (τobs) of [Ln2Ga8(shi)8(C-mip)4]2− MCs in solution at 298 K
| MC |
Solvent |
Q
LLn
, % |
τ
obs
, μs |
|
λ
exc = 420 nm.
λ
exc = 355 nm.
C = 50 μM.
C = 8.75 μM.
|
| [Nd2Ga8(shi)8(C-mip)4]2− |
DMSOc |
4.5(1) × 10−2 |
2.44(4) |
| H2O/DMSO 100:2d |
3.4(2) × 10−2 |
0.58(1) |
| [Er2Ga8(shi)8(C-mip)4]2− |
DMSOc |
4.8(3) × 10−3 |
7.63(5) |
| [Yb2Ga8(shi)8(C-mip)4]2− |
DMSOc |
2.12(3) × 10−2 |
53.2(6) |
Ligand-centered photophysical properties.
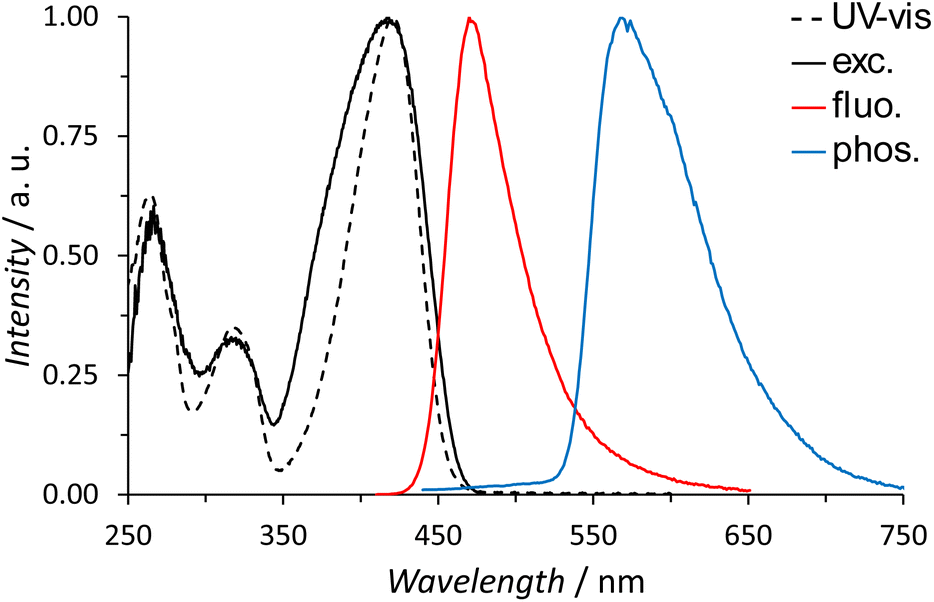
The lowest singlet (S1) and triplet (T1) energy levels of the C antenna in [Ln2Ga8(shi)8(C-mip)4]2− MCs were determined by recording the fluorescence and phosphorescence spectra of the GdIII analogue, respectively (Fig. 5). It was found that [Gd2Ga8(shi)8(C-mip)4]2− MC in DMSO solution displays a fluorescence band centered at λfluo = 470 nm, with a Stokes shift of 49 nm; the phosphorescence band is located in the range of 525–750 nm with the maximum at λphos = 568 nm.
 |
| | Fig. 5 UV-vis absorption (black dotted line), excitation (black line, λem = 500 nm), fluorescence (red line, λexc = 420 nm) and phosphorescence spectra (blue line, λexc = 420 nm, τdelay = 500 μs) of the [Gd2Ga8(shi)8(C-mip)4]2− MC, 50 μM in DMSO, 298 K and in frozen solution at 77 K (phosphorescence). | |
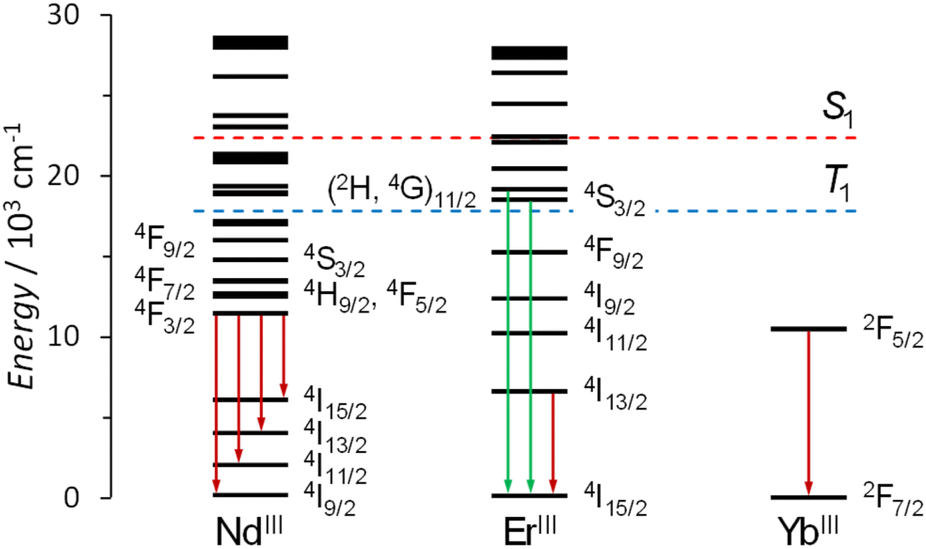
S1 was taken as the intersection of the absorption and the fluorescence spectra and lies at 22370 cm−1. T1 was determined as the 0–0 transition of the deconvoluted phosphorescence spectrum and equals 17825 cm−1 (Fig. S32, ESI†). Both S1 and T1 electronic levels are plotted on the energy diagram (Fig. 6), along with NdIII, ErIII and YbIII electronic states.78
 |
| | Fig. 6 Energies of the electronic levels of NdIII, ErIII and YbIII ions78 along with lowest singlet S1 (red dotted line) and triplet T1 states (blue dotted line) of C in [Gd2Ga8(shi)8(C-mip)4]2−. The vertical arrows represent the electronic transitions yielding to LnIII luminescence (in green: the green emission of ErIII in the visible range; in dark red: the emission of NdIII, ErIII and YbIII in the NIR domain). | |
As previously reported,79 an efficient intersystem crossing (ISC) from S1 to T1 in an organic sensitizer can be reasonably expected when the energy difference between these two excited states is around 5000 cm−1. Here, we observe a ΔE(S1 − T1) value of 4545 cm−1 which suggests a favorable ISC within the C-centered electronic states in [Ln2Ga8(shi)8(C-mip)4]2− MCs. The energy difference between the T1 state of the coumarin antenna and the 2F5/2 emissive electronic level of YbIII is large (ΔE = 7325 cm−1) and results in a relatively limited efficiency of this energy transfer. On the other hand, NdIII and ErIII possess multiple electronic levels located at higher energy that can be easily populated through the T1 state of the C antenna. These states can eventually undergo internal conversion to the lowest in energy main emissive electronic levels of NdIII (4F3/2) and ErIII (4I13/2).
NIR II luminescence imaging.
With this strategy, we have, for the first time, designed and synthesized NIR emitting MCs that can be excited using an additional organic sensitizer at a wavelength corresponding to lower energy, away from the UV range, opening unprecedented perspectives for cellular imaging in living cells. Indeed, UV excitation on living cells induces a high risk of toxicity and or/perturbation of the cellular system to be monitored. The luminescence of the three NIR-emitting [Ln2Ga8(shi)8(C-mip)4]2− MCs (Ln = NdIII, YbIII and ErIII) was first monitored in capillaries with the help of a macroscopic NIR imaging setup designed for small animal imaging. DMSO solutions of [Ln2Ga8(shi)8(C-mip)4]2− MCs were placed in quartz capillaries. The LnIII-centered luminescence was monitored upon excitation of the C antenna with light selected using a BP417/60 nm filter (Fig. 7).
 |
| | Fig. 7 Recorded luminescence images of [Ln2Ga8(shi)8(C-mip)4]2− MCs in DMSO at 298 K in quartz capillaries (CMC = 3.04 μM; A = 0.5 for a 1 cm optical path at λmax = 421.4 nm; λexc was selected with a BP417/60 nm filter). The NIR emission signal was recorded using a LP800 nm filter combined with additional BP filters depending on the nature of the LnIII considered. YbIII: BP996/70 nm; NdIII: BP1065/30 nm and BP1365/130 nm; ErIII: BP1530/30 nm. Exposition times: τexp = 0.5 to 1 s. | |
The collected images show that the LnIII-centered NIR emission in [Ln2Ga8(shi)8(C-mip)4]2− MCs obtained upon excitation of the C antenna (Fig. 7, top middle panel) allows the observation of the signal of each LnIII with an excellent signal-to-noise ratio. The specific emission signals of each of the three NIR-emitting LnIII can be easily discriminated using a narrow band interferential filter specific to the selected LnIII due to the ability of the corresponding [Ln2Ga8(shi)8(C-mip)4]2− MCs to emit enough photons. The low emission signal observed using a LP800 nm filter and arising from the [Gd2Ga8(shi)8(C-mip)4]2− MC (Fig. 7, top middle panel) is due to the residual fluorescence of the coumarin chromophore. It is worth noting that, while recording the signal arising from the YbIII 2F5/2 → 2F7/2 transition centered at 991 nm, a residual luminescence of the NdIII 4F3/2 → 4I11/2 transition with a maximum at 1063 nm was detected (Fig. 7, top right panel), being not fully blocked by the BP996/70 nm filter. Overall, this experiment is a direct way to show that solutions of [Ln2Ga8(shi)8(C-mip)4]2− emit a sufficient number of photons to ensure a sensitive and unambiguous detection with a biological NIR imaging setup.
Biological imaging
Cytotoxicity of the MCs.
To assess the biocompatibility of the [Ln2Ga8(shi)8(C-mip)4]2− MCs, HeLa cells were incubated with different concentrations of the MC for 24 h and the cytotoxicity was evaluated by the alamarBlue™ assay (Fig. 8).80
 |
| | Fig. 8 Results of HeLa cell viability tests performed using different concentrations of the [Ln2Ga8(shi)8(C-mip)4]2− MC (24 h incubation time, CMC = 0.2–100 μg mL−1). | |
In average, the HeLa cell viability remains above 0.95 within a MC concentration range of 0.2–40 μg mL−1 (0.04–8.75 μM). At a concentration of 100 μg mL−1, the MC starts to become cytotoxic and the viability drops to 0.86(2).
HeLa cell imaging.
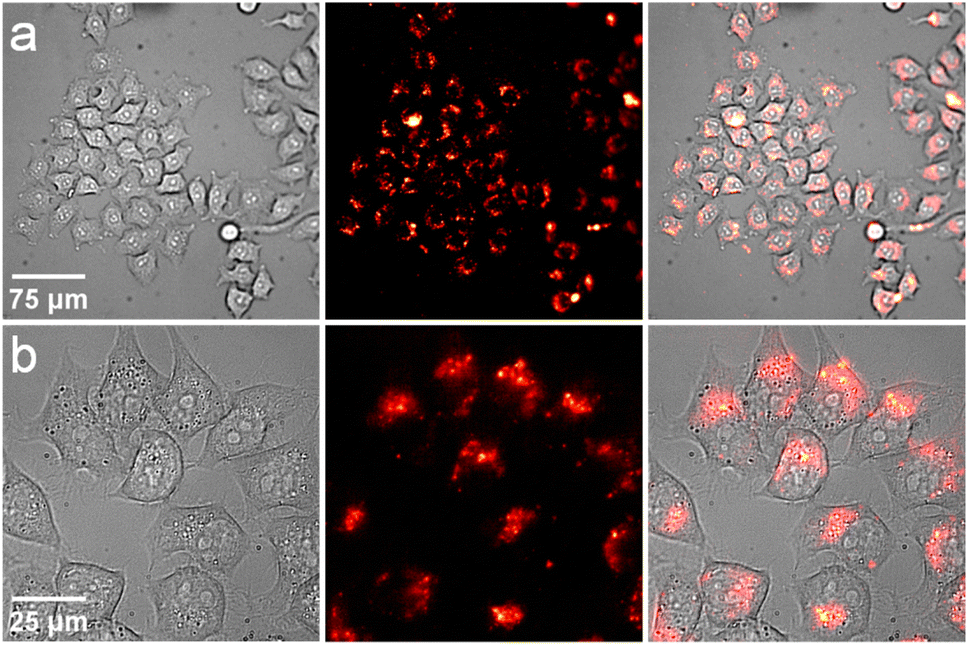
As the next step of the evaluation of this new family of MCs, the ability of [Nd2Ga8(shi)8(C-mip)4]2− to be used as a NIR imaging agent in a proof-of-concept experiment in living HeLa cells was tested. The NdIII MC was chosen as it emits the highest number of photons upon excitation of the C antenna based on its quantum yield value. HeLa cells were incubated with [Nd2Ga8(shi)8(C-mip)4]2− for 3 h at a non-cytotoxic concentration of 40 μg mL−1 (8.75 μM). At the end of the incubation time, the solution was removed and the HeLa cells were washed with 200 μL of Opti-MEM cell culture medium (2×). Epifluorescence microscopy images were collected upon excitation of the C sensitizer while monitoring the NdIII signal using a BP1065/30 nm filter (Fig. 9). The brightfield and NIR II epifluorescence images reveal all HeLa cells with healthy shapes, confirming the low cytotoxicity of these MCs at the selected concentration. The excellent quality of the collected NIR II epifluorescence images is due to the high signal to noise ratio, the absence of a fluorescence background caused by biomolecules in the NIR domain (autofluorescence) and the strong NIR II signal arising from NdIII in [Nd2Ga8(shi)8(C-mip)4]2− in living cells. This experiment also demonstrates that this MC does not dissociate under biological conditions (incubation in a cell culture medium and uptake by HeLa cells). In the case of dissociation of the MC, monitoring of the NIR signal of NdIII would not have been possible upon excitation at the wavelength corresponding to the population of the excited states of C. Indeed, the condition for an energy transfer to occur is that the C sensitizer and the emitting NdIII are in sufficiently close proximity,81 which is only satisfied if the MC remains intact. To confirm this conclusion, the NdIII signal corresponding to the 4F3/2 → 4I11/2 electronic transition (λem = 1063 nm) of the MC was recorded over time under the same experimental conditions used for cell imaging (Opti-MEM/DMSO 100:2, CMC = 8.75 μM, Fig. S34, ESI†).
 |
| | Fig. 9 Images obtained from epifluorescence microscopy experiments on living HeLa cells incubated with [Nd2Ga8(shi)8(C-mip)4]2− MC at 40 μg mL−1 (8.75 μM) for 3 h. From left to right: brightfield image (exposition time: 10 ms), NdIII NIR luminescence (λexc: BP417/60 nm; λem: BP1065/30 nm; exposition time: 5 s) and merged images. (a) 20× magnification objective. (b) 60× magnification objective. Images within the same row share the same scale bar. Control images of HeLa cells are provided in the ESI (Fig. S33†). | |
To ensure that the MCs are located inside of the living HeLa cells, laser scanning confocal microscopy experiments were performed. As the C moiety does not fully transfer its excitation energy to NdIII in the MC, some residual fluorescence can be monitored in the visible range from 475 to 600 nm. The collected images (Fig. S35, ESI†) show that the boundary between the cytoplasm and the nucleus is well defined. Overall, these experiments allow the monitoring of internal structure elements along the Z axis and confirm that the [Nd2Ga8(shi)8(C-mip)4]2− MC is entering living HeLa cells upon incubation.
Conclusion
LnIII-containing MCs possess very attractive luminescence properties, especially their ability to emit strong NIR II signals. One of their major limitations so far for biological applications lies in excitation wavelengths in the UV domain. This situation is due to the fact that, in most cases, the NIR-emitting LnIII are sensitized by the organic building blocks constituting these MCs. In this work, we present an innovative approach to address this drawback with a molecular design where organic chromophores/LnIII sensitizers are attached to the structure of the dimeric LnIII/GaIII MC. We carefully selected a highly absorbing, biocompatible chromophore, a coumarin derivative, which possesses appropriate singlet and triplet energy levels for the sensitization of NIR-emitting LnIII. The new [Ln2Ga8(shi)8(C-mip)4]2− MCs that we have designed and synthesized exhibit an additional excitation band located ≈104 nm (or 7700 cm−1) towards lower energy compared to the parent [Ln2Ga8(shi)8(mip)4]2− MCs. Moreover, the molar extinction coefficient of the C-centered band at 420 nm in [Ln2Ga8(shi)8(C-mip)4]2− is ≈3 times larger compared to that of the π → π* transitions in the UV range of the non-functionalized MC. Possessing NIR-emitting MC molecules that can be excited at lower energy, we have tested the [Nd2Ga8(shi)8(C-mip)4]2− for its ability to operate as an imaging agent in living HeLa cells. We observed that the use of this NdIII MC in a range of concentrations that does not induce cytotoxicity allows for highly sensitive NIR II detection inside of living HeLa cells. This series of MCs is (i) non-cytotoxic up to 8.75 μM (viability > 95%) and (ii) sufficiently robust in cell culture media and under biological conditions to be used as an imaging reporter as it does not dissociate. Our results also illustrate that, despite the significant distance between the C sensitizer and the LnIII metal ion, the [Ln2Ga8(shi)8(C-mip)4]2− MCs were able to emit enough photons to ensure highly sensitive NIR II detection in a NIR microscopy setup, validating the pertinence of our approach. Overall, this work opens unprecedented perspectives as the proposed molecular design allows a versatile choice of LnIII sensitizers.
Data availability
The data supporting the findings presented within this article are available as part of the ESI.†
Author contributions
T. L. – conceptualization, investigation, data curation, formal analysis, writing – original draft; J. B. – conceptualization, investigation, data curation, formal analysis, writing – review & editing; S. V. E. – conceptualization, investigation, data curation, formal analysis, funding acquisition, supervision, writing – review & editing; M. Z. – investigation, data curation, formal analysis, writing – review & editing; S. P. – conceptualization, funding acquisition, writing – review & editing; V. L. P. – conceptualization, funding acquisition, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by the National Science Foundation (NSF) under grants CHE-2102046 and CHE-2154116. The work in France was performed with financial support from Inserm Cancer (MultiNIR2), La Ligue Contre le Cancer (comités du Loiret, d’Eure-et-Loir, du Loir-et-Cher and de la Sarthe), the Réseau ‘Molécules Marines, Métabolisme & Cancer’ du Cancéropôle Grand Ouest and La Région Centre. V. L. P. thanks the Le Studium Loire Valley Institute for Advanced Studies, Orléans & Tours, France for Le Studium Professorship award. S. P. acknowledges support from Institut National de la Santé et de la Recherche Médicale (INSERM). The authors thank the MO2VING-MS and MO2VING-NMR facilities (Orléans, France).
Notes and references
- S. Yu, D. Tu, W. Lian, J. Xu and X. Chen, Sci. China Mater., 2019, 62, 1071–1086 CrossRef CAS.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- T. Wang, Y. Chen, B. Wang and M. Wu, Front. Physiol., 2023, 14, 1126805 CrossRef PubMed.
- F. Wang, Y. Zhong, O. Bruns, Y. Liang and H. Dai, Nat. Photonics, 2024, 18, 535–547 CrossRef CAS.
- T. Zhang, X. Zhu, C. C. W. Cheng, W.-M. Kwok, H.-L. Tam, J. Hao, D. W. J. Kwong, W.-K. Wong and K.-L. Wong, J. Am. Chem. Soc., 2011, 133, 20120–20122 CrossRef CAS PubMed.
- T. Wang, S. Wang, Z. Liu, Z. He, P. Yu, M. Zhao, H. Zhang, L. Lu, Z. Wang, Z. Wang, W. Zhang, Y. Fan, C. Sun, D. Zhao, W. Liu, J.-C. G. Bünzli and F. Zhang, Nat. Mater., 2021, 20, 1571–1578 CrossRef CAS PubMed.
- G.-Q. Jin, D. Sun, X. Xia, Z.-F. Jiang, B. Cheng, Y. Ning, F. Wang, Y. Zhao, X. Chen and J.-L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208707 CrossRef CAS PubMed.
- Y.-H. Yeung, H.-F. Chau, H.-Y. Kai, W. Zhou, K. H.-Y. Chan, W. Thor, L. J. Charbonnière, F. Zhang, Y. Fan, Y. Wu and K.-L. Wong, Adv. Opt. Mater., 2024, 12, 2302070 CrossRef CAS.
- A. T. Bui, A. Grichine, S. Brasselet, A. Duperray, C. Andraud and O. Maury, Chem.–Eur. J., 2015, 21, 17757–17761 CrossRef CAS PubMed.
- A. D’Aléo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2014, 53, 2927–2930 CrossRef CAS PubMed.
- I. Martinić, S. V. Eliseeva, T. N. Nguyen, V. L. Pecoraro and S. Petoud, J. Am. Chem. Soc., 2017, 139, 8388–8391 CrossRef PubMed.
- N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC.
- Q. Y. Zhang and X. Y. Huang, Prog. Mater. Sci., 2010, 55, 353–427 CrossRef CAS.
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CrossRef CAS.
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- M. S. Lah and V. L. Pecoraro, Comments Inorg. Chem., 1990, 11, 59–84 CrossRef CAS.
- I. Martinić, S. V. Eliseeva, T. N. Nguyen, F. Foucher, D. Gosset, F. Westall, V. L. Pecoraro and S. Petoud, Chem. Sci., 2017, 8, 6042–6050 RSC.
- C. C. Bădescu-Singureanu, A. S. Nizovtsev, V. L. Pecoraro, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2024, e202416101 Search PubMed.
- E. V. Salerno, J. Zeler, S. V. Eliseeva, M. A. Hernández-Rodríguez, A. N. C. Neto, S. Petoud, V. L. Pecoraro and L. D. Carlos, Chem.–Eur. J., 2020, 26, 13792–13796 CrossRef CAS PubMed.
- E. V. Salerno, A. N. C. Neto, S. V. Eliseeva, M. A. Hernández-Rodríguez, J. C. Lutter, T. Lathion, J. W. Kampf, S. Petoud, L. D. Carlos and V. L. Pecoraro, J. Am. Chem. Soc., 2022, 144, 18259–18271 CrossRef CAS PubMed.
- S. V. Eliseeva, E. V. Salerno, B. A. L. Bermudez, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2020, 142, 16173–16176 CrossRef CAS PubMed.
- E. V. Salerno, S. V. Eliseeva, S. Petoud and V. L. Pecoraro, Chem. Sci., 2024, 15, 8019–8030 RSC.
- C. Dendrinou-Samara, M. Alexiou, C. M. Zaleski, J. W. Kampf, M. L. Kirk, D. P. Kessissoglou and V. L. Pecoraro, Angew. Chem., Int. Ed., 2003, 42, 3763–3766 CrossRef CAS PubMed.
- C. M. Zaleski, E. C. Depperman, J. W. Kampf, M. L. Kirk and V. L. Pecoraro, Angew. Chem., Int. Ed., 2004, 43, 3912–3914 CrossRef CAS PubMed.
- C. M. Zaleski, S. Tricard, E. C. Depperman, W. Wernsdorfer, T. Mallah, M. L. Kirk and V. L. Pecoraro, Inorg. Chem., 2011, 50, 11348–11352 CrossRef CAS PubMed.
- P. Happ, C. Plenk and E. Rentschler, Coord. Chem. Rev., 2015, 289–290, 238–260 CrossRef CAS.
- X.-F. Jiang, M.-G. Chen, J.-P. Tong and F. Shao, New J. Chem., 2019, 43, 8704–8710 RSC.
- C. Y. Chow, R. Guillot, E. Rivière, J. W. Kampf, T. Mallah and V. L. Pecoraro, Inorg. Chem., 2016, 55, 10238–10247 CrossRef CAS PubMed.
- J. C. Lutter, T. T. Boron, K. E. Chadwick, A. H. Davis, S. Kleinhaus, J. W. Kampf, C. M. Zaleski and V. L. Pecoraro, Polyhedron, 2021, 202, 115190 CrossRef CAS.
- E. V. Salerno, J. W. Kampf, V. L. Pecoraro and T. Mallah, Inorg. Chem. Front., 2021, 8, 2611–2623 RSC.
- T. N. Parac-Vogt, A. Pacco, P. Nockemann, S. Laurent, R. N. Muller, M. Wickleder, G. Meyer, L. Vander Elst and K. Binnemans, Chem.–Eur. J., 2006, 12, 204–210 CrossRef CAS PubMed.
- M. A. Katkova, G. S. Zabrodina, M. S. Muravyeva, A. S. Shavyrin, E. V. Baranov, A. A. Khrapichev and S. Yu. Ketkov, Eur. J. Inorg. Chem., 2015, 2015, 5202–5208 CrossRef CAS.
- M. S. Muravyeva, G. S. Zabrodina, M. A. Samsonov, E. A. Kluev, A. A. Khrapichev, M. A. Katkova and I. V. Mukhina, Polyhedron, 2016, 114, 165–171 CrossRef CAS.
- J. Jankolovits, C. M. Andolina, J. W. Kampf, K. N. Raymond and V. L. Pecoraro, Angew. Chem., Int. Ed., 2011, 50, 9660–9664 CrossRef CAS PubMed.
- T. N. Nguyen, S. V. Eliseeva, I. Martinić, P. L. Carver, T. Lathion, S. Petoud and V. L. Pecoraro, Chem.–Eur. J., 2023, 29, e202300226 CrossRef CAS PubMed.
- T. N. Nguyen, C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, J. W. Kampf, I. Martinić, S. Petoud and V. L. Pecoraro, Chem.–Eur. J., 2018, 24, 1031–1035 CrossRef CAS PubMed.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef CAS PubMed.
- S. V. Eliseeva, T. N. Nguyen, J. W. Kampf, E. R. Trivedi, V. L. Pecoraro and S. Petoud, Chem. Sci., 2022, 13, 2919–2931 RSC.
- J. H. S. K. Monteiro, N. R. Fetto, M. J. Tucker and A. de Bettencourt-Dias, Inorg. Chem., 2020, 59, 3193–3199 CrossRef CAS PubMed.
- A. Picot, A. D'Aléo, P. L. Baldeck, A. Grichine, A. Duperray, C. Andraud and O. Maury, J. Am. Chem. Soc., 2008, 130, 1532–1533 CrossRef CAS PubMed.
- X. Wen, H. Li, Z. Ju, R. Deng and D. Parker, Chem. Sci., 2024, 15, 19944–19951 RSC.
- M. Starck, J. D. Fradgley, D. F. De Rosa, A. S. Batsanov, M. Papa, M. J. Taylor, J. E. Lovett, J. C. Lutter, M. J. Allen and D. Parker, Chem.–Eur. J., 2021, 27, 17921–17927 CrossRef CAS PubMed.
- C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, T. N. Nguyen, J. W. Kampf, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 5100–5109 CrossRef CAS PubMed.
- J. C. Lutter, B. A. L. Bermudez, T. N. Nguyen, J. W. Kampf and V. L. Pecoraro, J. Inorg. Biochem., 2019, 192, 119–125 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- M. Tegtmeier and W. Legrum, Arch. Pharm., 1998, 331, 143–148 CrossRef CAS PubMed.
- B. G. Lake, Food Chem. Toxicol., 1999, 37, 423–453 CrossRef CAS PubMed.
- J. Andres and A.-S. Chauvin, Phys. Chem. Chem. Phys., 2013, 15, 15981–15994 RSC.
- D. Kovacs, X. Lu, L. S. Mészáros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 CrossRef CAS PubMed.
- A. K. R. Junker, L. R. Hill, A. L. Thompson, S. Faulkner and T. Just Sørensen, Dalton Trans., 2018, 47, 4794–4803 RSC.
- A. Arauzo, L. Gasque, S. Fuertes, C. Tenorio, S. Bernès and E. Bartolomé, Dalton Trans., 2020, 49, 13671–13684 RSC.
- S. Di Pietro, D. Iacopini, A. Moscardini, R. Bizzarri, M. Pineschi, V. Di Bussolo and G. Signore, Molecules, 2021, 26, 1265 CrossRef PubMed.
- E. Mathieu, S. R. Kiraev, D. Kovacs, J. A. L. Wells, M. Tomar, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2022, 144, 21056–21067 CrossRef CAS PubMed.
- H. G. Ghalehshahi, S. Balalaie and A. Aliahmadi, New J. Chem., 2018, 42, 8831–8842 RSC.
- R. Maggi, F. Bigi, S. Carloni, A. Mazzacani and G. Sartori, Green Chem., 2001, 3, 173–174 RSC.
- T. P. Gustafson, G. A. Metzel and A. G. Kutateladze, Org. Biomol. Chem., 2011, 9, 4752–4755 RSC.
- S. Fiorito, V. A. Taddeo, S. Genovese and F. Epifano, Tetrahedron Lett., 2016, 57, 4795–4798 CrossRef CAS.
- A. A. Watrelot, D. T. Tran, T. Buffeteau, D. Deffieux, C. Le Bourvellec, S. Quideau and C. M. G. C. Renard, Appl. Surf. Sci., 2016, 371, 512–518 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
-
Apex3, v2019.1-0, SAINT V8.40A, Bruker AXS Inc., Madison (WI), USA, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
-
SHELXTL suite of programs, Version 6.14, 2000-2003, Bruker Advanced X-ray Solutions, Bruker AXS Inc., Madison (WI), USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
G. M. Sheldrick, SHELXL-2019, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 2019 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
-
CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, 95th edn, 2014 Search PubMed.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem.–Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- M. Melegari, V. Marzaroli, R. Polisicchio, D. Seletti, L. Marchiò, V. L. Pecoraro and M. Tegoni, Inorg. Chem., 2023, 62, 10645–10654 CrossRef CAS PubMed.
- K.-L. Wong, J.-C. G. Bünzli and P. A. Tanner, J. Lumin., 2020, 224, 117256 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 2003, 49, 4424–4442 CrossRef.
-
J.-C. G. Bünzli, K. A. Gschneider and V. K. Pecharsky, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1st edn, 2007, vol. 37 Search PubMed.
- R. D. Fields and M. V. Lancaster, Am. Biotechnol. Lab., 1993, 11, 48–50 CAS.
-
J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures for the synthesis of organic intermediates and their characterization (1H NMR and ESI-MS), characterization of the MCs (1H NMR, ESI-MS, UV-vis and IR), details about the [Nd2Ga8(shi)8(mip)4]2− X-ray structure (deposition number CCDC 2412013), and details about photophysical characterization (absorption and emission spectra, quantum yields and lifetimes). CCDC 2412013. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01320h |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Julie
Bourseguin
a,
Julie
Bourseguin