
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSegmented milli-fluidic crystallisation of paracetamol with in situ single-crystal X-ray diffraction†
Lois E.
Wayment
ab,
C. Daniel
Scott
 a,
Lucy K.
Saunders
b,
Pollyanna
Payne
a,
Lauren E.
Hatcher
ac,
Graeme
Winter
b,
Benjamin
Williams
b,
David R.
Allan
b,
Chick C.
Wilson
a,
Mark R.
Warren
*b and
Karen
Robertson
*ad
a,
Lucy K.
Saunders
b,
Pollyanna
Payne
a,
Lauren E.
Hatcher
ac,
Graeme
Winter
b,
Benjamin
Williams
b,
David R.
Allan
b,
Chick C.
Wilson
a,
Mark R.
Warren
*b and
Karen
Robertson
*ad
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, Bath and North East Somerset BA2 7AY, UK. E-mail: Karen.Robertson@nottingham.ac.uk
bDiamond Light Source (United Kingdom), Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
cSchool of Chemistry, Cardiff University, Park Place, Cardiff, Glamorgan CF10 3AT, UK. E-mail: mark.warren@diamond.ac.uk
dAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, University Park Campus, Nottingham, Nottinghamshire NG7 2RD, UK
First published on 19th June 2025
Abstract
Serial crystallography has revolutionalised our ability to analyse protein crystals; crystal structures can be uncovered by combining data from multiple crystals mitigating radiation damage through overexposure to the X-ray beam. With synchrotron sources becoming even brighter, radiation damage is ever more pertinent for small molecule crystals as well. Combining serial crystallography with flow crystallisation, we pave the way for exploring high-throughput screening and kinetic studies, with application to small molecule crystals up to mm-scale. Here we present the first known example of single crystal X-ray diffraction of small molecule crystals in a flow crystallisation environment. In situ single-crystal X-ray diffraction has been achieved on a series of growing singular crystals through the use of segmented flow cooling crystallisation, holding crystals in an X-ray beam for 4.2 s whilst they freely rotate whilst flowing inside tubing. Upon triggering of a passing slug at the analysis point, the tubing is moved in the opposite direction to the flow, enabling the crystal to remain within the X-ray beam whilst maintaining the free rotation of the crystal due to fluid movement. Structure solution of paracetamol form I has been achieved with 0.8 Å resolution using data combined from 13 crystals whilst unit cell information can be extracted from a single crystal.
Introduction
Flow crystallisation opens novel process windows for chemists in the same way that flow synthesis has exploded what we can do in a synthetic space. It offers superior control over crystallisation conditions due to the high surface to volume ratio and enhanced mixing regime with respect to batch conditions. This results in an increase in homogeneity and reproducibility, and ease in scale-up.1 In a tubular flow crystalliser, these enhanced mixing conditions result in what is termed ‘plug flow’ where there is a direct relationship between the crystalliser length and crystallisation time (irrespective of the experiment time) as there is no back-mixing of old and new solutions being pumped through the reactor. This opens up a novel operating space in which transient events, such as nucleation and crystal growth, can be explored for analysis periods longer than the event or species lifetime. Thus, by analysing at a given crystalliser length, all passing material will be at the same crystallisation time.The advent of serial crystallography has revolutionised protein crystallography. Radiation damage has always been a challenge for small molecule crystallography2 but with ever brighter sources this challenge is significantly increasing,3 and serial crystallography enables access to the excellent data quality obtained from a bright source without the penalty of radiation damage.4 Protein crystals are typically in the order of ∼10–50 μm (or smaller) and rapidly undergo radiation damage when exposed to X-rays. Serial crystallography is the method of rapidly analysing 100s–1000s of crystals via either a grid5,6 or slurry flow (through injection, tape delivery or microfluidics).5,7,8 In this case diffraction is obtained from a single orientation for each individual crystal with the full crystal structure compiled from the individual exposures at different orientations.8 A range of data analysis software packages have been developed to integrate and scale diffraction images from randomly orientated single crystals, compiling them into a single dataset.9–11 Both serial synchrotron crystallography (SSX) and serial femtosecond crystallography (SFX), performed at X-ray Free-Electron Laser (XFEL) sources, typically involve a pre-prepared crystal delivery to the radiation source. The preparation and inert delivery of these crystals can be limiting. Exploration of crystal structures for an actively growing crystal is more usually the province of earlier crystallisation stages such as XAS12 or SAXS/WAXS.13,14 The challenging nature of flow crystallisation, required to deliver the actively growing crystals in such environments has limited further application of dynamic crystallisation environments to serial crystallography.
In order to minimise the number of crystals required for full structure solution, Hadian-Jazi et al. used high viscosity carrier fluid to deliver a slow stream of crystal slurry which, coupled with a fast detector readout, therefore enables multiple hits on a single crystal.15 By evaluating the data for successive frames they have ascertained that radiation damage can be negligible for 8–15 successive hits of a lysozyme crystal (dose threshold of 0.38 MGy). Acquisition of reliable successive frames from a rotating single crystal significantly reduces the number of crystals required to obtain a suitable dataset for indexation.
Flow environments provide a unique opportunity to monitor transient events and to enable real-time feedback, expediting optimisation and discovery processes.16,17 We have already shown that it is possible to couple flow crystallisation with in situ powder X-ray diffraction (PXRD)18 and Raman spectroscopy.19 Through these methods, crystallisation profiles can be uncovered through analysis at different distances along the crystalliser (equivalent to crystallisation time) regardless of the experiment time.
Here we present atomic-scale structure solution of a highly symmetrical (few unique reflections per orientation) small molecule crystal system (paracetamol form I, monoclinic P21/n, a = 6.82, b = 8.37, c = 11.56, β = 99.32°, from HXACAN11) from SSX performed in a segmented flow active crystallisation apparatus (Fig. 1). Akin to the work of Hadian-Jazi et al. we exploit the natural movement of crystals in a flow environment to obtain multiple orientations from one crystal passing the beam. In order to maximise the number of orientations from each crystal, the crystal is artificially ‘held’ in the beam by moving the crystalliser tubing in the opposite direction to the flow at the same speed. Thus, the crystal appears to be rotating in front of the X-ray beam for sufficient time to acquire data from multiple orientations, without any alteration to the fluidic regime within the crystalliser. The diffraction data were then analysed through creep indexing using DIALS. Due to variability in the detector to sample distance (through spatial inconsistencies of the crystals within the tubing), the data acquired are not yet of sufficient quality to enable deposition of new crystal structures into the Cambridge Structural Database but are the first steps towards materials discovery with inline full structural analysis in one experimental set-up.
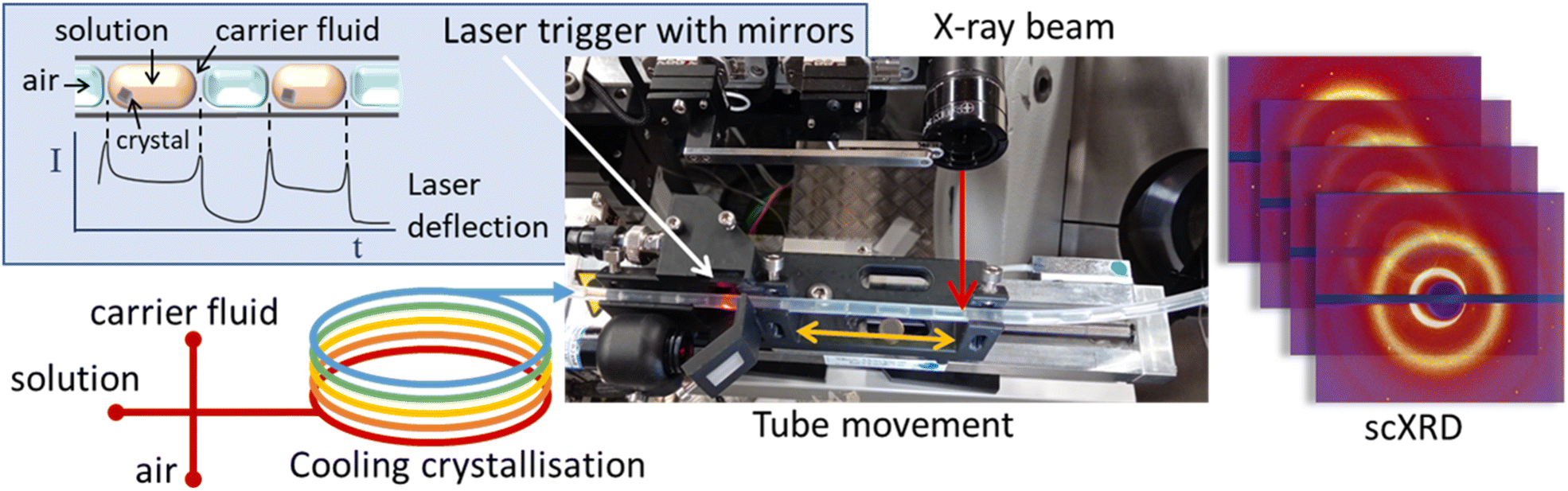 | ||
| Fig. 1 Schematic representation of the KRAIC-S concept highlighting laser triggering and obverse movement of tubing with solution flow in front of an X-ray beam. | ||
Methods
The KRAIC-S is a tri-segmented flow crystalliser based on the previously published KRAIC and KRAIC-D platforms.18,20 Three immiscible phases of feed solution, carrier fluid and air are combined in a segmentation section to produce the tri-segmented flow. All crystallisations reported here were from the same feed stock of 254 g L−1 paracetamol in H2O/IPA 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (v/v). Air and Galden SV110 were used as the segmenting gas and carrier fluid respectively. SF-10 peristaltic pumps (Vapourtec UK) were used for air (2.5 mL min−1) and solution (2.7 mL min−1) delivery, and an ASIA dual syringe pump (Syrris) was used for carrier fluid delivery (1 mL min−1). A PEEK cross-piece (thru-hole 1 mm, immersed in a water bath held at 40 °C) was employed for combination of the three immiscible flows. The crystalliser length employed 11 m of single extruded fluoroethylene propylene (FEP) tubing (1/8′′ internal diameter) with a room temperature of 23.5 ± 0.5 °C (supersaturation ratio: 1.58–1.52). During the feed delivery and segmentation, all feed lines were actively heated (see the ESI† for details); post-segmentation the temperature gradient was effected through air-cooling to induce crystallisation of paracetamol form I. At the analysis point (6.7 m, 24.2 °C, S = 1.51 or 8.7 m, 23.6 °C, S = 1.55) a custom carriage mounted on a translation stage moved the tubing at the same speed as the fluid flow through the tubing in an obverse direction, thus maintaining the slug within the X-ray beam during analysis. A laser diode and sensor mounted on the carriage 22 mm prior to the analysis point monitored the progression of slugs through the tubing. At a pre-determined interval after the rear edge of a slug was detected (see the ESI† for details), the X-ray shutter and carriage were activated simultaneously and diffraction data were acquired for the 4.2 s of carriage movement. After this the shutter is closed and the carriage returns to the origin. Data were acquired at two crystalliser distances, 6.7 m (8 min 12 s RT) and 8.7 m (10 min 54 s RT) to compare different growth stages.
:40 (v/v). Air and Galden SV110 were used as the segmenting gas and carrier fluid respectively. SF-10 peristaltic pumps (Vapourtec UK) were used for air (2.5 mL min−1) and solution (2.7 mL min−1) delivery, and an ASIA dual syringe pump (Syrris) was used for carrier fluid delivery (1 mL min−1). A PEEK cross-piece (thru-hole 1 mm, immersed in a water bath held at 40 °C) was employed for combination of the three immiscible flows. The crystalliser length employed 11 m of single extruded fluoroethylene propylene (FEP) tubing (1/8′′ internal diameter) with a room temperature of 23.5 ± 0.5 °C (supersaturation ratio: 1.58–1.52). During the feed delivery and segmentation, all feed lines were actively heated (see the ESI† for details); post-segmentation the temperature gradient was effected through air-cooling to induce crystallisation of paracetamol form I. At the analysis point (6.7 m, 24.2 °C, S = 1.51 or 8.7 m, 23.6 °C, S = 1.55) a custom carriage mounted on a translation stage moved the tubing at the same speed as the fluid flow through the tubing in an obverse direction, thus maintaining the slug within the X-ray beam during analysis. A laser diode and sensor mounted on the carriage 22 mm prior to the analysis point monitored the progression of slugs through the tubing. At a pre-determined interval after the rear edge of a slug was detected (see the ESI† for details), the X-ray shutter and carriage were activated simultaneously and diffraction data were acquired for the 4.2 s of carriage movement. After this the shutter is closed and the carriage returns to the origin. Data were acquired at two crystalliser distances, 6.7 m (8 min 12 s RT) and 8.7 m (10 min 54 s RT) to compare different growth stages.
Results and discussion
Slug triggering and crystal positioning
Under the cooling crystallisation conditions explored, singular crystals appeared in 3% of slugs, and one in twenty slug triggering events resulted in successful co-location of the beam and crystal during stage translation. With a flow velocity ranging between 3–11 mm s−1 and slug separation of 20 mm, the overall hit rate of 0.15% means that a successful run (typically a 10-degree slice of reciprocal space) is achieved every 20 minutes. A significantly higher hit rate can be achieved with higher concentration sample stocks as many acquisitions were obtained from slugs without crystals. However, this dramatically increased the number of multiple crystals, reducing the number of single crystals in the beam path. In slugs containing multiple crystals, for those where the fluid flow paths led to non-overlapping crystals (residing in different regions of the slug) single crystal diffraction data could also be indexed.During the experiment the position of the crystal can vary as the crystal tumbles in the slug. If the position varies in the plane normal to the X-ray path the crystal will move in/out of the beam and can easily be observed in the indexing vs. images plots (Fig. S6†). If only part of the run is affected the processing can automatically remove blank images. In the circumstances that the crystal position varies co-linear to the X-ray beam the sample to detector distance varies and therefore the unit cell dimensions vary. From processing where the unit cell dimensions are allowed to freely refine we can observe the drift in axis observed for the duration of the 4.2 s collection. This variance in the parameter over time can be attributed to the changes in the sample-detector distance as the crystal moves within the tubing (±1.59 mm).
Crystal rotation and habit
It has previously been shown that segmented flow results in recirculating vortices at the fore and rear of a slug, and the internal vortex will promote the crystal to move to the rear of the slug and then induce tumbling or rotation of crystals in that slug (Fig. 2c).21,22 As can be intuitively inferred, video evidence shows that the smaller the crystals are, the faster they will rotate. | ||
| Fig. 2 Images of paracetamol crystals recovered from flow crystallisation experiments showing (a) predominance of rhombohedral habit with a parallelopiped and agglomerations, (b) stress fractures and surface texturing; (c) schematic of a segmented flow showing eddies to the rear and fore of each solution slug. | ||
The crystals at 6.7 m (8 min, 12 s), on average, are smaller than later in the crystallisation process at 8.7 m (10 min, 54 s). Analysis of the total crystal rotation showed that that the smaller crystals at 6.7 m have a bigger discrepancy in the total rotation with a number of crystal rotations far exceeding that of the largest rotation at 8.7 m (Fig. 2). This rotation is entirely random, which is beneficial for merging datasets as it increases the likelihood of achieving a high percentage of completeness through fewer crystals.
Fig. 2 compares the overall rotation from 60 data sets at both crystalliser distances as the sample angular displacement from its original orientation. Here it can be seen that the highest rotation at 8.7 m is 33.4° whilst the highest rotation at 6.7 m is 58.6°. Out of the 16 data sets obtained at 6.7 m 12 data sets show rotation comparable to that found at 8.7 m; this can be attributed to the stochastic nature of crystallisation. However, it must be noted that the different crystalliser lengths are not considering the same single crystal, nor can we correlate the crystal shape with diffraction data at this time. Of note a further challenge in acquiring single crystal diffraction datasets in segmented flow is that, occasionally the crystal does not remain within the X-ray beam but due to random movement in the flow, can be pushed out and sometimes back into the X-ray beam (see the ESI† for details), and this behaviour is more pronounced for smaller crystals. Crystals maintained a relative position within the beam during collection for the majority of datasets obtained.
Whilst we primarily obtained platelet crystals of a rhombohedral habit, we also found some parallelopiped prismatic crystals which will have a different rotational effect from the fluid dynamics within the slug (see Fig. 3). The bolus flow within each slug creates eddies to the rear and fore of each slug which are responsible for the rotation of the crystals, and the aspect ratio and available surface area will dictate the overall rotation of each crystal. In Fig. 3b it can be seen that the corners of the rhombohedral platelet crystals suffer from stress fractures. It is presumed that this is a result of reduced rotation of the crystals after they have reached a critical size and they cannot rotate freely in the vortices due to the liquid–liquid boundary of the slug. This phenomenon is also presumed to be responsible for the wavy surface texturing seen on the dominant face of the recovered crystals.
 | ||
| Fig. 3 Left: example of a random rotation of a single crystal during data acquisition (taken from 8.7 m; top a-axis, middle b-axis, and bottom c-axis); right: overall rotation for crystals at 6.7 m and 8.7 m crystallisation stages. | ||
Structure solution
The crystals held in the X-ray beam are not rotating around a standard diffractometer axis and thus conventional processing routines cannot be utilised. Instead, the creep indexing technique in the DIALS software was employed. In creep indexing an individual frame is indexed and UB matrix (orientation of the crystal) is determined. The orientation of the crystal in the next frame is assumed to be related by a small rotation to the first and thus the orientation can be refined from the previous. This process can be repeated for the entire sequence. Due to the nature of the collection with a reduced parameter to data ratio the instrument model (from calibration, see ESI, S1.1†) and input unit cell are constrained. The full DIALS commands can be found in the ESI (S4 DIALS processing).† Merging the best 13 datasets together (acquired at 8.7 m) allows for completion to 56.8% and the structure can be solved and refined with restraints to 0.8 Å, as shown in Table 1 and (Fig. 4). The quality of the structure is poor and not of publication quality but does show that the technique has potential with further set-up optimisation. As expected under the crystallisation conditions employed, form I was found with unit cell parameters: a = 7.25(16), b = 9.40(2), c = 11.93(3), and β = 97.3(2)° in the P21/n space group. The major challenges for structure solution result from a reduction in data quality due to the movement of the crystal both in and out of the beam (perpendicular to the X-rays, causing varied intensity) and towards and away from the detector (normal to the X-rays, causing ambiguity in the position of reflections) during data collection.| Total reflections | 2138 |
| Unique reflections | 936 |
| Completeness (%) | 56.8 |
| Mean I/σ(I) | 7.37 |
| Min D | 0.8004 |
| Max D | 7.3619 |
| R int | 0.1859 |
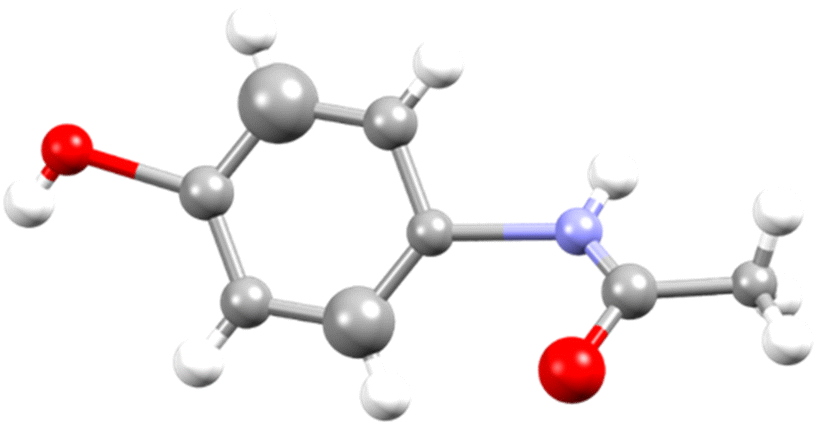 | ||
| Fig. 4 Isotropic image of an asymmetric unit for a merged structure using Olex2. C – grey, O – red, N – blue, and H – white. | ||
Conclusions
Segmented/slug flow crystallisers offer superior control over crystallisation conditions, increasing homogeneity, reproducibility and more efficient on-demand production. For this reason, pharmaceutical companies are paying close attention to the development of these techniques and the methods to monitor the process in situ.The segmented flow crystalliser can be used as alternative delivery methods for high-throughput crystallography where the crystal needs to be replenished due to being radiation sensitive, undergoes an irreversible transformation upon a stimulus or is unstable out of the crystallisation conditions.
In this paper, we describe the methods used to suspend a crystal in an X-ray beam to collect a partial dataset. This involves rotating the crystal over a small angular range, resulting in the collection of a limited segment (commonly referred to as a wedge) of reciprocal space. This wedge represents a small portion of the reciprocal lattice, which can be merged with additional wedges from other datasets to produce a complete dataset suitable for structure refinement. Applying this technique at different lengths along the crystalliser, we can monitor the crystallisation advancement through the apparatus which can be used to adjust the conditions to optimise the process.
The processing of crystals where the crystal rotates around a known axis is standard; however processing a randomly rotating crystal has not been explored previously. We have demonstrated the processing protocols for data reduction and how merging multiple runs helps to evaluate a refinable crystal structure. This processing routine paves the way for new crystallographic methods such as the processing of data collected on acoustically levitated crystals in the beam where the crystal will be randomly rotating.
The successful hit rate of acquiring diffraction data was low in the experiment (0.15%) and this can be improved through future upgrades to crystal tracking and linked stage movement.
Whilst there remain challenges in data quality stemming from crystal movement in and out of the detector and towards and away from the detector, we believe that this technique can be very powerful for both understanding the crystallisation behaviour of target compounds and for radiation sensitive materials due to the short exposure time required for each crystal.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lois E. Wayment: conceptualisation, methodology, formal analysis, writing – original draft, C Daniel Scott: methodology, Lucy K. Saunders: methodology, writing – review and editing, Pollyanna Payne: methodology, Lauren E. Hatcher: methodology, writing – review and editing, Graeme Winter: methodology, validation, Benjamin Williams: methodology, validation, David R. Allan: supervision, funding acquisition, Chick C. Wilson: conceptualisation, supervision, funding acquisition, Mark R. Warren: conceptualisation, methodology, formal analysis, writing – original draft, writing – review and editing, supervision, and Karen Robertson: conceptualisation, methodology, writing – original draft, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Diamond Light Source for awarding beamtime under proposal CY22497-1 and commissioning time MT21301-2. We would also like to thank the UK Engineering and Physical Sciences Research Council (EPSRC) Future Continuous Manufacturing and Advanced Crystallization Research Hub (EP/P006965/1, Wilson, Wayment, Payne), Diamond Light Source (Wayment), EPSRC Metastable Materials (EP/K004956/1, Hatcher, Robertson), and the EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies (EP/L016354/1, Scott) for funding.References
- A. Laybourn, K. Robertson and A. G. Slater, J. Am. Chem. Soc., 2023, 145(8), 4355–4365 CrossRef CAS PubMed.
- J. Christensen, P. N. Horton, C. S. Bury, J. L. Dickerson, H. Taberman, E. F. Garman and S. J. Coles, IUCrJ, 2019, 6, 703–713 CrossRef CAS PubMed.
- E. V. Boldyreva, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2024, 80, 1–3 CrossRef CAS.
- E. A. Schriber, D. W. Paley, R. Bolotovsky, D. J. Rosenberg, R. G. Sierra, A. Aquila, D. Mendez, F. Poitevin, J. P. Blaschke, A. Bhowmick, R. P. Kelly, M. Hunter, B. Hayes, D. C. Popple, M. Yeung, C. Pareja-Rivera, S. Lisova, K. Tono, M. Sugahara, S. Owada, T. Kuykendall, K. Y. Yao, P. J. Schuck, D. Solis-Ibarra, N. K. Sauter, A. S. Brewster and J. N. Hohman, Nature, 2022, 601, 360–365 CrossRef CAS PubMed.
- R. L. Owen, D. Axford, D. A. Sherrell, A. L. Kuo, O. P. Ernst, E. C. Schulz, R. J. D. Miller and H. M. Mueller-Werkmeister, Acta Acta Crystallogr. D Biol., 2017, 73, 373–378 CrossRef CAS PubMed.
- P. Mehrabi, H. M. Muller-Werkmeister, J. P. Leimkohl, H. Schikora, J. Ninkovic, S. Krivokuca, L. Andricek, S. W. Epp, D. Sherrell, R. L. Owen, A. R. Pearson, F. Tellkamp, E. C. Schulz and R. J. D. Miller, J. Synchrotron Radiat., 2020, 27, 360–370 CrossRef CAS PubMed.
- D. C. F. Monteiro, M. Vakili, J. Harich, M. Sztucki, S. M. Meier, S. Horrell, I. Josts and M. Trebbin, J. Synchrotron Radiat., 2019, 26, 406–412 CrossRef CAS PubMed.
- A. R. Pearson and P. Mehrabi, Curr. Opin. Struct. Biol., 2020, 65, 168–174 CrossRef CAS PubMed.
- T. A. White, Acta Crystallogr. D Biol. Crystallogr., 2019, 75, 219–233 CrossRef CAS PubMed.
- https://nxds.mr.mpg.de/ .
- https://dials.github.io/ .
- J. Brenker, K. Henzler, C. N. Borca, T. Huthwelker and T. Alan, Lab Chip, 2022, 22, 1214–1230 RSC.
- K. S. Pickering, S. Huband, K. L. Shafran and R. I. Walton, Chem.: Methods, 2022, 2(9), e202200033 CAS.
- H. G. Alison, R. J. Davey, J. Garside, M. J. Quayle, G. J. T. Tiddy, D. T. Clarke and G. R. Jones, Phys. Chem. Chem. Phys., 2003, 5, 4998–5000 RSC.
- M. Hadian-Jazi, P. Berntsen, H. Marman, B. Abbey and C. Darmanin, Crystals, 2021, 11, 49 CrossRef CAS.
- K. Robertson, P. H. Seeberger and K. Gilmore, React. Chem. Eng., 2023, 8, 77–83 RSC.
- K. C. Aroh and K. F. Jensen, React. Chem. Eng., 2018, 3, 94–101 RSC.
- M. A. Levenstein, L. Wayment, C. D. Scott, R. Lunt, P.-B. Flandrin, S. J. Day, C. C. Tang, C. C. Wilson, F. C. Meldrum, N. Kapur and K. Robertson, Anal. Chem., 2020, 92, 7754–7761 CrossRef CAS PubMed.
- A. R. Pallipurath, P. B. Flandrin, L. E. Wayment, C. C. Wilson and K. Robertson, Mol. Syst. Des. Eng., 2020, 5, 294–303 RSC.
- K. Robertson, P.-B. Flandrin, A. R. Klapwijk and C. C. Wilson, Cryst. Growth Des., 2016, 16, 4759–4764 CrossRef CAS.
- A. Günther, S. A. Khan, M. Thalmann, F. Trachsel and K. F. Jensen, Lab Chip, 2004, 4, 278–286 RSC.
- G. K. Kurup and A. S. Basu, Biomicrofluidics, 2012, 6(2), 022008 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08565e |
| This journal is © The Royal Society of Chemistry 2025 |