
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvation structure modulation via dipole–dipole interactions for high-rate lithium metal batteries exceeding 400 Wh kg−1†
Liwen
Zhang‡
ab,
Hongyu
Liu‡
ac,
Tingting
Wang
ab,
Hao
Wang
c,
Dong
Yan
c,
Min
Li
 c,
Xiaodi
Ren
b,
Hong
Li
ad and
Liping
Wang
*c
c,
Xiaodi
Ren
b,
Hong
Li
ad and
Liping
Wang
*c
aTianmu Lake Institute of Advanced Energy Storage Technologies, Changzhou 213300, China
bNano Science and Technology Institute, University of Science and Technology of China, Hefei 230026, China
cSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 611731, China. E-mail: lipingwang@uestc.edu.cn
dKey Laboratory for Renewable Energy, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
First published on 19th June 2025
Abstract
Electrolyte engineering is a key strategy to enhance the performance of high-voltage lithium-metal batteries (LMBs), with localized high-concentration electrolytes (LHCEs) emerging as a promising approach. However, the role of the diluent and its impact on the solvation structure remain unclear. In this study, we demonstrate that the dipole moment of the diluent is crucial for regulating diluent–solvent interactions. By modulating the dipole–dipole interactions between diluents and solvents, the coordination of solvents with Li+ has been successfully weakened, promoting the formation of a micelle-like electrolyte with anion-derived solvation. More importantly, the reduced desolvation energy barrier facilitates Li+ migration and improves the electrolyte's high-rate performance. The electrolyte achieves remarkable stability for 1500 cycles in a Li‖NCM811 cell at 10C with 76% capacity retention and a coulombic efficiency (CE) of 99.0% in a Li‖Cu cell. A prototype pouch cell (3.15 Ah) exhibits a high energy density of 403 Wh kg−1 with a lean electrolyte loading of only 1.9 g Ah−1. These findings provide critical insights into solvent–diluent interactions, advancing the development of fast-charging, high-voltage LMBs.
Introduction
The demand for energy from the development of contemporary society has greatly promoted the research and development of lithium metal batteries (LMBs), requiring LMBs with higher energy density, lower cost, and prolonged cycle lifespan.1–6 Recently, significant efforts have been made toward Ni-rich LiNixCoyMn1−x−yO2 (NCM) cathode materials (for example, LiNi0.8Co0.1Mn0.1O2, NCM811) because of their high operating voltage and excellent energy density.7–10 As is well-known, the structure and properties of electrolytes play a crucial role in the performance of batteries.11,12 Nevertheless, the development of such high-voltage LMBs is confronted with tremendous challenges due to the high reactivity of commercial electrolytes currently on the market with both the Ni-rich NCM cathode and the Li metal anode (LMA).8,13–15 Thus, it is widely accepted that the development of a compatible electrolyte is the key to solving the above issues related to high-voltage LMBs.16–18In recent years, high-concentration electrolytes (HCEs) and localized high-concentration electrolytes (LHCEs) have been widely reported.4,19,20 HCEs are typically composed of lithium salts at concentrations exceeding 3 M, which strongly bind most solvent molecules to lithium ions, reducing the presence of free (uncoordinated) solvent molecules. This helps expand the electrolyte's electrochemical window and promotes the formation of an anion-derived inorganic electrode–electrolyte interphase. However, HCEs face several challenges in practical applications, such as high viscosity, causing difficulties in injection of the electrolyte into cells, insufficient wetting of the electrolyte to the separator and thick electrodes, low ionic conductivity and high-cost issues due to the use of a large amount of expensive salts, etc.7 LHCEs are derived from HCEs, by introducing an inert diluent that cannot dissolve lithium salts, preserving the solvation structure of HCEs while reducing viscosity and cost. LHCEs have been proved to be one of the most promising systems to stabilize both high-voltage cathodes and the LMA for next-generation batteries.19,21–26 However, the interactions among different species in LHCEs are still convoluted, which hinders the further breakthrough on LHCE development.
Much work has been done to investigate the interaction between solvents and diluents in LHCEs.27–31 For example, Peng et al. investigated the influence of the diluent type and concentration on the inner solvation sheath of LHCEs, revealing that increasing the diluent content alters the solvation structure.32 Specifically, a higher diluent concentration leads to a simultaneous increase in free solvent molecules and the proportion of contact ion pairs (CIPs) relative to ionic aggregates (AGGs), showing that the diluent molecules not only create a nonsolvating medium but also disrupt the inner solvation structure through dipole–dipole interactions with solvent molecules. Ming et al. achieved highly reversible (de)intercalation of potassium ions in graphite electrodes in propylene carbonate (PC) based electrolytes by introducing a fluoroether diluent.28 It is found that fluorinated ethers can interact with PC solvents through dipole–dipole interactions, which can weaken the K+-PC interaction and reduce the desolvation energy, thereby promoting reversible K (de)intercalation. Ren et al. proposed a molecular anchoring strategy to suppress the side reactions of ether solvents at high voltage.33 It is suggested that there are interactions like hydrogen-bonding between diluents and solvents. Enabled by the solvent-anchor complex, a large number of diluent molecules in the electrolyte anchor the solvent molecules, preventing them from becoming free. This anchoring effect allows the ether-based electrolyte to remain stable and retain reliable cycling at a high voltage of 4.7 V. Obviously, diluents play an important role in the performance of electrolytes. However, the detailed mechanism and quantitative evaluation of diluents on solvation structure regulation have not been fully revealed.
In this work, the critical role of dipole moment in regulating diluent–solvent interactions has been revealed. Diluents with different dipole moments—hexafluorobenzene (HFB), toluene (TOL), and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE)—were introduced into an ether-based electrolyte to regulate the effect of dipole–dipole interactions on solvation structures. By adjusting dipole–dipole interactions between the solvent and diluent, the distribution of AGGs, CIPs, and solvent-separated ion pairs (SSIPs) in the electrolyte changed. The diluent with higher dipole moment exhibits a stronger dipole–dipole interaction with solvents, effectively lowering the Li+ desolvation energy barrier and improving Li+ transport kinetics, thereby significantly enhancing the electrolyte's high-rate performance. Consequently, the regulated electrolyte enables Li‖NCM811 cells to achieve stable cycling for 1500 cycles at 10C, exhibiting a capacity retention of 76%. Furthermore, the regulated electrolyte enabled stable operation of a 3.15 Ah lithium-metal pouch cell, delivering an energy density of 403 Wh kg−1 while minimizing electrolyte consumption to just 1.9 g Ah−1. These findings help highlight the significance of dipole moment in electrolyte design, offering fresh insights into electrolyte design for high-rate lithium metal batteries.
Results and discussion
Solvation structure regulation
A baseline electrolyte (denoted as Eb) was prepared by dissolving 2 M lithium bis(fluorosulfonyl)imide (LiFSI) in 1,2-dimethoxyethane (DME), with 1 wt% lithium nitrate (LiNO3) added as an additive.34,35 Then, three diluents of HFB, TOL, and TTE with the same volume as DME were introduced into Eb and named L-HFB, L-TOL, and L-TTE, respectively. LiNO3 was added to stabilize the cathode interface by regulating the electric double layer and suppressing solvent decomposition.36 All electrolyte formulae used in this work are shown in Table S1.† The optimization process of electrolytes is also presented in Fig. S1 and S2.† The formula in Fig. 1a illustrates the factors influencing the average potential energy of dipole–dipole interactions (Edipole–dipole).37,38 According to this formula, it can be found that Edipole–dipole is proportional to the square of the dipole moment (μ), indicating that stronger dipole moments lead to stronger dipole–dipole interactions. As shown in Fig. 1b, the dipole moments of the three diluents are displayed: HFB (0.00 D), TOL (0.41 D), and TTE (2.36 D). The selected diluents, covering a representative range of dipole moments while ensuring chemical inertness and miscibility with the electrolyte, enable a systematic investigation of the role of diluent dipole moment in governing the Li+ solvation structure, ionic transport, and interphase formation in LHCE. Table S2† also summarizes the physical and chemical properties of these diluents.30,39,40 To verify the relationship between dipole moment and dipole–dipole interaction strength, the binding energies between the solvent (DME) and different diluents were calculated using density functional theory (DFT) as shown in Fig. 1c and Table S3.†27,28 It is found that the strength of intermolecular binding is positively correlated with the dipole moment. TTE, having the largest dipole moment, exhibits the strongest binding energy with DME. Even at high lithium salt concentrations, the introduction of the TTE diluent does not trigger phase separation of the electrolyte (Fig. S3†). Based on the electrostatic potential (ESP) and charge distribution analysis (Fig. S4†), the potential interaction sites between DME and diluents were speculated. In HFB and TTE, the negatively charged F atoms are likely to interact with the positively charged H atoms on the methylene or terminal methyl groups of DME.41,42 Moreover, the electron-rich π-system of TOL generates a locally negative electrostatic potential above the benzene ring, which can attract the electron-deficient H atoms in DME, particularly those adjacent to the coordinated oxygen atoms. | ||
| Fig. 1 (a) The equation for describing dipole–dipole interactions (Edipole–dipole refers to the average potential energy of dipole–dipole interactions, μ refers to the dipole moment of solvent or diluent molecules, r refers to the distance between the centers of solvent and diluent molecules, T refers to absolute temperature, and ε and k refer to the vacuum dielectric constant and Boltzmann constant, respectively). (b) Dipole moments of different diluents. (c) The binding energy between diluents and DME. (d) Schematic diagram of the micelle structure in electrolytes. (e) Solvated cluster particle size measured using DLS and the Tyndall phenomenon. (f) The mechanism of diluents regulating the solvation structure. | ||
The presence of micelle-like structures in concentrated electrolytes has been reported to enable the controllable release of Li+ ions.43 As shown in Fig. 1d, in micelle-like electrolytes, Li+ is spatially confined within nanoscale salt-solvent clusters dispersed in a diluent-rich matrix, and under an applied electric field, Li+ gradually diffuses from the high-concentration cluster core to the electrode interface along a concentration gradient. This gradient-driven diffusion process effectively regulates the Li+ flux, preventing local accumulation and promoting uniform deposition. Additionally, the Tyndall phenomenon was observed in the three used electrolytes, indicating the presence of a micelle-like structure (Fig. 1e). The amount of diluent added in this work ensures the presence of micelle-like structures in the electrolyte and avoids low ion conductivity caused by exceeding the critical micelle concentration (CMC) mentioned in a previous study.44 The relative sizes of solvated clusters in the three electrolytes were measured using dynamic light scattering (DLS) to further investigate the microstructures. The size of solvent clusters measured by DLS matches the dipole moment of the diluent, but has no significant relationship with the molecular volume (Fig. S5†). The results showed that the solvation cluster size of L-TTE was the largest, while the solvation clusters in L-TOL and L-HFB decreased, respectively. The schematic diagram shown in Fig. 1f can help analyze this phenomenon. Diluent molecules (e.g., TTE) in the electrolyte system spontaneously interact with solvent molecules (DME) through dipole–dipole interactions, resulting in their partial displacement from the solvation sheath. This preferential attraction exerted by the diluent induces an increased spatial separation between DME molecules and Li+, thereby weakening the solvent's binding to Li+. The consequent structural rearrangement also enables FSI− anions to be preferentially incorporated into the coordination environment, promoting the development of an expanded solvation structure. The attenuated intermolecular forces between Li+ and solvent molecules significantly facilitate the desolvation process of lithium ions. This optimized desolvation behavior subsequently enhances the overall lithium-ion transport kinetics within the electrochemical system.
Li+ migration kinetics and solvation structure analysis
The physicochemical parameters of the three electrolytes were systematically investigated in detail to achieve effective electrolyte kinetic properties. To understand the electrode/electrolyte interphase kinetics, temperature-dependent electrochemical impedance spectroscopy (EIS) was employed to investigate the activation energy for charge transfer resistance (Table S4†).45 The resistance increases as the temperature drops (Fig. S6†), indicating the temperature-dependent electrochemical kinetics.46 As shown in Fig. 2a, the activation energy for charge transfer, relating to the desolvation process, is calculated to be 0.96 eV in L-TTE, which is lower than the other two. The reduced activation energy demonstrates that TTE weakens the binding between Li+ and DME, facilitating the desolvation of Li+ from the solvation sheath, which helps maintain the high-rate performance of LMBs.45,47 In addition, the constant potential polarisation is employed to determine the lithium-ion transference number of the three electrolytes.48 As shown in Fig. 2b and S7,† L-TTE has the highest Li+ transference number (0.78), which means that Li+ can migrate more rapidly in L-TTE, improving electrochemical kinetics. In addition, L-TTE also demonstrates satisfactory viscosity and ionic conductivity (Fig. S8 and S9†). At the same time, the change in the solvation structure of the electrolyte from a single solvent (DME) to binary solvents (DME-TTE, DME-TOL, and DME-HFB) was investigated by Raman spectroscopic characterization. The chemical shift differences observed in the Raman spectra of DME/diluent mixtures provide evidence for the presence of distinct intermolecular interactions between DME and the diluents (Fig. 2c). Compared with the DME single solvent, in the mixed DME/diluent solvent, the C–O–C bond has varying degrees of blue shift. Additionally, the chemical shift of the C–O–C bond was most pronounced in the DME–TTE mixture, indicating that the interaction between DME and TTE was the strongest, which is consistent with the binding energy results from previous DFT calculations. To further elucidate the solvation structure, Fig. 2d displays the Raman spectrum of the electrolyte. In the three electrolytes, the characteristic peaks of the diluent were consistent with those of the two-component solvent in the absence of lithium salts, indicating that there was no significant interaction between the diluent and the lithium salts (Fig. S10†). After the addition of lithium salt, part of DME is coordinated with Li+, and some free DME is still present in solution.31 For the purpose of identifying the specific solvation structures, peak fitting and analysis were performed on the peaks appearing in the range of 680–770 cm−1. As depicted in Fig. 2e and S11,† the peaks centered around 719 cm−1, 727 cm−1 and 743 cm−1 belong to SSIPs (free FSI−), CIPs (FSI− coordinating with one Li+), and AGGs (FSI− coordinating with more than one Li+), respectively.11 Obviously, DME with strong solvating ability is at an advantage when competing with FSI− for Li+ coordination, and due to the low concentration of lithium salts, the sum percentage of CIPs and AGGs is only 52.1%, while SSIPs dominate in Eb (47.9%). When the diluent is added, the proportion of SSIP significantly decreases, while the proportion of the CIP and AGG greatly increases. In L-TTE, the sum proportion of CIPs and AGGs further increases to 94.9%, possibly because TTE exhibits the strongest attraction to DME. As a result, DME molecules in the solvation cluster are positioned farther from Li+, allowing more FSI− to approach Li+ and form CIPs and AGGs. Due to the local lithium salt concentration of 2 M, more CIPs are formed in L-TTE than AGGs.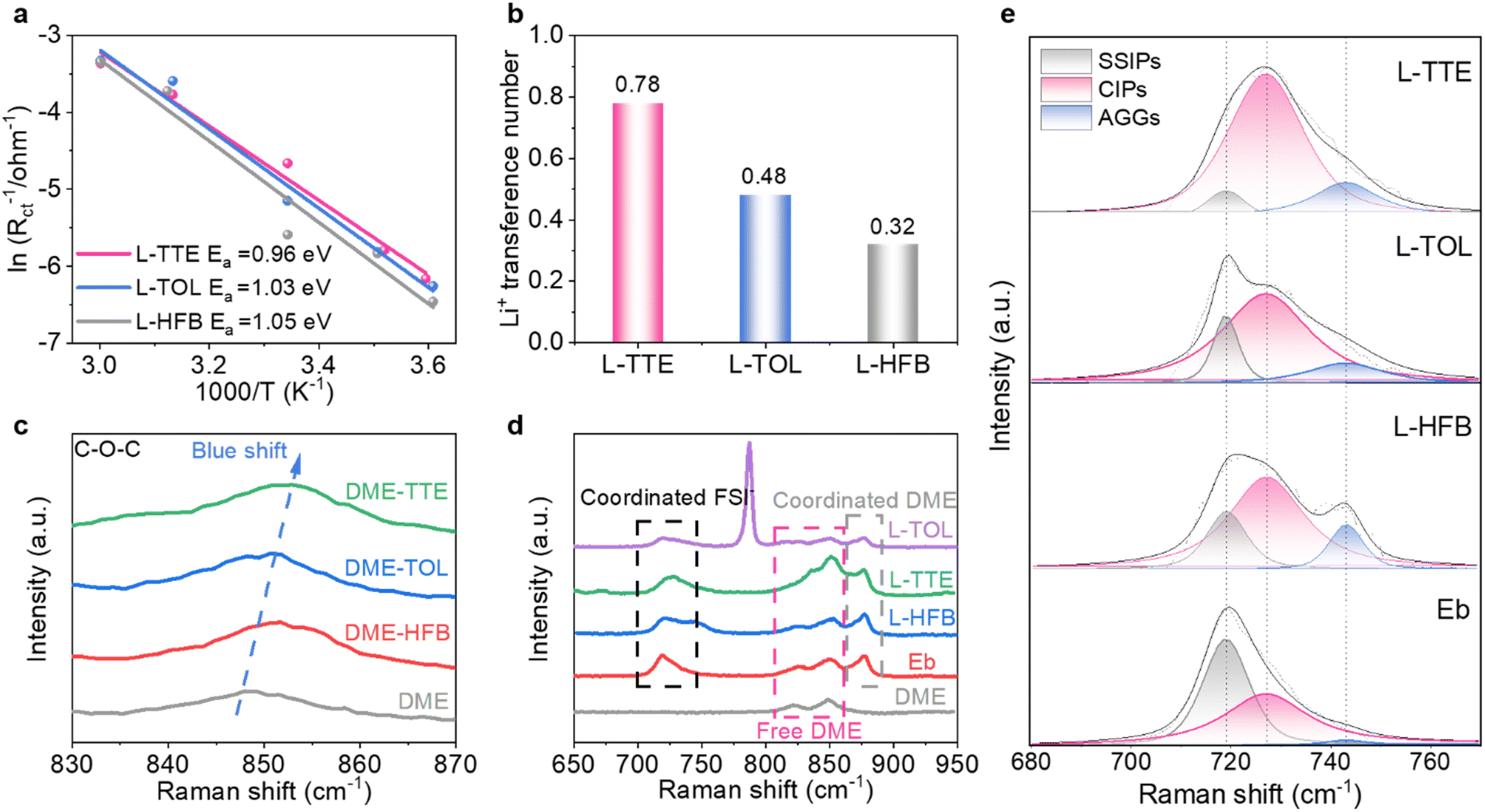 | ||
| Fig. 2 (a) Activation energies calculated from the Arrhenius equation. (b) Li+ transference numbers of different electrolytes. (c) Raman spectra of DME and DME/diluent. (d) Raman spectra of electrolytes. (e) Raman spectra and fitting results of electrolytes in the range of 680–770 cm−1. | ||
To evaluate the application potential of the three electrolytes in high energy density LMBs, coin cells coupling the NCM811 cathode (loading: 1.0 mAh cm−2) with the LMA were assembled. As shown in Fig. 3a, all cells were cycled at 2C charge/discharge rate after two precycles at 0.1C, and the Li‖NCM811 cell using the L-TTE electrolyte delivers an initial discharge capacity of 174 mAh g−1 at 2C along with the initial CE of 89.04%. After the activation process, the Li‖NCM811 cell in the L-TTE electrolyte operates stably over 650 cycles with a high-capacity retention of 72% and an average CE of 99.8%, while the cells in L-TOL and L-HFB electrolytes capacity decay quickly. The superior cycling performance of Li‖NCM811 cells in the L-TTE electrolyte was also confirmed from the charge/discharge curves by showing lower polarization and less capacity decay (Fig. 3b). Moreover, to minimize the influence of cell-to-cell variations on initial capacity, parallel cells were tested and their average values were reported to reduce experimental errors (Fig. S12–S14†). The electrochemical performance of the battery also confirms the previous conclusion that the stronger dipole–dipole interaction between TTE and DME weakens the bonding strength of Li+-DME in the solvent sheath of L-TTE, making Li+ desolvation easier and greatly improving high-rate performance of the electrolyte. To verify the rate performance of the electrolytes under more extreme conditions, the testing rate of Li‖NCM811 coin cells was increased to 10C. As shown in Fig. 3c and S15,† the NCM811 cathode using L-TTE shows superior cycling performance and still exhibits 76% capacity retention after cycling for 1500 cycles. It is worth noting that the single-crystal NCM811 cathode empowered by L-TTE can achieve a high specific capacity of 152 mAh g−1 at a rate of 10C. Additionally, the performance of the Li‖NCM811 cells at different current densities was also evaluated. As shown in Fig. 3d, consistent with the previous discussion, rate tests indicate that the Li‖NCM811 using L-HFB exhibits the poorest performance, with discharge specific capacities all lower than those of the cells using L-TOL and L-TTE at different rates. Notably, L-TTE exhibits outstanding rate performance and the most stable coulombic efficiency (CE), delivering specific discharge capacities of 190.95, 183.63, 165.77, 147.55, and 110.6 mAh g−1 at 0.5C, 1C, 5C, 10C, and 20C, respectively (Fig. S16†). This indicates that the rate performance of the battery is closely dependent on the interfacial kinetics of Li+ transportation. It is worth mentioning that the cycling performance of L-TTE is significantly better than most reports on electrolyte engineering (Fig. 3e).46,49–53
 | ||
| Fig. 3 (a) Cycling performance of Li‖NCM811 coin cells at 2C. (b) Charge–discharge curves of Li‖NCM811 coin cells at 2C. (c) Cycling performance of Li‖NCM811 coin cells at 10C. (d) Cycling performance of Li‖NCM811 coin cells at different current densities. (e) Electrochemical performance comparison of current studies. | ||
Interfacial chemistries of the NCM811 cathode and Li anode
To evaluate the stabilizing effect of L-TTE on NCM811 cathodes, the cycled cathode materials were characterized using scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM). Noticeable differences in surface morphology were observed on cathodes cycled in L-TTE, L-TOL, and L-HFB (Fig. S17†). Specifically, the cathode cycled in the L-TTE exhibited intact particles and smooth surfaces, while the electrodes cycled in L-TOL and L-HFB exhibited cracked particles or rough surfaces. This can indicate that L-TTE helps maintain the stability of the NCM811 electrode structure. The HR-TEM images of NCM811 cycled in different electrolytes are shown in Fig. 4a. The CEI formed in L-TTE was thin and uniform, approximately 5 nm in thickness. In contrast, the CEI in L-TOL was about 10 nm thick, while the CEI in L-HFB was highly non-uniform. Consistently, according to electrochemical impedance spectroscopy (EIS) results shown in Fig. S18,† the cycled L-TTE battery also exhibited the lowest impedance. These features are closely correlated with the cycling performance of the battery. | ||
| Fig. 4 (a) HR-TEM images of cycled NCM811 cathodes after 100 cycles. (b) The F 1s XPS spectra of the cycled NCM811 cathodes after 100 cycles. (c) Long-term cycling of Li‖Li symmetrical cells. (d) Modified Aurbach's measurement for Li‖Cu cells. (e) Tafel plots of Li‖Li cells before galvanostatic cycling. (f) SEM images of Li metal deposition on Cu (0.5 mA cm−2, 5 mAh cm−2) for different electrolytes. | ||
The CEI components were further systematically investigated through X-ray photoelectron spectroscopy (XPS) characterization, and the results are shown in Fig. 4b and S19.† Compared to the NCM811 cycled in L-TOL and L-HFB, the surface of the cathode cycled in L-TTE exhibits a reduced signal intensity for carbon-related species, including C–C/C–H (∼282.8 eV, C 1s) and CF2–CH2 (∼284.1 eV).7 The high-intensity LiF peak in the F 1s spectrum indicates that L-TTE forms a LiF rich CEI, which helps to suppress parasitic reactions on the cathode. The absence of the M − O (∼527.3 eV) signal in the O 1s spectrum also indicates that the L-TTE can inhibit the dissolution of transition metals, which helps to extend the cycle life of batteries.54
In order to investigate the compatibility of electrolytes with the LMA, Li‖Li and Li‖Cu cells were assembled for further analysis.50,55 The Li‖Li symmetric cells are cycled at a current density of 1 mA cm−2 with an area capacity of 1 mAh cm−2. As shown in Fig. 4c, the cells in the L-HFB electrolyte can only cycle stably for 350 hours. The cells in L-TOL experience a gradual increase in voltage polarization starting around 1050 hours, due to the depletion of electrochemically active lithium. In contrast, the cells in L-TTE show a stable charge–discharge profile for 1400 hours. In particular, a low voltage polarization of merely 40 mV is observed in L-TTE. In addition, Li‖Cu cells were tested according to the Aurbach method to evaluate the reversibility of Li plating and stripping on copper foil. L-TTE also showed a high average cycle efficiency of 99.33% (Fig. 4d).51 The larger exchange current densities calculated from the Tafel curves for L-TTE further confirm its faster kinetics compared to the other electrolytes (Fig. 4e).52 To confirm the morphology and interphase structure of Li metal, high-capacity Li deposition on Cu was conducted and examined by scanning electron microscopy (SEM). As shown in Fig. 4f and S20,† Li deposited on Cu foil in L-TTE is uniform and dense, without the formation of dendrites, which helps to improve the reversibility of Li plating and stripping.53 At the same time, the chemical components of the solid–electrolyte interphase (SEI) on Li anode surfaces in Li‖NCM811 batteries were comprehensively analyzed through XPS in Fig. S21.† The F 1s spectrum shows two peaks located at approximately 687.7 and 684.6 eV, corresponding to C–F and Li–F, respectively. The Li–F/C–F ratios in L-TTE are greater than those in L-TOL and L-HFB, indicating that L-TTE also contributes to the formation of a LiF-rich SEI.56 In summary, L-TTE forms a robust electrode/electrolyte interface rich in LiF on both the NCM811 cathode and Li anode to maintain long cycling stability of the battery.
A 3.15 Ah pouch cell with a high energy density of 403 Wh kg−1
In order to verify the potential of L-TTE, a 3.15 Ah pouch cell was assembled with a thin LMA (100 μm) and a high loading NCM811 cathode (4.7 mAh cm−2). It is worth mentioning that the amount of electrolyte in the pouch cell has been compressed to an extremely low value of 1.9 g Ah−1, far below the amount commonly used in commercial production (typically above 3 g Ah−1).57 Based on the total mass, the pouch cell can achieve a high energy density of 403 Wh kg−1 (Fig. 5a and Table S5†). The weight percentage of each part of the pouch cell is shown in Fig. 5b. As shown in Fig. 5c, the pouch cell stably operates for 20 cycles without significant capacity degradation, indicating great potential for practical applications of this electrolyte. The polarization demonstrated only a slight increase throughout the continuous cycling (Fig. 5d). Upon disassembling the failed pouch cell, a smooth cathode surface was observed with no active material detachment, and the lithium anode was also found to be devoid of severe dead lithium formation, indicating the favorable electrolyte compatibility with both electrodes. However, electrolyte depletion was identified within the cell and is concluded to be the primary failure mechanism (Fig. S22†). When compared to other studies with the Li‖NCM811 pouch cell, L-TTE can achieve higher energy density through a lean electrolyte, proving it to be a very promising candidate for high rate, high energy density LMB electrolytes (Fig. 5e).58–65 | ||
| Fig. 5 (a) Schematic illustration of pouch cell parameters. (b) Weight ratios of cell components. (c) Cycle performance of the 403 Wh kg−1 Li‖NCM811 pouch cell. (d) Capacity–voltage plots of the pouch cell. (e) Comparison of lithium-metal pouch cells in previous reports and this work. | ||
Conclusions
In this work, the dipole–dipole interactions between the solvent and diluent were systematically investigated, and the dipole moment was identified as a critical factor in solvation structure modulation. Specifically, the coordination of DME with Li+ can be effectively weakened by the high dipole moment of TTE, leading to an increased Li+ transference number of 0.78 and reduced activation energy for charge transfer, thereby enhancing electrochemical kinetics. This also promotes the formation of CIPs and AGG clusters, which contribute to the development of an inorganic-rich SEI/CEI. As a result, the NCM811 cathode enabled by L-TTE can achieve 152 mAh g−1 at a high rate of 10C, and retain 76% of its capacity over 1500 cycles. Furthermore, a 3.15 Ah pouch cell demonstrated an impressive energy density of 403 Wh kg−1 with a lean electrolyte of 1.9 g Ah−1. This study offers a promising approach for designing high-rate electrolytes via regulating solvent–diluent interactions.Data availability
Data are available from the authors on reasonable request.Author contributions
Liwen Zhang and Hongyu Liu: methodology, investigation, and writing – original draft. Tingting Wang, Hao Wang, Dong Yan and Min Li: validation and investigation. Xiaodi Ren and Hong Li: methodology, conceptualization, and supervision. Liping Wang: funding acquisition, conceptualization, supervision and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Sichuan, China (No. 2023NSFSC1914 and 2024NSFSC1013), Beijing National Laboratory for Condensed Matter Physics (2023BNLCMPKF015), and the National Natural Science Foundation of China (No. 22322903).References
- H. Liu, Z. Wu, H. Wang, X. Niu, H. Li and L. Wang, ACS Energy Lett., 2024, 9, 4666–4672 CrossRef CAS.
- L. Li, K. Ren, W. Xie, Q. Yu, S. Wu, H.-W. Li, M. Yao, Z. Jiang and Y. Li, Chem. Sci., 2025, 16, 7981–7988 RSC.
- S. Fazal, F. Ahmad, M. I. Khan, A. Shanableh and S. Manzoor, New J. Chem., 2024, 48, 14104–14127 RSC.
- F. Philippi, M. Middendorf, K. Shigenobu, Y. Matsuyama, O. Palumbo, D. Pugh, T. Sudoh, K. Dokko, M. Watanabe, M. Schönhoff, W. Shinoda and K. Ueno, Chem. Sci., 2024, 15, 7342–7358 RSC.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- Z. Y. Wang, Y. S. Wang, Y. H. Xin, Q. B. Zhou, X. Y. Ding, L. Liu, T. L. Song, F. Wu, Z. B. Wei and H. C. Gao, Chem. Sci., 2024, 15, 16669–16680 RSC.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. J. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- C. Liu, Z. Jiang, Y. Zhang, W. Xie, J. Zou, S. Wu, M. Sun and Y. Li, Chem. Sci., 2025, 16, 7847–7857 RSC.
- H. Zheng, S. Peng, S. Liang, W. Yang, C. Chen, C. Wang and R. Yu, Adv. Funct. Mater., 2024, 35, 2418274 CrossRef.
- F. Wu, J. Dong, L. Chen, G. Chen, Q. Shi, Y. Nie, Y. Lu, L. Bao, N. Li, T. Song, S. Chen and Y. Su, Energy Mater. Adv., 2023, 2023(4), 0007 CrossRef.
- S. Li, H. Y. Liu, L. J. Zheng, C. T. Ma, H. L. Yu, X. D. Wu, X. B. Niu and L. P. Wang, Nano Energy, 2024, 131, 110234 CrossRef CAS.
- P. T. Xiao, X. R. Yun, Y. F. Chen, X. W. Guo, P. Gao, G. M. Zhou and C. M. Zheng, Chem. Soc. Rev., 2023, 52, 5255–5316 RSC.
- Y. T. Dong, Y. M. Chen, Q. H. Zeng, J. Y. Feng, M. M. Fang, Z. Q. Shi, J. J. Liu, Y. L. Sheng, X. Y. Yue and Z. Liang, Energy Mater. Adv., 2024, 5(14), 0113 CrossRef CAS.
- P. F. Yan, J. M. Zheng, D. P. Lv, Y. Wei, J. X. Zheng, Z. G. Wang, S. Kuppan, J. G. Yu, L. L. Luo, D. Edwards, M. Olszta, K. Amine, J. Liu, J. Xiao, F. Pan, G. Y. Chen, J. G. Zhang and C. M. Wang, Chem. Mater., 2015, 27, 5393–5401 CrossRef CAS.
- P. T. Xiao, Y. Zhao, Z. H. Piao, B. H. Li, G. M. Zhou and H. M. Cheng, Energy Environ. Sci., 2022, 15, 2435–2444 RSC.
- S. S. Yan, N. Yao, H. Liu, Z. Zhang, Y. Lu, Z. Liu, W. H. Hou, P. Zhou, H. Y. Zhou, X. Chen, K. Liu and Q. Zhang, Energy Environ. Sci., 2025, 18, 1696–1706 RSC.
- Q. Xiong, X. Teng, J. Lou, G. Pan, X. Xia, H. Chi, X. Lu, T. Yang and Z. Ji, J. Energy Chem., 2020, 40, 1–6 CrossRef.
- A. Zhamu, G. R. Chen, C. G. Liu, D. Neff, Q. Fang, Z. N. Yu, W. Xiong, Y. B. Wang, X. Q. Wang and B. Z. Jang, Energy Environ. Sci., 2012, 5, 5701–5707 RSC.
- Z. Li, G. Zhou, S. Li, H. Liu, L. Wang and H. Li, Joule, 2023, 7, 2609–2621 CrossRef CAS.
- C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Gering, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J.-G. Zhang, E. J. Dufek, J. Xiao, X.-Q. Yang, J. Liu, Y. Qi and B. Li, Nat. Mater., 2023, 22, 1531–1539 CrossRef CAS PubMed.
- Y. Zhao, T. Zhou, L. P. H. Jeurgens, X. Kong, J. W. Choi and A. Coskun, Chem, 2023, 9, 682–697 CAS.
- Y. Liu, J. Li, X. Deng, S. S. Chi, J. Wang, H. Zeng, Y. Jiang, T. Li, Z. Liu, H. Wang, G. Zhang, Y. Deng and C. Wang, Small, 2024, 20, 2311812 CrossRef CAS PubMed.
- H. Y. Wu, X. Y. Chen, C. Y. Zhao, Y. K. Tian, X. F. Yang, R. C. Sun and X. J. Gao, Energy Mater. Adv., 2024, 5(11), 0130 CrossRef CAS.
- Y. Zhao, T. H. Zhou, T. Ashirov, M. El Kazzi, C. Cancellieri, L. P. H. Jeurgens, J. W. Choi and A. Coskun, Nat. Commun., 2022, 13, 2575 CrossRef CAS PubMed.
- J. Zheng, G. B. Ji, X. L. Fan, J. Chen, Q. Li, H. Y. Wang, Y. Yang, K. C. DeMella, S. R. Raghavan and C. S. Wang, Adv. Energy Mater., 2019, 9, 1803774 CrossRef.
- W. A. Zhao, K. Wang, R. Dubey, F. C. Ren, E. Brack, M. Becker, R. Grissa, L. Seidl, F. Pagani, K. Egorov, K. V. Kravchyk, M. V. Kovalenko, P. F. Yan, Y. Yang and C. Battaglia, Mater. Today Energy, 2023, 34, 101301 CrossRef CAS.
- H. Xie, H. Cheng, P. Kumar, Y. Wang, H. Liang, T. Cai, F. Zhao, Z. Cao, L. Cavallo, Z. Ma, Q. Li and J. Ming, ACS Nano, 2024, 18, 22503–22517 CrossRef CAS PubMed.
- H. Liang, P. Kumar, Z. Ma, F. Zhao, H. Cheng, H. Xie, Z. Cao, L. Cavallo, Q. Li and J. Ming, ACS Energy Lett., 2024, 9, 3536–3546 CrossRef CAS.
- H. R. Cheng, Z. Ma, P. Kumar, H. H. Liang, Z. Cao, H. L. Xie, L. Cavallo, H. Kim, Q. Li, Y. K. Sun and J. Ming, Adv. Energy Mater., 2024, 14, 2304321 CrossRef CAS.
- C. Li, Y. Li, Y. Wang, F. Bai, X. Chen and T. Li, J. Mater. Chem. A, 2024, 12, 8236–8243 RSC.
- Z. Ren, S. Li, H. Liu, H. Wang, X. Niu, Q. Yang and L. Wang, J. Colloid Interface Sci., 2024, 663, 203–211 CrossRef CAS PubMed.
- F. Ren, Z. Li, J. Chen, P. Huguet, Z. Peng and S. Deabate, ACS Appl. Mater. Interfaces, 2022, 14, 4211–4219 CrossRef CAS PubMed.
- Z. Cui, Z. Jia, D. Ruan, Q. Nian, J. Fan, S. Chen, Z. He, D. Wang, J. Jiang, J. Ma, X. Ou, S. Jiao, Q. Wang and X. Ren, Nat. Commun., 2024, 15, 2033 CrossRef CAS PubMed.
- X. Li, R. Zhao, Y. Fu and A. Manthiram, eScience, 2021, 1, 108–123 CrossRef.
- Y. Liang, W. Wu, D. Li, H. Wu, C. Gao, Z. Chen, L. Ci and J. Zhang, Adv. Energy Mater., 2022, 12, 2202493 CrossRef CAS.
- W. Zhang, Y. Lu, L. Wan, P. Zhou, Y. Xia, S. Yan, X. Chen, H. Zhou, H. Dong and K. Liu, Nat. Commun., 2022, 13, 2029 CrossRef CAS PubMed.
- X. Song, X. Liang, H. Kim and Y.-K. Sun, ACS Energy Lett., 2024, 9, 5576–5586 CrossRef CAS.
- M. Qin, Z. Zeng, Q. Wu, H. Yan, M. Liu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Energy Environ. Sci., 2023, 16, 546–556 RSC.
- M. Yang, K. Chen, H. Li, Y. Cao, H. Yang and X. Ai, Adv. Funct. Mater., 2023, 33, 2306828 CrossRef CAS.
- F. Hai, Y. Yi, Z. Xiao, J. Guo, X. Gao, W. Chen, W. Xue, W. Hua, W. Tang and M. Li, Adv. Energy Mater., 2024, 14, 2304253 CrossRef CAS.
- G.-X. Li, X. Lyu, A. Nguyen, R. Kou, C. George, S. Wu, R. Li, K. Wang, T. Li and D. Wang, Adv. Energy Mater., 2025, 15, 2405680 CrossRef CAS.
- Q. Sun, Z. Cao, Z. Ma, J. Zhang, H. Cheng, X. Guo, G.-T. Park, Q. Li, E. Xie, L. Cavallo, Y.-K. Sun and J. Ming, ACS Energy Lett., 2022, 7, 3545–3556 CrossRef CAS.
- Y. Deng, H. Wang, M. Fan, B. Zhan, L.-J. Zuo, C. Chen and L. Yan, J. Am. Chem. Soc., 2023, 145, 20109–20120 CrossRef CAS PubMed.
- C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Gering, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J.-G. Zhang, E. J. Dufek, J. Xiao, X.-Q. Yang, J. Liu, Y. Qi and B. Li, Nat. Mater., 2023, 22, 1531–1539 CrossRef CAS PubMed.
- Q. Liu, Z. Zeng, M. Qin, T. Yang, H. Ma, S. Ji, L. Huang, S. Cheng and J. Xie, Batteries Supercaps, 2024, 7, e202400034 CrossRef CAS.
- T. Yang, L. Li, J. H. Zou, Y. Q. Yao, Q. A. Zhang, Z. P. Jiang and Y. T. Li, Adv. Funct. Mater., 2024, 34, 2404945 CrossRef CAS.
- Y. R. Yang, Q. Li, H. Li, J. F. Ruan, F. M. Wang, Z. Y. Li, J. Y. Yang, J. Y. Zhang, U. Çaglayan, D. L. Sun, F. Fang, M. Kunduraci and F. Wang, Angew. Chem., Int. Ed., 2025, 64, e202419653 CrossRef CAS PubMed.
- M. Rafiee, D. J. Abrams, L. Cardinale, Z. Goss, A. Romero-Arenas and S. S. Stahl, Chem. Soc. Rev., 2024, 53, 566–585 RSC.
- M. L. Mao, X. Ji, Q. Y. Wang, Z. J. Lin, M. Y. Li, T. Liu, C. L. Wang, Y. S. Hu, H. Li, X. J. Huang, L. Q. Chen and L. M. Suo, Nat. Commun., 2023, 14, 1082 CrossRef CAS PubMed.
- Y. C. Liu, Y. S. Lin, Z. L. Yang, C. X. Lin, X. X. Zhang, S. J. Chen, G. L. Hu, B. S. Sa, Y. Q. Chen and Y. N. Zhang, ACS Nano, 2023, 17, 19625–19639 CrossRef CAS PubMed.
- A.-M. Li, O. Borodin, T. P. Pollard, W. Zhang, N. Zhang, S. Tan, F. Chen, C. Jayawardana, B. L. Lucht, E. Hu, X.-Q. Yang and C. Wang, Nat. Chem., 2024, 16, 922–929 CrossRef CAS PubMed.
- Y. Zhang, F. Li, Y. Cao, M. Yang, X. Han, Y. Ji, K. Chen, L. Liang, J. Sun and G. Hou, Adv. Funct. Mater., 2024, 34, 2315527 CrossRef CAS.
- Y. Chen, M. Li, Y. Liu, Y. Jie, W. Li, F. Huang, X. Li, Z. He, X. Ren, Y. Chen, X. Meng, T. Cheng, M. Gu, S. Jiao and R. Cao, Nat. Commun., 2023, 14, 2655 CrossRef CAS PubMed.
- Y. Liu, Z. Jin, Z. Liu, H. Xu, F. Sun, X. Q. Zhang, T. Chen and C. Wang, Angew. Chem., Int. Ed., 2024, 63, e202405802 CrossRef CAS PubMed.
- G. X. Lu, J. Nai, D. Y. Luan, X. Y. Tao and X. W. Lou, Sci. Adv., 2023, 9, eadf1550 CrossRef CAS PubMed.
- Y. P. Guo, H. Q. Li and T. Y. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- X. Wang, J. Zhu, X. Wei, D. Wang, W. Xu, Y. Jin and H. Dai, Energy Storage Mater., 2024, 65, 103160 CrossRef.
- Q. Zhao, N. W. Utomo, A. L. Kocen, S. Jin, Y. Deng, V. X. Zhu, S. Moganty, G. W. Coates and L. A. Archer, Angew. Chem., Int. Ed., 2022, 61, e202116214 CrossRef CAS PubMed.
- Y. Qiao, H. J. Yang, Z. Chang, H. Deng, X. Li and H. S. Zhou, Nat. Energy, 2021, 6, 653–662 CrossRef CAS.
- Y. L. Gao, F. H. Qiao, J. Y. You, Z. Y. Ren, N. Li, K. Zhang, C. Shen, T. Jin and K. Y. Xie, Nat. Commun., 2022, 13, 5 CrossRef CAS PubMed.
- L. H. Zhang, F. Q. Min, Y. Luo, G. J. Dang, H. T. Gu, Q. Y. Dong, M. H. Zhang, L. M. Sheng, Y. B. Shen, L. W. Chen and J. Y. Xie, Nano Energy, 2022, 96, 107122 CrossRef CAS.
- B. Y. He, W. Deng, Q. G. Han, W. H. Zhu, Z. Y. Hu, W. Fang, X. F. Zhou and Z. P. Liu, J. Mater. Chem. A, 2021, 9, 25558–25566 RSC.
- Z. Q. Liu, D. C. Guo, W. T. Fan, F. L. Xu and X. Y. Yao, ACS Mater. Lett., 2022, 4, 1516–1522 CrossRef CAS.
- P. Y. Zhao, Y. Li, S. J. Chen, H. Fan, Y. Y. Feng, L. L. Hu, Y. H. Zhang, Q. N. Nie, H. J. Pei, C. Yang, J. K. Deng, C. G. Bao and J. X. Song, Adv. Energy Mater., 2022, 12, 2200568 CrossRef CAS.
- Q. K. Zhang, X. Q. Zhang, J. Wan, N. Yao, T. L. Song, J. Xie, L. P. Hou, M. Y. Zhou, X. Chen, B. Q. Li, R. Wen, H. J. Peng, Q. Zhang and J. Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02700d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |