
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBayesian molecular optimization for accelerating reverse intersystem crossing†
Taehyun
Won
a,
Naoya
Aizawa
 *abcd,
Yu
Harabuchi
*de,
Reo
Kurihara
a,
Mitsuharu
Suzuki
a,
Satoshi
Maeda
def,
Yong-Jin
Pu
c and
Ken-ichi
Nakayama
a
*abcd,
Yu
Harabuchi
*de,
Reo
Kurihara
a,
Mitsuharu
Suzuki
a,
Satoshi
Maeda
def,
Yong-Jin
Pu
c and
Ken-ichi
Nakayama
a
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
bCenter for Future Innovation, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cRIKEN Center for Emergent Matter Science (CEMS), Wako, Saitama 351-0198, Japan
dInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido 001-0021, Japan
eJST, ERATO Maeda Artificial Intelligence in Chemical Reaction Design and Discovery Project, Sapporo, Hokkaido 060-0810, Japan
fDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan
First published on 15th April 2025
Abstract
Spin conversion in molecular excited states is crucial for the development of next-generation optoelectronic devices. However, optimizing molecular structures for rapid spin conversion has relied on time-consuming experimental trial-and-error, which limits the elucidation of the structure–property relationships. Here, we report a Bayesian molecular optimization approach that accelerates virtual screening for rapid triplet-to-singlet reverse intersystem crossing (RISC). One of the molecules identified through this virtual screening exhibits a fast RISC rate constant of 1.3 × 108 s−1 and a high external electroluminescence quantum efficiency of 25.7%, which remains as high as 22.8% even at a practical luminance of 5000 cd m−2 in organic light-emitting diodes. Post-hoc analysis of the trained machine learning model reveals the impact of molecular structural features on spin conversion, paving the way for informed and precise materials development.
Introduction
Excitonic spin conversion is a key process driving innovation in various optoelectronic applications.1–5 In the case of organic light-emitting diodes (OLEDs), the internal quantum efficiency is limited by inherent spin statistics that dictate a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of singlet to triplet excitons from electron–hole recombination, resulting in a maximum efficiency of 25%. This limitation is due to the fact that triplet excitons in most organic molecules are dark states, which are spin-forbidden from decaying radiatively to the ground states. Researchers have explored the use of reverse intersystem crossing (RISC), where triplet excitons are converted into emissive singlet excitons to overcome this issue.6–18 RISC is driven by ambient thermal energy, resulting in thermally activated delayed fluorescence (TADF) and allowing OLEDs to achieve internal quantum efficiencies approaching 100%.
:3 ratio of singlet to triplet excitons from electron–hole recombination, resulting in a maximum efficiency of 25%. This limitation is due to the fact that triplet excitons in most organic molecules are dark states, which are spin-forbidden from decaying radiatively to the ground states. Researchers have explored the use of reverse intersystem crossing (RISC), where triplet excitons are converted into emissive singlet excitons to overcome this issue.6–18 RISC is driven by ambient thermal energy, resulting in thermally activated delayed fluorescence (TADF) and allowing OLEDs to achieve internal quantum efficiencies approaching 100%.
The time constants of RISC are generally in the order of microseconds,19–22 which presents a challenge in the form of competing bimolecular recombination processes, such as triplet–triplet annihilation. Such detrimental processes become increasingly dominant at higher current densities in OLEDs, resulting in an appreciable decrease in device efficiency, a phenomenon known as efficiency roll-off.23 Thus, accelerating RISC and reducing efficiency roll-off are important hurdles to overcome for the practical use of TADF molecules in high-performance OLEDs.
Traditionally, TADF materials have been discovered through time-intensive experimental approaches; however, the advent of high-throughput virtual screening has offered a significant change in the materials design process.24–29 By employing computationally inexpensive density functional theory (DFT) and machine learning techniques, researchers can extensively explore the relevant molecular space for potential TADF materials with a small singlet–triplet energy gap (ΔEST). However, despite these advances, calculating ΔEST alone remains an inefficient way to estimate the rate constant of RISC (kRISC) and narrow down the most promising candidate molecules.
In the simplest physical picture, RISC is driven by a spin–orbit coupling at the crossing seam between the potential energy surfaces of singlet and triplet excited states. In cases of weak spin–orbit coupling in organic molecules, kRISC is expected to follow the Marcus-type non-adiabatic rate expression:
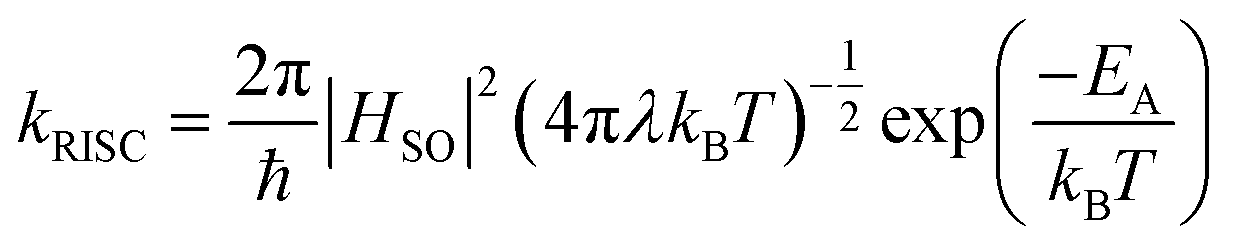 | (1) |
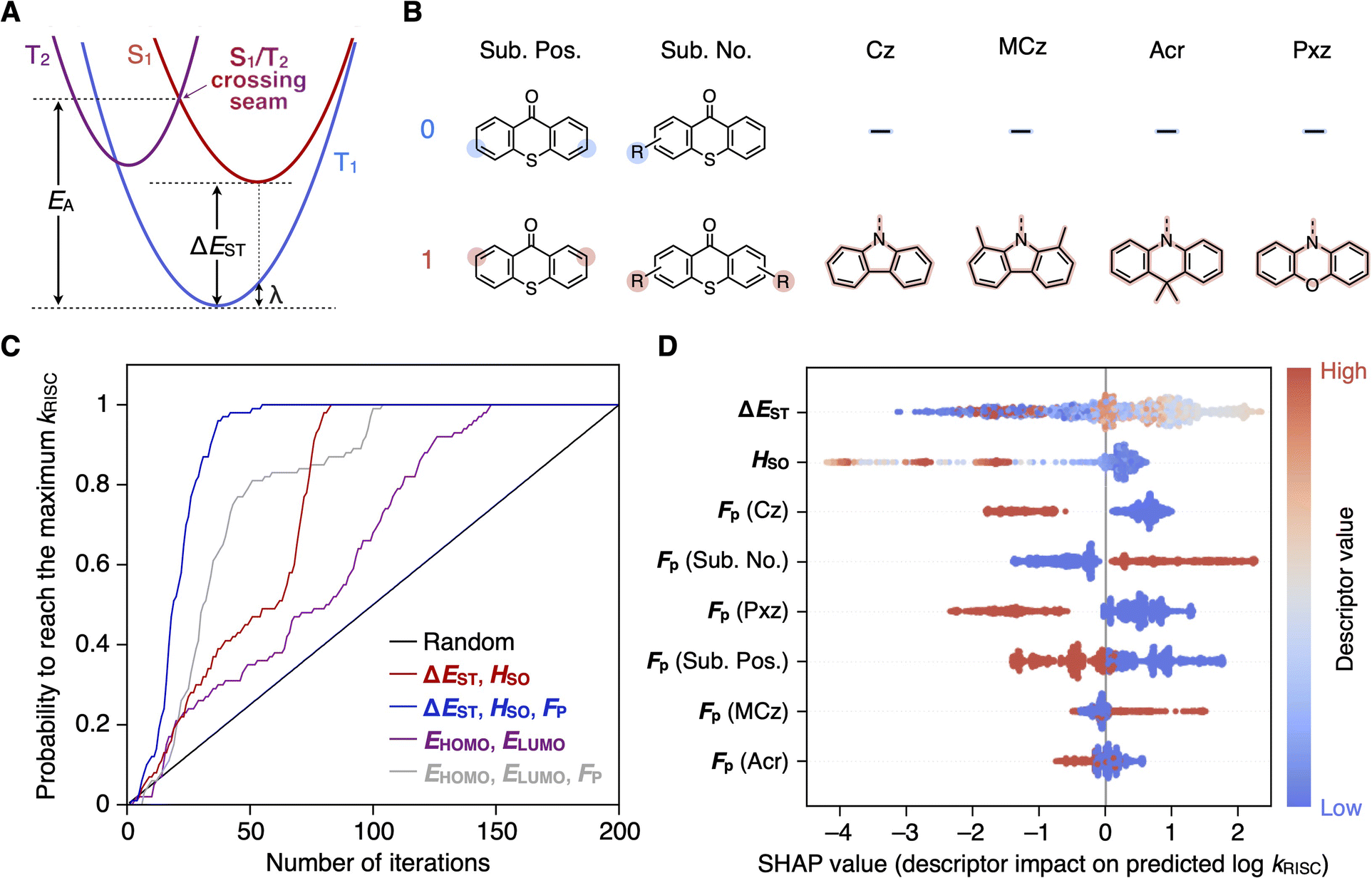 | ||
| Fig. 1 (A) Schematic potential energy surfaces of excited states associated with RISC. (B) Binary molecular fingerprints FP for classifying candidate molecules in a six-dimensional space. (C) Comparison of Bayesian molecular optimization using different descriptor combinations and uniform random sampling for identifying the molecule with the highest kRISC among 200 candidates for pre-training. Each line shows the probability of successful identification after a given number of iterations. (D) Beeswarm plots of the SHAP values for each descriptor, showing their impact on the model output (log10kRISC). A high Fp (Sub. Pos.) value corresponds to substitutions at the 2,7-positions (meta-positions) of thioxanthone, while a low Fp (Sub. Pos.) value corresponds to substitution at the 3,6-positions (para-positions). | ||
Here we implement a machine learning technique called Bayesian optimization to computationally screen for TADF molecules with high kRISC. While conventional machine learning models for virtual screening require large number of training datasets, Bayesian optimization iteratively trains a probabilistic surrogate model with a limited number of datasets, strategically selecting the next data points to evaluate based on both exploration of uncertain space and exploitation of known space.37–42 This dual focus allows Bayesian optimization to rapidly identify the optimal molecules with a minimized number of high-fidelity excited-state calculations.
In the following section, we describe the development of our Bayesian optimization-based virtual screening that identified a TADF molecule with experimentally determined kRISC of 1.3 × 108 s−1. The external electroluminescence quantum efficiencies of the identified molecule reached 25.7%, which remained as high as 22.8% even at a practical luminance of 5000 cd m−2.
Results and discussion
Search space generation
Typical TADF molecules contain donor and acceptor units that mediate low-lying singlet and triplet charge-transfer excited states (1CT and 3CT) between the spatially separated highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO); such CT states result in a low singlet–triplet exchange energy, enabling RISC with ambient thermal energy. Spin flipping in RISC is driven by spin–orbit coupling at the singlet–triplet crossing seam, where an orbital transition between 1CT and a triplet local excitation state (3LE) induces a relativistic interaction capable of spin flipping. To further enhance spin–orbit coupling and accelerate RISC, one can advantageously exploit the heavy atom effect of the group 16 and 17 atoms such as S, Se, Br, and I.43–51 With these principles in mind, we generated a search space of 1.4 thousand molecules, all sharing a common thioxanthone acceptor unit with vastly different donor units. To ensure synthetic feasibility, we employed a donor–acceptor structural design (Dn–A, n = 1 or 2) that adheres to a set pattern dictating the substitution positions and symmetry (Fig. 1B). Each molecule incorporates a single type of donor unit, classifiable into four categories: carbazole (Cz), 1,8-dimethylcarbazole (MCz), 9,9-dimethylacridane (Acr), and phenoxazine (Pxz). The molecular weight of the candidates was limited to less than 1200 g mol−1 due to the need for thermal evaporation in OLED fabrication. All molecular structures in the search space are listed in ‘ESI†’.For each molecule, four quantum chemical properties were represented as descriptors for a vector search space: EHOMO, ELUMO, ΔEST, and HSO, derived from low-cost DFT calculations with the 6-31G basis set without diffuse and polarization functions, as detailed in ‘Methods’. We also used in-house binary molecular fingerprints FP as descriptors, which explicitly represent the number, positions, and categories of donor units (i.e., Cz, MCz, Acr, or Pxz) to classify candidate molecules in a six-dimensional space (Fig. 1B).
Pre-training
To identify the most effective combinations of descriptors for search performance, we tested Bayesian optimization with different descriptor sets on a search space consisting of 200 molecules; the kRISC values of these molecules were pre-computed and stored in a database, using high-fidelity excited-state calculations including geometry optimization of the singlet–triplet crossing seam described in ‘Methods’. Beginning with a randomly selected molecule, the corresponding kRISC value was retrieved from the database to train a Gaussian process surrogate model;52 an acquisition function was then used to select a molecule in the search space based on the expected improvement criterion for the next evaluation. This procedure continued iteratively until the molecule with the highest kRISC in the 200 molecules was found.Fig. 1C compares the search performance of the Bayesian molecular optimization with different descriptor sets to that of uniform random sampling. This comparison evaluates the cumulative probability of finding the molecule with the highest kRISC as a function of the number of iterations, derived from 100 independent trials of the Bayesian optimization procedures, each with a different starting molecule. Essentially, Bayesian optimization outperforms uniform random sampling; however, its performance is highly dependent on the descriptor sets. In all 100 independent Bayesian optimization trials, the descriptor set (ΔEST, HSO) consistently found the highest kRISC within 82 iterations, whereas (EHOMO, ELUMO) required up to 148 iterations. Superficially, the superior performance of (ΔEST, HSO) is not surprising, given their physical relationship to kRISC. What is surprising, however, is that the DFT calculations of these descriptors rely on overly broad approximations to remain cost-effective, whose results may not even qualitatively align with those of the high-fidelity calculations for kRISC. Such circumstances suggest that the surrogate model captures the complex correlation between the descriptors and kRISC.
Combining FP with the descriptors further improves the performance of Bayesian optimization, locating the maximum point within only 55 iterations using (ΔEST, HSO, FP). Incorporating FP offers an additional advantage of chemical interpretability, allowing us to quantify the impact of molecular features on kRISC based on the trained surrogate model, as will be discussed later.
Shortlisting and analysis
Having established the effective descriptor set (ΔEST, HSO, FP), we proceeded to examine the full search space of 1.4 thousand candidate molecules. Starting with the pre-trained data for the 200 molecules, 100 additional iterations of Bayesian optimization led to the identification of 78 candidate molecules with quantum-chemically calculated kRISC over 107 s−1, an order of magnitude higher than that of the seminal TADF molecule 2,4,5,6-tetra(carbazol-9-yl)isophthalonitrile (4CzIPN).6 At the end of the optimization, the trained surrogate model achieved a coefficient of determination (R2) of 0.91 and a root mean squared error (RMSE) of 0.88 in predicting log10kRISC, as verified through tenfold cross-validation. The trained model suggests 129 molecules with predicted kRISC exceeding 108 s−1, corresponding to 9% of all the candidate molecules. We then assessed the synthetic feasibility of these molecules and selected MCz-p2 for further experimental evaluation, as will be discussed later. We note that the model was able to reidentify 3,6-bis(1,3,6,8-tetramethylcarbazol-9-yl)thioxanthone (MCz-TXT),46 previously reported with an exceptionally high kRISC of 1.1 × 108 s−1.To extract chemical knowledge from the trained machine learning model, we assessed the descriptor importance using Shapley additive explanations (SHAP). This method assigns a SHAP value to each descriptor, quantifying its contribution to the model output by fairly distributing the difference between a given output and the average across all descriptors.53Fig. 1D presents a series of beeswarm plots of the SHAP values for each descriptor in the trained model. A positive SHAP value indicates that a specific descriptor value, represented by the color, increases the model output (i.e., log10kRISC) above the average, whereas a negative SHAP value indicates that it decreases the output below the average. A wider spread of the SHAP values for ΔEST and HSO in Fig. 1D suggests that these two descriptors are the most influential. However, the relationship between the descriptor values and the SHAP values appears to be nonlinear, implying that a smaller ΔEST and a larger HSO do not necessarily equate to a higher kRISC. These results highlight the limitations of the low-cost DFT outputs, and the importance of accounting for the spin-vibronic effects through the high-fidelity calculations of singlet–triplet crossing seams in predicting kRISC. For FP, a simpler pattern emerges in the SHAP plots: the presence of MCz positively correlates with the output, whereas the other descriptors (Cz, Pxz, and Acr) tend to correlate negatively with the output. The SHAP plots for FP also indicate that two substitutions, especially at the 3,6-positions (para-positions) of thioxanthone, are more beneficial for enhancing kRISC than one substitution or those at the 2,7-positions (meta-positions).
To understand the mechanism responsible for the trends in the importance of FP for kRISC, excited-state energy landscapes were established for seven model molecules by the high-fidelity TD-DFT calculations (Fig. 2A and S1† for relevant FP). Four molecules, MCz-m1, MCz-m2, MCz-p1, and MCz-p2, share common MCz donor units, but possess different substitution numbers and positions. The calculated adiabatic ΔEST of MCz-p1 is smaller than that of MCz-m1 by increasing the substitution number from one to two and changing the meta-substitution to para-substitution, resulting in a lower EA (the activation energy to reach the minimum-energy singlet–triplet crossing seam from equilibrium T1) and thus a higher kRISC of MCz-p2 (Table 1). Furthermore, the singlet–triplet crossing seam of MCz-p2 comprises more distinct orbital characters (1CT and 3LE) compared to MCz-m2 (1CT and 3CT + 3LE) (Fig. S2†), which is responsible for a higher HSO of MCz-p2, consistent with El-Sayed's rule.54 Additional calculations reveal that the 3,6-positions of thioxanthone are located at the nodes of its HOMO (Fig. 2B), leading us to interpret that the para-substitution of donor units suppresses π-conjugation and thus maintain a relatively pure 3LE on the thioxanthone unit, as in MCz-p2, resulting in higher kRISC.
 | ||
| Fig. 2 (A) Molecular structures of seven thioxanthone derivatives with their relative excitation energies of S1, T1, and T2 calculated at the optimized S1 geometry, T1 geometry, and S1/T2 crossing seam where the two states are equal in energy and relativistically coupled by a weak HSO. (B) HOMO and LUMO of thioxanthone at C2v symmetry. | ||
| Molecule | E S1 (eV) | E T1 (eV) | ΔESTc (eV) | E A (eV) | λe (eV) | H SO (cm−1) | k RISC (s−1) |
|---|---|---|---|---|---|---|---|
| a Vertical S0–S1 excitation energy at the optimized S1 geometry. b Vertical S0–T1 excitation energy at the optimized T1 geometry. c Adiabatic energy gap between S1 and T1 at their respective optimized geometries. d Activation energy from the optimized T1 state to the minimum-energy singlet–triplet crossing seam. e Reorganization energy from the optimized T1 state to the optimized S1 state. f Spin–orbit coupling matrix element between S1 and T2 at the minimum-energy singlet–triplet crossing seam. g Rate constant of RISC calculated by using eqn (1). | |||||||
| MCz-m1 | 2.550 | 2.341 | 0.262 | 0.264 | 0.227 | 2.32 | 1.10 × 105 |
| MCz-m2 | 2.334 | 2.254 | 0.161 | 0.191 | 0.134 | 1.44 | 9.20 × 105 |
| MCz-p1 | 2.724 | 2.518 | 0.261 | 0.269 | 0.169 | 3.96 | 3.05 × 105 |
| MCz-p2 | 2.524 | 2.257 | 0.110 | 0.121 | 0.0953 | 6.11 | 2.94 × 108 |
| Cz-p2 | 2.614 | 2.305 | 0.380 | 0.452 | 0.235 | 3.74 | 1.96 × 102 |
| Acr-p2 | 2.374 | 2.308 | 0.0424 | 0.100 | 0.0292 | 0.599 | 1.13 × 107 |
| Pxz-p2 | 1.993 | 1.994 | 0.0181 | 0.0896 | 0.00669 | 0.385 | 1.47 × 107 |
The effect of the structural modification of the donor units is also evaluated in MCz-p2, Cz-p2, Acr-p2, and Pxz-p2 with the same substitution numbers and positions (Fig. 2A). Removal of the electron-donating methyl groups from MCz-p2 results in a blue-shifted S1 of Cz-p2, which explain its larger ΔEST between S1 (1CT) and T1 (3LE) and lower kRISC (Table 1, Fig. 2A and 3 ESI†). In contrast, Acr-p2 and Pxz-p2, with more electron-rich donor units, possess a red-shifted S1 (1CT) and T1 (3CT) with a smaller ΔEST, due to weak electron exchange coupling associated with the spatially separated orbitals comprising these CT excited states. However, as the 1CT and 3CT undergo a red shift, their energy separation with the higher-lying 3LE increases, and thus the minimum-energy singlet–triplet crossing seam comprises CT characters, rather than LE, for both singlet and triplet states (Fig. S2†). The resultant lack of orbital change at the crossing seam suppresses spin–orbit coupling, as corroborated by the lower HSO and kRISC of Acr-p2 and Pxz-p2 than that of MCz-p2 (Table 1). These quantum chemical interpretations of the seven model molecules are in good agreement with the general trends in the FP importance shown in Fig. 1D.
Experimental validation
To experimentally validate the virtual screening results, MCz-p2 with a quantum-chemically calculated kRISC of 2.94 × 108 s−1 and a model-predicted kRISC of 1.11 × 108 s−1 was synthesized. The photophysical properties of MCz-p2 were evaluated in a N2-purged toluene solution. Under a steady-state photoexcitation condition, MCz-p2 exhibited a sky-blue emission with a maximum peak wavelength of 485 nm (2.56 eV) (Fig. 3A and Table 2), as consistent with the S1 excitation energy (ES1 = 2.52 eV) calculated by TD-DFT. The emission is characterized by a broad, unstructured spectrum originating from the predominantly 1CT state. The photoluminescence (PL) quantum yield was measured to be 82%, which was reduced to 63% when measured in air. This reduction indicates the contribution of triplet excited states to the emission since the triplet states can be at least partially quenched by atmospheric O2. | ||
| Fig. 3 (A) Steady-state absorption and PL spectra of MCz-p2 in a toluene solution (10−5 M). The inset in (A) is a photograph of the MCz-p2 solution exposed to UV light. (B) Transient PL decays of MCz-p2 measured with picosecond pulsed excitation at 375 nm. The blue line in (B) shows the fit of the transient PL decay to eqn (2) in ‘Methods’. The inset in (B) is the log–log representation. (C) Transient PL decays of MCz-p2 measured at varying temperatures. The inset in (C) represents the temperature-dependence of kISC and kRISC with their fits to the Arrhenius equation. (D–F) Current density–voltage–luminance characteristics (D), EL spectra (E), and external quantum efficiency-luminance characteristics (F) of the fabricated OLEDs. The inset in (F) is a photograph of an OLED using MCz-p2 working at high luminance (>5000 cd m−2). | ||
| λ PL (nm) | Φ PL (%) | τ PF (ns) | τ DF (ns) | k r (s−1) | k nr (s−1) | k ISC (s−1) | k RISC (s−1) | ΔEST (eV) |
|---|---|---|---|---|---|---|---|---|
| a Photoluminescence (PL) peak wavelength. b PL quantum yield. c Time constant of prompt fluorescence. d Time constant of delayed fluorescence. e Rate constant of radiative decay of the S1 state to the S0 state. f Rate constant of non-radiative decay of the S1 state to the S0 state. g Rate constant of intersystem crossing (ISC) of the S1 state to the T1 state. h Rate constant of reverse intersystem crossing (RISC) of the T1 state to the S1 state. i Energy gap between the S1 and T1 states, estimated from the kRISC/kISC ratio, assuming the equilibrium between the S1 state and the three T1 sublevels. j Energy gap between the S1 and T1 states, estimated as the difference between activation energies of RISC and ISC. | ||||||||
| 485 | 82 | 1.4 | 852 | 5.3 × 106 | 1.2 × 106 | 5.8 × 108 | 1.3 × 108 | 0.010i/0.050j |
Transient PL decay measurements of MCz-p2 confirmed both prompt and delayed fluorescence components, with time constants (τPF and τDF) of 1.4 ns and 852 ns, respectively (Fig. 3B and Table 2). In comparison to this sub-microsecond τDF of MCz-p2, typical TADF materials exhibit τDF in the microsecond to millisecond time range, e.g. 2.8 μs for the seminal TADF molecule 4CzIPN.6 Further kinetic analysis of the transient PL decay (described in ‘Methods’) revealed that the RISC of MCz-p2 is indeed on the order of nanoseconds with an exceptionally high kRISC of 1.3 × 108 s−1, in good quantitative agreement with the model-predicted kRISC of 1.11 × 108 s−1. We note that the rapid RISC of MCz-p2 can be retained in a solid-state host matrix 3,3′-bis(carbazol-9-yl)biphenyl (mCBP) (Table S1 and Fig. S4†), though the emission is slightly red-shifted to a maximum peak wavelength of 495 nm, indicating a stabilization of a 1CT state in a higher dielectric environment of mCBP than toluene.
We measured the temperature-dependent transient PL decays of MCz-p2 in toluene (Fig. 3C). The decay kinetics suggest that the delayed fluorescence is attributed to a thermally activated process, as evidenced by the increase of τDF with decreasing temperature. Using the Arrhenius equation, the corresponding activation energies of ISC and RISC were estimated to be 0.001 eV and 0.051 eV, respectively. The difference between these activation energies gives ΔEST of 0.050 eV. An estimate of ΔEST was also obtained from the kRISC/kISC ratio, yielding a value of 0.010 eV (Table 2). We note that the former estimate is reasonably closer to the calculated value of 0.110 eV (Table 1).
The electroluminescence properties of MCz-p2 were also evaluated to demonstrate the practical implications of high kRISC in OLED performance (Fig. 3D–F). The details of the OLED structures and fabrication procedures are described in ‘Methods’. The fabricated OLEDs were confirmed to have MCz-p2 as the only source of electroluminescence with a maximum peak wavelength of 495 nm (Fig. 3E). The measured external quantum efficiency reached a maximum of 25.7% (Fig. 3F), corresponding to an internal quantum efficiency of 85.7%, assuming a light-outcoupling efficiency of 30% for a bottom-emission OLED. Crucially, a high external quantum efficiency of 22.8% was retained even at a practically high luminance of 5000 cd m−2. The suppression of efficiency roll-off was further quantified by the critical current density (J80%) at which the external quantum efficiency decreases to 80% of its maximum. A J80% of 19 mA cm−2 was recorded for the MCz-p2 device, indeed higher than 8.2 mA cm−2 observed for a device using 4CzIPN as an emitter. These results are attributed to the higher kRISC of MCz-p2, which maintains a comparatively low triplet exciton concentration and thus suppresses bimolecular recombination processes at high luminance.
Conclusions
We have shown that the coupling of Bayesian optimization and quantum chemical calculations provides a promising tool for identifying novel TADF molecules capable of rapid triplet-to-singlet conversion. A relevant molecule has been experimentally demonstrated to exhibit a high kRISC of 1.3 × 108 s−1. Furthermore, the post-hoc interpretation of the machine-learning model reveals how the molecular structures influence excited-state energies, spin–orbit couplings, and ultimately kRISC, offering valuable chemical insights for future materials design.Bayesian optimization is poised to become an empowering tool for materials scientists in both academic and industrial settings by mitigating costly evaluations. Its efficient and scalable nature can further expedite the development of spin-conversion materials for organic optoelectronics, as demonstrated in this study, as well as in broader applications such as photocatalysis, fluorescent bioimaging, and photodynamic therapy.
Methods
Quantum chemical calculations
All of the quantum chemical calculations were performed in vacuum. For the low-cost calculations of the descriptors, the geometries of T1 were optimized for each molecule using spin-unrestricted DFT with the LC-BLYP functional and the 6-31G basis set, as implemented in the Gaussian16 program.55 Using the optimized T1 geometries, single-point TD-DFT calculations were performed with the LC-BLYP functional and the 6-31G basis set within the Tamm–Dancoff approximation to obtain descriptors: EHOMO, ELUMO, and ΔEST. Based on these low-cost TD-DFT calculations, a descriptor HSO was obtained using the Breit–Pauli spin–orbit Hamiltonian with an effective charge approximation, as implemented in the PySOC program.56 The range-separation parameter of the LC-BLYP functional was set to 0.15 bohr−1 to balance the Hartree–Fock exchange with the DFT exchange for the CT states in the donor–acceptor systems.For the high-fidelity calculations of kRISC using eqn (1), the geometries of the S1, T1, and singlet–triplet crossing seam were optimized for each molecule using the TD-DFT with the LC-BLYP functional and the 6-31+G(d) basis set within the Tamm–Dancoff approximation, as implemented in the GRRM17 program,57 which refers to the energies and gradients calculated by the Gaussian16 program. The minimum-energy singlet–triplet crossing seam was calculated between S1 and T2 for all molecules except for mol-00003 (its molecular structure is given in ‘ESI†’), where the calculations converged between S1 and T3. The range-separation parameter of the LC-BLYP functional was non-empirically optimized for each molecule to minimize the differences between the HOMO energies and the ionization potentials in both the neutral and radical anion systems.58 Geometry optimization calculations for the seven model molecules were carried out, considering relevant conformers.
Machine learning
The logarithmic scale values of the calculated kRISC were used for the training data to facilitate balanced learning from the entire dataset, including both low and high kRISC values. A Gaussian process surrogate model was used for Bayesian optimization, as implemented in the COMBO library.38 Hyperparameters of the Gaussian kernels were optimized by maximizing the type-II likelihood, allowing the model to adapt to the data at each iteration of Bayesian optimization. Expected improvement was used as the acquisition function for balanced exploration and exploitation to select the next molecule for evaluation. SHAP values were calculated by the SHAP version 0.34.053 for the Gaussian process model trained on the dataset obtained through the Bayesian optimization procedures.
Synthesis
MCz-p2 was synthesized by palladium-catalyzed Buchwald–Hartwig amination of 3,6-dibromothioxanthone with 1,8-dimethyl-3,6-diphenyl-9H-carbazole. The detailed synthetic procedure and characterization data are given in ‘ESI†’.Photophysical evaluation
Steady-state PL spectra were recorded on an Edinburgh Instruments FS5 spectrofluorometer with 375 nm photoexcitation from a Xe arc lamp. The absolute PL quantum yields were determined using a Hamamatsu Photonics C9920 integrated sphere system with 375 nm excitation from a Xe arc lamp. Transient PL decay measurements were performed by time-correlated single photon counting using an Edinburgh Instruments FS5 spectrofluorometer equipped with a 375 nm pulsed diode laser (pulse width 60 ps).The differential rate equation of the S1 and T1 populations (eqn (2)) was employed in the numerical fitting of the transient PL decays to obtain the rate constants of each excited-state transition, assuming negligible radiative and non-radiative decays from T1 to S0.
 | (2) |
OLED fabrication and evaluation
OLEDs were fabricated on glass substrates covered with indium tin oxide (ITO) (sheet resistance = 15 Ω sq−1). The substrates were sequentially cleaned by sonication in detergent, deionized water, acetone, and 2-propanol, followed by UV-ozone treatment. Phosphomolybdic acid (PMA) in acetonitrile (0.25 g L−1) was spin-coated on the substrates and annealed at 120 °C on a hot plate for 10 min in ambient conditions. The organic layers and a LiF/Al cathode were sequentially deposited on the substrates under vacuum (<8 × 10−4 Pa) at a deposition rate of <0.9 nm s−1 through a shadow mask defining a pixel size of 4.0 mm2. The deposition rate and layer thicknesses of each layer were monitored using a quartz crystal microbalance. The fabricated OLEDs consist of the following layer sequences: glass/ITO (50 nm)/PMA (5 nm)/TAPC (70 nm)/CCP (10 nm)/12 wt% Emitter:mCBP (30 nm)/PPF (10 nm)/B3PyPB (40 nm)/LiF (1 nm)/Al (100 nm). EL spectra were recorded using a Hamamatsu Photonics PMA-12 photonic multichannel analyzer. Current density–voltage–luminance characteristics were measured using a Konica Minolta CS-200 luminance meter and a Keithley 2400 source meter. The viewing-angle dependence of luminance was confirmed to follow the Lambertian distribution.Data availability
All data including the input and output data used with the machine learning model are present in the paper and/or the ESI.†Author contributions
N. A. and Y. H. conceived the project and performed the computational work. T. W., R. K., and N. A. synthesized the compound and characterized the photophysical properties. M. S., S. M., Y.-J. P., and K. N. supervised the research. T. W. and N. A. wrote the paper. All authors contributed to the discussion and the preparation of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by JST FOREST (JPMJFR221J for N. A. and JPMJFR2221 for Y. H.), JSPS KAKENHI (21H05413, 22H02051, and 23H03958 for N. A.), and NEDO (JPNP20004 for N. A.). T. W. thanks Sumitomo Electric Group CSR Foundation for their financial support.Notes and references
- M. R. Wasielewski, et al. , Nat. Rev. Chem, 2020, 4, 490–504 CrossRef CAS PubMed.
- M. Einzinger, et al. , Nature, 2019, 571, 90–94 CrossRef CAS PubMed.
- A. Rao, Nature, 2013, 500, 435–439 CrossRef CAS PubMed.
- S. L. Bayliss, et al. , Science, 2020, 370, 1309–1312 CrossRef CAS PubMed.
- S. Izawa, et al. , Nat. Commun., 2023, 14, 5494 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- T. L. Wu, et al. , Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- L.-S. Cui, et al. , Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- Z. Tu, G. Han, T. Hu, R. Duan and Y. Yi, Chem. Mater., 2019, 31, 6665–6671 CrossRef CAS.
- S. Dey, et al. , J. Phys. Chem. C, 2022, 126, 5649–5657 CrossRef CAS.
- J. Bian, et al. , Adv. Mater., 2022, 34, 2110547 CrossRef CAS PubMed.
- X.-C. Fan, et al. , Nat. Photonics, 2023, 17, 280–285 CrossRef CAS.
- X. Li, et al. , J. Phys. Chem. C, 2024, 128, 6612–6620 CrossRef CAS.
- L. Zhu, et al. , J. Phys. Chem. C, 2024, 128, 15763–15777 CrossRef CAS.
- A. Sevilla-Pym, W. L. Primrose, B. T. Luppi, K. Bergmann and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2024, 16, 46133–46144 CrossRef CAS PubMed.
- T. Huang, Q. Wang, H. Zhang, Y. Xin, Y. Zhang, X. Chen, D. Zhang and L. Duan, Nat. Mater., 2024, 23, 1523–1530 CrossRef CAS PubMed.
- W. Whitaker, I. V. Sazanovich, Y. Kwon, W. Jeon, M. S. Kwon and A. J. Orr-Ewing, J. Phys. Chem. A, 2023, 127, 10775–10788 CrossRef CAS PubMed.
- W. Qiu, et al. , Nat. Commun., 2023, 14, 2564 CrossRef CAS PubMed.
- L. Wan, Z. Cheng, F. Liu and P. Lu, Mater. Chem. Front., 2023, 7, 4420–4444 RSC.
- A. N. Stuart, K. Bergmann, I. Cho, W. J. Kendrick, Z. M. Hudson, W. W. Wong and G. Lakhwani, Chem. Sci., 2024, 15, 14027–14036 RSC.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, et al. , Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- R. Pollice, et al. , Acc. Chem. Res., 2021, 54, 849–860 CrossRef CAS PubMed.
- K. Zhao, Ö. H. Omar, T. Nematiaram, D. Padula and A. Troisi, J. Mater. Chem. C, 2021, 9, 3324–3333 RSC.
- K. H. Lin, G. J. A. Wetzelaer, P. W. Blom and D. Andrienko, Front. Chem., 2021, 9, 800027 CrossRef CAS PubMed.
- P. Li, Z. Wang, W. Li, J. Yuan and R. Chen, J. Phys. Chem. Lett., 2022, 13, 9910–9918 CrossRef CAS PubMed.
- Y. Bu and Q. Peng, J. Phys. Chem. C, 2023, 127, 23845–23851 CrossRef CAS.
- N. Aizawa, Y. Harabuchi, S. Maeda and Y. J. Pu, Nat. Commun., 2020, 11, 3909 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- C. M. Marian, J. Phys. Chem. C, 2016, 120, 3715–3721 CrossRef CAS.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- H. Imahori, Y. Kobori and H. Kaji, Acc. Mater. Res., 2021, 2, 501–514 CrossRef CAS.
- K. Shizu and H. Kaji, Nat. Commun., 2024, 15, 4723 CrossRef CAS PubMed.
- F. Coppola, M. Hussain, J. Zhao, A. M. El-Zohry and M. Pastore, J. Phys. Chem. C, 2024, 128, 11998–12009 CrossRef CAS.
- K. Terayama, M. Sumita, R. Tamura and K. Tsuda, Acc. Chem. Res., 2021, 54, 1334–1346 CrossRef CAS PubMed.
- T. Ueno, T. D. Rhone, Z. Hou, T. Mizoguchi and K. Tsuda, Mater. Discovery, 2016, 4, 18–21 CrossRef.
- D. M. Packwood and T. Hitosugi, Appl. Phys. Express, 2017, 10, 065502 CrossRef.
- B. J. Shields, et al. , Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- X. Li, et al. , Nat. Chem., 2024, 16, 1286–1294 CrossRef CAS PubMed.
- N. H. Angello, et al. , Nature, 2024, 633, 351–358 CrossRef CAS PubMed.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- B. H. Drummond, et al. , J. Phys. Chem. C, 2020, 124, 6364–6370 CrossRef CAS.
- N. Aizawa, A. Matsumoto and T. Yasuda, Sci. Adv., 2021, 7, 5769 CrossRef PubMed.
- Y. Ren, Y. Wada, K. Suzuki, Y. Kusakabe, J. Geldsetzer and H. Kaji, Appl. Phys. Express, 2021, 14, 071003 CrossRef CAS.
- S. Goto, et al. , J. Mater. Chem. C, 2021, 9, 13942–13953 RSC.
- Y. Hu, et al. , Angew. Chem., Int. Ed., 2023, 62, 202302478 CrossRef PubMed.
- K. Shizu, Y. Ren and H. Kaji, J. Phys. Chem. A, 2023, 127, 439–449 CrossRef CAS PubMed.
- X. Lv, J. Song, X. Fu, S. Guo, J. Gu, L. Meng and C. Z. Lu, J. Phys. Chem. A, 2024, 128, 1611–1619 CrossRef CAS PubMed.
- H. Miranda-Salinas, J. Wang, A. Danos, T. Matulaitis, K. Stavrou, A. P. Monkman and E. Zysman-Colman, J. Mater. Chem. C, 2024, 12, 1996–2006 RSC.
- C. E. Rasmussen and H. Nickisch, J. Mach. Learn. Res., 2010, 11, 3011–3015 Search PubMed.
- S. M. Lundberg and S.-I. Lee, Adv. Neural Inf. Process. Syst., 2017, 30, 4765–4774 Search PubMed.
- M. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- X. Gao, et al. , J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- S. Maeda, et al. , J. Comput. Chem., 2018, 39, 233–251 CrossRef CAS PubMed.
- R. Baer, E. Livshits and U. Salzner, Annu. Rev. Phys. Chem., 2010, 61, 85–109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Table S1, ESI method, materials, synthesis, note, and data for machine learning. See DOI: https://doi.org/10.1039/d5sc01903f |
| This journal is © The Royal Society of Chemistry 2025 |