
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase effect of TiO2 on surface hydrogen adsorption/desorption in controlling photocatalytic methane conversion†
Jiakang
You
 a,
Ardeshir
Baktash
a,
Dazhi
Yao
b,
Yanzhao
Zhang
a,
Shanshan
Ding
a,
Jingwei
Hou
a,
Guangyu
Zhao
b,
Yonggang
Jin
b,
Zhiliang
Wang
*a and
Lianzhou
Wang
*a
a,
Ardeshir
Baktash
a,
Dazhi
Yao
b,
Yanzhao
Zhang
a,
Shanshan
Ding
a,
Jingwei
Hou
a,
Guangyu
Zhao
b,
Yonggang
Jin
b,
Zhiliang
Wang
*a and
Lianzhou
Wang
*a
aNanomaterials Centre, School of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Queensland 4072, Australia. E-mail: zhiliang.wang@uq.edu.au; l.wang@uq.edu.au
bCSIRO Mineral Resources, 1 Technology Court, Pullenvale, QLD 4069, Australia
First published on 8th May 2025
Abstract
Identifying the rate-determining step is crucial for designing an effective photocatalytic system. The surface adsorption/desorption behaviour of reactants has received much less attention in photocatalyst design because the charge separation and transfer in the bulk is commonly regarded as a more sluggish process. In this work, we investigate photocatalytic methane (CH4) conversion (PMC) on various titanium oxide (TiO2) surfaces, including rutile and anatase, and reveal that the influence of surface CH4 adsorption can outweigh the photogenerated charge separation and transfer. Specifically, the rutile TiO2 surface is totally inert for CH4 activation. Further theoretical calculations reveal the significance of the hydrogen-adsorption/desorption process during the initial C–H bond cleavage on the TiO2 surface. A reversible hydrogen adsorption/desorption process with a small Gibbs free energy not only enables the activation of the first C–H bond in CH4 but also ensures a timely clearance of surface-adsorbed species, leading to a continuous PMC process. The findings of the phase effect study on the interaction between the photocatalyst surface and hydrogen atoms provide new insights into the rational design of efficient photocatalysts towards PMC. It also highlights the gap in transferring the knowledge of photocatalytic water splitting into PMC.
Photocatalytic methane conversion (PMC) has received increasing attention due to its potential to mitigate the greenhouse effect of methane (CH4) and upgrade it into high-value products.1–6 As an emerging research frontier, a comprehensive understanding of the photocatalyst design principle is critical for a highly selective and efficient PMC process. Currently, the knowledge from photocatalytic water splitting is used to guide photocatalyst design.7–9 However, the PMC process is obviously different from the water-splitting system, making some of the knowledge accumulated from water-splitting reactions invalid.10–13 There is a clear need to explore the key selection criteria for CH4 conversion photocatalysts.
It is commonly known that photocatalysis is a complicated multi-step process, dominated by three key steps: light harvesting, charge separation and transfer (CST), and surface reaction.14–18 Identifying the rate-determining step is crucial for designing an effective photocatalytic system. In the photocatalytic water splitting process, the CST efficiency generally bottlenecks the whole process.19–21 Therefore, materials engineering has been implemented to create a higher CST driving force, with the surface properties of the photocatalysts being dramatically changed.22–25 However, the majority of PMC studies are conducted in a gas–solid reaction system, making this process sensitive to the photocatalyst surface.26 The experience from the conventional thermocatalytic CH4 conversion process has recognized the significance of surface properties in methane activation.27–29 Therefore, an in-depth understanding of how methane interacts with the photocatalyst surface will guide the future photocatalyst design for PMC.
The CH4 molecule possesses high symmetric order, exhibiting a highly stable structure with strong C–H bonds.30 Activating CH4, i.e. scissoring the first C–H bond, is regarded as the most challenging step in the PMC process. Numerous researchers have reported that lattice oxygen plays a significant role in CH4 activation, wherein CH4 is cracked on the surface of metal oxide photocatalysts and C–C coupling occurs on the supported metal cocatalysts.31–33 Despite the fact that many surface metal cocatalysts have been developed, only very limited metal oxides, including TiO2 and ZnO, were reported to achieve a successful PMC process.34–38 Taking the extensively studied TiO2 as an example, various photoactive phases, including anatase, rutile, and brookite, have been reported to be active in water splitting,39–43 and the formation of mixed-phase junctions has been confirmed to be an effective strategy for CST improvement.44–46 Nevertheless, current TiO2-based photocatalysts for PMC reactions are dominated by the anatase phase,2,47,48 while a fundamental understanding of the phase effect of photocatalysts on the interaction of CH4 on the catalyst surface is still limited.
In this work, we investigate the CH4 activation on different surfaces of TiO2, including rutile and anatase, and discover that the rutile TiO2 surface is completely inert for CH4 activation. In comparison, the anatase TiO2 exhibits a photocatalytic oxidative coupling of methane (POCM) to ethane (C2H6) activity of 2.1 mmol g−1 h−1. Further investigations into the anatase/rutile mixed phase system indicate that the presence of rutile TiO2 seriously deteriorates the CH4 conversion despite the improved CST capability from the phase junction. Further theoretical calculation suggests that the behaviour of hydrogen (H) adsorption and desorption after the first C–H bond cleavage on TiO2 plays a crucial role in the overall catalytic process. On rutile TiO2, the H is strongly bonded to the surface and blocks the CH4 activation pathway. In comparison, the H bonded to the anatase surface exhibits mild binding strength. Interestingly, it is discovered that the bond is sufficiently strong to activate CH4 molecules, yet weak enough to be removed, thereby exposing active sites for the subsequent methane reaction. These findings highlight the surface sensitivity during methane activation and indicate that the H-adsorption strength is an important parameter in designing new photocatalysts for efficient methane conversion.
A series of TiO2 photocatalysts containing different ratios of anatase and rutile phases were prepared via thermal annealing of commercial anatase TiO2 nanoparticles (Fig. S1†) in the temperature range of 700–1000 °C in a muffle furnace (samples denoted as T-700–1000). Synchrotron-based X-ray diffraction (XRD) patterns (Fig. 1a and S2†) proved the gradual crystal phase transition from anatase to rutile. The XRD peaks at 25.3° and 27.4° correspond to anatase (101) and rutile (110) TiO2, respectively,49 based on which the ratio of these two phases is quantitatively determined (Table S1†). This indicates that the TiO2 maintains the anatase phase below 700 °C, while the proportion of rutile TiO2 increases with elevated temperature and is fully converted into rutile at 1000 °C. Raman spectral analysis further confirms this gradual crystal phase transition with Raman shifts at 398, 517 and 641 cm−1 corresponding to anatase, while the shifts at 449 and 611 cm−1 are assigned to rutile (Fig. 1b).50,51
 | ||
| Fig. 1 The change in the TiO2 photocatalyst at different temperatures (from 700 °C to 1000 °C), as determined from (a) XRD patterns and (b) Raman spectra. (c) POCM performance of different TiO2-based photocatalysts (orange bar: C2H6; green bar: CO2). (d) The CH4 conversion rate (blue dot line) and the surface area change (black dot line) of different TiO2-based photocatalysts prepared at different temperatures. | ||
To evaluate the methane conversion rate, the as-prepared TiO2 photocatalysts were first loaded with gold (Au) nanoparticles as cocatalysts, which have been confirmed to be a good surface catalyst for ethane production.32,52 The Au cocatalyst can enhance the C2H6 selectivity by facilitating the desorption of methyl species and suppressing CH4 overoxidation to CO2.32,52 Inductively coupled plasma atomic emission spectroscopy (ICP-AES) confirms the Au amount to be around 3.8 wt% for all the samples. A stainless-steel flow reactor was used to evaluate the POCM performance with a flow rate of 126 mL min−1 (CH4/O2 gas ratio of 125/1) and a 300 W Xe lamp (full spectrum, Perfectlight Technology Co., Ltd) as the light source. As shown in Fig. 1c, T-700 shows an ethane production rate of 2.15 mmol g−1 h−1 together with a methane conversion rate of 5.60 mmol g−1 h−1. The corresponding apparent quantum yield of methane conversion is 8.89% (±0.32%) at the wavelength of 350 nm (see the ESI† for experimental details). Upon increasing the annealing temperature, a gradual decrease in the ethane production rate is observed. Eventually, when the sample is heated at 1000 °C, it shows no photoactivity at all. Meanwhile, a significant change in surface area is observed after the annealing process, as illustrated in Fig. 1d. As heating has changed several features (i.e., phase and surface area) of TiO2, we need to further study what dominated the key changes in the PMC process. Additionally, blank experiments were performed under identical conditions, with ultra-high-purity argon (Ar, 99.95%) used as the purge gas instead of CH4. When Ar was employed in place of CH4, no production of C2H6 was observed. This result confirms that the formation of C2H6 originates from the PMC. A long-term stability test was conducted using T-700, demonstrating stability over 20 hours (Fig. S3, see more discussion in Fig. S4†). These results safely exclude the impact of carbon contamination on PMC.53,54
Scanning electron microscopy (SEM) was conducted to observe the morphology of the as-prepared TiO2-based photocatalysts. As shown in Fig. 2a, the particles start to sinter together when the annealing temperature increases over 800 °C. Further combining with the phase change results in Fig. 1a and d, it is revealed that large particles should possess the rutile phase with a reduced surface area. According to previous studies and our observations, during the phase transformation in TiO2 nanoparticles, the rutile phase initially forms at the interfaces where anatase particles come into contact and undergo sintering (scheme in Fig. 2a).55,56 UV-visible light absorption spectra reveal a slightly extended light absorption range with increased heating temperatures (Fig. 2b), corresponding to the phase transfer from anatase (3.18 eV) to rutile (2.98 eV).57 We then applied the time-resolved photoluminescence (TRPL) decay to investigate the CST process of the photocatalysts. As shown in Fig. 2c and Table S2,† the formation of a mixed-phase junction extends the charge carrier lifetime from 1.54 ns to 7.65 ns. However, further increasing the rutile content leads to a reduction in the carrier lifetime to 4.49 ns for pure rutile TiO2.
 | ||
| Fig. 2 (a) Schematic illustration of TiO2 particle growth with the corresponding SEM images; (b) UV-vis spectra of commercial anatase annealed at different temperatures having different ratios of the rutile phase; (c) the TRPL decay curves of the as-prepared TiO2 photocatalysts. | ||
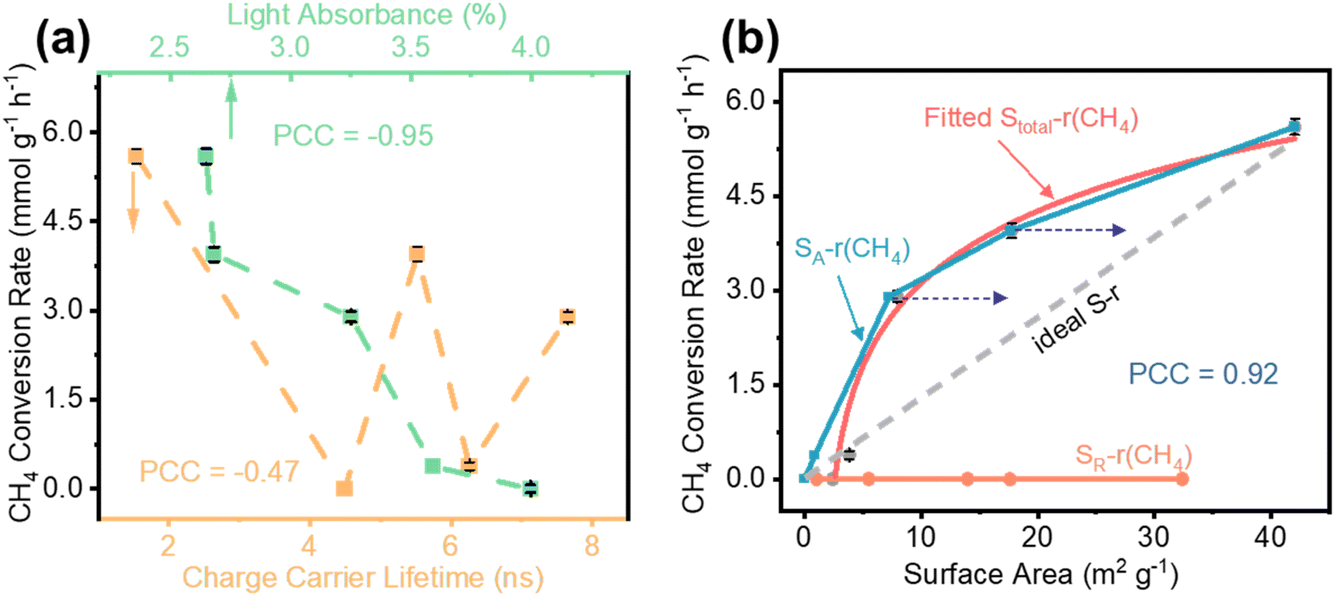
To analyze the correlation between light absorbance and CST with the CH4 conversion rate (r(CH4)), a Pearson Correlation Coefficient (PCC) analysis was performed (Table S3†). An absolute PCC value close to one indicates a strong correlation, while the sign of the value (positive or negative) indicates whether the correlation is positive or negative. It is generally accepted that extended light absorption ability and CST would benefit a photocatalytic process; therefore, we can expect a positive PCC if any of these two factors plays a determining role. Since the applied photocatalysts are loaded with Au nanoparticles, they show obvious localized surface plasmon resonance absorption at around 550 nm (Fig. S5†). However, the POCM test with a 420 nm cutoff filter shows no detectable product, which indicates that the LSPR effect cannot directly activate CH4. Therefore, the light absorbance was calculated based on the pure TiO2 samples (see the ESI† for calculation details). According to the light absorbance–r(CH4) relationship in Fig. 3a, the negative PCC (−0.95) means that better light harvesting will result in a worse performance, which is contradictory to the consensus of light harvesting influence on photocatalysis. This result suggests that light-harvesting is not the key feature in determining the CH4 conversion rate.
 | ||
| Fig. 3 CH4 conversion rate (r(CH4)) plotted against (a) the charge carrier lifetime (orange) and light absorbance (green) and (b) surface area. | ||
Moreover, the PCC of r(CH4)-τ is −0.47, indicating that the charge carrier lifetime is less determinative for the CH4 conversion process. In particular, the 850 °C annealing treatment has extended the photocatalyst lifetime by 5 times but reduced the CH4 conversion rate by ca. 50% compared to the samples treated at 700 °C. This finding is significantly different from the knowledge of photocatalytic water splitting wherein a longer lifetime usually leads to a better performance.58,59 Nevertheless, when taking the as-prepared samples for a photoelectrocatalytic water splitting test, it still shows that the sample with a longer lifetime (e.g., T-850) possesses higher photocurrent density (Fig. S6†). The distinguishable performance dependence on carrier lifetime for water splitting and POCM indicates an unexplored mechanism during the POCM process.
Since the CH4 reaction process occurs on the surface of catalysts in the gas phase, the surface area will play a critical role. We further analysed the change in the CH4 conversion rate as a function of surface areas of the TiO2 nanoparticles. As indicated in Fig. 3b, the surface area plays a significant role with a close correlation of the CH4 conversion. Using the TiO2 phase composition and surface area (S), a rough evaluation of the anatase surface area (SA) can be conducted, assuming a uniform phase mixture. A strong correlation is observed between the two variables, SA and r(CH4) (Fig. 3b, PCC = 0.92). When SA exceeds a threshold, the r(CH4) exhibits a minimal change. For example, despite the pristine anatase TiO2 having a much larger surface area than that of the 700 °C treated sample (retaining the anatase phase), they present nearly the same r(CH4), as shown in Fig. S7.† Meanwhile, according to previous studies, when phase transition happens in TiO2 nanoparticles, the rutile phase first emerges at the interfaces when anatase particles are sintered.55,56 It means that the majority of the TiO2 surface remains as the anatase phase, despite the emergence of rutile inside the particles. Taking this into consideration, the SA–r(CH4) relationship should closely resemble the ideal first-order reaction indicated by the grey dashed line in Fig. 3b. For comparison, we have prepared a series of rutile-based photocatalysts with different surface areas (see the ESI† for experimental details). None of these samples show POCM activity, as shown in Fig. 3b.
In particular, when comparing the rutile TiO2 nanoparticle-based photocatalyst with the T-850 sample, even though the rutile sample shows a higher surface area (Table S4†) and similar particle size (Fig. S8†) and Au loading features (Fig. S9†), it is completely inactive for POCM (Fig. 3b). The strong dependence of r(CH4) on the anatase surface area and the nonactivity of rutile photocatalysts indicate that anatase is the only photoactive phase for methane activation. The surface properties play a more dominating role compared with the CST process, which is clearly different from the photocatalytic water splitting process. Considering that the anatase and rutile TiO2 possess very similar features, it is worth further investigation on how such a tiny change can lead to a significant difference in methane activation.
Previous studies have revealed that methane activation starts from CH4 cracking on the metal oxide surface with the participation of lattice oxygen.60,61 Since oxygen can be consumed during methane activation, it will lead to the formation of oxygen vacancies (VOs). We thus further applied in situ electron paramagnetic resonance (EPR) measurements to trace the VO formation and investigate the interaction of CH4 on the rutile and anatase surfaces. The EPR spectra for anatase- and rutile-based samples were measured under illumination without (Fig. S10a) and with (Fig. S10b†) methane (see the ESI† for experimental details). Methane activation dependence on different phases is revealed via the differential spectra shown in Fig. 4a. A significant increase of VO (g = 2.003) and associated Ti3+ signals (g = 1.98) can be observed on anatase when exposed to a CH4 atmosphere, wherein a much weaker VO signal and undetectable Ti3+ signals are observed on the rutile phase. This result further confirms our hypothesis that CH4 activation on anatase is much more favourable.
 | ||
| Fig. 4 (a) In situ EPR spectra of anatase and rutile; (b) computed Gibbs free energy of methane activation steps on anatase (101) and rutile (110) surfaces; (c) TPD-CH4-MS spectra of as-prepared TiO2 photocatalysts; (d) schematic illustration of surface adsorption behaviour of CH4 on rutile and anatase, respectively. | ||
To acquire a deeper understanding of this methane activation process, a theoretical calculation on the first C–H bond activation with the following process (1) on rutile and anatase surfaces was performed:
| CH4 → *CH4 → *H + CH3 | (1) |
In this theoretical calculation, Gibbs free energy is applied to evaluate this process at room temperature. The anatase (101) and rutile (110) surfaces are used for the calculation due to the significant diffraction peaks of these facets (Fig. 1a) and the widely accepted choice in previous research.66–68 As shown in Fig. 4b, the Gibbs free energy is 0.49 eV on anatase TiO2, while it is −1.13 eV on the rutile TiO2. Furthermore, the H adsorption energy (E(Hads)) on rutile and anatase was calculated to be −1.40 eV and 0.21 eV, respectively. This result indicates that H adsorption on rutile is very stable, making it eventually impossible to be desorbed from the rutile TiO2 surface. In comparison, H adsorption on anatase TiO2 is moderately endothermic, which can be easily overcome by external stimulation (such as light excitation) to enable process (1). Meanwhile, the small Gibbs free energy also facilitates the H desorption process.
The H adsorption/desorption behaviour is significant for the continuous conversion of CH4. Although process (1) is important for CH4 activation, the H desorption is critical in clearing the TiO2 surface and allowing continuous CH4 activation. The dramatic negative Gibbs free energy on rutile makes process (1) irreversible, terminating the CH4 activation process, whereas process (1) on anatase is reversible, enabling the continuous CH4 conversion. To further investigate the H desorption feature, temperature-programmed desorption mass spectrometry of CH4 (TPD-MS-CH4) experiments were carried out. An obvious H2 molecular signal in Fig. 4c indicates the cleavage of CH4 molecules on the catalyst. Moreover, the rutile phase TiO2 shows much higher H2 desorption temperatures than the anatase phase, indicating a stronger H bonding energy on rutile TiO2. Fig. 4d schematically explains the difference in CH4 activation on rutile and anatase surfaces. On the rutile surface, strong bonding between hydrogen atoms and surface sites leads to higher energy barriers for desorption, whereas on the anatase surface, weaker interactions facilitate desorption more easily. This variation in desorption capability can significantly tune the PMC performance.
In conclusion, we demonstrated that the rutile TiO2 surface is inert during CH4 activation, and the influence of surface CH4 adsorption can outweigh the CST improvement when a phase junction is formed between anatase and rutile. Further investigation on CH4 activation reveals the significant impact of the H-adsorption/desorption process during C–H bond breaking. A reversible H-adsorption/desorption with a small Gibbs free energy not only enables the activation of the first C–H bond in CH4 but also ensures the removal of surface adsorbed species to facilitate the continuous process of photocatalytic CH4 conversion. This research highlights the critical role of H desorption for continuous CH4 activation and provides a new understanding of the design of efficient photocatalysts for methane conversion to produce value-added chemicals.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. W. and Z. W. proposed the research direction, designed the project, and guided the project. J. Y., L. W., and Z. W. designed various experiments. A. B. performed the theoretical calculation. J. Y., D. Y., Y. Z., and S. D. performed the experiments (synthesis, characterisations, catalysis, mechanism) and analysed the data. The overall manuscript was written by J. Y. and Z. W. Everyone commented on the manuscript. G. Z., Y. J., and J. H. participated in editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the Australian Research Council through its Future Fellowship (FT230100251), DECRA (DE210100930), Discovery (DP200101900) and Laureate Fellowship (FL190100139) schemes. This work used the Queensland node of the NCRIS-enabled Australian National Fabrication Facility (ANFF). This work was performed in part at the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and microfabrication facilities for Australia's researchers. The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. The authors acknowledge access to computational resources at the NCI National Facility through the National Computational Merit Allocation Scheme supported by the Australian Government. This research was undertaken on the PD beamline at the Australian Synchrotron, part of ANSTO. J. Y. acknowledges scholarship support from UQ Graduate School.References
- S. Shoji, X. Peng, A. Yamaguchi, R. Watanabe, C. Fukuhara, Y. Cho, T. Yamamoto, S. Matsumura, M.-W. Yu, S. Ishii, T. Fujita, H. Abe and M. Miyauchi, Nat. Catal., 2020, 3, 148–153 CrossRef CAS
.
- Z. Liu, B. Xu, Y.-J. Jiang, Y. Zhou, X. Sun, Y. Wang and W. Zhu, ACS Environ. Au, 2023, 3, 252–276 CrossRef CAS PubMed
- Q. Li, Y. Ouyang, H. Li, L. Wang and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202108069 CrossRef CAS
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617–632 CrossRef CAS
- J. F. Kasting, K. J. Zahnle and J. C. G. Walker, Precambrian Res., 1983, 20, 121–148 CrossRef CAS
- J. You, Y. Bao, Y. Zhang, M. Konarova, Z. Wang and L. Wang, EES Catal., 2024, 2, 1210–1227 RSC
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- W. Cao, W. Zhang, L. Dong, Z. Ma, J. Xu, X. Gu and Z. Chen, Exploration, 2023, 3, 20220169 CrossRef CAS
- Y. Jiang, Y. Fan, S. Li and Z. Tang, CCS Chem., 2023, 5, 30–54 CrossRef CAS
- X.-Y. Lin, J.-Y. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Catal. Commun., 2021, 159, 106346 CrossRef CAS
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, ACS Catal., 2014, 4, 3013–3019 CrossRef
- Z. Zhu, W. Guo, Y. Zhang, C. Pan, J. Xu, Y. Zhu and Y. Lou, Carbon Energy, 2021, 3, 519–540 CrossRef CAS
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC
- R. Qian, H. Zong, J. Schneider, G. Zhou, T. Zhao, Y. Li, J. Yang, D. W. Bahnemann and J. H. Pan, Catal. Today, 2019, 335, 78–90 CrossRef CAS
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS
- Z. Wang, Y. Gu, L. Zheng, J. Hou, H. Zheng, S. Sun and L. Wang, Adv. Mater., 2022, 34, 2106776 CrossRef CAS
- Y. Qi, S. Chen, J. Cui, Z. Wang, F. Zhang and C. Li, Appl. Catal., B, 2018, 224, 579–585 CrossRef CAS
- C. Cheng, B. He, J. Fan, B. Cheng, S. Cao and J. Yu, Adv. Mater., 2021, 33, 2100317 CrossRef CAS
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed
- S. Odabasi Lee, S. K. Lakhera and K. Yong, Adv. Energy Sustainability Res., 2023, 4, 2300130 CrossRef CAS
- R. Yanagi, T. Zhao, D. Solanki, Z. Pan and S. Hu, ACS Energy Lett., 2022, 7, 432–452 CrossRef CAS
- Y. Jiang, S. Li, Y. Fan and Z. Tang, Angew. Chem., Int. Ed., 2024, 63, e202404658 CrossRef CAS
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS
- S. Song, H. Song, L. Li, S. Wang, W. Chu, K. Peng, X. Meng, Q. Wang, B. Deng, Q. Liu, Z. Wang, Y. Weng, H. Hu, H. Lin, T. Kako and J. Ye, Nat. Catal., 2021, 4, 1032–1042 CrossRef CAS
- P. Wang, R. Shi, Y. Zhao, Z. Li, J. Zhao, J. Zhao, G. I. N. Waterhouse, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304301 CrossRef CAS PubMed
- M. Xiao, L. Wang, H. Wang, J. Yuan, X. Chen, Z. Zhang, X. Fu and W. Dai, Catal. Sci. Technol., 2023, 13, 4148–4155 RSC
- J. Lang, Y. Ma, X. Wu, Y. Jiang and Y. H. Hu, Green Chem., 2020, 22, 4669–4675 RSC
- W. Zhang, C. Fu, J. Low, D. Duan, J. Ma, W. Jiang, Y. Chen, H. Liu, Z. Qi, R. Long, Y. Yao, X. Li, H. Zhang, Z. Liu, J. Yang, Z. Zou and Y. Xiong, Nat. Commun., 2022, 13, 2806 CrossRef CAS
- X. Li, J. Xie, H. Rao, C. Wang and J. Tang, Angew. Chem., Int. Ed., 2020, 59, 19702–19707 CrossRef CAS
- X. Li, C. Wang, J. Yang, Y. Xu, Y. Yang, J. Yu, J. J. Delgado, N. Martsinovich, X. Sun, X.-S. Zheng, W. Huang and J. Tang, Nat. Commun., 2023, 14, 6343 CrossRef CAS PubMed
- X. Li, C. Li, Y. Xu, Q. Liu, M. Bahri, L. Zhang, N. D. Browning, A. J. Cowan and J. Tang, Nat. Energy, 2023, 8, 1013–1022 CrossRef CAS
- R. Li, Y. Weng, X. Zhou, X. Wang, Y. Mi, R. Chong, H. Han and C. Li, Energy Environ. Sci., 2015, 8, 2377–2382 RSC
- R. Abe, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 16052–16061 CrossRef CAS
- K. Maeda, Chem. Commun., 2013, 49, 8404–8406 RSC
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS
- W. Kim, T. Tachikawa, G.-h. Moon, T. Majima and W. Choi, Angew. Chem., Int. Ed., 2014, 53, 14036–14041 CrossRef CAS
- Y. Ma, X. Wang and C. Li, Chin. J. Catal., 2015, 36, 1519–1527 CrossRef CAS
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS
- J. Chen, T. Chen, Q. Fang, C. Pan, O. U. Akakuru, W. Ren, J. Lin, A. Sheng, X. Ma and A. Wu, Exploration, 2022, 2, 20220014 CrossRef CAS PubMed
- N. Feng, H. Lin, H. Song, L. Yang, D. Tang, F. Deng and J. Ye, Nat. Commun., 2021, 12, 4652 CrossRef CAS
- Y. Jiang, W. Zhao, S. Li, S. Wang, Y. Fan, F. Wang, X. Qiu, Y. Zhu, Y. Zhang, C. Long and Z. Tang, J. Am. Chem. Soc., 2022, 144, 15977–15987 CrossRef CAS PubMed
- W.-K. Wang, J.-J. Chen, X. Zhang, Y.-X. Huang, W.-W. Li and H.-Q. Yu, Sci. Rep., 2016, 6, 20491 CrossRef CAS PubMed
- C. R. Aita, Appl. Phys. Lett., 2007, 90(21), 213112 CrossRef
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912 CrossRef CAS
- D. Hu, C. Dong, S. Belhout, S. Shetty, H. Ng, P. Brasseur, L. S. Bezerra, K. Ben Tayeb, P. Simon, A. Addad, M. Virginie, R. Wojcieszak, V. V. Ordomsky, P. H. C. Camargo and A. Y. Khodakov, Mater. Today Energy, 2023, 36, 101358 CrossRef CAS
- J. You, M. Xiao, S. Liu, H. Lu, P. Chen, Z. Jiang, W. Shangguan, Z. Wang and L. Wang, J. Mater. Chem. A, 2023, 11, 10149–10154 RSC
- Y. Zhang, D. Yao, B. Xia, M. Jaroniec, J. Ran and S.-Z. Qiao, ACS Energy Lett., 2022, 7, 1611–1617 CrossRef CAS
- J. Zhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927–935 CrossRef CAS
- J. Zhang, Q. Xu, M. Li, Z. Feng and C. Li, J. Phys. Chem. C, 2009, 113, 1698–1704 CrossRef CAS
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef CAS PubMed
- R. Li, T. Takata, B. Zhang, C. Feng, Q. Wu, C. Cui, Z. Zhang, K. Domen and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202313537 CrossRef CAS
- G. Zhai, L. Cai, J. Ma, Y. Chen, Z. Liu, S. Si, D. Duan, S. Sang, J. Li, X. Wang, Y.-A. Liu, B. Qian, C. Liu, Y. Pan, N. Zhang, D. Liu, R. Long and Y. Xiong, Sci. Adv., 2024, 10, eado4390 CrossRef CAS PubMed
- Z. Li, P. Chen, J. Feng, M. Zhao, Z. Zhao, Y. Zhang, X. Xu, H. Huang, Z. Zou and Z. Li, Angew. Chem., Int. Ed., 2024, e202409876 CAS
- A. Ishikawa and Y. Tateyama, ACS Catal., 2021, 11, 2691–2700 CrossRef CAS
- X. Wang, N. Luo and F. Wang, Chin. J. Chem., 2022, 40, 1492–1505 CrossRef CAS
- Q. Zhan, Y. Kong, X. Wang and L. Li, Chem. Commun., 2024, 60, 2732–2743 RSC
- J. Zhang, J. Shen, D. Li, J. Long, X. Gao, W. Feng, S. Zhang, Z. Zhang, X. Wang and W. Yang, ACS Catal., 2023, 13, 2094–2105 CrossRef CAS
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H. M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed
- Y. Chen, L. Soler, C. Cazorla, J. Oliveras, N. G. Bastús, V. F. Puntes and J. Llorca, Nat. Commun., 2023, 14, 6165 CrossRef CAS
- L. Li, J. Yan, T. Wang, Z.-J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00888c |
| This journal is © The Royal Society of Chemistry 2025 |