
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mechanistic continuum of nucleophilic aromatic substitution reactions with azole nucleophiles†
Harrison W.
Toll
 a,
Xiaoyi
Zhang
a,
Tong
Gao
a,
Guilherme
Dal Poggetto
b,
Mikhail
Reibarkh
b,
Joshua J.
Lee
a,
Katherine J.
Yang
a,
Eugene E.
Kwan
*c and
Amanda K.
Turek
*a
a,
Xiaoyi
Zhang
a,
Tong
Gao
a,
Guilherme
Dal Poggetto
b,
Mikhail
Reibarkh
b,
Joshua J.
Lee
a,
Katherine J.
Yang
a,
Eugene E.
Kwan
*c and
Amanda K.
Turek
*a
aDepartment of Chemistry, Williams College, 47 Lab Campus Drive, Williamstown, Massachusetts 01267, USA. E-mail: akt2@williams.edu
bAnalytical Research and Development, MRL, Merck & Co., Inc., Rahway, New Jersey 07065, USA
cMerck & Co., Inc., 33 Avenue Louis Pasteur, Boston, Massachusetts 02115, USA
First published on 30th April 2025
Abstract
Nucleophilic aromatic substitution (SNAr) is a broadly used method for generating structural complexity in pharmaceuticals. Although SNAr reactions were long assumed to be stepwise, recent kinetic isotope effect (KIE) studies have shown that many SNAr reactions are actually concerted. However, it remains unclear how variations in substrate structure affect whether a reaction is stepwise, concerted, or borderline. In this paper, we show that reactions between indole and moderately electron-deficient aryl fluorides proceed by a borderline mechanism and are subject to general base catalysis. These findings are consistent with density functional theory (DFT) calculations, which also predict that borderline mechanisms are operative for a broad range of industrially relevant SNAr reactions involving azole nucleophiles. The predicted transition structures vary smoothly independent of the mechanism, suggesting that these SNAr reactions exist on a mechanistic continuum. The findings of widespread general base catalysis and a mechanistic continuum will guide future efforts to devise general models of SNAr reactivity.
Introduction
Nucleophilic aromatic substitution (SNAr) reactions are a crucial synthetic tool for the amination of electron-deficient arenes.1,2 Classically, SNAr reactions were presumed to be stepwise, due to early mechanistic studies that focused on electrophiles capable of generating stable anionic σ-complexes, such as polynitroarenes and aza-aromatics.3 The highly electron-deficient nature of these substrates makes them easy to study, but also renders their σ-complexes (“Meisenheimer complexes”) unusually stable.4 Because these substrates are not representative, the conventional picture of most SNAr reactions as stepwise processes is an overgeneralization (Scheme 1a).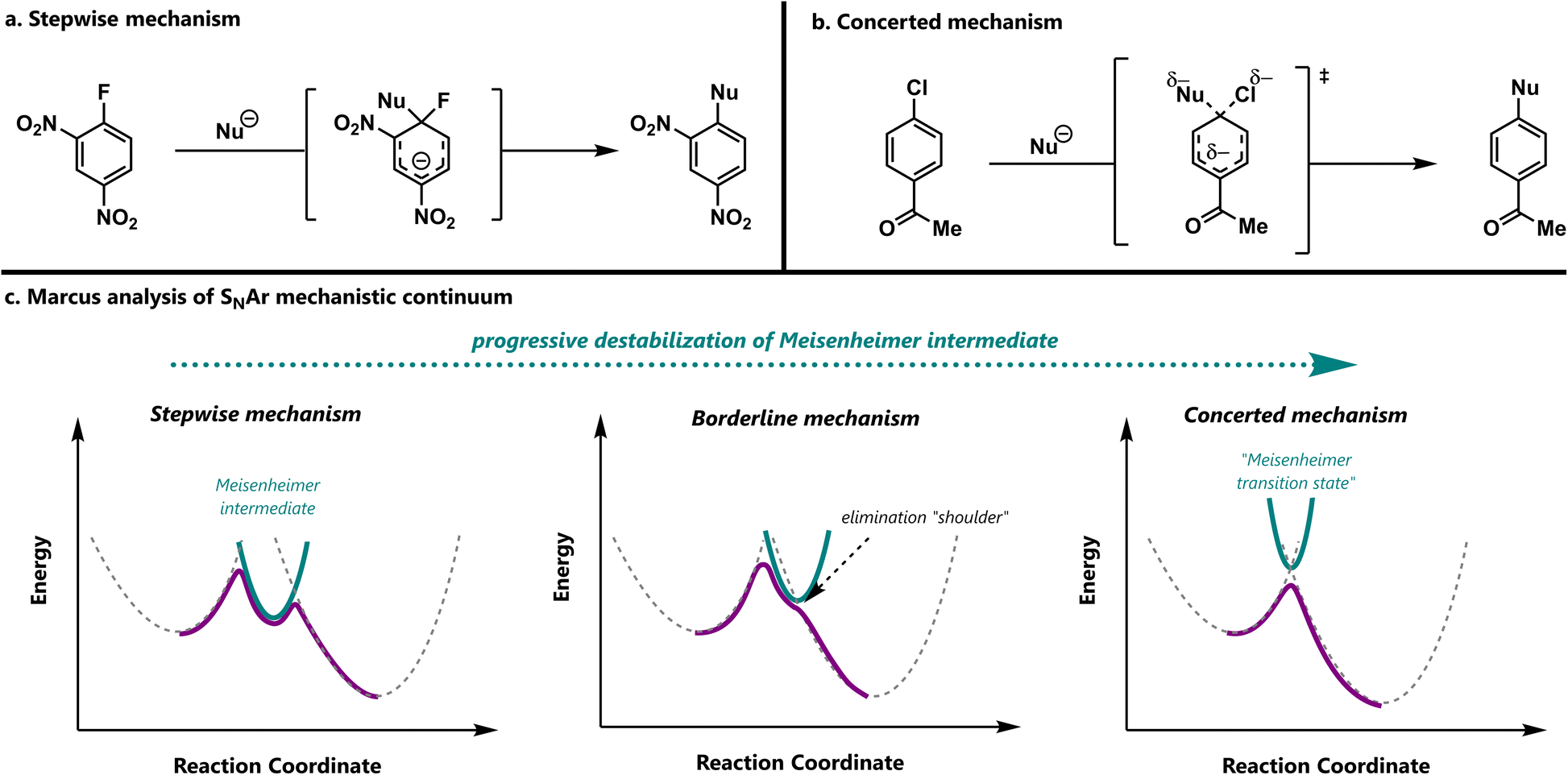 | ||
| Scheme 1 (a) A representative stepwise SNAr reaction with a highly activated electrophile and poor leaving group. (b) A representative concerted SNAr reaction with a less activated electrophile and better leaving group. (c) A qualitative Marcus analysis applied to SNAr mechanisms. | ||
Indeed, mechanistic studies of SNAr reactions have recognized the possibility of concerted pathways (Scheme 1b),5–8 with many such proposals arising from density functional theory (DFT) predictions.9 The value of DFT for predicting whether a given SNAr reaction will be stepwise or concerted was recently confirmed by both coupled-cluster-quality benchmark calculations and kinetic isotope effect (KIE) measurements on a series of halogen-exchange reactions.10 In general, concerted pathways are predicted to be broadly operative in SNAr reactions with typical, modestly activated electrophiles.
The stepwise and concerted mechanisms for SNAr represent limiting topographies of the free energy surface. Stepwise reactions involve distinct addition and elimination transition states that are connected by a Meisenheimer intermediate, which lies in an energetic minimum and has an appreciable lifetime. In contrast, concerted reactions proceed via a single transition state that avoids an inaccessible Meisenheimer region that is high in both energy and energetic gradient.
As these previous studies make clear, the topography of a given SNAr reaction depends on the structure of the starting materials. One way to conceptualize this influence is to consider the relative barriers of addition and elimination. When the arene is very electron-deficient, it is highly reactive and the addition step is fast. However, once the σ-complex is formed, the leaving group is tightly held and the elimination step is slow. As a result, the reaction occurs in two steps.
Concerted mechanisms can appear when the Meisenheimer intermediate is less stable. For example, when the leaving group is changed from fluoride (σm = 0.34) to bromide (σm = 0.39),11 the electron demand of the arene increases such that addition is modestly accelerated. However, because bromide (pKa = −9)12 is a much better leaving group than fluoride (pKa = −3), the elimination step becomes barrierless. Accordingly, the reaction proceeds through a single transition state. While the foregoing analysis might suggest that there are two distinct clusters of SNAr mechanisms, it is also possible that the mechanism changes smoothly between the stepwise and concerted extremes. Such a mechanistic continuum is familiar in the context of aliphatic substitution reactions,13,14 and distinct clusters are generally proposed for acyl substitution.15
In fact, a simple Marcus analysis16,17 predicts that the transition between the stepwise and concerted regimes will be smooth. In this Marcus view, the minimum energy path for an SNAr reaction results from the intersection of diabatic curves for the reactants, Meisenheimer intermediate, and products (Scheme 1c). When the intermediate is relatively stable, its diabatic curve forms part of the minimum energy path, and the reaction is stepwise with a clear intermediate and two transition states. As the curve corresponding to the Meisenheimer intermediate is destabilized, its diabatic curve rises above the starting material and product curves. Correspondingly, the rate of elimination increases, resulting in a concerted mechanism with a single transition state. When the Meisenheimer curve is moderately stable, it barely touches the minimum energy path, and a “borderline” mechanism results. Thus, the stepwise and concerted mechanisms can be considered as the extremes of a mechanistic continuum, connected by the “borderline” region.
Crucially, the borderline SNAr mechanism does not represent a competition between stepwise and concerted reactions with similar rate constants. Rather, it is a reaction path that shares features of the stepwise and concerted mechanisms. In the borderline mechanism, the Meisenheimer region is stable enough to influence the minimum energy path as an energetic shoulder,18 but not stable enough to create a true intermediate. When the energy is plotted as a function of the forming and breaking bond lengths, this shoulder appears as a shallow trough. Accordingly, reactive trajectories may linger there for several bond vibrations before proceeding to product. Therefore, borderline reactions can be considered to be formally concerted on the potential energy surface, but stepwise on the free energy surface.
Although Marcus theory readily predicts the influence of electrophile structure on mechanism, the effect of nucleophile structure is less clear. Furthermore, many mechanistic studies have only considered anionic nucleophiles, despite the fact that many SNAr reactions are carried out with protonated nucleophiles. The prior studies of protic amine nucleophiles that do exist have largely examined highly activated arenes and found stepwise mechanisms with rate-limiting addition,19 elimination,20,21 and proton transfer22–28 with different modes of acid and base catalysis. In the specific case of SNAr with indoles, the only studies reported have focused on highly electrophilic species.29–31
It is difficult to predict whether protic nucleophiles favour the stepwise or concerted mechanism. One possibility is that the increased positive charge on the nucleophile reduces the charge transferred to the arene in the addition step, increasing the stability of the Meisenheimer intermediate, and thus favouring the stepwise regime. However, this picture either requires a zwitterionic intermediate or partial deprotonation of the nucleophile in a general-base-catalysed addition step. General base catalysis is known to occur in both the addition32 and deprotonation33 steps in SNAr mechanisms with highly activated electrophiles. However, the effect of the proton on the stability of the Meisenheimer intermediate could not be elucidated under these conditions, because the highly activated electrophiles used already strongly favour the stepwise pathway. Alternatively, protic species, being inherently weak nucleophiles, might simply prefer to react in anionic form via specific base catalysis. Subsequently, the conjugate acid might then decrease the elimination barrier through hydrogen bonding, thus favouring a concerted mechanism.34,35
In this study, we elucidate the effect of the proton on the mechanism of SNAr reactions between aryl fluorides and indole nucleophiles. We show that this reaction proceeds with general base catalysis, and that the mechanism is neither stepwise nor concerted, but actually borderline (Scheme 2). DFT studies of related azole nucleophiles predict that borderline mechanisms are commonplace and confirm the Marcus picture of a mechanistic continuum in which the transition state geometries vary continuously across all three mechanisms. Because these transition structures can be viewed as linear interpolations, various reaction parameters exhibit straightforward correlations with ground state properties. This phenomenon provides a convenient basis for the future development of a general model for SNAr reactivity.
 | ||
| Scheme 2 Prototypical SNAr reaction between indoles and aryl fluorides, examined in this study and shown to proceed through a borderline mechanism that is subject to general base catalysis. | ||
Absolute rates do not distinguish between mechanisms
A kinetic analysis of the K3PO4-promoted reaction between indole and 4-fluorobenzonitrile was carried out using initial rates. The reaction was studied using DMA, a polar aprotic solvent, due to the elevated temperatures and relative insolubility of the base.‡ At low concentrations, the process is first-order in both the aryl fluoride and indole. At higher concentrations, the rate plateaus, presumably due to due to rate-limiting phase transfer of the base in this heterogeneous reaction (Fig. 1). While the pKa of K3PO4 in DMA is unknown, phosphate anion is likely the only sufficiently basic species in the reaction mixture, as experiments with K2HPO4 result in no reaction. | ||
| Fig. 1 Plots of initial rates vs. initial concentrations of starting materials, indicating that both starting materials affect reaction rate at low concentrations. (a) A plot of kobs as [ArF]0 is varied (0.5–0.025 M, [indole]0 = 0.25 M). (b) A plot of kobs as [indole]0 is varied (0.175–0.01 M, [ArF]0 = 0.25 M). | ||
While these data show that single equivalents of the aryl fluoride and indole are both present in the rate-limiting transition state, they cannot distinguish between concerted, borderline, or stepwise processes. This limitation becomes clear when the corresponding rate laws for the potential mechanisms are examined, considering both general and specific base catalysis.
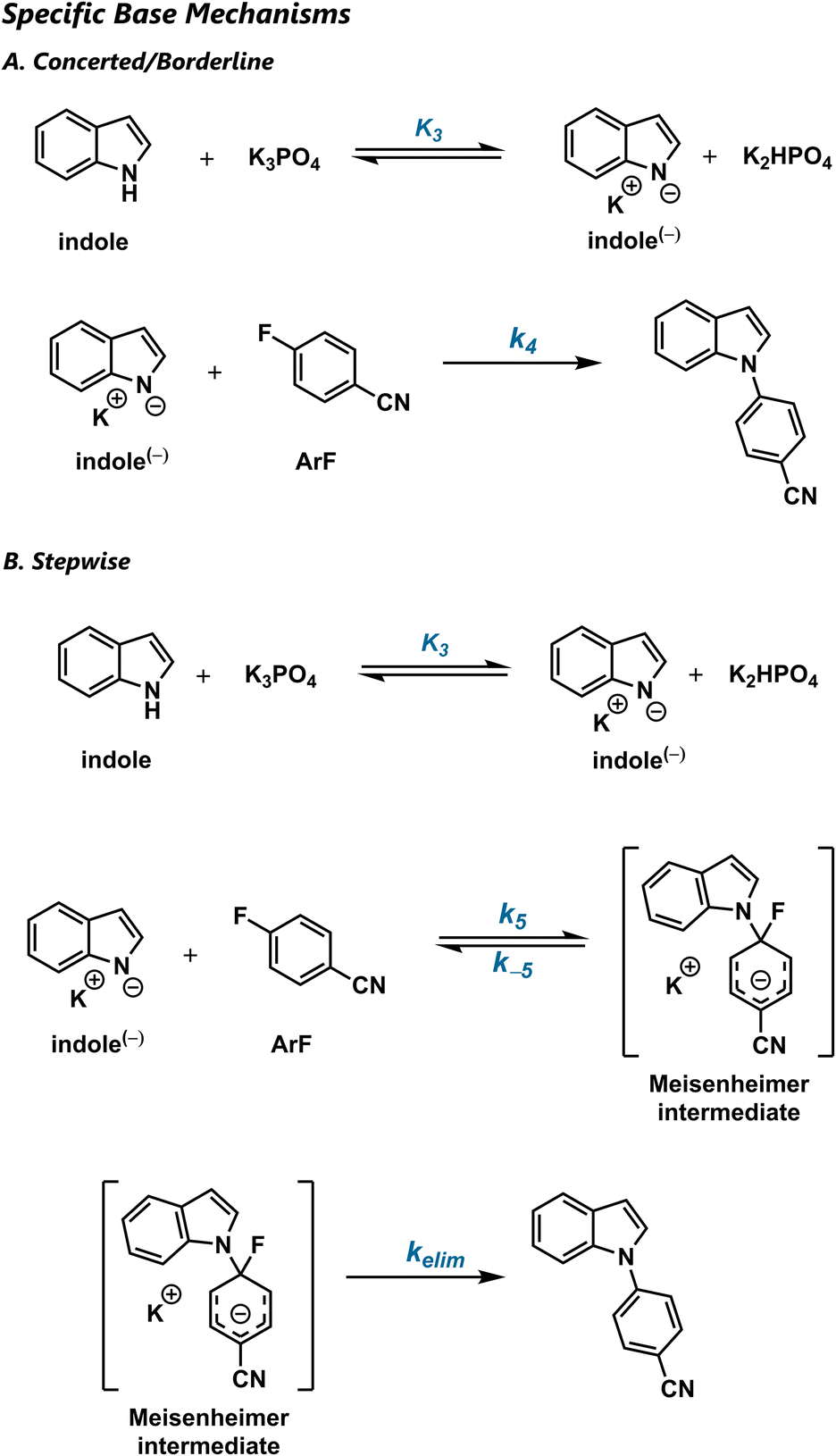
If SNAr occurs in a single elementary step, the rate will depend on the concentrations of both the indole and the aryl fluoride (Scheme 3a). Because the concerted and borderline mechanisms both involve high-barrier addition and low-barrier elimination, they are kinetically indistinguishable. If general base catalysis is involved, the rate law will be first-order in base:
| Rate = kobs[ArF][indole][K3PO4] | (1) |
 | ||
| Scheme 3 Mechanisms for general-base-catalysed concerted/borderline and stepwise SNAr. | ||
Stepwise addition–elimination results in the same rate law, although the microscopic composition of the macroscopic rate constant becomes more complex. Applying the steady-state approximation to the general-base-catalysed scenario (Scheme 3b) gives:
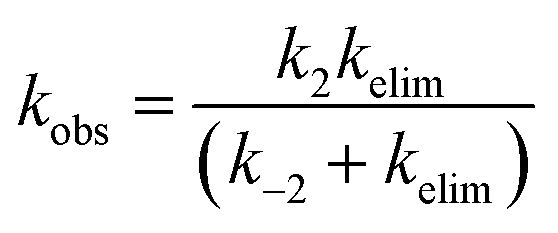 | (2) |
However, if addition becomes so fast that it is essentially instantaneous, then elimination can no longer consume the intermediate quickly enough for the steady state approximation to hold. In this case, most of the reaction time course will be dominated by zero-order decay of the intermediate. While such behaviour would unambiguously implicate a stepwise process, this possibility is clearly ruled out by our data, which show that the rate does depend on starting material concentration when phase transfer is not rate-limiting.
The expected kinetic behaviour is analogous for specific-base-catalysed reactions. However, the rate law now depends on the pKa of the base rather than its concentration:
| Rate = kobs[ArF][indole] | (3) |
Once again, the various mechanistic scenarios are indistinguishable (Scheme 4). For a concerted or borderline reaction, kobs = k3k4/ka. In the stepwise mechanism:
 | (4) |
 | ||
| Scheme 4 Mechanisms for specific-base-catalysed concerted/borderline and stepwise SNAr. | ||
Thus, the absolute rates analysis shown in Fig. 1 is compatible with any of these mechanistic possibilities. Furthermore, it cannot determine the mode of base catalysis.
Moderate negative charge buildup
Relative rates offer a different approach for distinguishing the possible mechanisms. In particular, the modularity of the aryl fluoride and indole starting materials allowed us to conduct Brønsted and Hammett linear free energy relationship (LFER) studies. When substituents on the aryl fluoride were varied, we found that the reaction was moderately accelerated by electron-withdrawing substituents (Fig. 2).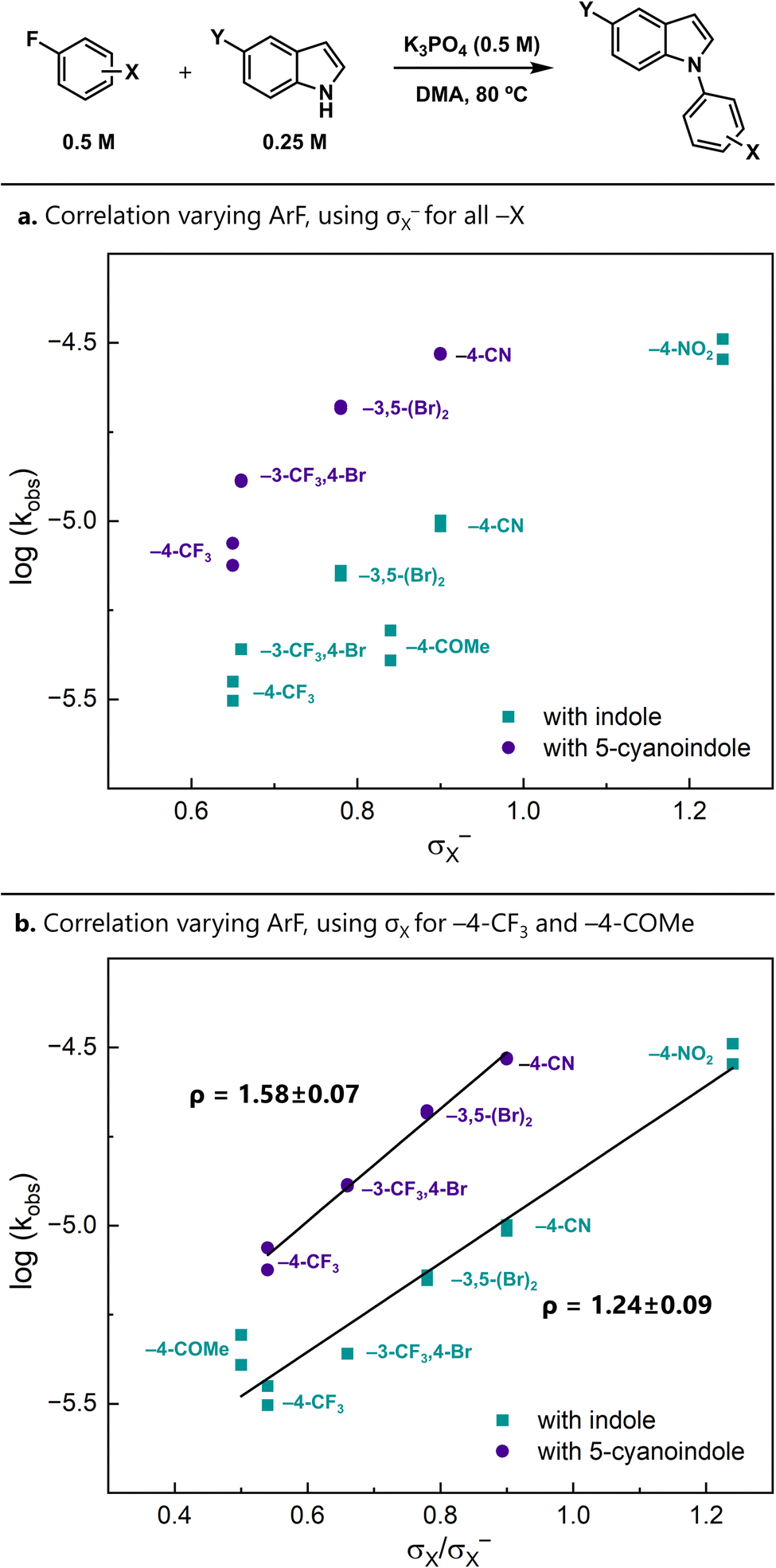 | ||
| Fig. 2 (a) LFER analysis varying the aryl fluoride, correlated with σX−. (b) A different analysis of the same data, using σX for −4–CF3 and −4–COMe. In both plots, σ values were used for substituents with either no σ− value or a nearly identical σ− value. Indole (Y = H) and 5-cyanoindole (Y = CN) were used as the nucleophiles. | ||
When σ− is used for all substituents, the resulting Hammett correlation is relatively scattered (Fig. 2a).¶ An alternative approach to analysing the data uses σ for less electron-withdrawing substituents (−4–CF3 and −4–COMe) and σ− for the more electron-withdrawing substituents (−4–CN and −4–NO2). The intervening substituents have nearly identical σ and σ− values. The resulting correlations are linear and give moderate ρ values of 1.24(9) and 1.58(7) with indole and 5-cyanoindole as the nucleophile, respectively (Fig. 2b). Importantly, these ρ values are much smaller than those that are expected for SNAr reactions proceeding through a Meisenheimer intermediate (ρ = 7–8)36,37 and are more consistent with those observed in concerted reactions.38–40
The observation that less electron-withdrawing substituents are best described by σ (as opposed to σ−) may reflect a difference in charge delocalization in the transition states. Stepwise-like reactions place more delocalized negative charge in the transition state. Therefore, σ− parameters, which have been determined in model systems involving significant resonance,41 better represent substituent effects in such cases. In contrast, concerted-like mechanisms localize charge at the ipso position, and thus the substituent effects are better represented by σ. Correspondingly, one interpretation is that this plot is consistent with a changing transition state over the series of substituents, from a stepwise-like transition state to a concerted-like one.
Using 4-fluorobenzonitrile as the electrophile, we also examined the effects of the indole electronics and observed a ρ value of 1.06(12) using σm (Fig. 3a). This also indicates negative charge buildup on the indole nitrogen in the rate-limiting transition state. This diminished sensitivity compared to that observed on the arene could be due to the increased distance between the substituent and the indole nitrogen.
 | ||
| Fig. 3 (a) LFER analysis varying the indole, correlated with σY(m), indicating a moderate degree of negative charge on the indole in the rate-limiting transition state. (b) A pseudo-Brønsted analysis of the same data, showing partial deprotonation of the indole in the rate-limiting transition state. | ||
An alternative analysis treats these indole electronic effects as a pseudo-Brønsted42 correlation between kobs and the pKa of the indole.43 This view provides additional insight into the degree of proton transfer between the nucleophile and base in the transition state (Fig. 3b). For a reaction with complete deprotonation in the rate-limiting transition state, the pKa differences in the nucleophiles should be fully reflected in the reaction rate and thus the slope of the plot would be −1.|| Similarly, rate-limiting transition states with no proton transfer at all would result in a slope of 0. In this reaction, the slope of the pseudo-Brønsted plot is −0.38(4), which is consistent with partial deprotonation of the indole in the transition state in a general-base-catalysed mechanism.
General base catalysis is operative
While these LFER data indicate a modest degree of negative charge buildup in the transition state that is inconsistent with a stepwise mechanism, further experiments were needed to distinguish between the concerted or borderline mechanisms. Thus, we sought additional evidence in the form of a 12C/13C kinetic isotope effect (KIE) at the ipso carbon because this measurement is sensitive to both the forming and breaking bond lengths. Since DFT calculations have been shown to reproduce both ab initio benchmarks and experimental KIEs in other SNAr reactions,10 the experimental KIEs can be directly compared to those predicted across the mechanistic continuum.We measured a KIE of 1.035(4) via nuclear magnetic resonance (NMR) at natural abundance for the reaction between indole and 4-fluorobenzonitrile.44,45 In contrast, the predicted KIE is much larger at 1.047 for a concerted mechanism involving indole anion as a nucleophile (Fig. 4a). Similarly large KIEs were obtained when modelling a specific base mechanism, and were unaffected by the presence or absence of a counterion.
 | ||
| Fig. 4 Predicted transition structures for the SNAr reaction between indole and 4-fluorobenzonitrile. (a) Anionic transition state (TSanionic) with no base. (b) Specific base transition state (TSSB) with dihydrogenphosphate (H2PO4−) base. (c) General base transition state (TSGB) with dihydrogenphosphate (H2PO4−) base. All calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMF). | ||
We also considered the general-base-catalysed mechanism, but were unable to locate one using phosphate as the base. However, DFT does predict both general- and specific-base mechanisms (Fig. 4b and c) for weaker bases (DFT pKa < 13)** such as dihydrogenphosphate (DFT pKa = 3.4), chloride (DFT pKa = −3.6), tetrazole (DFT pKa = 4.6), and cyanide (DFT pKa = 12.8). While the specific-base-catalysed transition states are similar in structure to the anionic transition state, the general-base catalysed transition states are later, with more advanced C–N bond formation. Accordingly, the predicted KIEs are reduced.
Remarkably, the predicted geometries and KIEs are largely independent of base structure and primarily depend on whether the general base or specific base mechanism is operative (Fig. 5). Each mode of base catalysis exhibits a characteristic KIE prediction: specific base transition states give predicted KIEs of 1.05, whereas general base transition states give predicted KIEs of 1.03, which is consistent with our experimental value.
 | ||
| Fig. 5 DFT-predicted KIEs plotted against predicted transition state geometries, defined by the length of the forming C–N bond. Transition states are calculated for the reaction between 4-fluorobenzonitrile and indole, using a series of bases. Calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMF). | ||
Despite the very different geometries and charge distributions of the two base-catalysed transition states, they are predicted to have very similar energies. This prediction is implausible and highlights the limitations of implicitly solvated DFT calculations. Nonetheless, the insensitivity of the KIEs to base structure, coupled with the consistency between the predicted and experimental KIEs, allows us to reasonably conclude that general base catalysis is operative in this SNAr reaction.
Partial deprotonation results in a borderline mechanism
This KIE analysis also reveals the influence of the proton on the mechanism. When the KIEs for the anionic reaction are plotted as a function of geometry, the concerted regime is seen to give the largest KIEs (Fig. 6). The transition states for the anionic (TSanionic) and specific base (TSSB) reactions lie on the verge of the concerted, high KIE region. In contrast, the general base transition state (TSGB) is shifted later along the addition coordinate, and out of the high KIE region. | ||
| Fig. 6 DFT-predicted KIEs for the anionic reaction as a function of geometry. The predicted geometries for the anionic transition state, TSSB, and TSGB are marked, showing that TSGB lies outside of the high KIE regime. Calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMF). | ||
This shift of TSGB lands it squarely in the “Meisenheimer region,” which encompasses structures with advanced C–N bond formation and little C–F bond cleavage. In contrast, TSanionic and TSSB lie earlier on the addition coordinate. The potential energy surface for the anionic reaction (Fig. 7) further shows that only a single transition state is expected along the addition coordinate, with no subsequent intermediate or elimination transition states being predicted.
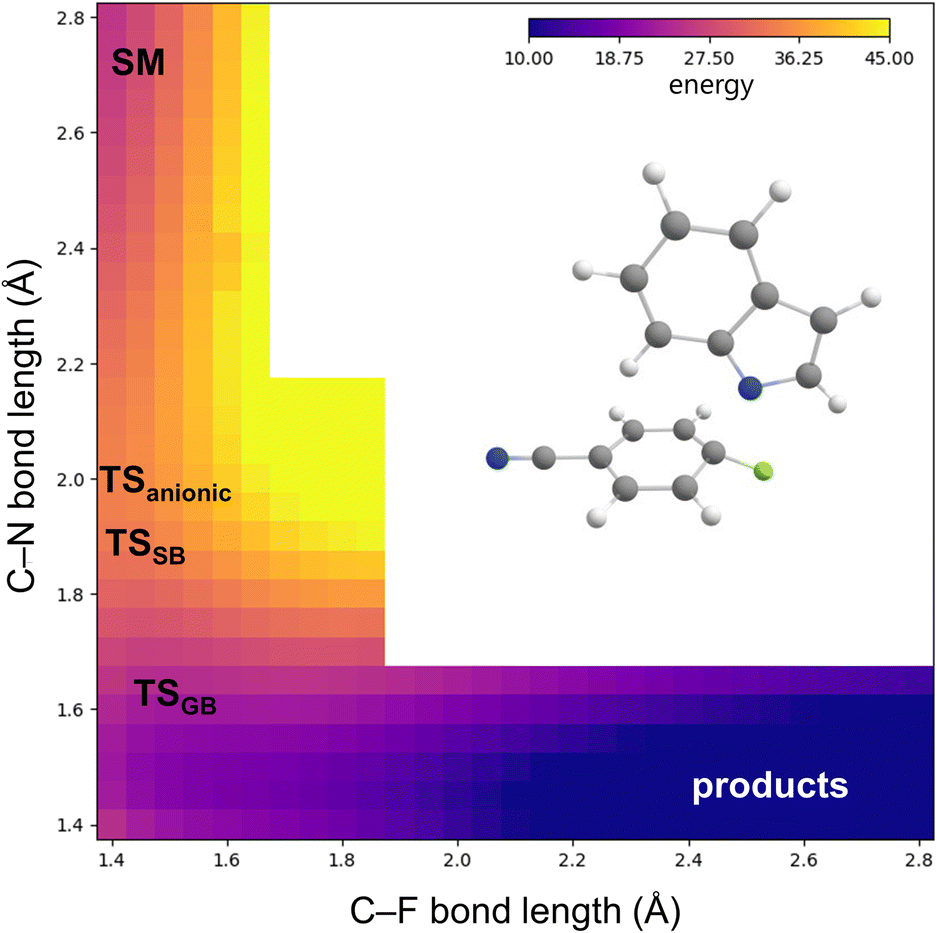 | ||
| Fig. 7 Calculated potential energy surface for the anionic reaction, showing a borderline mechanism. The predicted geometries for the anionic transition state, TSSB, and TSGB are marked. Calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMF). | ||
Given the structural similarities between TSGB and a hypothetical Meisenheimer intermediate, we might expect that TSGB should bear significant negative charge on the arene. However, because a proton is now present, coordinates beyond the forming C–N and breaking C–F bond distances must be considered. While one might also consider a deprotonation coordinate, multi-dimensional representations are challenging to interpret.
Instead, we chose to conduct a Hirsheld population analysis46 of the Meisenheimer intermediate and different transition structures (Table 1). At one extreme, the highly nucleophilic indole anion generates an early transition state. Correspondingly, there is only a minor degree of negative charge buildup on the arene (entry 2). Interestingly, the late structure TSGB experiences a similarly small degree of negative charge buildup (entry 3) because some of the charge is absorbed by the proton. This diminished degree of negative charge in TSGB is also consistent with the modest ρ values observed in the LFER experiments. At the other extreme, TSSB (entry 4), which does not have a proton available, has a degree of charge buildup more comparable to that of the Meisenheimer intermediate (entry 5), despite falling earlier on the addition coordinate relative to TSGB.
| Entry | Structure | Arene chargeb | C–F (Å) | C–N (Å) | Base–H (Å) | KIEc |
|---|---|---|---|---|---|---|
| a Calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMF). b Sum of charges at the positions ortho and para to the leaving group. c DFT-predicted KIE. d TS for the reaction between indole anion and 4-fluorobenzonitrile. e Calculated using tetrazole anion as the base. f Optimised structure constraining C–N and C–F at 1.4 Å. | ||||||
| 1 | 4-Fluorobenzonitrile | −0.171 | n/a | n/a | n/a | n/a |
| 2 | Anionic TSd | −0.317 | 1.39 | 2.00 | n/a | 1.047 |
| 3 | General base TSe | −0.340 | 1.51 | 1.62 | 1.62 | 1.031 |
| 4 | Specific base TSe | −0.379 | 1.41 | 1.89 | 1.06 | 1.052 |
| 5 | Meisenheimer intermediatee,f | −0.429 | 1.40 | 1.40 | 1.00 | n/a |
Thus, although a concerted mechanism might be expected for this reaction based solely on the structure of the electrophile, the presence of the proton sufficiently stabilizes the Meisenheimer region to create a borderline mechanism. Still, the stabilization is insufficient to render the reaction fully stepwise.47 Importantly, the poor leaving group ability of fluoride anion is the key factor that allows for observation of this mechanistic transition from concerted to borderline. For starting materials bearing better leaving groups, the reaction would almost certainly remain in the concerted regime.
Further, the stabilization conferred by general base catalysis is likely what allows this reaction to occur at all and is reflected in the smaller ρ value observed in the Hammett plot. General base catalysis decreases the negative charge transferred to the arene and makes strongly electron-withdrawing substituents unnecessary. Consequently, ρ decreases and the reaction scope becomes more general.48
Visualizing the mechanistic continuum
We were interested to learn how the insights we gained into this SNAr reaction with indole would relate to a broader range of azole nucleophiles and aryl fluoride electrophiles. To study this, we calculated the potential energy surfaces for 72 reactions spanning a range of azole nucleophiles and aryl fluoride electrophiles. Across this set of reactions, we observe a smooth transition from stepwise to concerted mechanisms, through the borderline regime. A subset of these calculations is shared here (Fig. 8a), and the full set is shared in the ESI.† | ||
| Fig. 8 (a) Ensemble of potential energy surfaces calculated for 20 anionic SNAr reactions involving aryl fluoride electrophiles and azole nucleophiles. Electrophile range from highly activated (left) to deactivated (right); nucleophiles range from weakly acidic (top) to strongly acidic (bottom). The colour scale represents the relative energy for each individual potential energy surface. The complete set of 72 reactions can be found in the ESI.† (b) Transition structure geometries for all 72 SNAr reactions. Inset plot: locations of transition states plotted on the same addition–elimination coordinate, showing clustering around the addition arm. The same points are shown in the larger plot, which has been zoomed-in. Points of the same colour represent transition states involving the same electrophile. Calculations carried out at B3LYP-D3(BJ)/6-31+g*/CPCM(DMA). | ||
As electrophilicity decreases across the grid from left to right, the Meisenheimer region (short C–N and C–F distances, bottom left corner) becomes progressively less stable and the addition transition state shifts earlier. For stepwise reactions with very reactive electrophiles, the local minimum corresponding to the Meisenheimer intermediate can be observed, flanked by discrete addition and elimination transition states. Concerted reactions, on the right side of the grid, show a minimum energy path involving only a single transition state, and the Meisenheimer region is very unstable. In between, the minimum energy path for borderline reactions proceeds through the Meisenheimer region, with a gradual decrease in energy from the transition state to the products. Remarkably, the electrophile solely determines whether the mechanism is predicted to be stepwise, borderline, or concerted, as seen when considering any individual column in the grid of potential energy surfaces. Changes in nucleophilicity simply shift the location of the transition state along the addition coordinate.
Across the mechanistic continuum for this reaction, the transition structures are similar and vary smoothly (Fig. 8b). In the inset plot for Fig. 8b, the locations of the DFT-predicted transition structures for each reaction in Fig. 8a are plotted on the same addition–elimination coordinate. All of the transition structures are located along the addition arm of the reaction coordinate, with some variation in C–N distance but very little in C–F distance. Thus, regardless of whether the mechanism is stepwise, borderline, or concerted, the rate-limiting step involves predominantly addition and essentially no elimination. The mechanisms are instead distinguished only by the relative energies of the addition, elimination, and Meisenheimer structural regimes. The smooth changes in transition state geometry are also predicted by Marcus theory and are consistent with the gradually changing charge character as observed in the Hammett plot described above. In contrast, many reactions that can proceed through competing mechanisms exhibit clear delineations between those mechanisms.13,15
This theoretical prediction is also mirrored by literature measurements of Eyring parameters for SNAr reactions. When these values are aggregated, we see that the entropies of activation do not neatly separate into negative and positive groups for associative and dissociative rate-limiting steps, respectively.49 Rather, there is significant enthalpy-entropy compensation and the entropy values vary smoothly from −60 e.u. to +60 e.u. across a wide range of substrates that likely span a mechanistic range from stepwise to concerted. This experimental observation is consistent with our DFT prediction that the SNAr transition structures located in our computational grid also vary smoothly in geometry, irrespective of mechanism. We measured the entropy of activation for the indole/4-fluorobenzonitrile reaction and obtained ΔS‡ = −44(1) e.u., consistent with a highly organized rate-determining transition structure. It also falls within the previously reported range for SNAr. In the context of SNAr, this finding could be consistent with the associative step in any of the stepwise, borderline, or concerted regimes, but is inconsistent with rate-determining elimination in a stepwise mechanism.
Color-coding the transition states according to electrophile reveals sets of linear correlations between the C–N and C–F distances within homologous series of electrophiles (Fig. 8b). This suggests that the ensemble of DFT-predicted potential energy surfaces has a simple structure that lends itself to straightforward modelling. A linear regression analysis of the constituent transition state geometries shows that both the C– N and C–F distances can be linearly correlated with the pKa of the nucleophile, the σ value of the electrophile substituent, and an intercept. That is, these distances can be predicted with high accuracy based only on these two ground-state parameters. Similarly, the energies of the Meisenheimer complexes and transition states can be modelled using the same parameters.
Given the importance of SNAr as a method for functionalizing aromatic rings, there has been considerable interest in general methods for estimating the feasibility of unknown substitutions. An appealing strategy for prediction of SNAr feasibility thus far has been to generate machine learning surrogates for DFT surfaces,50–52 and the simple structures of the DFT surfaces generated here do lend themselves to this approach. However, future work in this area will need to account for the possibility of competing modes of base catalysis, given that DFT cannot accurately estimate the relative energies of these pathways. Failure to account for general base catalysis could lead to erroneous predictions of some SNAr reactions as infeasible, when in fact the general base pathway would enable the reaction due to the stabilization and generality conferred by partial deprotonation. Future experimental studies across a broader range of chemical space will be required before a general model of SNAr reactivity can be constructed.
Conclusions
We have demonstrated here that SNAr reactions between indoles and aryl fluorides proceed through a general-base catalysed borderline mechanism and that these reactions broadly appear to lie on a mechanistic continuum. The Meisenheimer region is stabilized by partial deprotonation of the nucleophile, but not to a degree that allows for a stepwise process. As a result, elimination is fast, and the rate-determining step involves primarily addition. Our conclusions are supported by LFER studies, which find moderate degrees of negative charge on both the electrophile and nucleophile, and are reinforced by DFT predictions. A computational survey of 72 SNAr reactions further reveals the mechanistic continuum, as transition structure geometries change smoothly between the stepwise and concerted extremes. The changing charge delocalization observed in the experimentally obtained LFER plot further corroborates the existence of this mechanistic continuum. It remains to be seen whether the existence of general-base catalysis is widespread, and whether the mechanistic continuum observed here translates to a broader scope of nucleophiles and electrophiles.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. K. T. conceived of and initiated the study. H. W. T., T. G., J. J. L., and A. K. T. performed and analysed kinetics experiments. K. J. Y. participated in the early development of the project. G. D. P., M. R., and E. E. K. performed NMR analysis of the KIE experiments. X. Z. and E. E. K. carried out the DFT calculations. A. K. T. supervised the project. The manuscript was written by A. K. T., including discussions with E. E. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Dr Nathan Cook and Dr Silas Brown (Williams College) and Dr Robert Samples (Smith College) for assistance with instrumentation, and Professor Joseph Gair (Michigan State University) for guidance and assistance with computations. This work was partially supported by the donors of the ACS Petroleum Research Fund under Undergraduate New Investigator Grant 65288-UNI4.Notes and references
- S. D. Roughley and A. M. Jordan, The Medicinal Chemist's Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?, J. Med. Chem., 2016, 59, 4443 CrossRef CAS PubMed.
- F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, 2013, and references therein Search PubMed.
- W.-L. Zeng, Q. Xia, C.-Q. Li, M.-Y. Wang, W.-Y. Jin, H. Ding and W. Li, Bench-Stable Meisenheimer Complexes: Synthesis, Characterization, and Divergent Reactivity for Dearomatization, J. Am. Chem. Soc., 2024, 146, 30764 CrossRef CAS PubMed.
- A. Hunter, M. Renfrew, J. A. Taylor, J. M. J. Whitmore and A. Williams, A single transition state in nucleophilic aromatic substitution: reaction of phenolate ions with 2-(4-nitrophenoxy)-4,6-dimethoxy-1,3,5-triazine in aqueous solution, J. Chem. Soc., Perkin Trans. 2, 1993, 1703 Search PubMed.
- A. Hunter, M. Renfrew, D. Rettura, J. A. Taylor, J. M. J. Whitmore and A. Williams, Stepwise versus Concerted Mechanisms at Trigonal Carbon: Transfer of the 1,3,5-Triazinyl Group between Aryl Oxide Ions in Aqueous Solution, J. Am. Chem. Soc., 1995, 117, 5484 CrossRef CAS.
- J. Shakes, C. Raymond, D. Rettura and A. Williams, Concerted displacement mechanisms at trigonal carbon: the aminolysis of 4-aryloxy-2,6-dimethoxy-1,3,5-triazines, J. Chem. Soc., Perkin Trans. 2, 1996, 1553 RSC.
- N. R. Cullum, D. Rettura, J. M. J. Whitmore and A. Williams, The aminolysis and hydrolysis of N-(4,6-diphenoxy-1,3,5-triazin-2-yl) substituted pyridinium salts: concerted displacement mechanism, J. Chem. Soc., Perkin Trans. 2, 1996, 1559 RSC.
- S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Concerted Nucleophilic Aromatic Substitution Reactions, Angew. Chem., Int. Ed., 2019, 58, 16368 CrossRef CAS PubMed.
- E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Concerted nucleophilic aromatic substitutions, Nat. Chem., 2018, 10, 917 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- A. V. Levanov, U. D. Gurbanova, O. Y. Isaikina and V. V. Lunin, Dissociation Constants of Hydrohalic Acids HCl, HBr, and HI in Aqueous Solutions, Russ. J. Phys. Chem. A, 2019, 93, 93 CrossRef CAS.
- A. R. Katritzky and B. E. Brycki, The Mechanisms of Nucleophilic Substitution in Aliphatic Compounds, Chem. Soc. Rev., 1990, 19, 83 RSC.
- Some aliphatic substitution pathways involve competing SN1 and SN2 mechanisms, T. B. Phan, C. Nolte, S. Kobayashi, A. R. Ofial and H. Mayr, Can One Predict Changes from SN1 to SN2 Mechanisms?, J. Am. Chem. Soc., 2009, 131, 11392 CrossRef CAS PubMed.
- M. L. Bender, Mechanisms of Catalysis of Nucleophilic Reactions of Carboxylic Acid Derivatives, Chem. Rev., 1960, 60, 1 CrossRef.
- R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- T. P. Silverstein, Marcus theory: thermodynamics can control the kinetics of electron transfer reactions, J. Chem. Educ., 2012, 89, 1159–1167 CrossRef CAS.
- J. F. Bunnett and C. Bernasconi, Kinetics of Reactions of Piperidine with Ethers of 2,4-Dinitrophenol in 10% Dioxane-90% Water. Dependence of Base Catalysis on the Group Displaced, J. Am. Chem. Soc., 1965, 87, 5209 CrossRef CAS.
- H. Raissi, I. Jamaoui, R. Goumont and T. Boubaker, Kinetic Studies on SNAr Reactions of Substituted Benzofurazan Derivatives: Quantification of the Electrophilic Reactivities and Effect of Amine Nature on Reaction Mechanism, Int. J. Chem. Kinet., 2017, 49, 837 CrossRef.
- J. A. Orvik and J. F. Bunnett, Kinetics of the Separately Observable Formation and Decomposition of the Intermediate Complex in Aromatic Nucleophilic Substitution. Reactions of 2,4-Dinitro-1-naphthyl Ethyl Ether with n-Butyl- and t-Butylamine in Dimethyl Sulfoxide Solution, J. Am. Chem. Soc., 1970, 92, 2417 CrossRef CAS.
- I.-H. Um, M.-Y. Kim, T.-A. Kang and J. M. Dust, Kinetic Study on SNAr Reaction of 1-(Y-Substituted-phenoxy)-2,4-dinitrobenzenes with Cyclic Secondary Amines in Acetonitrile: Evidence for Cyclic Transition-State Structure, J. Org. Chem., 2014, 79, 7025 CrossRef CAS PubMed.
- C. F. Bernasconi, Kinetic Behavior of Short-Lived Anionic σ Complexes, Acc. Chem. Res., 1978, 11, 147 CrossRef CAS.
- I.-H. Um, S.-W. Min and J. M. Dust, Choice of Solvent (MeCN vs. H2O) Decides Rate-Limiting Step in SNAr Aminolysis of 1-Fluoro-2,4-dinitrobenzene with Secondary Amines: Importance of Brønsted-Type Analysis in Acetonitrile, J. Org. Chem., 2007, 72, 8797 CrossRef CAS PubMed.
- I.-H. Um, L.-R. Im, J.-S. Kang, S. S. Bursey and J. M. Dust, Mechanistic Assessment of SNAr Displacement of Halides from 1-Halo-2,4-dinitrobenzenes by Selected Primary and Secondary Amines: Brønsted and Mayr Analyses, J. Org. Chem., 2012, 77, 9738 CrossRef CAS PubMed.
- C. F. Bernasconi and C. L. Gehriger, Intermediates in Nucleophilic Aromatic Substitution. XI. Spiro Meisenheimer Complex of N,N’-Dimethyl-N-picrylethylenediamine. Partially Rate-Limiting Proton Transfer of Complex Formation. A Temperature-Jump Study, J. Am. Chem. Soc., 1974, 96, 1092 CrossRef CAS.
- C. F. Bernasconi and F. Terrier, Intermediates in Nucleophilic Aromatic Substitution. XIV. Spiro Meisenheimer Complexes Derived from N,N’-Dimethylethylenediamine. Kinetics in Aqueous Dimethyl Sulfoxide, J. Am. Chem. Soc., 1975, 97, 7458 CrossRef CAS.
- E. Buncel and W. Eggimann, Base Catalysis of σ-Complex Formation between 1,3,5-Trinitrobenzene and Aniline in Dimethyl Sulfoxide. Rate Limiting Proton Transfer, J. Am. Chem. Soc., 1977, 99, 5958 CrossRef CAS.
- C. F. Bernasconi, M. C. Muller and P. Schmid, Rate-Limiting Proton Transfer in the Formation of Meisenheimer Complexes between 1,3,5-Trinitrobenzene and Amines. The Effect of Dimethyl Sulfoxide on Proton-Transfer Rates. Relative Leaving-Group Abilities of Amines and Alkoxide Ions, J. Org. Chem., 1979, 44, 3189 CrossRef CAS.
- F. Terrier, M.-J. Pouet, J.-C. Halle, S. Hunt, J. R. Jones and E. Buncel, Electrophilic Heteroaromatic Substitutions: Reactions of 5-X-Substituted Indoles with 4,6-Dinitrobenzofuroxan, J. Chem. Soc., Perkin Trans. 2, 1993, 1665 RSC.
- F. Terrier, S. Lakhdar, T. Boubaker and R. Goumont, Ranking the Reactivity of Superelectrophilic Heteroaromatics on the Electrophilicity Scale, J. Org. Chem., 2005, 70, 6242 CrossRef CAS PubMed.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, Nucleophilic Reactivities of Indoles, J. Org. Chem., 2006, 71, 9088 CrossRef CAS PubMed.
- R. H. de Rossi and A. Veglia, General-Base Catalysis in the Reaction of Water with Activated Aromatic Substrates. The Hydrolysis of 3-Methyl-1-picylimidazolum Ion, J. Org. Chem., 1983, 48, 1879 CrossRef CAS.
- C. F. Bernasconi, R. H. de Rossi and P. Schmid, Changing Views on the Mechanism of Base Catalysis in Nucleophilic Aromatic Substitution. Kinetics of Reactions of Nitroaryl Ethers with Piperidine and with n-Butylamine in Aqueous Dioxane, J. Am. Chem. Soc., 1977, 99, 4090 CrossRef CAS.
- J. Kim, Y. Hayashi, S. Badr, K. Okamoto, T. Hakogi, H. Furukawa, S. Yoshikawa, H. Nakanishi and H. Sugiyama, Mechanistic Insights into Amination via Nucleophilic Aromatic Substitution, React. Chem. Eng., 2023, 8, 2060 RSC.
- C. F. Bernasconi and J. R. Gandler, Multiple Structure-Reactivity Relationships in the Acid-Catalyzed Breakdown of Meisenheimer Complexes, J. Am. Chem. Soc., 1978, 100, 8117 CrossRef CAS.
- J. Miller, Electrophilic and nucleophilic substitution in the Benzene ring and the Hammett equation, Aust. J. Chem., 1956, 9, 61 CrossRef CAS.
- J. Miller and W. Kai-Yan, The SN Mechanism in Aromatic Compounds. Part XXIX. Some para-Substituted Chlorobenzenes, J. Chem. Soc., 1963, 3492 RSC.
- S. E. Fry and N. J. Pienta, Effects of Molten Salts on Reactions. Nucleophilic Aromatic Substitution by Halide Ions in Molten Dodecyltributylphosphonium Salts, J. Am. Chem. Soc., 1985, 107, 6399 CrossRef CAS.
- C. N. Neumann, J. M. Hooker and T. Ritter, Concerted nucleophilic aromatic substitution with 19F− and 18F−, Nature, 2016, 534, 369 CrossRef CAS PubMed.
- D. J. Leonard, J. W. Ward and J. Clayden, Asymmetric α-arylation of amino acids, Nature, 2018, 562, 105 CrossRef CAS PubMed.
- A. I. Biggs and R. A. Robinson, The ionisation constants of some substituted anilines and phenols: a test of the Hammett relation, J. Chem. Soc., 1961, 388 RSC.
- E. W. Lewis, Rate-Equilibrium LFER Characterization of Transition States: The Interpretation of α, J. Phys. Org. Chem., 1990, 3, 1 CrossRef CAS.
- M. A. Muñoz, P. Guardado, J. Hidalgo, C. Carmona and M. Balón, An Experimental and Theoretical Study of the Acid-base Properties of Substituted Indoles, Tetrahedron, 1992, 48, 5901 CrossRef.
- G. Dal Poggetto, J. V. Soares and C. F. Tormena, Selective Nuclear Magnetic Resonance Experiments for Sign Sensitive Determination of Heteronuclear Couplings: Expanding the Analysis of Crude Reaction Mixtures, Anal. Chem., 2020, 92, 14047 CrossRef CAS PubMed.
- C. Mycroft, G. Dal Poggetto, T. M. Barbosa, C. F. Tormena, M. Nilsson, G. A. Morris and L. Castañar, Rapid Measurement of Heteronuclear Coupling Constants in Complex NMR Spectra, J. Am. Chem. Soc., 2023, 145, 19824 CrossRef CAS PubMed.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases, J. Chem. Theory Comput., 2012, 8, 527 CrossRef CAS PubMed.
- W. P. Jencks, When Is an Intermediate Not an Intermediate? Enforced Mechanisms of General Acid-Base Catalyzed, Carbocation, Carbanion, and Ligand Exchange Reactions, Acc. Chem. Res., 1980, 13, 161 CrossRef CAS.
- M. G. Kung, P. Onnuch and R. Y. Liu, Rapid and General Amination of Aryl Boronic Acids and Esters Using O-(Diphenylphosphinyl)hydroxylamine (DPPH), Org. Lett., 2024, 26, 9847 CrossRef CAS PubMed.
- V. M. Vlasov, Energetics of bimolecular nucleophilic reactions in solution, Russ. Chem. Rev., 2006, 75, 765 CrossRef CAS.
- K. Jorner, T. Brinck, P.-O. Norrby and D. Buttar, Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies, Chem. Sci., 2021, 12, 1163 RSC.
- T. Stuyver and S. Shaik, Promotion Energy Analysis Predicts Reaction Modes: Nucleophilic and Electrophilic Aromatic Substitution Reactions, J. Am. Chem. Soc., 2021, 143, 4367 CrossRef CAS PubMed.
- J. Lu, I. Paci and D. C. Leitch, A broadly applicable quantitative relative reactivity model for nucleophilic aromatic substitution (SNAr) using simple descriptors, Chem. Sci., 2022, 13, 12681 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and initial rates plots, kinetic isotope effect methods, computational methods and coordinates, characterization of reaction products, NMR spectra, and raw data files (ZIP). See DOI: https://doi.org/10.1039/d5sc01856k |
| ‡ The reaction can also be carried out in DMF and DMSO. |
| § General base catalysis likely also would proceed through a mechanism involving pre-association of the base and the indole, and not through a termolecular elementary step. However, this microscopic pre-association step would also only lead to a change in the composition of kobs, and is therefore also indistinguishable from the other scenarios. |
| ¶ We chose to use σ− as a reference, recognizing that resonance delocalization might play an important role. Using Hammett σ as a reference gave a scattered correlation (see ESI section II-E†). |
| || This is a “pseudo-Brønsted” analysis because we examined the pKa of the nucleophile rather than the pKa of the base. This results in a slope of −1 for a mechanism involving full deprotonation; if the rate were correlated with the pKa of the base, the maximum slope would be 1. |
| ** These pKa values are DFT-predicted and linearly scaled according to the experimental pKa values in DMSO (see ESI section IV-C†). |
| This journal is © The Royal Society of Chemistry 2025 |