
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging techniques and scenarios of scanning electrochemical microscopy for the characterization of electrocatalytic reactions
Jinming
Xu
,
Ran
Chen
 *,
Juanxian
Song
,
Songqin
Liu
,
Yanfei
Shen
and
Yuanjian
Zhang
*,
Juanxian
Song
,
Songqin
Liu
,
Yanfei
Shen
and
Yuanjian
Zhang
Jiangsu Province Key Laboratory of Critical Care Medicine, Jiangsu Engineering Laboratory of Smart Carbon-Rich Materials and Device, Jiangsu Province Hi-Tech Key Laboratory for Bio-Medical Research, School of Chemistry and Chemical Engineering, Southeast University, Nanjing, 211189, China. E-mail: rchen@seu.edu.cn
First published on 13th May 2025
Abstract
To fulfill the evergrowing energy consumption demands and the pursuit of sustainable and renewable energy, electrocatalytic reactions such as the water electrocatalysis reaction, the O2 reduction reaction, the N2 reduction reaction (NRR), the CO2 reduction reaction (CO2RR), etc., have drawn a lot of attention. Scanning electrochemical microscopy (SECM) is a powerful technique for in situ surface characterization, providing critical information about the local reactivity of electrocatalysts and unveiling key information about the reaction mechanisms, which are essential for the rational design of novel electrocatalysts. There has been a growing trend of SECM-based studies in electrocatalytic reactions, with a major focus on water splitting and O2 reduction reactions, and relying mostly on conventional SECM techniques. Recently, novel operation modes of SECM have emerged, adding new features to the functionality of SECM and successfully expanding the scope of SECM to other electrocatalytic reactions, i.e., the NRR, the NO3− reduction reaction (NO3RR), the CO2RR and so on, as well as more complicated electrolysis systems, i.e. gas diffusion electrodes. In this perspective, we summarized recent progress in the development of novel SECM techniques and recent SECM-based research studies on the NRR, NO3RR, CO2RR, and so on, where quantitative information on the reaction mechanism and catalyst reactivity was uncovered through SECM. The development of novel SECM techniques and the application of these techniques can provide new insights into the reaction mechanisms of diverse electrocatalytic reactions as well as the in situ characterization of electrocatalysts, facilitating the pursuit of sustainable and renewable energy.
 Jinming Xu | Jinming Xu received his BS in chemistry from Yangzhou University in 2023 and is currently a master's student under the supervision of Prof. Ran Chen and Prof. Yuanjian Zhang. He is currently focusing on reactive oxygen species detection using nanopipet-supported interfaces between two immiscible electrolytes (nano-ITIES) and scanning electrochemical microscopy, and his research interests include ultramicroeletrodes, the oxygen reduction reaction and molecular sensing. |
 Ran Chen | Ran Chen received his BS from Nanjing University in 2010, and completed his PhD in Chemistry at the University of Pittsburgh in 2017 under the supervision of Prof. S. Amemiya. After graduation, he worked as a postdoctoral research associate in Prof. M. Shen's group at the University of Illinois at Urbana-Champaign from 2017 to 2020. He is currently an associate professor at the School of Chemistry and Chemical Engineering in Southeast University, China. His research interest focused on the electrochemical characterization of reaction mechanisms using ultramicroelectrodes. |
 Yuanjian Zhang | Yuanjian Zhang received his BS from Nanjing University in 2002 and completed his PhD at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 2007. Subsequently, he joined the Max-Planck Institute of Colloids and Interfaces (Germany) as a postdoctoral researcher. From 2009–2012, he worked at the National Institute for Materials Science (Japan) as an ICYS researcher. He joined the faculty at Southeast University (China) in 2012 as a Professor of Chemistry. He is interested in carbosensing (carbon matter-derived optoelectronic sensors), and has expertise in electroanalytical chemistry, electrochemiluminescence, electrocatalysis, photocatalysis and photoelectrochemistry. |
Introduction
The pollution issues and greenhouse effects caused by fossil fuels as well as the ever-growing demand for energy have become a worldwide concern.1–4 Thus, the pursuit of sustainable and renewable energy, and the utilization of this energy, has drawn lots of attention.5–10 Electrocatalytic reactions driven by electricity generated through wind power, tidal power, solar power, etc., play crucial roles in the field of sustainable and renewable energy generation and utilization.11–14 For example, the water electrolysis reaction, one of the most-studied electrocatalytic reactions, is vital for hydrogen power generation.15–17 The carbon dioxide reduction reaction (CO2RR) can convert CO2 in the atmosphere into organic products.18–20 The nitrate reduction reaction (NO3RR) helps to convert the nitrogen pollutant in industrial and agriculture wastewater into value-added ammonia products.21–23 As a result, several core electrocatalytic reactions have triggered a great deal of research.24–28Comprehensive knowledge of various complicated processes at the catalyst interface during an electrocatalytic reaction, including the adsorption and desorption process, the electron transfer process, solvation and desolvation behaviors, electrostatic interactions, etc., is crucial for the engineering and optimization of electrocatalytic systems as well as for the rational design of novel electrocatalysts. The structural and chemical characteristics of electrocatalysts can be examined using various characterization methods, including scanning electron microscopy-energy dispersive X-ray spectrometry (SEMEDS), X-ray photoelectron spectroscopy (XPS), UV-vis fluorescence microscopy, Raman spectroscopy and so on.29 On the other hand, conventional electrochemical techniques such as cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) are widely used in the study of electrocatalytic reactions, capturing the average electrochemical properties of the catalysts.30,31 However, since electrocatalysts are often spatially heterogeneous due to surface features at the microscopic level, i.e., surface defects, various crystalline facets and phases, non-uniform catalyst distribution, etc.,32–35 an electrochemical technique with high spatial resolution is critical for characterizing the local reactivity at different microscopic regions and unraveling the complicated reaction processes during electrolysis.
Scanning electrochemical microscopy (SECM) is a powerful and versatile technique for in situ surface characterization with high spatial and temporal resolution.36–39 Using nanoelectrodes as the probe, a lateral spatial resolution ∼10 nm and a horizontal tip–substrate distance of a few nanometers on flat substrates have been achieved, enabling the detection of short-lived intermediates and the analysis of ultrafast reaction kinetics near the substrate.40–43 Due to its high spatial resolution, in situ operation modes, and versatile functionality, SECM has also been widely used in various fields, such as studying complicated reaction mechanisms, characterizing the local morphology and reactivity at the electrode surface, probing cellular activity, etc.36,44–48 There is a growing trend of SECM-based studies in electrocatalytic reactions, where critical information on the local reactivity at different microscopic regions on an electrocatalytic material was revealed using SECM.43,49,50 The majority of these studies focused on water splitting and O2 reduction reactions,51–60 and traditional SECM operation techniques, namely, the feedback mode and the generation/collection mode, were used to quantify the localized reactivity.61–63 Additionally, since a flat electrode surface was required for positioning the probe close and carrying out accurate SECM measurements, these SECM-based studies were often limited to two-dimensional planar materials.64,65
Recently, novel operation modes of SECM have emerged, adding new features to the functionality of SECM.66–72 For example, the surface interrogation mode enabled the quantification of active sites on catalytic surfaces,66,67 and sequential voltammetric SECM (SV-SECM) permits the simultaneous identification of numerous species under complicated working conditions and provided the capability for mapping facet-dependent products selectively,68 while shear-force-based and capacitance-based approach curves allowed for the positioning of probes near non-flat surfaces.69–72 These new techniques helped to expand the scope of SECM to other electrocatalytic reactions, i.e., the N2 reduction reaction (NRR), NO3RR, CO2RR, and so on, as well as more complicated electrolysis systems, such as the gas diffusion electrode (GDE). Thus, in this perspective, the recent development of novel SECM techniques was first summarized, followed by the recent SECM-based research on the NRR, NO3RR, CO2RR, and so on, where quantitative information on the reaction mechanism and catalyst reactivity was uncovered through SECM.
Recent progress in novel SECM operative modes
Surface interrogation mode
Although first introduced in 2008,73,74 the surface interrogation (SI) mode of SECM has regained a lot of interest in the field of electrocatalysis recently,59,66,75,76 as it allowed for the study of active site densities and reaction kinetics where adsorbents are involved.The working principle of the SI mode is shown in Fig. 1a–d. An UME serves as the probe to ‘interrogate’ the substrate surface coated with electrocatalysts by electrochemical titration. A redox mediator O is dispersed in solution, which can be reduced to R through a simple electron transfer reaction O + e → R at the biased UME surface, and when the substrate is under open circuit potential (OCP), R cannot convert back to O on the substrate. However, with the species A adsorbed at the active sites on the electrocatalyst surface, R can react with A to regenerate O, leading to a feedback current on UME, until all of the A species have reacted with R (Fig. 1b).77 This way, the amount of A on the electrocatalyst surface can be electrochemically titrated by monitoring the feedback current on the UME, leading to a quantification of the active site density on the electrocatalyst surface. Additionally, if there are other competing processes on the active sites, such as the adsorption of another species B (Fig. 1c), the surface concentration of A will change with time. By varying the delay time before the titration, the time-dependent surface concentration of A can be quantified, leading to kinetic information of the adsorption rate of B (Fig. 1d). Note that a substrate comparable in size to the UME is usually required for accurate measurement of adsorbates to minimize the possible interference from lateral charge transfer effects.
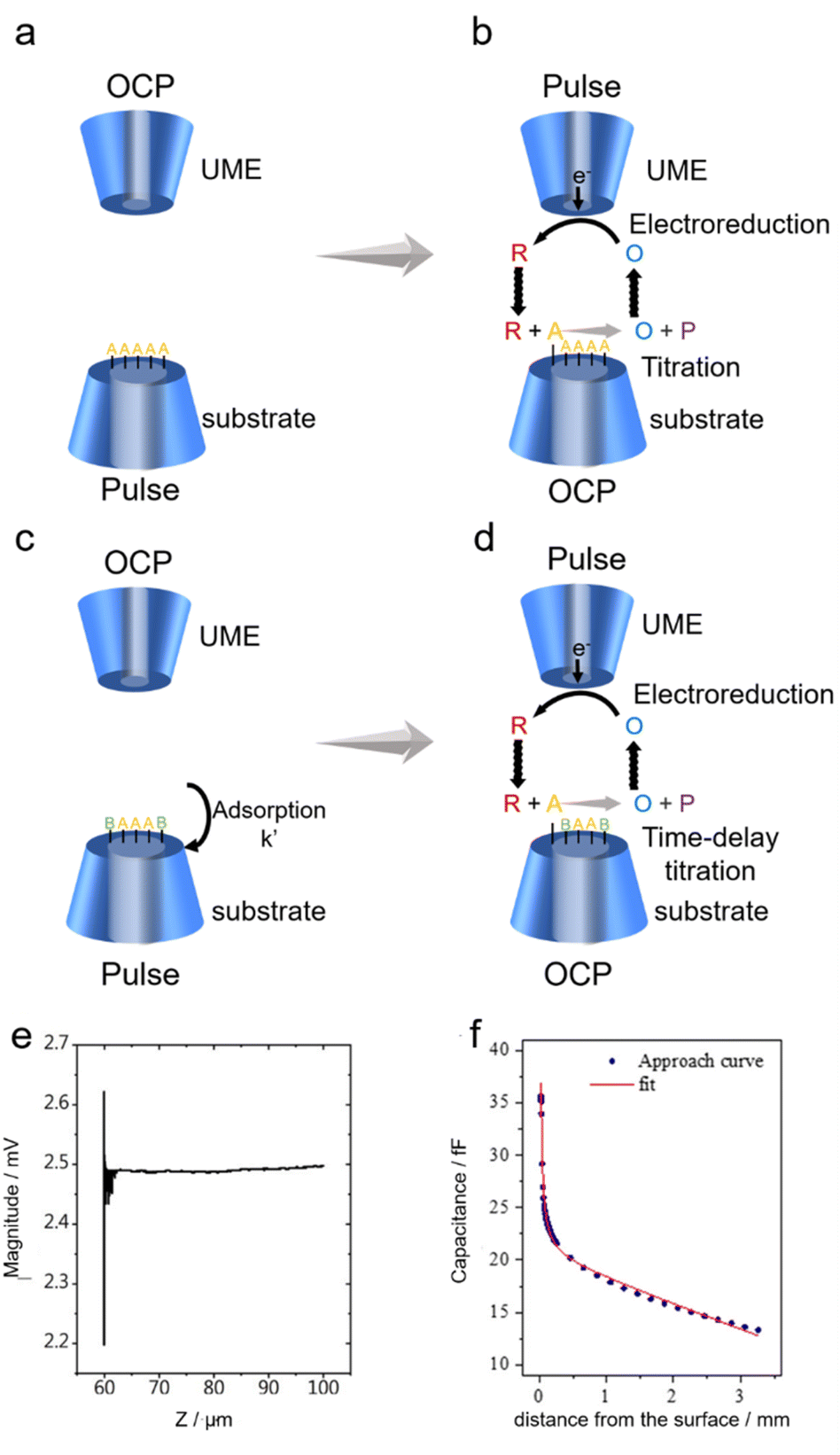 | ||
| Fig. 1 (a–d) Schematic diagrams of SECM operation in SI mode. P represents the byproduct of the reaction between R and A. B is a substance that competes with A for adsorption on the substrate. (e) Shear forced based approach curve. Reproduced with permission from ref. 69, Royal Society of Chemistry, 2021. (f) Capacitive approach curve obtained (blue circles) with its fit to the theoretical result (red line). Reproduced with permission from ref. 71, American Chemical Society, 2019. | ||
Novel principles for carrying out approach curves
Prior to SECM measurements, the SECM probe (also known as the tip) was positioned near the substrate surface through the approach curve. Conventionally, the approach curve relies on the feedback effect where the diffusion towards the tip is interfered with by the substrate as the tip–substrate distance gets shorter, i.e. around the same magnitude as the size of the tip. The conventional approach curves prefer substrates with superior flatness, and are conducted in the aqueous phase, limiting the application of SECM when a rough substrate coated with catalysts or a solid/gas interface is under investigation. Recently, new principles for carrying out the approach curves have been proposed that address the above issues,69–72 namely shear-force-based and capacitance-based approach curves.Shear force, a type of short-range hydrodynamic force that exists only a few hundred nanometers away from solid surfaces,78 lays the foundation for shear-force-based approach curves. During the shear-force-based approach curves, a resonantly oscillating tip is moving towards the substrate by a piezo with the oscillating magnitude monitored. When far from the substrate, the oscillating magnitude on the tip barely changes, yet once the tip–substrate distance is within the range of the shear force, the oscillation characteristics of the tip are modulated by the force, indicating a successful approach to the substrate (Fig. 1e). Since the shear-force is present regardless of the surface flatness, the shear-force based approach curve could work on electrodes with non-ideal flatness.
Another alternative method to carry out the approach curve relies on capacitance measurements in air.70–72 By applying an AC voltage (typically 10 kHz, 1.4 VRMS) on the substrate, a capacitive current can be generated on the tip. Considering the UME near a substrate as a system with a point charge positioned in front of a parallel-plate, the tip–substrate distance-dependent capacitance can be described by using eqn (1):
 | (1) |
Novel SECM technique-based study of electrocatalytic reactions
SI-SECM-based NRR study
Over the past century, the Haber–Bosch process, which contributes annually to over 90% of the world's ammonia production, has powered the chemical synthesis of ammonia and fed billions of people. Due to its heavy reliance on fossil fuels, this process actually consumes around 2% of the total anthropogenic energy and emits 400 million tons of CO2 annually.79–81 The synthesis of ammonia by electrocatalysis offers the potential to operate on renewable electricity under ambient conditions with no carbon footprint, and has garnered lots of attention recently.82–85 NH3 synthesis through the electrochemical NRR is becoming a viable substitute for the traditional Haber–Bosch fertilizer industry.86–88 Vast amounts of efforts have been devoted to the development of stable and effective catalysts for the NRR, particularly earth-abundant non-precious transition metal (TM)-based materials such as metallic oxides, sulphides, nitrides, and carbides.89–91 However, under acidic conditions, the abundance of protons in the solution causes an increased competition with the hydrogen evolution reaction (HER), posing a problem of reaction selectivity.A novel type of TiO2 nanoreactor with surface strain was proposed by Li and colleagues, which allowed for the synthesis of ammonia with high selectivity.92 TiO2 nanotube arrays (TiO2 NTs) were first created by anodizing Ti plates twice, followed by annealing to form anatase TiO2. The electrochemical intercalation of Li ions and the follow-up delithiation created lattice strain (s-TiO2 NTs). The structural and chemical environments of TiO2 nanoreactors were examined using X-ray diffraction (XRD) and high-resolution transmission electron microscopy (TEM) to confirm the presence of lattice strain (Fig. 2a–d). Then the authors studied the NRR activity of the TiO2 NTs by applying −0.5 V on the TiO2 nanoreactors for 2 hours, and monitored the NH3 production through UV/vis absorption using the indophenol blue indicator. As shown in Fig. 2e, compared to TiO2 NTs, the tailored s-TiO2 NTs delivered improved performance for the conversion of nitrogen to ammonia. It was discovered that s-TiO2 exhibited 26% higher faradaic efficiency compared to TiO2 NTs. To better understand this strain-induced increase in NRR efficiency, the active site densities on TiO2 and s-TiO2 were quantified using the SI-SECM approach (Fig. 2f). Two identical 50 μm-diameter glass-sealed platinum UMEs served as the tip and substrate electrodes. Using a tip-generated titrant (ferrocenium, Fc+), the amount of the active species, Ti3+ on the surface of the catalyst was analyzed. More precisely, the substrate was first biased at a reduction potential Esubstrate for 20 s to convert all the Ti4+ at the active sites to Ti3+. Then the substrate was biased at OCP, the tip was biased to oxidize Fc to Fc+, and the chronoamperometric curve on the tip was recorded. At the substrate, Ti3+ on the catalyst surface would reduce Fc+ to Fc, which was then oxidized on the tip, causing feedback current signals on the tip until all of the Ti3+ was used up. Thus, by integrating the charges in the chronoamperometric curve, the precise amount of Ti3+ generated at Esubstrate was determined. Similarly, the reaction rate constants for the H+ and N2
and N2 binding on TiO2 NTs and s-TiO2 NTs were determined, by adding a delay time td before the titration, and tracking the remaining Ti3+ after Ti3+ had reacted with N2 and H+ for a duration of td. The outcome indicated that both
binding on TiO2 NTs and s-TiO2 NTs were determined, by adding a delay time td before the titration, and tracking the remaining Ti3+ after Ti3+ had reacted with N2 and H+ for a duration of td. The outcome indicated that both  and
and  exhibited characteristics of a pseudo-first-order process. Specifically,
exhibited characteristics of a pseudo-first-order process. Specifically,  for s-TiO2 NTs (0.62 s−1) was almost twice that on TiO2 NTs (0.32 s−1), suggesting that N2 adsorption on s-TiO2 NTs was more advantageous than on TiO2 NTs. Additionally, the
for s-TiO2 NTs (0.62 s−1) was almost twice that on TiO2 NTs (0.32 s−1), suggesting that N2 adsorption on s-TiO2 NTs was more advantageous than on TiO2 NTs. Additionally, the  adsorption on s-TiO2 was weaker compared with
adsorption on s-TiO2 was weaker compared with  explaining the good selectivity towards NH3 production observed on s-TiO2. The difference in the
explaining the good selectivity towards NH3 production observed on s-TiO2. The difference in the  and
and  observed on TiO2 and s-TiO2 was also supported by density functional theory (DFT) calculations (Fig. 2g).
observed on TiO2 and s-TiO2 was also supported by density functional theory (DFT) calculations (Fig. 2g).
 | ||
| Fig. 2 (a and b) XRD patterns of TiO2 NTs and s-TiO2 NTs. (c and d) TEM images and the corresponding fast Fourier transform (FFT) patterns (the inset) of TiO2 NTs (c) and s-TiO2 NTs (d). (e) UV/vis absorption spectra of the electrolyte with an indophenol indicator after 2 h of electrolysis at −0.5 V. (f) Illustration of SI-SECM for determining the reaction rate constants of H+ and N2 adsorption on the Ti3+ site in TiO2 NTs and s-TiO2 NTs. (g) DFT-calculated pathways of the NRR through the distal mechanisms on pristine and strained TiO2 (101). Reproduced with permission from ref. 92, John Wiley and Sons, 2020. | ||
Besides the SI-SECM technique, conventional SECM modes like SG/TC and SECM imaging have also been utilized in NRR study. Park et al. evaluated NRR electrocatalysis on a Fe–CuS/C electrode by in situ detection of NH3 using SECM for the first time.93 Conventionally, the activity of the NRR catalyst was estimated by the ex situ detection of NH3 using spectrophotometric methods including Nessler reagent, indophenol, salicylic acid reactions, etc. Additionally, liquid chromatography, ion chromatography, and a rotating ring-disc electrode (RRDE) were also commonly used for ex situ NH3 detection.94 To enable the in situ detection of NH3, the authors fabricated a polycrystalline Pt UME through thermochemical deposition of Pt on an unmodified Pt UME (d = 25 μm), and carried out CV measurements of the ammonia oxidation reaction (AOR) in solutions with different NH3 concentrations (Fig. 3a). It was found that the modified Pt UME showed an enhanced tip current towards the AOR, which was due to the easier AOR kinetics and increased surface area caused by the deposited Pt nanoparticles, and a calibration curve of peak current at 0.71 VRHEversus NH3 concentration was established (Fig. 3b). The calibration curve was compared with the ex situ colorimetrical measurements using the indephenol method, and consistent results were obtained. Furthermore, the authors showed that the stability of NRR catalysts can be evaluated through real-time NH3 detection using SECM, as presented in Fig. 3c. By locating the modified Pt UME close to the Fe–CuS/C electrode, the authors detected the NH3 generated at the electrode in situ and found that during repetitive CV scans, the amount of NH3 produced decreased, indicating a degrading process of the Fe–CuS/C electrode (Fig. 3d).
 | ||
Fig. 3 (a) CVs of NH3 oxidation at the modified Pt UME in 1 M KOH solution containing 0 to 20 mM NH3. Scan rate: 20 mV s−1. (b) The relationship between NH3 concentration and the peak current of the AOR in 1 M KOH solution. (c) A schematic diagram of the study of the NRR on the Fe–CuS/C-loaded carbon paper with a Pt UME in the SECM SG-TC mode. The inset shows an actual image of the experimental setup. (d) Repetitive CVs of the AOR were collected at the Pt UME when the UME was positioned close to the Fe–CuS/C-loaded carbon paper. The carbon paper was biased at −0.2 V vs. RHE. Scan rate: 20 mV s−1. Reproduced with permission from ref. 93, Elsevier, 2023. (e and f) Three-dimensional SECM image showing the local catalytic activity of Cu–Ni4B3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The tip was biased at a potential of 1.1 V in (e) under an Ar atmosphere to quantify the HOR on the tip, and in (f) under a N2 atmosphere to detect the products of both the HER and NRR. Reproduced with permission from ref. 95, Royal Society of Chemistry, 2023. :2). The tip was biased at a potential of 1.1 V in (e) under an Ar atmosphere to quantify the HOR on the tip, and in (f) under a N2 atmosphere to detect the products of both the HER and NRR. Reproduced with permission from ref. 95, Royal Society of Chemistry, 2023. | ||
Nagaiah et al. prepared a Cu–Ni4B3 catalyst and investigated the competition between the NRR and HER using SECM.95 The Cu–Ni4B catalyst with a grape bunch-like morphology was synthesized by a one-step sonochemical reduction method, with Cu added to the surface of nickel boride in an effort to change the electronic structure and increase the activity of the electrocatalytic NRR. Electrocatalytic NRR activity measurement with a catalyst-coated glass carbon plate by chronoamperometry demonstrated that Cu–Ni4B3 (1:2) exhibited superior NRR activity in 0.1 M H2SO4 electrolyte compared to catalysts of other proportions. Nevertheless, the catalyst faced fierce competition from the HER in acidic environments, making it challenging to determine the absolute NRR activity using traditional ex situ methods like gas chromatography and the RRDE. In order to quantify the NRR activity of the Cu–Ni4B3 (1:2) catalyst and investigate the competition from the HER, the in situ SG/TC mode of SECM was applied (Fig. 3e and f). The experiments were carried out in 0.1 M H2SO4 saturated under a N2 and Ar atmosphere, respectively, with 1.1 V vs. RHE applied on the tip and −0.3 V applied on the substrate. At this tip potential, both the HOR and AOR could occur on the tip surface. Under an Ar atmosphere, the concentration of N2 in the solution was negligible, so that the current signal corresponded to the HER on Cu–Ni4B3 (1:2) (Fig. 3e). However, when under a N2 atmosphere, larger tip currents were observed, which was due to the AOR current as a result of the NRR on Cu–Ni4B3 (1:2) (Fig. 3f), leading to solid proof of the competition between the HER and NRR.
SI-SECM-based study of the NO3RR
Apart from the NRR, the electroreduction of nitrate to ammonia (NO3RR) has been proposed as an alternative method to synthesize NH3, considering the abundant nitrate (NO3−) in waste water.96,97 Compared to the Haber-Bosch process, the NO3RR is more effective and more efficient, opening the door to waste recycling and sustainable nutrient recovery. Because of this, this approach has garnered a lot of interest, and numerous SECM-based studies have been conducted on the reaction mechanisms and selectivity of the NO3RR,98–103 which also suffers from the competition from the HER in a similar manner to the NRR.Yu's group constructed a new type of Cu single-atom-modified gel (Cu-SAG) for the NO3RR and the nitrite-to-ammonia reduction reaction (NO2RR) with reduced competition from the HER.98 The Cu-SAGs were synthesized through pyrolysis of the hydrogel-based precursors, namely, supramolecularly cross-linked polypyrrole (PPy)–copper(II) phthalocyanine tetrasulfonate (CuPcTs). Afterwards, Brunauer–Emmett–Teller (BET) surface area analysis and scanning electron microscopy (SEM) were used to confirm the porous structure of the gel. Atomic resolution scanning transmission electron microscopy (STEM) was used to further identify the metallic substances. Afterwards, the electrochemical properties of Cu-SAGs were characterized by linear sweep voltammetry (LSV). The onset potential for the HER on Cu-SAGs was found to be around −0.7 V vs. RHE, while the onset potential for NO3− reduction to NO2− and NO2− reduction to NH4+ was found to be around −0.5 V and −0.8 V, respectively. An enhanced Faraday efficiency for NH3 (∼78%) over H2 (∼20%) was observed, with a yield rate of ∼440 μg cm−1 h−1 for ammonia production. At more negative potentials (−0.9 V), the reduction of NO2− to NH4+ had a faradaic efficiency close to 100%. Additionally, a noteworthy enhancement in the yield rate of NO2− converting to NH4+ was also observed, reaching a maximum of 10.5 μg mL−1 at −0.9 V. This value was about three times greater than that of the NO3RR under the same circumstances. These findings indicated a different path for the NO2RR with minimal influence from the HER, considerably enhancing the faradaic efficiency and yield rate. To gain insight into the mechanism of the NO3RR and NO2RR, the authors investigated the adsorption rate constants of NO3−, NO2− and H2O on Cu sites using the SI-SECM technique, and discovered a much higher rate constant for NO2− adsorption (1.98 s−1) than that for NO3− (0.83 s−1) and H2O (0.06 s−1). This result explained the enhanced activity of NO3− reducing to NO2− compared with the HER, and it was also consistent with the molecular dynamics (MD) and DFT simulations. Based on this, a pulse electrolysis method was designed, where a higher potential of −0.5 V was first applied for 1.0 s to accumulate NO2− near the catalyst surface to overwhelm the H2O adsorption, and then a potential of −0.8 V was applied to reduce NO2− to NH4+. Through this pulse electrolysis method, the competition from the HER was heavily suppressed.
Yu and associates created a Cu single-atom alloy oxide nanowire (Ni1Cu SAAO NW) catalyst modified with Ni-sites for converting nitrate to ammonia with high efficiency.99 Cu(OH)2 nanowires were first grown on copper mesh and then treated with a Ni ion exchange process to form Ni1–Cu(OH)2 NWs. The Ni1–Cu(OH)2 NWs were then annealed and electrochemically reduced to form Ni1Cu SAAO (Fig. 4a), which was inspected by aberration-corrected high-angle annular dark-field scanning TEM (AC-HAADF-STEM) to acquire a more in-depth understanding of the Ni status in Ni1Cu SAAO (Fig. 4b). A 3D topographic atom imaging analysis of the pixel intensity, as presented in Fig. 4c, made it evident that Ni atoms were located between Cu atoms and exhibited a single-atom distribution on the substrate. Significantly, LSV measurements showed that compared to Cu2O NWs, the onset potential for the NO3RR on Ni1Cu SAAO was substantially less negative, suggesting that the NO3RR was more advantageous on the Ni1Cu SAAO catalyst (Fig. 4d). Furthermore, a clear peak of NO2− reduction was observed at 0.1 V vs. RHE in Cu2O NWs, yet this behavior was not observed on Ni1Cu SAAO. This suggested that the Ni single atoms enabled the relay electrocatalysis of the NO2− intermediate product through the Ni site-generated active hydrogen species (*H). Additionally, as the cell temperature increased, the reaction rates of both the NO3RR and HER on Ni1Cu SAAO were enhanced, leading to an increase in NH3 production and Faraday efficiency (Fig. 4e). This was due to the utilization of thermal energy to surmount the thermodynamic barriers in the reaction pathways. Furthermore, the reaction rate for the formation of *H on Ni single atoms was quantified through in situ SI-SECM measurements to explain the increased selectivity of the Ni1Cu SAAO catalyst. More specifically, the authors used the catalyst-loaded substrate to determine the *H concentration when the Cu2O NWs and Ni1Cu SAAO were biased at different potentials. The catalytic substrate was first biased at a reducing potential Esubstrate to generate *H. After this, the system was restored to open-circuit conditions so that *H was neither generated nor consumed, and the redox reaction of the redox mediator FcMeOH (Fc) in the solution could not occur on the substrate. Then an oxidizing potential was biased on the UME to oxidize Fc to Fc+. Fc+ can be reduced to Fc by *H on the substrate, generating a positive feedback current on the UME until all *H were consumed by Fc+, and the surface concentration of *H could be obtained from the charges transferred during the process. As Esubstrate became more negative in the first step, more *H were generated on the substrate (Fig. 4f). Remarkably, the concentration of *H on the Ni1Cu SAAO catalyst was four times greater than that on Cu2O NWs when Esubstrate was −0.4 V vs. RHE. Moreover, *H production signals were seen in Ni1Cu SAAO at a more positive potential of around 0.05 V vs. RHE, indicating a considerable increase in water dissociation upon the addition of Ni single atoms. These results were also supported by the results of DFT calculations and in situ Raman spectroscopy, confirming that the addition of single-atom Ni sites greatly increases the hydrogen adsorption capacitance of Ni1Cu SAAO.
 | ||
| Fig. 4 (a) Schematic representation of the Ni1Cu SAAO catalyst preparation process. (b) Atomic-resolution HAADF-STEM image of Ni1Cu SAAO. (c) Local 3D topographic atom images for the region in (b). (d) LSVs of Ni1Cu SAAO and Cu2O NWs with and without adding NO3− in the electrolytes (top panel) as well as the LSVs at different cell temperatures (bottom panel). (e) Faradaic efficiency and yield rate of Ni1Cu SAAO and Cu2O NWs for the reduction of nitrate to ammonia at different cell temperatures at a potential of 0.1 V vs. RHE. (f) Plots of tip-titration charges against substrate potential using SI-SECM for quantifying the concentration of surface-active hydrogen species on Cu2O NWs and Ni1Cu SAAO in 0.01 M KOH and 1.0 mM Fc. Reproduced with permission from ref. 99, American Chemical Society, 2024. | ||
SECM-based study of the CO2RR using alternative approach curves
One environmentally friendly way to achieve the goal of carbon neutrality is the electrochemical reduction of CO2 (CO2RR) into chemicals with added value using electricity from renewable sources. This process also addresses the intermittent nature of renewable electricity generation.104–108 An additional benefit is that the electrochemical CO2 reduction process operates at room temperature and pressure and may be adjusted by varying the applied potential,109 making the reaction conditions environmentally friendly. These factors enable the use of this technology in scale-up applications. SECM has been widely used in the field of the CO2RR at present,68,69,72,110–112 and alternative approach curves played crucial roles in these studies where the conventional approach curves fell short.Recently, with the development of UME-based pH-sensors, the quantitative analysis of the local pH during the CO2RR has been achieved using SECM, providing insight into the CO2RR mechanism.72,110 For instance, Koper et al. designed a UME-based voltammetric pH sensor to measure the pH in the diffusion layer during CO2 reduction through SECM.72 The Au UME was functionalized with the 4-hydroxylaminothiophenol (4-HATP)/4-nitrosothiophenol (4-NSTP) redox couple. Since a proton was involved in the redox reaction of 4-HATP/4-NSTP, according to the Nernst equation, the formal potential of the redox reaction obtained from CV (Fig. 5a) had a linear relationship with pH, and the calibration curve was obtained in solutions with different pH values (Fig. 5b). Up to pH 3.45, the calibration curves under the argon and CO2 atmospheres overlapped, while the calibration curve under a CO2 atmosphere reached a plateau at higher pH. This was due to the formation of CO2-saturated solutions under a CO2 atmosphere, with a stable pH of about 4. To position the Au UME near the electrode surface, the capacitance-based approach curve was applied, and the Au UME was positioned 80 ± 2 μm from the surface of a polycrystalline gold disc. The local pH during the CO2RR was measured using tip voltammetry at 4 s/data point while the reaction on the gold disc electrode was switched ‘on’ and ‘off’ by stepping the potential from −0.5 to −0.9 V vs. Ag/AgCl in 50 mV steps, with a potential of 0 V applied during potential steps. The authors focused on the interfacial pH recovery profile once the reaction was turned ‘off’ by stepping the potential on the gold disc back to 0 V (Fig. 5c), where no electrochemical reactions occurred. The monitored pH recovery was heavily affected by the hindered diffusion of OH− with an UME positioned in the diffusion layer, as illustrated in Fig. 5d. According to the finite element method (FEM) simulation, when the Au UME was positioned close to the substrate (L = 3.4, L being the tip–substrate distance), the OH− concentration at the Au UME surface was significantly higher than the case when the Au UME was positioned far from the substrate (L = 50). Additionally, a good fit between the experimental result and the simulated pH profile demonstrated that the diffusion hindrance caused by the UME was well-accounted for. Additionally, another UME-based voltammetric pH-sensor was used as the SECM probe to detect the local pH changes during the CO2RR from the CuxOyCz nanostructured electrocatalysts.110 A Pt UME (diameter < 1 μm) was positioned ∼100 nm from the CuxOyCz-coated GDE surface by the shear-force-based SECM approach curve, and the local OH− concentration near the catalyst surface was monitored as the formal potential of the redox reaction of Pt/PtO on the UME had a linear correlation with OH− concentration.
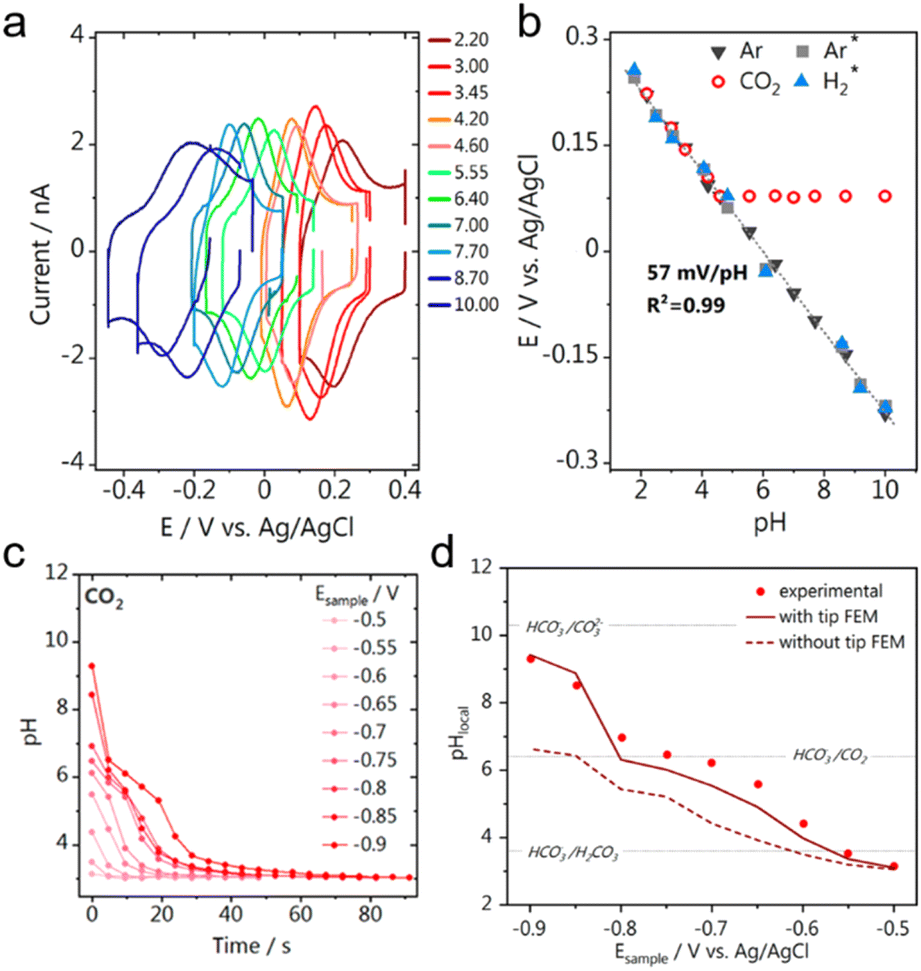 | ||
| Fig. 5 (a) CVs on the Au-UME in 0.1 M Li2SO4 solutions with different pH values. Scan rate: 200 mV s−1. (b) Calibration curves between pH and the formal potential recorded from the Au UME pH sensor under different atmospheres. The calibration curves from previous work (grey square for Ar and blue triangle for H2) are also shown for comparison. (c) The pH recovery profile recorded on the Au UME after the CO2RR was turned “off” by setting the potential to 0 V vs. Ag/AgCl. Before the CO2RR on the substrate was turned off, the substrate was biased at different sample potentials Esample (vs. Ag/AgCl). (d) Experimental (red dots) and simulated (red lines) local pH values at the Au-UME after the substrate was biased at different Esample to drive the CO2RR. The local pH values when the Au-UME was positioned close to the substrate (L = 3.4, dark red solid line) and when the Au-UME was positioned far from the substrate (L = 50, dark red dashed line) were both simulated. Bulk CO2 concentration was fixed at 10 mM. L is the normalized tip–surface separation. Reproduced with permission from ref. 69, American Chemical Society, 2021. | ||
Monteiro et al. devised a technique to probe the local activity of GDEs under operando conditions utilizing SECM and Au UME (r = 1 μm) (Fig. 6a and b).69 They examined the effects of CO2 back-pressure and catalyst loading on the local activity of Au-GDEs (3 cm2 in size) at various applied potentials. Catalysts were loaded onto GDEs with a loading gradient, so that a GDE surface with low to high catalyst-loading densities was used as the substrate, and the Au UME was positioned close to 100 nm from the GDE using the shear force-based approach curve, which was otherwise impractical using conventional approach curves due to the surface roughness of the GDE. The CO2RR activity on the GDE was probed using the SG/TC mode of SECM, where CO was produced on the GDE and oxidized at the Au UME, and by moving the Au UME laterally above the GDE, the activity at locations with different loading densities could be mapped. In order to maximize the performance of GDEs, the applied sample potential and the CO2 back-pressure were adjusted simultaneously, and the interaction between catalyst loading and CO2 back-pressure was assessed from the current signal on the Au UME. It was found that higher catalyst loadings lead to enhanced CO2RR activity in the presence of sufficient CO2, while the situation was more complicated for the CO2 back-pressure. An optimal CO2 back-pressure was required to reach maximum CO2RR activity, which was dependent on the catalyst loading density. Additionally, Fig. 6c–e display the activity maps at various GDE potentials, with more CO generated at more negative potentials. It is worth noting that significant variations in the GDE activity over the scanned region were observed, indicating the inhomogeneity of the lateral response throughout the GDEs. This should not be caused by a variation in the catalyst loading density as the catalyst gradient was produced over a significantly larger length of the GDE (1.7 cm) than the length of the lateral scan in Fig. 6c–e (30 μm). As observed by SEM, a considerable number of larger holes existed on the surface of the GDE, which might contribute to the notable variations in activity. The varying results demonstrated that, in addition to a high catalyst loading, sufficient CO2 supply and a uniform distribution of GDE pores accessible to CO2 were necessary to produce a three-phase reaction boundary.
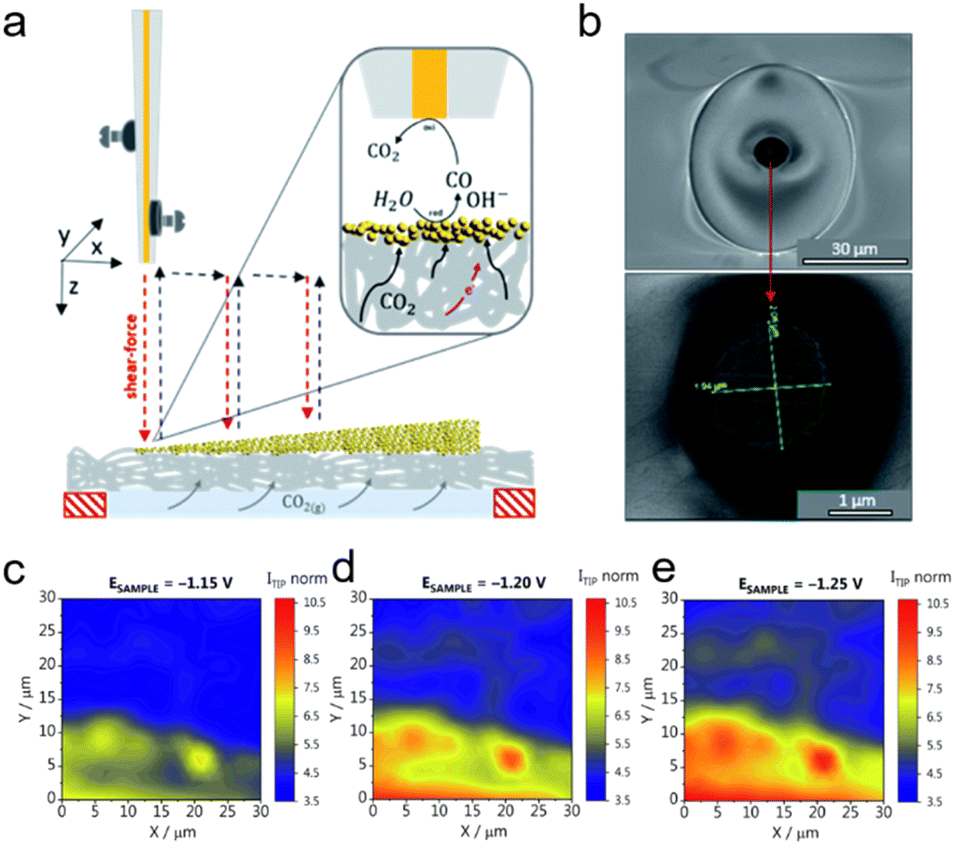 | ||
| Fig. 6 (a) Schematic representation of the SECM experimental setup, with the Au UME mounted on the piezo, approaching the GDE coated with catalysts using the capacitance-based approach curve. After the Au UME was positioned near the GDE, the CO2RR activity was characterized in the SG-TC mode, as shown in the inset. (b) SEM micrographs of the Au UME. (c–e) Activity maps recorded for the GDE at a CO2 back-pressure of 0.7 mbar. The GDE potentials were reported versus Ag/AgCl/3 M KCl in 1 M KHCO3. The tip current (Itip norm) was normalized to the double-layer charging current recorded at −0.6 V. Reproduced with permission from ref. 69, Royal Society of Chemistry, 2021. | ||
Koper et al. used CV measurements to study the CO2RR on 5 mm diameter gold disc electrodes using the hanging meniscus configuration, and found that the CO2RR did not occur in a pure H2SO4 electrolyte in the absence of a metal cation.111 To confirm this and to extend the study to silver and copper electrodes, the CO2RR near the substrate with and without the presence of metal cations was monitored using the SG/TC mode of SECM with a Pt UME (r = 6.5 ± 0.07 μm) (Fig. 7a). As shown in Fig. 7b, on the gold substrate only hydrogen oxidation (H2ox) occurred under an argon atmosphere. In comparison, under a CO2 atmosphere, a strong peak owing to the oxidation of CO (COox) appeared when Cs+ was introduced into the electrolyte, while this peak was not observed in the absence of Cs+. Similar outcomes were observed on silver and copper electrodes, where the CO2RR occurred only in the presence of Cs+ under a CO2 atmosphere (Fig. 7c and d). In conclusion, on gold, silver, or copper electrodes, CO was only identified under a CO2 atmosphere, and only after the electrolyte had been supplemented with Cs+. Through DFT calculations, the authors found that Cs+, as well as other alkaline metal ions, were partially desolvated in the Au–H2O–M+ system (M+ being the metal ion), and had three promotional effects for the CO2RR. Namely, the existence of Cs+ helped stabilize the CO2 adsorption on the catalyst through the electrostatic interaction between Cs+ and an oxygen atom in CO2. The partially desolvated Cs+ also decreased the O–C–O angle from 180° to 140°, activating the CO2 molecule. Lastly, the partially desolvated Cs+ enhanced the electron transfer from the catalytic surface to CO2. Overall, using CV and SECM, the authors demonstrated that positively charged species from the electrolyte were essential for the CO2RR, and the underlying mechanism was explained by DFT calculations.
 | ||
| Fig. 7 (a) Schematic representation of the SECM experiment. (b) CV on the Pt UME obtained directly after CO2 reduction on gold electrodes. (c) CV on the Pt UME obtained directly after CO2 reduction on silver electrodes. (d) CV on the Pt UME obtained directly after CO2 reduction on copper electrodes. The CVs on the Pt-UME before applying any potential to the sample are shown in light grey for reference. Reproduced with permission from ref. 111, Nature, 2021. | ||
SECM-based detection of short-lived reactive oxygen species
Reactive oxygen species (ROS) refer to a group of chemically active substances containing oxygen atoms,113–116 including singlet oxygen (1O2), hydrogen peroxide (H2O2), hydroxyl radicals (˙OH), and superoxide anion radicals (O2˙−). ROS participate in a variety of chemical and biological processes, such as energy metabolism, cell signalling, and antioxidant defence.117,118 Nonetheless, detection of certain ROS, such as 1O2, ˙OH, and O2˙− remains challenging due to their short lifetime and high reactivity.119–129Known for its high reactivity, strong oxidative properties, and short lifetime, it is challenging to detect ˙OH. Traditionally, ˙OH can be detected using electron spin resonance (ESR) with the help of spin traps such as 5,5-dimethyl-1-pyrroline N-oxide (DMPO), yet this method suffers from an unsatisfactory sensitivity and is inconvenient to measure the generation process of ˙OH in real time. To solve these issues, Rodríguez-López's group proposed a novel method for detecting ˙OH at operating electrodes.123 The authors discovered that the [DMPO–OH]˙ adduct, formed by the reaction of ˙OH with DMPO, was stable in the aqueous solution and was redox-active with a formal potential of 0.85 V vs. Ag/AgCl. Electrodes of various materials were tested to electrochemically generate ˙OH and the boron-doped diamond (BDD) was found to be the most effective one. Thus, using SECM, the freshly generated [DMPO–OH]˙ adduct from a BDD electrode surface was detected with a UME (12.5 μm in radius) positioned 10 μm from the BDD electrode using SECM (Fig. 8a).
 | ||
| Fig. 8 (a) Detection of the [DMPO–OH]˙ adduct formed from spin trapping of ˙OH radicals generated on BDD electrodes at different applied potentials (EBDD) using SECM. Reproduced with permission from ref. 123, American Chemical Society, 2022. (b) The radical detection response collected at the gold tip as the substrate potential was swept at 10 mV s−1 with 25 mM DMPO in solution. (c) The H2O2 detection response collected at a Pt tip as the substrate potential was swept at 10 mV s−1. (d) ESR spectra of 50 mM DMPO after two minutes of electrolysis at the pyrolyzed Fe–N–C catalyst substrate at increasingly reducing potentials. Reproduced with permission from ref. 130, Royal Society of Chemistry, 2024. | ||
Furthermore, the Rodríguez-López group studied ROS formation from a Fe–N–C catalyst during the ORR in real-time using the SG/TC mode of SECM.130 Specifically, the [DMPO–OH]˙ adduct generated near the Fe–N–C catalyst was detected using Au UMEs with a diameter of 25 μm at a tip–substrate distance of approximately 10 μm, while H2O2 generated near the catalysts was collected using Pt UMEs (d = 25 μm). As the substrate was biased at more and more negative potentials, more ˙OH was generated, yet H2O2 production reached a peak when the substrate was biased at ∼0.55 V vs. Ag/AgCl (Fig. 8b and c). The different behaviors of ˙OH and H2O2 with varying substrate potential were due to ˙OH being the predominant radical species, as confirmed by ex situ ESR measurements (Fig. 8d). Overall, real-time measurements of difficult-to-observe free radical intermediates and by-products using SECM were helpful in identifying the mechanistic variations among various electrocatalytic materials for the ORR and other processes.
Conclusions and outlook
In this perspective, the recent advances in the development of SECM operation modes were summarized. Using shear-force-based and capacitance-based approach curves, SECM tips could be positioned near the surface of non-flat electrodes, benefiting the characterization of catalysts under operating conditions. The SI-SECM technique proves to be a powerful tool for detecting the surface active site densities, as well as determining the reaction rates of the surface adsorption process. Also, the development of potentiometric SECM probes opens up the opportunity to study local pH near the substrate at the microscopic level. All of these expanded the functionality of SECM, and SECM-based progress has been made in new scenarios, such as electrocatalytic reactions including the NO3RR, NRR, CO2RR and so on, quantifying the catalytic properties of the catalysts and providing insights into the reaction mechanisms.Despite recent advancements in SECM-based studies of electrocatalytic reactions, critical challenges persist in achieving quantitative kinetic information and unveiling the reaction mechanisms. The first issue is obtaining a precise tip–substrate distance when the substrate is not ideally flat, which is often the case when the substrate is loaded with nanoparticles of electrocatalysts. In SECM studies, the mass transfer towards the tip is greatly controlled by the tip–substrate distance. Thus, the current signal on the tip in SG/TC modes is not only a function of the reaction kinetics, but also largely affected by the tip–substrate distance. A precise and quantitative control of the tip–substrate distance is crucial for the quantitative study of catalytic reaction kinetics. When the substrate is flat, the tip–substrate distance can be well-controlled by comparing the approach curves with the theory, yet the approach curve over a non-flat surface such as a GDE is much more complicated and usually cannot be analyzed quantitatively. Even though UMEs could be brought to a few hundred nanometers above a rough substrate using the shear-force-based approach curves, a quantitative tip–substrate distance is missing, making the analysis of reaction kinetics difficult.
The second issue is spatial resolution, which is limited by the UMEs. Although smaller UMEs have been developed, a large amount of work still prefers UMEs with 10–25 μm diameter so far. Typically, electrocatalysts tens to hundreds of nanometers in size were used, which were far beyond the spatial resolution provided by these UMEs. Usually, a smaller probe size leads to a better spatial resolution. However, experiments with smaller UMEs get much more challenging, and special care must be taken to properly handle the tip and the potentiostat,131,132 as well as to minimize thermal drift caused by temperature fluctuations near the tip.133
The third challenge arises from the complicated mechanisms of multi-electron transfer reactions, including the NRR, NO3RR, and CO2RR. These processes involve intertwined proton-coupled electron transfer (PCET) steps, competitive adsorption of intermediates, and potential-dependent selectivity bifurcations, and have multiple potential products. A lot of effort is required to acquire the selectivity towards different reaction pathways and the kinetics of each step. Moreover, the adsorption processes may play critical roles in these reactions. Using SECM, complicated reaction mechanisms involving adsorption and multiple electron transfer steps have been unveiled,134 yet the adsorption process is barely considered in current SECM-based electrocatalysis studies.
The further development of SECM-related theories, instrumental innovation and coupling SECM with other analytical methods would be helpful to tackle these challenges. Finite element simulation combined with machine learning might help to provide a numerical solution to the theoretical approach curve over a non-flat substrate, enabling a quantitative positioning of UME on operando electrodes like the GDE. Multiscale modeling combining DFT-calculation, finite element simulations of intermediate surface concentration, and microkinetic analysis of PCET steps would shed light on the complicated reaction mechanisms during multi-electron transfer reactions like the NRR, NO3RR, and CO2RR. The development of smaller UMEs as well as protocols of using these UMEs will help to greatly improve the spatial resolution of SECM-based studies.135 And coupling in situ electrochemical measurements with other analytical techniques would lead to more information about reaction mechanisms. For example, by coupling a rotating disk electrode with Raman spectroscopy and infrared spectroscopy, information on the adsorption of protons on noble metals has been revealed.136,137 Coupling CV with mass spectrometry provided structural information of the intermediates during electron transfer reactions, and can help to identify the reaction pathways.138 Overall, progress in the above-mentioned fields will boost our understanding of the electrocatalytic reactions and facilitate the pursuit for sustainable and renewable energy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Writing-original draft, J. X.; writing-review & editing, J. S., R. C. and Y. Z.; funding acquisition, R. C. and Y. Z.; supervision, Y. S. and Y. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (22102025 and 22174014) and the ZhiShan Young Scholar Program of Southeast University.References
- J. Lelieveld, K. Klingmüller, A. Pozzer, R. T. Burnett, A. Haines and V. Ramanathan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7192–7197 CrossRef CAS
.
- K. Kohse-Höinghaus, Chem. Rev., 2023, 123, 5139–5219 CrossRef PubMed
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS
- A. A. Lacis, G. A. Schmidt, D. Rind and R. A. Ruedy, Science, 2010, 330, 356–359 CrossRef CAS PubMed
- J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 15298–15312 CrossRef CAS
- C. Long, J. Han, J. Guo, C. Yang, S. Liu and Z. Tang, Chem Catal., 2021, 1, 509–522 CrossRef CAS
- H. Zhang, M. Zhu, O. G. Schmidt, S. Chen and K. Zhang, Adv. Energy Sustainability Res., 2021, 2, 2000090 CrossRef CAS
- A. Kumar, V. K. Vashistha, D. K. Das, S. Ibraheem, G. Yasin, R. Iqbal, T. A. Nguyen, R. K. Gupta and M. Rasidul Islam, Fuel, 2021, 304, 121420 CrossRef CAS
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H. Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruña, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed
- C. A. Campos-Roldán, D. J. Jones, J. Rozière and S. Cavaliere, ChemCatChem, 2022, 14, e202200334 CrossRef PubMed
- G. Russo, Nature, 2014, 513, 478–480 CrossRef CAS PubMed
- A. K. Sleiti, Renewable Sustainable Energy Rev., 2017, 69, 435–441 CrossRef
- M. Roeb and H. Müller-Steinhagen, Science, 2010, 329, 773–774 CrossRef CAS PubMed
- H. Zhang and J. Yan, Joule, 2022, 6, 1142–1144 CrossRef
- H. Zhao and Z. Yuan, Adv. Energy Mater., 2023, 13, 2300254 CrossRef CAS
- J. T. Ren, L. Chen, H. Y. Wang, W. W. Tian and Z. Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC
- Z. Chen, S. Yun, L. Wu, J. Zhang, X. Shi, W. Wei, Y. Liu, R. Zheng, N. Han and B. J. Ni, Nano-Micro Lett., 2023, 15, 4 CrossRef CAS
- T. Li, P. Wang, M. He, T. Zhang, C. Yang and Z. Li, Coord. Chem. Rev., 2024, 521, 216179 CrossRef CAS
- B. A. Yusuf, W. Yaseen, S. Meng, J. Xie, F. O. Fapohunda, R. Nankya, A. I. Muhammad, M. Xie and Y. Xu, Coord. Chem. Rev., 2023, 492, 215273 CrossRef CAS
- J. Zhang, J. Ding, Y. Liu, C. Su, H. Yang, Y. Huang and B. Liu, Joule, 2023, 7, 1700–1744 CrossRef CAS
- H. Zhang, K. Fang, J. Yang, H. Chen, J. Ning, H. Wang and Y. Hu, Coord. Chem. Rev., 2024, 506, 215723 CrossRef CAS
- F. Y. Chen, A. Elgazzar, S. Pecaut, C. Qiu, Y. Feng, S. Ashokkumar, Z. Yu, C. Sellers, S. Hao, P. Zhu and H. Wang, Nat. Catal., 2024, 7, 1032–1043 CrossRef CAS
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS PubMed
- H. F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC
- Y. Lin, Y. Dong, X. Wang and L. Chen, Adv. Mater., 2023, 35, 2210565 CrossRef CAS PubMed
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS
- J. Li, S. U. Abbas, H. Wang, Z. Zhang and W. Hu, Nano-Micro Lett., 2021, 13, 216 CrossRef CAS
- X. Zhao, G. Hu, G. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2207650 Search PubMed
- Z. Zhang, J. Liu, Y. Xu, C. Xie, S. Wang and X. Yao, Chem. Soc. Rev., 2024, 53, 10620–10659 RSC
- P. Sebastián-Pascual and M. Escudero-Escribano, ACS Energy Lett., 2020, 5, 130–135 CrossRef
- W. Ge, Y. Chen, Y. Fan, Y. Zhu, H. Liu, L. Song, Z. Liu, C. Lian, H. Jiang and C. Li, J. Am. Chem. Soc., 2022, 144, 6613–6622 CrossRef CAS PubMed
- Z. Kou, X. Li, L. Zhang, W. Zang, X. Gao and J. Wang, Small Sci., 2021, 1, 2100011 CrossRef CAS PubMed
- E. P. Alsaç, N. Bodappa, A. W. H. Whittingham, Y. Liu, A. D. Lazzari and R. D. L. Smith, Chem. Phys. Rev., 2021, 2, 031306 CrossRef
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS
- D. Polcari, P. Dauphin-Ducharme and J. Mauzeroll, Chem. Rev., 2016, 116, 13234–13278 CrossRef CAS PubMed
- M. Shen, R. Ishimatsu, J. Kim and S. Amemiya, J. Am. Chem. Soc., 2012, 134, 9856–9859 CrossRef CAS PubMed
- R. Chen, P. Pathirathna, R. J. Balla, J. Kim and S. Ameyiya, Anal. Chem., 2024, 96, 10765–10771 CrossRef CAS PubMed
- Y. Li, X. Ning, Q. Ma, D. Qin and X. Lu, TrAC, Trends Anal. Chem., 2016, 80, 242–254 CrossRef CAS
- T. Kai, M. Zhou, S. Johnson, H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2018, 140, 16178–16183 CrossRef CAS PubMed
- M. Zhou, Y. Yu, K. Hu and M. V. Mirkin, J. Am. Chem. Soc., 2015, 137, 6517–6523 CrossRef CAS PubMed
- T. Kai, M. Zhou, Z. Duan, G. A. Henkelman and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 18552–18557 CrossRef CAS PubMed
- Y. Yang, Y. Xiong, R. Zeng, X. Lu, M. Krumov, X. Huang, W. Xu, H. Wang, F. J. DiSalvo, J. D. Brock, D. A. Muller and H. D. Abruña, ACS Catal., 2021, 11, 1136–1178 CrossRef CAS
- M. A. Bhat, N. Nioradze, J. Kim, S. Amemiya and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 15891–15899 CrossRef CAS PubMed
- R. Chen, S. Liu and Y. Zhang, Mater. Horiz., 2023, 10, 52–64 RSC
- K. Barman, X. Wang, R. Jia and M. V. Mirkin, J. Am. Chem. Soc., 2021, 143, 8547–8551 CrossRef CAS PubMed
- J. Clausmeyer and W. Schuhmann, TrAC, Trends Anal. Chem., 2016, 79, 46–59 CrossRef CAS
- J. Zhang, Y. Liu, Y. Li, T. Zhu, J. Qiu, F. Xu, H. Zhang and F. Li, Small Methods, 2016, 6, 2200689 CrossRef PubMed
- J. Timoshenko and B. R. Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed
- M. Rüscher, A. Herzog, J. Timoshenko, H. S. Jeon, W. Frandsen, S. Kühl and B. R. Cuenya, Catal. Sci. Technol., 2022, 12, 3028–3043 RSC
- H. J. Niu, Y. Yan, S. Jiang, T. Liu, T. Sun, W. Zhou, L. Guo and J. Li, ACS Nano, 2022, 16, 11049–11058 CrossRef CAS PubMed
- A. Djire, X. Wang, C. Xiao, O. C. Nwamba, M. V. Mirkin and N. R. Neale, Adv. Funct. Mater., 2020, 30, 2001136 CrossRef CAS
- G. Li, T. Sun, H. Niu, Y. Yan, T. Liu, S. Jiang, Q. Yang, W. Zhou and L. Guo, Adv. Funct. Mater., 2023, 33, 2212514 CrossRef CAS
- Z. Wang, R. Liu, T. Sun, M. Li, N. Ran, D. Wang and Z. Wang, Anal. Chem., 2024, 96, 7618–7625 CrossRef CAS PubMed
- H. Niu, C. Huang, T. Sun, Z. Fang, X. Ke, R. Zhang, N. Ran, J. Wu, J. Liu and W. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202401819 CrossRef CAS PubMed
- C. Iffelsberger and M. Pumera, J. Mater. Chem. A, 2021, 9, 22072–22081 RSC
- Z. Wang, T. Sun, C. HuangFu, S. Jiang, C. Gu, L. Jiao and Z. Wang, Nano Res., 2023, 16, 10011–10017 CrossRef CAS
- S. Jiang, T. Sun, C. Gu, Y. Ma, Z. Wang, D. Wang and Z. Wang, Nano Res., 2023, 16, 8902–8909 CrossRef CAS
- L. Lan, Y. Wu, Y. Pei, Y. Wei, T. Hu, D. Lützenkirchen-Hecht, K. Yuan and Y. Chen, Adv. Mater., 2025, 37, 2417711 CrossRef CAS PubMed
- S. Kaur, K. Garg and T. C. Nagaigh, ACS Energy Lett., 2025, 10, 1430–1438 CrossRef
- J. L. Fernández and A. J. Bard, Anal. Chem., 2004, 76, 2281–2289 CrossRef PubMed
- C. M. Sánchez-Sánchez, J. Rodríguez-López and A. J. Bard, Anal. Chem., 2008, 80, 3254–3260 CrossRef PubMed
- B. B. Katemann, A. Schulte and W. Schuhmann, Chem.–Eur. J., 2003, 9, 2025–2033 CrossRef CAS PubMed
- J. Park, J. H. Lim, J. H. Kang, J. Lim, H. W. Jang, H. Shin and S. H. Park, J. Energy Chem., 2024, 91, 155–177 CrossRef CAS
- T. B. Clarke, L. E. Krushinski, K. J. Vannoy, G. Colón-Quintana, K. Roy, A. Rana, C. Renault, M. L. Hill and J. E. Dick, Chem. Rev., 2024, 124, 9015–9080 CrossRef CAS PubMed
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC
- Y. Nam, S.-E. Cho and H. S. Ahn, ACS Catal., 2024, 14, 17084–17089 CrossRef CAS
- M. C. O. Monteiro, S. Dieckhöfer, T. Bobrowski, T. Quast, D. Pavesi, M. T. M. Koper and W. Schuhmann, Chem. Sci., 2021, 12, 15682–15690 RSC
- M. C. O. Monteiro, L. Jacobse and M. T. M. Koper, J. Phys. Chem. Lett., 2020, 11, 9708–9713 CrossRef CAS PubMed
- M. C. O. Monteiro, L. Jacobse, T. Touzalin and M. T. M. Koper, Anal. Chem., 2020, 92, 2237–2243 CrossRef CAS PubMed
- M. C. O. Monteiro, A. Mirabal, L. Jacobse, K. Doblhoff-Dier, S. C. Barton and M. T. M. Koper, JACS Au, 2021, 1, 1915–1924 CrossRef CAS PubMed
- J. Rodríguez-López, M. A. Alpuche-Avilés and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16985–16995 CrossRef PubMed
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef CAS PubMed
- X. Tang, Y. Zhang, S. Tang, D. Lützenkirchen-Hecht, K. Yuan and Y. Chen, ACS Catal., 2024, 14, 13065–13080 CrossRef CAS
- W. Wu and Y. Wang, J. Am. Chem. Soc., 2025, 147, 11662–11666 CrossRef CAS PubMed
- J. Xu, H. Gao, F. Wang and M. Zhou, Curr. Opin. Electrochem., 2023, 39, 101299 CrossRef CAS
- M. Nebel, K. Eckhard, T. Erichsen, A. Schulte and W. Schuhmann, Anal. Chem., 2010, 82, 7842–7848 CrossRef CAS PubMed
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef
- X. Xin, Q. Qu, I. E. Khalil, Y. Huang, M. Wei, J. Chen, W. Zhang, F. Huo and W. Liu, Chin. Chem. Lett., 2024, 35, 108654 CrossRef CAS
- J. Zhang, M. Sun, J. Ren, R. Zhang, M. Ma, Q. Xue and J. Tian, Chin. Chem. Lett., 2025, 36, 110491 CrossRef CAS
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC
- X. Yang, B. Xu, J. G. Chen and X. Yang, ChemSusChem, 2023, 16, e202201715 CrossRef CAS PubMed
- K. N. Dinh, Q. Liang, C. F. Du, J. Zhao, A. I. Y. Tok, H. Mao and Q. Yan, Nano Today, 2019, 25, 99–121 CrossRef CAS
- Z. J. Huba, M. D. Donakowski and A. Epshteyn, Chem. Mater., 2017, 29, 1467–1471 CrossRef CAS
- P. Li, Z. Jin, Z. Fang and G. Yu, Angew. Chem., Int. Ed., 2020, 59, 22610–22616 CrossRef CAS PubMed
- J. Kong, H. Kim and H. S. Park, Appl. Catal., B, 2023, 338, 123019 CrossRef CAS
- M. Ferrara, M. Bevilacqua, C. Tavagnacco, F. Vizza and P. Fornasiero, ChemCatChem, 2020, 12, 6205–6213 CrossRef CAS
- D. Gupta, A. Kafle, M. Singh, D. Dahare and T. C. Nagaiah, J. Mater. Chem. A, 2023, 11, 24812–24822 RSC
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed
- K. Liu, H. Li, M. Xie, P. Wang, Z. Jin, Y. Liu, M. Zhou, P. Li and G. Yu, J. Am. Chem. Soc., 2024, 146, 7779–7790 CrossRef CAS PubMed
- H. Li, S. Li, R. Guan, Z. Jin, D. Xiao, Y. Guo and P. Li, ACS Catal., 2024, 14, 12042–12050 CrossRef CAS
- P. Li, L. Liao, Z. Fang, G. Su, Z. Jin and G. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305489120 CrossRef PubMed
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC
- C. Park, M. Y. Seo, T. Kwon, J. Kim, K. M. Nam, Y. K and J. Chang, J. Am. Chem. Soc., 2025, 147, 687–700 CrossRef PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- S. Pacala and R. Socolow, Science, 2004, 305, 968–972 CrossRef CAS PubMed
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 6438 CrossRef PubMed
- X. Wang, Y. Wang, L. Cui, W. Gao, X. Li, H. Liu, W. Zhou and J. Wang, Chin. Chem. Lett., 2024, 35, 110031 CrossRef CAS
- X. Jiang, Y. Zhao, Y. Kong, J. Sun, S. Feng, X. Lu, Q. Hu, H. Yang and C. He, Chin. Chem. Lett., 2025, 36, 109555 CrossRef CAS
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed
- N. Sikdar, J. R. C. Junqueira, S. Dieckhöfer, T. Quast, M. Braun, Y. Song, H. B. Aiyappa, S. Seisel, J. Weidner, D. Öhl, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 23427–23434 CrossRef CAS PubMed
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS
- Y. K. Yesudas, M. Balamurugan, K. T. Nam, B. Gopal and A. S. Kumar, J. Mater. Chem. A, 2024, 12, 31135–31144 RSC
- Y. Zhang, M. Dai and Z. Yuan, Anal. Methods, 2018, 10, 4625–4638 RSC
- J. Wu, Y. Zhao, K. Li, S. Muhammad, M. Ju, L. Liu, Y. Huang, B. Wang, W. Ding, B. Shen and H. Huang, TrAC, Trends Anal. Chem., 2022, 157, 116734 CrossRef CAS
- F. Yan, Y. Zang, J. Sun, Z. Sun and H. Zhang, TrAC, Trends Anal. Chem., 2020, 131, 116009 CrossRef CAS
- N. Kwon, D. Kim, K. M. K. Swamy and J. Yoon, Coord. Chem. Rev., 2021, 427, 213581 CrossRef CAS
- Y. Su, H. Song and Y. Lv, Microchem. J., 2019, 146, 83–97 CrossRef CAS
- D. Wang, L. Zhao, L. H. Guo and H. Zhang, Anal. Chem., 2014, 86, 10535–10539 CrossRef CAS PubMed
- W. Yu and L. Zhao, TrAC, Trends Anal. Chem., 2021, 136, 116197 CrossRef CAS
- C. Iffelsberger, T. Raith, P. Vatsyayan, V. Vyskočil and F. M. Matysik, Electrochim. Acta, 2018, 281, 494–501 CrossRef CAS
- X. Zhao, S. Lam, J. Jass and Z. Ding, Electrochem. Commun., 2010, 12, 773–776 CrossRef CAS
- F. P. Filice and Z. Ding, Analyst, 2019, 144, 738–752 RSC
- J. S. Barroso-Martínez, A. I. B. Romo, S. Pudar, S. T. Putnam, E. Bustos and J. Rodríguez-López, J. Am. Chem. Soc., 2022, 144, 18896–18907 CrossRef PubMed
- A. Preet and T. E. Lin, Catalysts, 2021, 11, 594 CrossRef CAS
- C. G. Zoski, J. Electrochem. Soc., 2016, 163, H3088–H3100 CrossRef CAS
- P. Bertoncello, Energy Environ. Sci., 2010, 3, 1620 RSC
- J. Zhang, Y. Liu, Y. Li, T. Zhu, J. Qiu, F. Xu, H. Zhang and F. Li, Small Methods, 2022, 6, 2200689 CrossRef CAS PubMed
- A. Latus, J. M. Noël, E. Volanschi, C. Lagrost and P. Hapiot, Langmuir, 2011, 27, 11206–11211 CrossRef CAS PubMed
- T. Wu, X. Ning, Q. Xiong, F. Zhang and P. He, Electrochim. Acta, 2022, 403, 139638 CrossRef CAS
- S. T. Putnam and J. Rodríguez-López, Chem. Sci., 2024, 15, 10036–10045 RSC
- N. Nioradze, R. Chen, J. Kim, M. Shen, P. Santhosh and S. Amemiya, Anal. Chem., 2013, 85, 6198–6202 CrossRef CAS PubMed
- J. Kim, B. Kim, S. K. Cho and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 8173–8176 CrossRef CAS
- J. Kim, M. Shen, N. Nioradze and S. Amemiya, Anal. Chem., 2012, 84, 3489–3492 CrossRef CAS PubMed
- M. A. Bhat, N. Nioradze, J. Kim, S. Amemiya and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 15891–15899 CrossRef CAS PubMed
- J. Kim, C. Renault, N. Nioradze, N. Arroyo-Currás, K. C. Leonard and A. J. Bard, Anal. Chem., 2016, 88, 10284–10289 CrossRef CAS PubMed
- S. Zhu, X. Qin, F. Xiao, S. Yang, Y. Xu, Z. Tan, J. Li, J. Yan, Q. Chen, M. Chen and S. Shao, Nat. Catal., 2021, 4, 711–718 CrossRef CAS
- S. Zhu, X. Qin, Y. Yao and S. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef
- X. Zhang, W. Lu, C. Ma, T. Wang, J. Zhu, R. N. Zare and Q. Min, Chem. Sci., 2022, 13, 6244–6253 RSC
| This journal is © The Royal Society of Chemistry 2025 |