
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRethinking boron's role in intramolecular charge transfer: from an acceptor to a donor–acceptor regulator
Jiaqi
Dong†
,
Lingjuan
Chen†
and
Deng-Tao
Yang
 *
*
Xi'an Key Laboratory of Hybrid Luminescent Materials and Photonic Device, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, China. E-mail: dtyang@nwpu.edu.cn
First published on 9th May 2025
Abstract
In-depth exploration of charge transfer contributes directly to the comprehension of the microscopic mechanisms underlying life processes while accelerating progress in the cutting-edge fields of organic electronics. At the molecular level, the boron atom with its unique empty p-orbital has been widely exploited to construct intramolecular charge transfer (ICT) molecules. This perspective seeks to thoroughly examine the types and emerging mechanisms of ICT in both tricoordinate and tetracoordinate organoboron-based ICT molecules (OBCTs), thereby clarifying boron's role in the ICT process. With respect to three-coordinated OBCTs, organoboron molecules with distinct CT pathways and distances are discussed in terms of their development history, CT mechanisms and structure–property relationships, which can provide guidance for designing highly sought-after molecules. For four-coordinated OBCTs, emerging CT mechanisms and the role of coordination in modulating CT properties are discussed, indicating substantial opportunities for the development of CT in these systems. In addition, the development of novel CT mechanisms or the integration of multiple CT processes holds promise for overcoming existing limitations in current OBCTs. Coupling the advancement of CT mechanisms with the discovery of innovative application scenarios is poised to propel the future progression of OBCTs.
1. Introduction
“Life is nothing but an electron looking for a place to rest”—Albert Szent-Györgyi. Nearly all life forms on Earth rely, directly or indirectly, on charge transfer (CT) processes for survival, which are one of the critical steps in photosynthesis, biological signal transduction, and various forms of energy conversion.1,2 At the molecular level, intramolecular charge transfer (ICT) involves electron transfer from an electron-rich segment to an electron-deficient segment within the same molecule.3 ICT has been extensively studied as a foundational mechanism in molecular photophysics and photochemistry3,4 and is integral to applications in organic dyes,5 biotechnology,6,7 organic optoelectronic devices,8 nonlinear optics (NLO),9 and sensing.10,11 A common approach to enhance ICT is the incorporation of an electron-donating (D) unit and an electron-accepting (A) unit, typically linked by a conjugated linker. The ICT process inherently involves the separation of positive and negative charges, whereas the ideal condition of complete charge separation is referred to as the charge-separated (CS) state (D+–A−).1,3 The CS state can be considered a pure charge-transfer state, comprising a radical cation (D˙+) and a radical anion (A˙−). In D–π–A molecules, the CS state can be represented by resonance structures (i.e., the molecular dipolar forms), which offer an intuitive and convenient way to analyze the excited-state electron distribution with strong CT character (Fig. 1a). Selection of appropriate donor and acceptor units is crucial to define the expected ICT properties. | ||
| Fig. 1 (a) CS state of organoboron compounds upon excitation. (b) Role of tricoordinate boron as an electron acceptor. (c) Similar bonding characteristics of boron and carbon. | ||
Boron atoms with vacant p-orbitals rank among the most unique ICT acceptors.12–18 Compared to other electron-deficient or highly electronegative acceptors such as pyridine, cyano and fluoride, boron provides distinctive features: (a) tricoordinate boron acts as a strong π-electron acceptor with its empty p-orbital prone to being filled by electron delocalization in an electron-rich D–A system, resulting in a moderate acceptor. Additionally, the low electronegativity of boron enables it to act as a σ-electron donor. These characteristics allow the boron atom to effectively separate frontier molecular orbitals (FMOs) and avoid a strong CT process that can lead to transition-forbidden states and low oscillator strength (Fig. 1b), (b) boron exhibits similar bonding characteristics to carbon (Fig. 1c), allowing it to integrate into organic skeletons.19–22 The dual role of boron as both an acceptor and a structural component of the molecular backbone offers a versatile platform for designing organoboron compounds, and (c) lastly, boron possesses a Lewis-acidic empty p-orbital that enables coordination with lone-pair-electron-bearing ligands to form tetracoordinate boron compounds, facilitating the fine-tuning of photophysical properties (Fig. 1c).23,24 Consequently, organoboron compounds are widely applied across fields such as organic light-emitting diodes (OLEDs),25,26 organic field-effect transistors (OFETs),27 organic solar cells (OSCs),28 stimuli-responsive materials,29 biomedicine,30 and catalysis.31
Boron atoms typically act as acceptors in organoboron-based ICT molecules (OBCTs). The systematic classification of OBCTs enables a deeper understanding of boron's acceptor roles (Fig. 2a), facilitating the exploration of additional functions of boron beyond its role as an acceptor. OBCTs can be classified based on CT pathways into through-bond charge transfer (TBCT) molecules, where CT occurs via a π-electron bridge,32 and through-space charge transfer (TSCT) molecules, where CT occurs directly through space.33,34 Structurally, OBCTs are D–A or D–π–A systems, involving the transfer of π or n-electrons to the boron-containing segment. OBCTs can be further subdivided based on CT distance into long-range charge transfer (LR-CT) and short-range charge transfer (SR-CT) molecules (Fig. 2a).35 LR-CT typically occurs in different segments involved in electron transitions, as seen in twisted intramolecular charge transfer (TICT)36 and TSCT molecules. By contrast, SR-CT refers to charge transfer occurring within the same segment, as exemplified by multiple-resonance (MR) molecules.37 In LR-CT molecules, strong donor–acceptor interactions can lead to long-wavelength absorption and emission, making them suitable for near-infrared (NIR) materials.38–40 The substantial separation of FMOs also results in a small ΔEST, which is defined as the energy difference between the lowest singlet (S1) and triplet (T1) excited states.41,42 However, the small ΔEST is usually accompanied by a transition-forbidden nature that could reduce oscillator strength (f) of transition. Additionally, drastic changes in the electronic structure increase vibrational relaxation in the excited state, broadening the emission peak and yielding a large Stokes shift. In contrast, MR molecules exhibit narrow-band and bright emission due to the alternating distribution of FMOs and a more rigid molecular framework. The moderate donor–acceptor interactions in MR molecules make them ideal candidates for blue emitters.43–47 The combination of a locally excited (LE) state with LR-CT can be achieved in D–π–A structures, where the π-segment enhances oscillator strength and allows for a moderate ΔEST. Some molecules with this structure exhibit a hybridized local and charge-transfer (HLCT) excited-state, also known as “hot exciton” compounds,48 which can potentially resolve the transition-forbidden issue in thermally activated delayed fluorescence (TADF). Correspondingly, hybridized multi-resonance and charge transfer (HMCT) molecules combining SR-CT with LR-CT can enhance the CT of MR molecules to achieve bathochromic-shifted emission.49
 | ||
| Fig. 2 Types of (a) tricoordinate and (b) tetracoordinate OBCTs. | ||
Finally, based on boron's coordination number, OBCTs can be classified as either three-coordinated or four-coordinated systems. Four-coordinated boron provides flexible coordination, imparting stimuli-responsive properties to the molecule.29 However, the role of tetracoordinate boron in OBCTs remains unclear. Based on our group's recent report, this perspective systematically discusses the role of boron in tetracoordinate OBCTs, followed by a summary of two unique coordination-regulated charge transfer (CR-CT) processes (Fig. 2b): (a) coordination-enhanced charge transfer (CE-CT): boron's vacant orbitals coordinate with electron-deficient ligands while covalently bonding to donor segments. This arrangement simultaneously enhances donor and acceptor strengths, leading to red-shifted emission. (b) Coordination-quenched charge transfer (CQ-CT): this perspective further analyzes the ICT variations in organoboron compounds under the coordination of different ligands. For D–π–B molecules, coordination with electron-rich ligands can hinder charge transfer, resulting in the blue-shifted emission.
Extensive OBCTs have been developed around boron's unique electronic structure. However, a systematic overview of their intrinsic ICT mechanisms is still lacking, and boron's role is not limited merely to acting as an electron acceptor. This perspective provides a detailed classification of boron-dominated ICT types and reconsiders the roles of boron in organoboron molecules. Tricoordinate boron molecules are discussed in the context of TBCT and TSCT. TBCT molecules are further categorized into D–A, TICT, D–π–A, HLCT and MR systems. We discuss the fundamental principles of each process and explore their relationship. For instance, most D–A molecules rely on a π-bridge for effective CT, making them inherently D–π–A molecules. Similarly, MR molecules are actually a subset of the D–π–A molecules. Accordingly, the D–π–A molecules mentioned in this perspective are those in which π-bridges consist of alkenes or multiple aromatic rings. Unlike three-coordinated boron, four-coordinated boron acts as donor–acceptor regulator that influences molecular ICT through coordination with electron-rich or electron-deficient ligands. This perspective focuses on the emerging CE-CT and other intramolecular interactions, exhibiting significant development potential for tetracoordinate boron-domain CT compounds. The primary aim of this perspective is to summarize and analyze the ICT mechanisms in organoboron compounds, with a focus on the relationships between molecular structures and their corresponding ICT behaviors. While we include examples of both tri- and tetra-coordinated boron compounds, our selection of molecules was deliberate, chosen to illustrate specific ICT pathways and mechanisms rather than to provide a comprehensive catalog all organoboron compounds. A well-founded understanding of ICT design strategies and structure–function relationships will continue to advance design and application of OBCTs.
2. LE in organoboron compounds
LE represents an excited state without significant charge separation, predominantly occurring in π–π* transitions. The overlap of FMOs in LE ensures that electron transitions are allowed, resulting in an enhanced molecular oscillator strength when the LE fraction is increased. Direct bonding between boron and electron-rich heteroatoms (e.g., N or O) is a common strategy for constructing organoboron compounds due to its synthetic simplicity and this approach also promotes electron delocalization across the entire molecule, achieving high oscillator strength.50–57 For instance, a three-center four-electron (3c-4e) N–B–N unit provides electron-rich characteristics and high photoluminescence quantum efficiency (PLQY), where the boron p-orbital can undergo further coordination to fine-tune the optical properties.58–60 However, the isoelectronic relationship between the C–C unit and B–N unit leads to an LE-predominated character, reducing boron's electron-accepting capacity. Similarly, integrating three-coordinated boron into electron-rich systems serves as a powerful approach to tuning optoelectronic properties of polycyclic aromatic hydrocarbons (PAHs), accompanied by weakened electron-accepting ability of boron (Fig. 3a). To achieve effective ICT with three-coordinated boron, it is advisable to avoid direct bonding between boron and heteroatoms or embedding boron into a large planar conjugated system. If direct bonding cannot be avoided, increasing the number of boron atoms or increasing the molecular twisting degree also promotes desirable CT properties, as seen in TICT-type molecules (Section 3.2). | ||
| Fig. 3 (a) The p-orbitals of boron atoms filled with π-electrons in electron-rich systems via electron delocalization. D: electron-rich atoms (e.g., N or O). (b) Stable radical 1 and its spin density distribution. Copyright 2017, American Chemical Society. (c) Stable radical 2 with a bulky boron substituent and electron transition from the SOMO to the LUMO. Copyright 2022, John Wiley & Sons. The red arrow indicates the direction of electron transition.2 | ||
In contrast, the presence of LE in OBCTs is not entirely detrimental. The appropriate incorporation of LE character into ICT systems can enhance molecular oscillator strength (Section 3.3) or enable highly excited-state conversion pathways such as HLCT (Section 3.4). Furthermore, the boron p-orbital can stabilize the reactive neutral carbon radical in a planar structure due to electron delocalizing properties of boron (Fig. 3b).61 Spin density analysis of 1 indicates that the unpaired electron is efficiently delocalized over the planar molecular framework, with a significant contribution from boron. This delocalization provides compound 1 with sufficient stability for separation via column chromatography and the formation of single crystals detectable by X-ray diffraction. Boron also provides the LUMO for radicals 1 and 2, enabling the rare radical electron transition from the singly occupied molecular orbital (SOMO) to the LUMO (Fig. 3c).62 As a result, compound 1 exhibits intense red fluorescence with a high fluorescence quantum yield (ΦF) of 0.78 in toluene.
3. TBCT in tricoordinate boron molecules
TBCT in tricoordinate boron molecules is the most extensively studied type of OBCT. We subdivide TBCT molecules into five categories: D–A, TICT, D–π–A, HLCT and MR. For D–A molecules, we outline representative structural design strategies. TICT molecules are discussed in terms of their development and classification based on the presence or absence of rotational freedom. D–π–A molecules are categorized by their different π-bridges, while HLCT is separately discussed as a representative of D–π–A. MR molecules—recently developed and well-reviewed—are examined solely in the context of their CT process.3.1 D–A OBCTs
Boron as an electron acceptor in D–A OBCTs can be divided into unbridged and bridged triarylboryl acceptors (Fig. 4).23,63 The singly bridged (Fig. 4b), doubly bridged (Fig. 4c), and triply bridged (Fig. 4d) electron acceptors are common in TADF molecules and generally serve as weakened electron acceptors, which will be separately discussed in later sections. This section focuses only on the classic unbridged triarylboryl acceptors (Fig. 4a). | ||
| Fig. 4 (a) Unbridged, (b) single bridged, (c) doubly bridged, and (d) triply bridged triarylboryl acceptors in D–A OBCTs. X = N, O, S, CMe2 and so on. | ||
Triarylborane as an acceptor in D–A molecules has been extensively studied since its initial discovery in 1885 (Fig. 5).64 To prevent nucleophilic attacks from H2O or O2, bulky aryl groups like 2,4,6-trimethylphenyl (Mes) or 2,4,6-triisopropylphenyl (Tipp) are often used to improve kinetic stability. Building on a prior study on photostability of dimesitylphenylborane (Mes2BPh),65 Williams et al. investigated a series of D–A type molecules with different substituents on the phenyl ring of Mes2BPh.66 Their findings show that para-positioned donor groups (e.g., NMe2 or NPh2) on the phenyl ring of Mes2BPh significantly increased the PLQY (88% for 3 in cyclohexane), while electron-withdrawing groups (CN or Br) result in decreased PLQYs. Additionally, the fluorescence of Mes2BPh with electron-donating substituents exhibited a solvent-polarity-dependent red shift. This is a typical ICT process that highly polar solvents can stabilize the more polar CT state compared to the ground state, resulting in red-shifted emission. This also indicates that the introduction of D–A CT could enhance PLQY. A trigonal D–A–D molecule (4) with a triarylboryl core was introduced,67 which exhibits a symmetry-broken state due to carbazole group rotation and this molecule shows a unique blue-shifted absorption sensitive to solvent polarity attributed to dipole moment inversion upon photoexcitation.
 | ||
| Fig. 5 Representative D–A OBCTs with unbridged triarylboryl acceptors. | ||
Müllen et al. reported dendrimers 5a and 5b with different donor–acceptor ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2, respectively).685a exhibits a 26 nm red-shifted emission and a twofold increase in concentration-dependent emission compared to 5b. This is attributed to the additional BMes2 acceptor at the para-position of nitrogen in 5b, resulting in reduced nitrogen-donor strength. 5b with six boron atoms displays a red-shifted emission upon the addition of three equivalents of fluoride, while further fluoride titration resulted in a blue-shifted emission. The former exhibits a stronger D(B)–D–A CT due to the formation of partial B–F coordination, whereas the latter quenches the ICT process. The detailed mechanism of this phenomenon will be discussed in Section 5.2. In addition to para-D–A molecules, a series of meta-D–A molecules were reported including 6a, 6b and 6c with phenoxazine (PXZ), phenothiazine (PTZ), and 9,10-dihydro-9,9-dimethylacridine (DMAC) aselectron donors, respectively.69 The donor and acceptor units, positioned perpendicular to the central benzene ring, minimize electron delocalization and result in separated FMOs. This results in a small ΔEST value (∼30 meV). To improve the PLQY of previously reported D–A–D molecules,70 Yang et al. constructed two D–B–A-type compounds, 7a and 7b, which showed a 5.6-fold increase (from 0.12 to 0.67) and a 1.6-fold increase (from 0.47 to 0.75) in PLQY compared to the parent D–A–D molecules.71 These enhancements in PLQY occur despite the full charge separation in the FMOs of 7a and 7b. Similarly, D–A–B-type molecules using BMes2 acceptors combined with triazine or sulfonyl acceptors achieve greater HOMO–LUMO separation without increasing molecular twist. 8a and 8b feature small ΔEST values (0.037 eV and 0.013 eV, respectively) and high PLQYs.72 Recently, an A–D–A molecule 9 was reported to enable strong n → p or n → π* transitions from N and Se atoms to two BMes2 acceptors.73 Combined with the heavy atom effect, pure green phosphorescence was observed in both the solution and doped solid film of 9 at 298 K with a PLQY of 78%. OLEDs with an external quantum efficiency (EQE) of 18.2% and a luminance of 3000 cd m−2 were obtained. Triarylboryl acceptors can flexibly assemble with donor units in a Lego-like manner to construct D–A OBCTs. Further tuning of the donor-to-acceptor ratio, altering arrangement patterns, and incorporating functional atoms or fragments will enable the enhancement of charge-transfer properties.
:1 and 1:2, respectively).685a exhibits a 26 nm red-shifted emission and a twofold increase in concentration-dependent emission compared to 5b. This is attributed to the additional BMes2 acceptor at the para-position of nitrogen in 5b, resulting in reduced nitrogen-donor strength. 5b with six boron atoms displays a red-shifted emission upon the addition of three equivalents of fluoride, while further fluoride titration resulted in a blue-shifted emission. The former exhibits a stronger D(B)–D–A CT due to the formation of partial B–F coordination, whereas the latter quenches the ICT process. The detailed mechanism of this phenomenon will be discussed in Section 5.2. In addition to para-D–A molecules, a series of meta-D–A molecules were reported including 6a, 6b and 6c with phenoxazine (PXZ), phenothiazine (PTZ), and 9,10-dihydro-9,9-dimethylacridine (DMAC) aselectron donors, respectively.69 The donor and acceptor units, positioned perpendicular to the central benzene ring, minimize electron delocalization and result in separated FMOs. This results in a small ΔEST value (∼30 meV). To improve the PLQY of previously reported D–A–D molecules,70 Yang et al. constructed two D–B–A-type compounds, 7a and 7b, which showed a 5.6-fold increase (from 0.12 to 0.67) and a 1.6-fold increase (from 0.47 to 0.75) in PLQY compared to the parent D–A–D molecules.71 These enhancements in PLQY occur despite the full charge separation in the FMOs of 7a and 7b. Similarly, D–A–B-type molecules using BMes2 acceptors combined with triazine or sulfonyl acceptors achieve greater HOMO–LUMO separation without increasing molecular twist. 8a and 8b feature small ΔEST values (0.037 eV and 0.013 eV, respectively) and high PLQYs.72 Recently, an A–D–A molecule 9 was reported to enable strong n → p or n → π* transitions from N and Se atoms to two BMes2 acceptors.73 Combined with the heavy atom effect, pure green phosphorescence was observed in both the solution and doped solid film of 9 at 298 K with a PLQY of 78%. OLEDs with an external quantum efficiency (EQE) of 18.2% and a luminance of 3000 cd m−2 were obtained. Triarylboryl acceptors can flexibly assemble with donor units in a Lego-like manner to construct D–A OBCTs. Further tuning of the donor-to-acceptor ratio, altering arrangement patterns, and incorporating functional atoms or fragments will enable the enhancement of charge-transfer properties.
3.2 TICT OBCTs
The electron donor and acceptor units in D–A OBCTs are commonly linked through a freely rotatable single bond, enabling a conformational transformation of the molecular excited state accompanied by a dual emission phenomenon. This process is known as TICT and was first observed in 1959, when Lippert and colleagues synthesized a dye molecule DMABN, exhibiting dual emission peaks in solution.74 The long-wavelength peak of DMABN exhibits red-shifted emission as solvent polarity increases, while the short-wavelength peak remains unaffected. The short-wavelength emission peak is believed to originate from the LE state, whereas the long-wavelength peak is due to an ICT state formed upon excitation. This ICT theory provides an initial explanation for TICT phenomenon. The unique phenomenon sparked substantial debates on the essential mechanism until Grabowski in 1973 proposed the TICT theory to explain this dual fluorescence.75 According to the TICT theory (Fig. 6a), the TICT molecule emits from an LE/ICT state (state 2) upon excitation and then the excited molecule rotates around the single bond into a twisted conformation where the donor and acceptor are nearly perpendicular (90°) to each other. This results in a distinct TICT state (state 3) that emits fluorescence at a longer wavelength. The twisted TICT conformation is highly polar and can be stabilized in polar solvents, leading to solvent-dependent emission behaviors. Dual emission is terminated and replaced by a single emission peak when the rotation of donor or acceptor is restricted, due to limitations on excited-state structural changes (Fig. 6b). This is similar to the emission process from a fixed-plane ICT molecule (Fig. 6c). Notably, TICT OBCTs with freely rotating donor or acceptor units (as shown in Fig. 6a) may lack dual emission. This is because two distinct excited-state conformers cannot always coexist: (1) environmental variations influence the degree of rotational freedom. For instance, temperature changes can cause one conformer to disappear entirely. (2) In some molecules, there is no energy barrier between the LE and TICT states, resulting in rapid structural relaxation of the excited state to the TICT state without any LE emission. Additionally, TICT OBCTs with twisted rigid molecular frameworks (Fig. 6b) exhibit separated FMOs, making them extensively used in TADF applications. In summary, TICT molecules are characterized by a twisted excited-state molecular structure and can be classified as flexible and rigid TICT systems depending on the flexibility of single-bond rotation.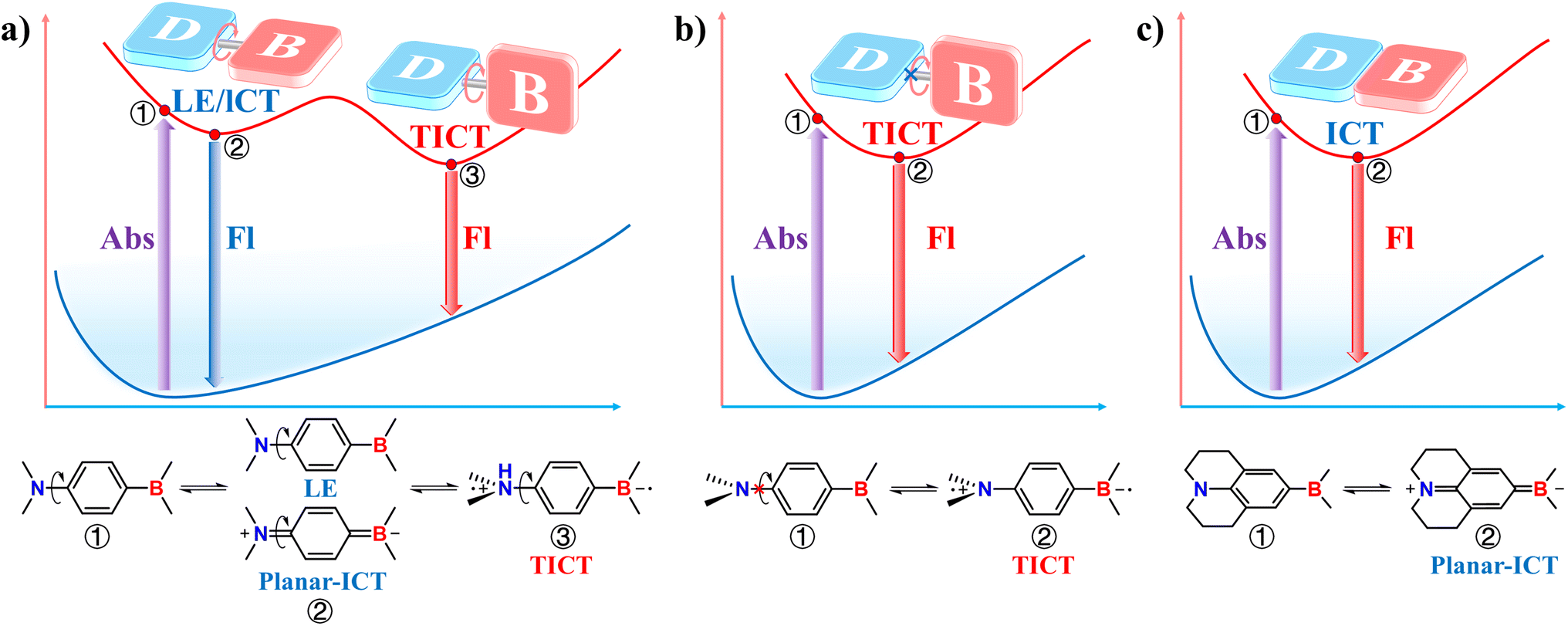 | ||
| Fig. 6 (a) Dual emission in flexible TICT OBCTs. (b) Single emission in rigid TICT OBCTs. (c) ICT in planar molecules. | ||
Flexible TICT OBCTs undergo significant structural changes upon excitation, resulting in large Stokes shifts (Fig. 7). Yang and colleagues reported a fluorescent thermometer (10a/10b) operating over a temperature range of −50 °C to 100 °C (Fig. 7a).76 The pyrene segments in 10a enable flexible rotation at higher temperatures, which favors blue emission from the LE-state. In contrast, the rotation becomes restricted at lower temperatures, leading to a twisted conformation 10b that features TICT-state green emission. This finding highlights the significant impact of temperature on the LE to TICT emission ratio. Later, they introduced another D–A–D-type temperature indicator 11 that displayed temperature-driven LE and TICT transitions in both liquid solvents and solid-state polymers.77
 | ||
| Fig. 7 Flexible TICT OBCTs. (a) Fluorescent thermometer based on temperature-dependent LE/TICT conversion and photographs showing a fluorescence color change from 10a to 10b. Copyright 2011, John Wiley & Sons. (b) Photo-induced TICT structural changes in OBCTs with direct B–N linkages, calculated molecular structures and fragment dihedral angles for 12b and 12c, and their experimental Stokes shifts. Copyright 2013, The Royal Society of Chemistry. (c) B–N linked TICT OBCTs with different donor and acceptor segments. | ||
The weakened D–A strength arising from direct B–N bonding can be restored in TICT molecules. Aminoborane TICT molecules 12a and 12b with direct B–N linkage were reported (Fig. 7b).7812a demonstrates a substantial increase in BN bond twisting upon excitation, with the corresponding dihedral angle increasing from 28.6° to 62.4°. 12b features flexible rotation around both the thiophene–pyrrole and B–N bonds, exhibiting a 308 nm Stokes shift in THF (absorption maximum at 349 nm and fluorescence at 657 nm). The thiophene–pyrrole segment in S1 of 12b becomes nearly planar while the BN fragment further twists upon excitation, which enhances donor strength and forms a TICT state. Ghosh and colleagues described another series of phenothiazine-based TICT OBCTs 13a–13c with direct B–N bonding (Fig. 7c), which exhibit large Stokes shifts (up to 312 nm), aggregation-induced emission (AIE), and mechano-fluorochromism upon grinding.79 The mechanoluminescence in these compounds is ascribed to the flexible phenothiazine structure. Spiro-functionalized acridine as a donor was used to further enhance FMO separation in aminoboranes, resulting in the development of first N-borylated emitters 14 with TADF. A green OLED device based on 14 exhibits an EQE of 19.2%.80 Subsequently, methoxy-substituted carbazoles as donors were exploited to achieve efficient blue N-borylated emitters.81 π-Conjugation extensions in both the carbazole (15)82 and the mesityl of the boron group (16)83 were also investigated. It was found that extending the donor enhanced π-conjugation and steric hindrance, resulting in a red-shifted emission and a smaller Stokes shift in 15.82 Compound 16 exhibits solvatochromism in its emission spectra, with its emission peak shifting from 448 nm in hexane to 495 nm in acetonitrile. It also shows circularly polarized luminescence (CPL) with a dissymmetry factor (glum) of approximately 3 × 10−4 due to its axial chirality.83
Rigid TICT molecules are frequently utilized in achieving TADF (Fig. 8). Adachi reported the first boron-substituted TADF molecules 17 with phenoxaborin as the acceptor.84 The dimethylacridine donor and boron acceptor of 17 are almost orthogonal (89.61°), resulting in excellent FMO separation (Fig. 8a) with a ΔEST of 0.10 eV and enabling a blue OLED with a high EQE of 21.7%. Among structurally similar D–A molecules, 18a and 18b, the dihedral angles between the phenylene bridge and the donors differ greatly (54.7° in 18aversus 88.9° in 18b) due to the smaller steric hindrance of the carbazole donor in 18a (Fig. 8b). As a result, only 18b displays TADF with a ΔEST of 0.013 eV, whereas 18a shows no TADF with a ΔEST of 0.35 eV.85 This indicates that large steric hindrance between the donor and acceptor is crucial for effectively separating the FMOs, thereby achieving TADF in rigid TICT OBCTs. Compound 19 with a blocked triarylborane acceptor, also exhibits TADF behavior due to its near-orthogonal donor–acceptor dihedral angle (87.3°), leading to a green OLED based on 19 with 22.8% EQE.86 Incorporation of a boron-sulfur unit into molecule 20 increases the rate of reverse intersystem crossing (RISC) through enhancing spin–orbit coupling (SOC), thereby shortening the TADF lifetime to ≈1 μs.87 In addition, 20 with a donor–acceptor torsion angles of 87.1° restricts relaxation from S1 to the ground state (S0), producing a narrow full-width at half-maximum (FWHM) of 0.32 eV and enabling TADF-OLEDs based on 20 to achieve a maximum external quantum efficiency (ηext) of 25.3% at 503 nm. Adjusting donor components can further achieve efficient blue TADF-OLEDs.88,89
 | ||
| Fig. 8 Rigid TICT OBCTs with TADF properties. (a) Classic TICT boron-based TADF molecule and its FMO distribution. Copyright 2015, The Royal Society of Chemistry. (b) TADF TICT OBCTs affected by donor–acceptor steric hindrance. (c) Dual-boron-doped TADF TICT OBCTs and a schematic representation of the spin-converting RISC mechanism induced by SOC for 22a and 22b. Copyright 2020, American Chemical Society. (d) TICT TADF OBCTs with different boron acceptors and their corresponding TADF-OLED performance. | ||
Doping more than one boron atom into molecules can enhance TADF performance. Cheng and co-workers reported twisted D–A–D configuration molecules, 21a and 21b, with borylanthracene as the acceptor.90 This arrangement effectively separates FMOs, resulting in small ΔEST values of 22–33 meV and achieving up to 37.8% EQE with small efficiency roll-off featuring only a 3%drop at 1000 cd m−2 for green OLEDs. π-Extended ladder-type oxaborin and thiaborin acceptors have been exploited to construct 22a and 22b.91 Introducing sulfur atoms endows compound 22b with a kRISC that is 3.5 times higher than that of 22a (8.8 × 10−6 s−1vs. 2.5 × 10−6 s−1). NTO analyses and SOC matrix element calculations reveal that RISC occurs from the locally excited T2 state rather than T1 to the charge-transfer S1 state (Fig. 8c). Furthermore, 〈S1|ĤSO|T2〉 of 22b is over 30 times larger than that of 22a (2.93 cm−1vs. 0.09 cm−1), attributed to the heavy-atom effect of sulfur. The combination of acridine derivatives as donors and highly hindered triarylborane as the central acceptor has been used to construct star-shaped blue TADF emitters.92 Upon replacing the donor with a dimethylacridine derivative, a star-shaped molecule achieved 38.8% EQE.93 TICT molecule 23 achieved yellow TADF-OLED emission at 556 nm, owing to its B-heterotriangulene acceptors bearing electron-deficient carbonyl groups that lower the LUMO level (Fig. 8d).94 OLEDs based on 23 exhibit very low efficiency roll-off, achieving a maximum EQE of 28.4% and maintaining EQEs of 21.5% at 5000 cd m−2 and 17.7% at 10000 cd m−2. Replacing the carbonyl group with an electron-rich oxygen atom yields weaker acceptors, resulting in blue TADF.63 A linear D–A molecule 24 was synthesized by Wang et al., featuring a weak spiro-donor and spiro-acceptor separated by a bulky π-spacer.95 The rigid framework enables an OLED doped with 24 (30 wt% in mCBP) to exhibit high-purity emission at 444 nm with CIEy < 0.06. This deep-blue device also shows great resistance to efficiency roll-off, achieving a maximum EQE of 25.4% and retaining 20.0% at 1000 cd m−2. Recently, TICT molecules 25 with a rigid structure exhibiting a small ΔEST of 0.1 eVenabled a sky-blue OLED with 32.6% EQE.96 Compared to the extensively studied TICT molecules with para-positioned acceptors and donors, molecules with meta-positioned triarylboryl acceptors have also been used to fabricate TADF-OLEDs.97
3.3 D–π–A OBCTs
D–π–A OBCTs can be divided into two categories: those with single bond linked (Fig. 9 and 10) and fused aromatic (Fig. 11 and 12) π-bridges. Additionally, single bond linked D–π–A OBCTs including macrocyclic and pillar[5]arene D–π–A OBCTs are also included in this section (Fig. 10). Numerous linear D–π–A OBCTs have been developed since the first D–π–A OBCTs reported by Williams et al. in 1972.66 Shirota reported boron-containing linear D–π–A molecules with a thiophene π-bridge, fabricating blue-green and green OLEDs.98 In contrast to linear D–π–A configurations, non-linear D–π–A molecules (26a–26c) that incorporate boryl groups as side chains exhibit twisted molecular geometries and intense solid-state fluorescence in films.9926a displays a more intense red-shifted fluorescence than 26b (562 nm, ΦF = 0.90 vs. 555 nm, ΦF = 0.86) in films, while an ethenylbenzene π-bridge in compound 26c enables a further red-shifted emission up to 596 nm (ΦF = 0.73). These observations imply that regulating π-bridges can achieve red–NIR light emissions by influencing the CT process. Marder's team combined a dithienyl π-bridge and a strong donor–acceptor pair to obtain compound 27, which exhibited near-infrared (NIR) emission at 745 nm with a PLQY of 48% in acetonitrile. This is the first example of a three-coordinated organoboron compound showing efficient NIR emission.100 Subsequently, D–π–A molecules with unsymmetrically substituted triarylboryl acceptors were developed.101 This indicates that steric hindrance from substituents on boron could prevent the excited-state from becoming planar, thereby enhancing the molecules' photostability. | ||
| Fig. 9 D–π–A OBCTs with linked aromatic π-bridges. | ||
 | ||
| Fig. 10 Macrocyclic and pillar[5]arene D–π–A OBCTs. | ||
 | ||
| Fig. 11 D–π–A OBCTs with fused aromatic π-bridges. | ||
 | ||
| Fig. 12 (a) NIR emissive OBCTs with fused aromatic π-bridges based on an electron push–pull effect. (b) Illustration showing the orbital interaction of FMOs and the push–pull effect in 43a. Copyright 2021, John Wiley & Sons. | ||
Introducing the tetraphenylethene (TPA) π-bridge and the TPA-like fused selenophenothiophene π-bridge into D–π–A OBCTs affords compounds 28 (ref. 102) and 29,103 respectively, both of which exhibit notable TICT-driven solvatochromism and AIE behavior. Star-shaped D–π–A molecule 30 with asymmetric architecture, has the HOMO primarily concentrated on the more electron-rich diphenylamine segment, enabling blue phosphorescent OLEDs.104 In fact, ICT primarily occurs on fragments comprising the strongest donor and acceptor parts in molecules. Ozturk presented a D–π–A molecule with a fused thiophene π-bridge, exhibiting a 200 nm solvent-dependent red shift (412 nm in hexane to 611 nm in acetonitrile).105 Recently, Yamaguchi introduced olefin-borane acceptors to construct D–π–A molecule 32a, which exhibited pronounced solvatochromism.106 The unique olefin structure in 32a allows the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to weakly coordinate with boron. The addition of a Lewis base to 32a triggers frustrated Lewis pair (FLP)-type interactions and forms 32b rather than direct coordination with the boron atom, resulting in significantly altered absorption and emission properties.
C double bond to weakly coordinate with boron. The addition of a Lewis base to 32a triggers frustrated Lewis pair (FLP)-type interactions and forms 32b rather than direct coordination with the boron atom, resulting in significantly altered absorption and emission properties.
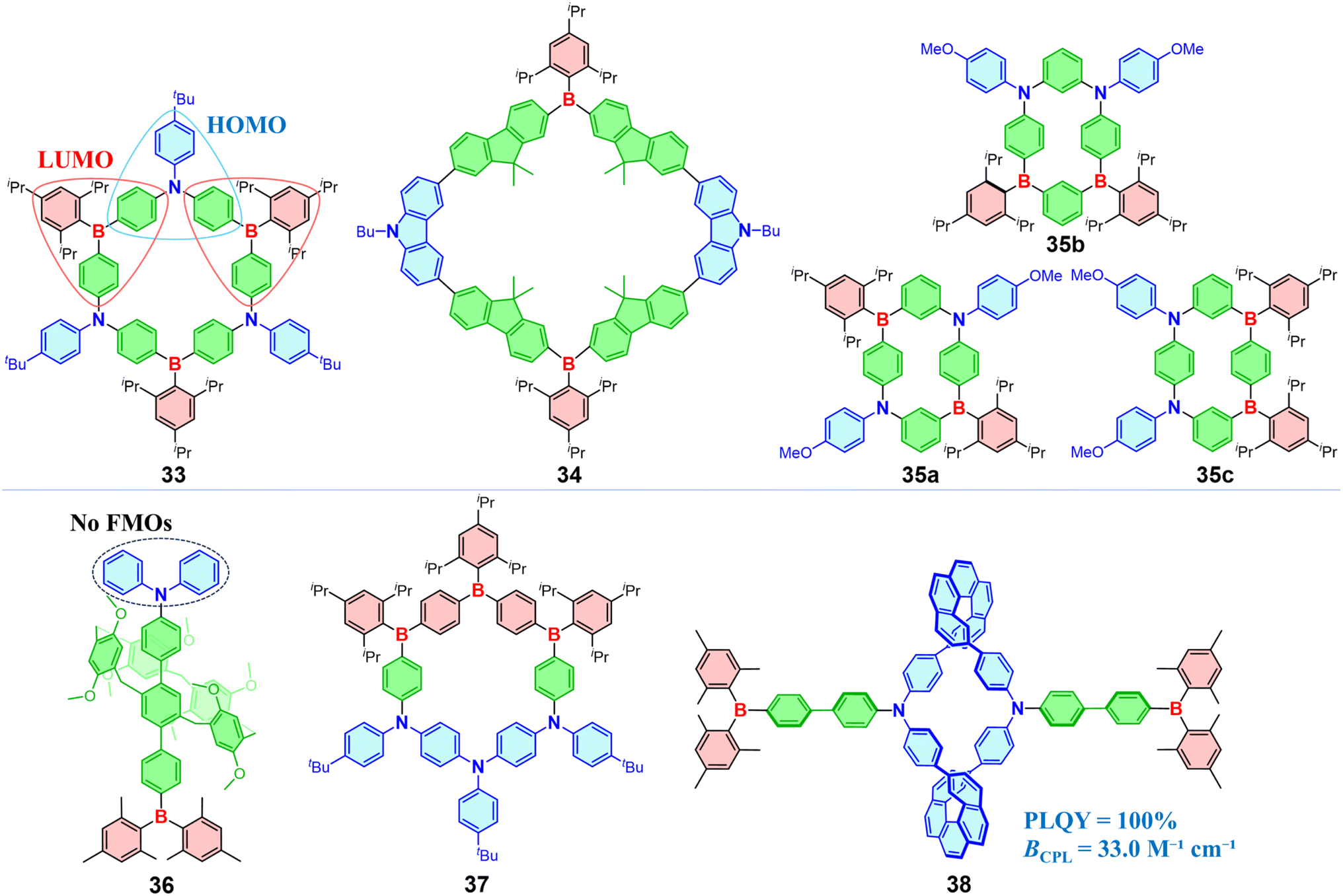
Design strategies of D–π–A are also applied in constructing macrocyclic and pillar[5]arene OBCTs (Fig. 10). In 2012, Jäkle introduced the seminar ambipolar macrocycle with a boron–π–nitrogen (B–π–N) structure (33), exhibiting intense blue fluorescence.107 Notably, the HOMO of 33 only distributed across one of the nitrogen donors and bridging phenylene rings, while the LUMO primarily localizes on two boron acceptors adjacent to the nitrogen donor where the HOMO is located, with minor contributions from the exocyclic phenyl rings. Additionally, macrocycle 34 with fluorene as the π-bridge exhibits significant D–π–A transitions, with the overlapping region of FMOs located on the fluorene moiety. Consequently, molecule 34 displays a pronounced bathochromic shift as the solvent polarity increases.108 Among B–π–N-embedded cyclophanes, 35a–35c, the arrangement of 35c exhibits the smallest HOMO–LUMO gap and clearly separated FMOs, resulting in fluorescence quenching of the S1 state.109 Interestingly, 35a exhibits a red-shifted emission and a rise in ΦF from 0.16 to 0.40 with increasing solvent polarity, which violates the energy-gap law as opposed to compound 35b. Incorporating a B–π–N structure into pillar[5]arenes achieves a PLQY of 99% in solution and a PLQY of 57% in solid for 36, compared to parent pillar[5]arenes with low PLQY (<0.01).110 Notably, DFT calculations indicate that the HOMO responsible for the S1 transition of 36 localizes on the pillar[5]arene structure instead of nitrogen, because the electron-donating effect of the methoxy group allows the pillar[5]arene framework to act as a donor. Compound 37 exhibits a red-shifted emission at 612 nm compared to 33, where the donor and acceptor of 37 are spatially separated on opposite sides of a π-extended ring system.111 Further exploration examines the impact of low symmetry and the B/N ratio on CT, which indicates that single-boron macrocycles exhibit stronger CT properties and smaller HOMO–LUMO energy gaps than their highly symmetric aza/boracyclophane analogues.112 Chen et al. synthesized organoborane macrocycles 38 using chiral [5]helicenes combined with a B–π–N structure, which displayed strong emissions with PLQYs of up to 100% in solution and 34% in solid.113 The CPL brightness (BCPL = ε × ΦPL × glum/2) of the figure-of-eight macrocycle 38 is 33.0 M−1 cm−1, ranking among the highest values reported for [n]helicene-based small organic molecules at that time. Introducing boron atoms to tailor charge transfer in macrocyclic molecules is an efficient strategy for significantly boosting their luminescence performance.
As one of the representative fused aromatic π-bridges, pyrene was used to construct OBCTs 39a and 39b (Fig. 11).11439a exhibits a more twisted structure than 39b due to larger steric hindrance between donor/acceptor and pyrene in 39a. This results in a more red-shifted solvatochromism for 39a, with a maximum emission at 658 nm in acetone. Notably, 39b shows an increase in PLQY from 0.07 to 0.36 as the emission shifts from 458 nm in n-hexane to 561 nm in CH2Cl2, which obeys the energy-gap law. Similarly structured molecule 40 with a fixed electron donor exhibits a high radiative transition rate.115 Axially chiral D–π–A molecules 41a and 41b were reported with naphthalene as the π-bridge.116 Compound 41a exhibits dual emissions of 485 nm and 535 nm at 20 °C. Upon heating, the intensity of the short-wavelength emission peak increases rapidly, whereas upon cooling from 20 °C to −50 °C, the long-wavelength emission peak originating from the CT state undergoes a 31 nm bathochromic shift. The transition of S1 is from the dimethylaminonaphthyl group of one D–π–A subunit to the LUMO on the dimesitylborylnaphthyl moiety of other subunit, while S2, responsible for short-wavelength emission, primarily involves transitions within the same subunit (Fig. 11). 41b exhibits an intense CPL response, achieving tunable CPL properties via fluoride titration and a high luminescence glum of 1.6 × 10−2. Compared to the star-shaped D–π–A OBCTs, molecules 42 with bulky 9,10-anthrylene as the π-bridge shows a pronounced D–π–A transition from the anthrylamine moiety to the anthrylborane moiety.117 The large steric hindrance of the π-bridge in 42 imparts significant resistance to aggregation-caused quenching (ACQ), exhibiting solid-state emission at 643 nm with a PLQY of 31%.
In addition, Yamaguchi introduced a push–pull effect by fusing an aromatic π-bridge with boron to generate narrow NIR emitting materials 43a and 43b (Fig. 12a).11843a exhibits a narrow emission at 791 nm, with a Stokes shift of 21 nm (345 cm−1), a FWHM of 49 nm (771 cm−1), and a PLQY of 8.9%. The crystal structure of 43a reveals a quinone-like π-bridge that facilitates conjugation between the empty p-orbital on boron and the π/π* orbitals of the butadiene structure (Fig. 12b). This arrangement enables an effective electron push–pull interaction between boron and the distal diphenyl ether donor, resulting in NIR emission. Furthermore, replacing the central benzene with an electron-rich thienothiophene unit yields compound 43b, which exhibits an even narrower NIR emission at 952 nm, with a Stokes shift of 7 nm (78 cm−1), a FWHM of 28 nm (309 cm−1) and a PLQY of 3%. Integrating boron-dominated CT with a fused aromatic π-bridge is an effective strategy for inducing red-shifted emission in tricoordinate boron systems, particularly enabling near-infrared emission.
3.4 HLCT OBCTs
In addition to the typical upconversion from T1 to S1, reverse intersystem crossing (RISC) can also occur between higher triplet states (Tm, m > 1) and singlet states (Sn, n ≥ 1), a process known as high-lying reverse intersystem crossing (hRISC).48,119 This can be achieved by HLCT molecules that feature a hybrid LE state and CT state, ensuring a high radiative decay rate from S1 to S0 and facilitating hRISC from Tn to Sn.120,121 HLCT molecules enable efficient triplet exciton utilization and typically possess a D–π–A structure. Boron-based receptors, which readily introduce LE character, are ideal for constructing HLCT molecules. However, HLCT OBCTs have been relatively less reported (Fig. 13). | ||
| Fig. 13 HLCT OBCTs. The inset shows S0 → S1 and S0 → S2 NTO analysis of 47. Copyright 2024, John Wiley & Sons. | ||
Introducing cyano groups to modify the triarylborane acceptor endows 44 with strong electron-accepting characteristics (Fig. 13).122 It exhibits oxygen-sensitive PLQYs and delayed fluorescence phenomena. Lower temperatures result in shortened prompt fluorescence decay and lengthened delayed fluorescence decay for 44, probably due to the hot-exciton process. The weakened boron acceptor, such as DOBNA, is more common in HLCT OBCTs in order to ensure a higher LE ratio. 45 was constructed by combining DOBNA as a weak acceptor and anthracene as a weak donor.123 The LE state in compound 45 is located on the central anthracene unit, whereas the CT state occurs from the anthracene to DOBNA. A significant ΔEST of 1.33 eV was observed for 45 while the S1/S2 and T4/T5 energy levels were very close, promoting efficient hRISC processes. Devices doped with compound 45exhibit deep-blue emissions with a maximum EQE of 10.1%. Recently, Yan and co-workers reported a structurally similar HLCT OBCT, based on which a non-doped device achieved deep-blue emission with minimal efficiency roll-off, from an EQE of 8.4% at 1000 cd m−2 to 5.14% at 10899 cd m−2.124 D–π–A molecule 46, which exhibits near ultraviolet (UV) emission at 414 nm, was generated by incorporating DOBNA and tetrahedral tetraphenylsilane (TPS) with large steric hindrance, featuring a LE state located on DOBNA and a CT process occurring from DOBNA to the phenyl rings of TPS.125 Additionally, 46 exhibits a 0.52 eV energy gap between the S1 and T1 states while the energy gap between S1 and T3 is only 0.04 eV, indicating the HLCT properties of 46. Yang et al. exploited fluorene as the π-bridge to construct a D–π–A molecule, 47.126 Weak hybridization is observed between the S2 and S1 states of 47, wherein a high PLQY is achieved via an LE-like HLCT S1 with higher oscillator strength, while a ‘‘hot-exciton’’ channel is primarily formed from the nearly pure CT S2 state (Fig. 13). DFT calculations further indicate that hRISC occurs between S1 and T6 as well as between S2 and T5 in 47.
Weak boron acceptors can facilitate an effective HLCT process with short-wavelength emission. Double BO-embedded PAHs 48a and 48b exhibit narrow UV and violet-blue emission (FWHMs of 25 and 37 nm, respectively) in toluene, where the directly bonded B–O atoms act as weak acceptors, imparting the molecules with LE-dominated HLCT properties (Fig. 14a).127 TD-DFT calculations of 48a and 48b reveal that S1 has small energy gaps and large SOC matrix elements with T3–T6, whereas it exhibits opposite characteristics with T1 (Fig. 14b). This suggests the efficient utilization of hot excitons via hRISC processes, as evidenced by oxygen-sensitive PLQYs. The 48a- and 48b-based UV and UV-blue OLEDs exhibit both high color purity and high efficiency. In particular, the 48a device features emission at 394 nm, with a CIEy of 0.021, a FWHM of 48 nm, and a EQE of 11.3%, making it one of the best performing UV OLEDs reported with CIEy < 0.05.
 | ||
| Fig. 14 (a) HLCT OBCTs with a directly bonded B–O atom acceptor, and their corresponding OLED performances. (b) TD-DFT-calculated singlet and triplet energy levels, along with SOC matrix elements between S1 and Tn states, for 48a and 48b. Copyright 2024, American Chemical Society. | ||
3.5 MR OBCTs
LR-CT involves substantial vibrational relaxation processes in the excited state, resulting in broadened emissions and reduced PLQYs in TADF molecules. In 2016, Hatakeyama and co-workers introduced the SR-CT mechanism of MR-TADF, utilizing the opposing resonance effects of boron and nitrogen atoms to achieve an alternant HOMO/LUMO arrangement in the central phenyl ring (Fig. 15a).128 The efficient separation of FMOs enables a small ΔEST with TADF characteristics while minimizing excited-state electron-structure changes. Additionally, the rigid framework decreases reorganization energy, leading to a narrow FWHM and high PLQY. Two classic MR-TADF molecules named DOBNA128 and DABNA37 have been successively reported. Furthermore, Duan129 and Wang130 developed a carbazole-based molecule 49, featuring a more rigid structure and red-shifted emission (Fig. 15b). Boron-based MR-TADF molecules have made rapid progress in recent years and have been systematically summarized.131–133 Therefore, this section focuses on strategies for tuning MR-TADF properties involving CT. | ||
| Fig. 15 (a) MR-TADF design principles. (b) Representative MR-TADF OBCT structures. (c) B–π–B and N–π–N structures reported by Yasuda. (d) Effects of atomic arrangements on ΔEST and donor–acceptor characteristics of MR-TADF molecules. | ||
Most of the MR-TADF molecules emit exclusively in the blue to green region. By expanding the framework of 49 based on carbazole and its derivatives, Yasuda et al. synthesized MR-TADF OBCTs 50a and 50b with opposite shifts in emission wavelength.13450a with para-arranged B–π–N units displays a blue-shifted emission due to weakened D–A interactions, while 50b with para-arranged B–π–B and N–π–N units produces red emission at 615 nm due to strengthened D–A interactions (Fig. 15c). This strategy of combining B–π–B and N–π–N is widely applied to achieve longer-wavelength emission for MR-TADF molecules.132,135 Additionally, the non-bonding nature of MR-TADF which limits conjugation expansion is generally accepted as a factor restricting the red shift of emission for MR-TADF molecules.136 Considering that donor–acceptor strengths directly affect the energy gap of molecular transitions, the influence of different MR-arrangements on donor–acceptor interactions are discussed as follows.
As shown in Fig. 15d, the ortho D–π–B configuration facilitates charge separation within the central phenyl ring, resulting in a smaller ΔEST. However, opposite resonance and inductive effects between the donor and boron mutually weaken their strengths. Similarly, in the para-D–π–B configuration, donor–acceptor strengths are diminished without an inductive effect. Therefore, MR-TADF essentially involves a weak CT process. For meta-B–π–B/D–π–D structures, the opposite resonance effects that act on the same atoms of phenyl ring strengthen the MR in OBCTs. When these configurations are combined, they are expected to yield blue-shifted TADF emission. In contrast, the para-B–π–B/D–π–D configurations, in which the same resonance effects alternately locate on the central phenyl ring against MR effects, result in enhanced donor or acceptor strength. Similar influences on the donor or acceptor are observed in the ortho-B–π–B/D–π–D configuration, while an additional inductive effect further enhances donor or acceptor strength. In the meta-D–π–B configuration, opposite resonance effects on the same atoms of the central phenyl ring cancel each other out and increase the ΔEST of the molecules. MR-TADF molecules with these two configurations are anticipated to exhibit red-shifted emission, in which the central phenyl ring shows π–π* LE characteristic.
Integrating the MR effect with LR-CT also enables red-shifted emissions (Fig. 16). Duan et al. introduced electron-withdrawing substituents at the para-position of the boron-attached benzene ring in 49 to generate molecules 51. The LUMO of 51 extends onto the fluorophenyl substituent, conferring CT characteristics and resulting in blue-green TADF at 494–499 nm.129 Then, the concept of HMCT was proposed to construct molecule 52 by extending the π system with an electron-withdrawing aza-phenanthrene moiety.49 The enhanced ICT character of 52 reduces the HOMO–LUMO energy gap, leading to a sharp green emission at 522 nm (FWHM = 28 nm) and an extremely high PLQY of 99.7%.
 | ||
| Fig. 16 TADF molecules utilizing hybrid LR/SR-CT. | ||
Applying the HMCT strategy into D–π–A systems formed the hybrid LR/SR-CT molecule 53, in which D-MR-A design resulted in a high oscillator strength with a smaller ΔEST.35 In addition, symmetric DOBNA units on both sides of 53 exhibit dual CT pathways, doubling the oscillator strength (from 0.1426 to 0.3498) compared to the molecule with single-sided DOBNA and achieving an OLED with a high EQE of 40.4%. Recently, Hatakeyama and co-workers developed a donor/acceptor ‘‘core–shell’’ molecule 54 by introducing an additional boron atom into a deep-blue MR matrix.137 The highly localized tri-boron region with a strong electron-withdrawing effect effectively deepens the LUMO level, inducing LR-CT between the B core and electron-donating MR shell fragments in the rigid structure. As a result, compound 54 exhibits an emission at 613 nm with a narrow FWHM of 31 nm, representing a 172 nm red shift compared to the MR-matrix shell compound. The 54-based OLEDs achieve pure-red emission up to 624 nm with an EQE of 23.3% and demonstrate excellent electroluminescence stability, with a maximum LT99 (time to 99% of the initial luminance) exceeding 400 h at an initial luminance of 1000 cd m−2.
4. TSCT in tricoordinate boron molecules
TSCT occurs in π-stacked systems where two or more aromatic units are close but not covalently bonded.33,34 TSCT OBCTs can be classified based on the type of π-spacer: rotatable and fused. A single-bond-connected spacer is rotatable with greater flexibility, enabling parallel D–A configurations (Fig. 17), while a fused π-spacer imparts rigidity to molecule frameworks, which is ideal for constructing TSCT TADF emitters (Fig. 18–20). | ||
| Fig. 17 (a) U-shaped and V-shaped TSCT OBCTs with a rotatable π-spacer. Insets showing the fluorescence change of 55 before and after fluoride titration. Copyright 2006, John Wiley & Sons. (b) TSCT OBCTs with a smaller spacer. Insets showing the fluorescence change ofr 57a before and after fluoride titration. Copyright 2011, American Chemical Society. | ||
 | ||
| Fig. 18 (a) TADF-emissive TSCT molecules with triarylboron acceptors. (b) The calculated HOMO/LUMO distributions, ΔEST and the TSCT/CT ratio (ηTSCT/CT) for 61. Copyright 2021, The Royal Society of Chemistry. | ||
In 2006, Wang and colleagues reported the first TSCT OBCT 55 where the donor N(Ph)(1-naphthyl) and acceptor BMes2 are linked to rotatable biphenyl groups bridged by a naphthalene unit.138 The U-shaped geometry of 55 allows TSCT from the amine to the boron unit, which can be blocked by fluoride titration, resulting in a fluorescence change from green to brighter blue (Fig. 17a), serving as a “turn-on” sensor for fluoride. Subsequently, V-shaped TSCT molecule 56 with an organosilicon spacer was developed, offering increased rotational freedom between donor and acceptor groups and increased sensitivity to fluoride.139 Introduction of a dimethylarylamine donor created a TSCT molecule featuring a similar structure to 55 and dual responsiveness to fluoride and protons, in which fluorescence shifts to sky blue upon fluoride titration while protonation of the amine shifts emission to purple.140 A smaller spacer can also be applied to construct TSCT molecules. A rotatable biphenyl spacer was used to construct molecules 57a and 57b with BMes2 and NPh2 at adjacent positions. These molecules exhibit bright TSCT fluorescence and undergo significant blue shifts upon fluoride titration due to the inhibition of the ICT process (Fig. 17b).141 Then, they introduced a new family of triarylborane-based [2,2]paracyclophane derivatives 58a and 58b, enabling efficient TSCT fluorescence and TADF.142 Remarkably, the enantiomerically pure form of 58a exhibits strong CPL with a glum of up to 4.24 × 10−3.
The twisted TSCT structures can effectively separates FMOs and avoid ACQ of emission (Fig. 18a), which have been widely utilized in designing efficient TADF emitters. The TSCT molecules with TADF are typically constructed by arranging the donor and acceptor at adjacent positions on the fused π-spacer. This can shorten the π–π interaction distance between the donor and acceptor, resulting in an enhanced transition dipole moment and reduced vibrational relaxation. The bulky triarylboron acceptors can improve molecular rigidity of TSCT OBCTs to enhance TADF performance. Compound 59 with a BMes2 acceptor has intrinsic steric ‘‘locking’’ that enables a highly twisted conformation, resulting in a small ΔEST of 0.15 eV and an efficient blue TADF at 463 nm.143 The electron-donating groups substituted at boron's para position of 59 induce a blue shift in emission,144 while electron-withdrawing groups cause a red shift.145 The highly separated HOMO and LUMO of 60 are localized primarily on the carbazole and BMes2 units, respectively, indicating that charge transfer occurs through both the aryl linker and through space. This dual-mode CT gives rise to a small ΔEST of 0.05 eV alongside a large transition dipole moment.146 Moreover, the twisted framework of 60 effectively suppresses intermolecular π–π stacking, enabling blue non-doped OLEDs to achieve an exceptional EQE of 19.1%. ortho-D–A–D TSCT OBCT 61 features distinct CT from two carbazole units to the triarylboryl unit (Fig. 18b).147 NTO analysis of the hole–particle overlap reveals that TSCT contributes up to 84.49% of the total CT character. The OLEDs incorporating 61 exhibit green electroluminescence with a maximum EQE of 27.5%.
Planar MR boron acceptors, such as DOBNA, are also widely used for constructing TSCT OBCTs with TADF properties, which require larger fused aromatic rings as spacers. Yang et al. employed tert-butyl carbazole as a rigid spacer to build the TSCT molecule 62, where the planar donor and DOBNA acceptor exhibited strong intramolecular π–π interactions in a face-to-face orientation (Fig. 19a).148 This increases the radiative decay rate of S1 and suppresses non-radiative decay, resulting in a PLQY up to 0.99 in thin films doped with 62. A device with 30 wt% of 62 achieves maximum EQE/current efficiency/power efficiency values of 23.96%/76.74 cd A−1/65.63 lm W−1, respectively. A TSCT dendrimer with blue TADF characteristics was developed by incorporating three DOBNA acceptors. The CT process in this dendrimer mainly arises from TSCT, with intrafragment charge transfer (IFCT) analysis revealing a contribution ratio of 92.2%.149 Gradual planar skeleton modification of phenyl acridine from 63a through 63b to 63c leads to progressively closer intermolecular donor–acceptor stacking, which diminishes vibrational relaxation and produces increasingly narrow FWHMs from 57 nm to 35 nm (Fig. 19b). 63a and 63b show ultrapure-blue TADF, while 63c exhibits a large ΔEST of 0.29 eV without TADF due to the weak carbazole donor strength.150 Notably, introducing an additional tert-butylcarbazole substituent at the para position of carbazole in 63c induced TADF emission by enhancing the carbazole donor strength, reducing ΔEST to 0.09 eV.151
 | ||
| Fig. 19 (a) TSCT OBCTs with a planar donor and DOBNA acceptor in a face-to-face orientation. Single crystal structure of 62 showing the strong intramolecular π–π interactions. Copyright 2020, John Wiley & Sons. (b) TSCT OBCTs with different skeleton-modified phenyl acridine donors, as well as the relationship between structures and HOMO levels of phenyl acridine and its derivatives based on DFT calculations. Copyright 2022, John Wiley & Sons. | ||
The MR segment DABNA can also act as an acceptor to construct TSCT OBCTs (Fig. 20). 64a and 64b display a sandwiched structure, with the DABNA acceptor playing dual roles as both a TADF emitter and an electron acceptor.152 The RISC process of 64b occurs from the TSCT-type T2 to the MR-type S1, with a large SOC matrix element 〈1MRCT|ĤSOC|3TSCT〉 of 0.33 cm−1 (Fig. 20), resulting in fast delayed fluorescence with a lifetime of 10.6 μs. A 3 wt% doped device with 64b reaches a maximum EQE of 31.7%. Recently, Xiao and co-workers combined MR and TSCT strategies to design a high-performance green narrowband OLED, achieving an EQE of 32.3%.153 Another planar 9,10-diboraanthracene acceptor was used to construct 65, which exhibited strong red TADF and produced a red OLED with an EQE of 10.1% at 615 nm.154 In addition, molecules 66a and 66b were obtained using a highly planar benzene-fused boracycle.155 The distance between B and N in 66a and 66b decreases when the donor is switched from carbazole to electron-rich phenothiazine, indicating stronger π–π interactions between the boron acceptor and phenothiazine. Notably, 66a exhibits no TADF properties, likely due to the weak carbazole donor. These compounds display low PLQYs in solution and high PLQYs at low temperatures or in doped thin films.
 | ||
| Fig. 20 TSCT OBCTs with different planar boron acceptors. The illustration showing calculated hole (green) and electron (blue) distributions and energy gaps of the 1,3MRCT and 1,3TSCT excited states for 64b. Copyright 2023, John Wiley & Sons. | ||
5. ICT in tetracoordinate boron molecules
Tetracoordinate boron molecules with sp3 hybridized boron atom exhibit a stable and blocky octet electron configuration compared to tricoordinate boron molecules, forming a three-dimensional architecture with ACQ resistance.26,156 Additionally, the three-dimensional form also allows greater flexibility in designing tetracoordinate boron compounds. In terms of electronic properties, the filled p-orbital in tetracoordinate boron eliminates its electron-withdrawing capability, giving it a distinctly different role in the CT process compared to tricoordinate boron. To date, research on the CT mechanism of tetracoordinate boron compounds is still relatively limited. This section provides an in-depth analysis of the roles of tetracoordinate boron in the CT process.5.1 Chelation of tetracoordinate boron
Tetracoordinate boron is commonly exploited as an acceptor fragment in charge-transfer molecules, which can be categorized into four types based on the chelated donor atoms: O,O-chelate, N,C-chelate, N,O-chelate, and N,N-chelate (Fig. 21a).157 Strong electron-withdrawing groups like F, CF3, and C6F5 are usually incorporated into these compounds as substituents of boron. Understanding the impact of chelation at the tetracoordinate boron center on the ICT process is crucial for advancing the design of functional tetracoordinate compounds. | ||
| Fig. 21 (a) Chelation types of tetracoordinate boron as an acceptor fragment. (b) Resonance hybrid involving B–N and B→N bonding (c). Hyperconjugation effect in the tetracoordinate boron molecules. (d) Tetracoordinate boron TADF molecules. | ||
Liu and co-workers analyzed single-crystal structures of BODIPY and observed that the two B–N bonds in the N–B–N unit had equivalent bond lengths of approximately 1.55 Å, suggesting the existence of a resonance hybrid involving both B–N and B→N bonding (Fig. 21b). Accordingly, a novel tetracoordinate boron molecule 67 was developed to exhibit a resonance hybrid of N–B–N bonds.158 The single-crystal structure of 67 reveals similar B–N bond lengths (around 1.54–1.55 Å), despite the two nitrogen atoms in the N–B–N unit differing in their chemical environments. Notably, the HOMO distribution of 67 is skewed toward the dihydrophenazine moiety, while the LUMO is slightly preferentially localized on the thiazole segment. This is likely due to the influence of more dominant resonance forms on FMOs. Furthermore, molecules 68a and 68b were synthesized, featuring a hyperconjugation effect that influences the electronic structure and stabilizes different redox states (Fig. 21c).159 This highlights that σ–π hyperconjugation can occur in tetracoordinate boron compounds without disrupting conjugation, thus facilitating the CT process. The covalent and coordination bond lengths were measured to be 1.56 and 1.64 Å in the single crystal of 68a, respectively, indicating that bulky phenyl substituents on boron lengthen the B–N bonds and weaken the coordination bond, resulting in a reduced resonance effect.
Chou et al. utilized tetracoordinate boron as the linkage unit to generate molecule 69, which revealed that the HOMO and LUMO were well-separated and that tetracoordinate boron contributed to a rigid molecular framework (Fig. 21d).160 They subsequently fabricated the first TADF OLED based on tetracoordinate boron molecule 69, achieving a maximum EQE of 13.5%. Recently, spirocyclic NIR emitters 70a and 70b were introduced by incorporating a tetracoordinate boron acceptor, a dimethylacridine (DMAC) substituent and either chiral binaphthol, or octahydro-binaphthol donors.161 The LUMOs of 70a and 70b are primarily localized on the acceptor unit of the β-diketone-coordinated boron and the adjacent benzene ring, indicating a robust OBO resonance effect, while the HOMOs exhibit different distributions depending on the electron-donating strength of the chiral units. Solution-processed, non-doped OLEDs based on these emitters show NIR emission peaking at 716 nm, with a maximum EQE of 1.9% and a high exciton utilization efficiency of 86%.
5.2 Coordination-enhanced/quenched charge transfer
The thriving development of OBCTs has been closely linked to the innovative exploration of the CT process. In contrast to the well-established π-acceptor roles of tricoordinate OBCTs, tetracoordinate boron compounds with a filled p-orbital present a conundrum regarding clarification of their underlying CT process, an understanding of which will facilitate the diverse advancement of tetracoordinate boron OBCTs. In 2022, our group reported a tetracoordinate boron compound 72a, featuring intriguing TADF properties.162 Compared to its parent tricoordinate boron molecule 71,16372a exhibits a red-shifted emission exceeding 100 nm, reaching 557 nm. In addition, 72b with a fused heptagon displays a deep-red emission at 660 nm and shows significant AIE behavior (Fig. 22). | ||
| Fig. 22 Tri- and tetra-coordinate boron molecules with NBN fragments and their maximum emission wavelengths in 10−5 M THF solution. The inset shows the photograph showing the emission intensity increase of 72b with the increasing H2O fraction. Copyright 2022, The Royal Society of Chemistry. | ||
Recently, we constructed a family of tetracoordinate boron-based twisted helicenes 73a–73d, achieving red-shifted emission with narrower FWHM by combining an alternating D–π–A framework with boron-induced CE-CT (Fig. 23).16473a–73d designed using the CE-CT strategy, utilize the electron-withdrawing pyridine ligand to coordinate with the empty p-orbital of boron, enhancing the pyridine acceptor strength. The four-coordinated boron atom in 73a–73d features low electronegativity and an electron-octet structure, which pushes σ-electrons to the carbazole donor fragments. The boron atom in the CE-CT process serves as a bridge for the electron shuttling, resulting in a push–pull effect with enhanced CT. In addition, these compounds display red-shifted emissions up to 753 nm at higher concentrations. Cis/trans configurational isomers of 73c were separated and exhibit nearly identical photophysical properties. Subsequently, IFCT and Huang–Rhys factor calculations clarified the phenomenon of narrowing red-shifted emission. The CE-CT strategy offers a solution to the challenge of achieving red-shifted emission in OBCTs.
 | ||
| Fig. 23 (a) Coordination-enhanced charge transfer mechanism. Copyright 2024, John Wiley & Sons. (b) A family of tetracoordinate boron helicenes and their maximum emission wavelengths and FWHMs in 10−5 M toluene solution. | ||
Inspiringly, the principle of CE-CT can also elucidate the effect of coordination on the CT process of tricoordinate boron compounds. Different ligands could lead to opposite CT effects during the coordination process, in which the ligands can be categorized into two types: (1) electron-rich donor-type ligands, which include various anions like fluoride and saturated amine groups such as NMe2 and NPh2, and (2) electron-deficient acceptor-type ligands, including heteroaromatics (e.g., pyridine and benzothiadiazole) and carbonyl (e.g., ketone and aldehyde). Two tricoordinate boron theoretical models are proposed in this perspective, D–π–B and A–π–B systems, to investigate the impact of coordination with different ligands on CT process, aided by in-depth analysis of CE-CT (Fig. 24). Coordination with an electron-rich ligand in the D–π–B system will reduce the electron-donor ability of the ligand and form a D–π–D(B) system, which commonly exhibits an enlarged energy gap due to the D(B)-quenched CT process (Fig. 24a). Notably, weak D(B) usually does not participate in FMOs and the above process can be termed as coordination-quenched charge transfer (CQ-CT). During this process, coordination with an electron-rich ligand inverts the original boron acceptor into a weak donor unit (D(B)), so CQ-CT can also be termed acceptor-inversion coordination-quenched charge transfer (iCQ-CT). The proposed CQ-CT has several potential applications: (1) the change in emission properties upon coordination with specific ligands can be exploited for sensing applications. For instance, the quenching of emission in response to the presence of certain anions or biomolecules can serve as a selective and sensitive detection mechanism. This can be particularly useful in biological sensing, where the detection of specific ions or molecules is crucial. (2) CQ-CT can be utilized to design switchable materials whose optical properties can be modulated by external stimuli. By controlling the coordination environment, one can switch between states with different emissive properties, which can be applied in smart windows, displays, and other optoelectronic devices that require dynamic control of light transmission or emission. (3) In energy transfer systems, CQ-CT can be used to regulate the efficiency of energy transfer. By designing systems where the charge transfer is quenched upon coordination, one can control the flow of energy in a molecular circuit, which can be beneficial in solar cells and other photovoltaic applications.
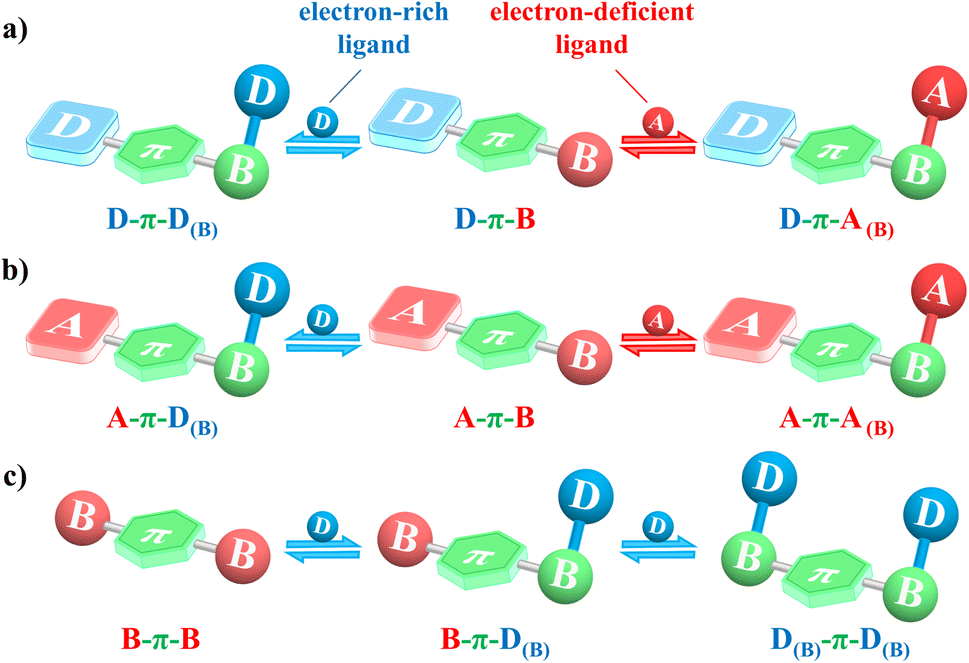 | ||
| Fig. 24 The effect of coordination on the CT process of (a) D–π–B, (b) A–π–B and (c) B–π–B systems, D: electron-donor, A: electro-acceptor, and B: boron atom. | ||
In contrast, the D–π–B system coordinating with an electron-deficient ligand could enhance electron-acceptor ability of the ligand and form a new D–π–A(B) system, resulting in a red-shifted absorption and emission if the new CT is stronger than that of the D–π–B system (Fig. 24a). This process involves CE-CT similar to that in 73a–73d. In the A–π–B system, coordination with an electron-rich ligand forms an A–π–D(B) system, which typically enhances molecular CT and leads to a red-shifted emission due to the generated D–π–A architecture (Fig. 24b). This process, which inverts the boron acceptor into a donor unit (D(B)) to enhance CT, is termed acceptor-inversion coordination-enhanced CT (iCE-CT). In addition, coordination with an electron-deficient ligand in the A–π–B system yields an A–π–A(B) system.
When the electron-acceptor A is the boron atom (B), the A–π–B can be also represented by B–π–B (Fig. 24c). Recently, boron-based multi-helicene 74a with superior PLQYs and excellent chiroptical properties was reported by our group (Fig. 25). Upon fluoride titration, the emission of 74a first bathochromically shifts from green (522 nm) to orange (567 nm) with the formation of mono-fluorinated helicene 74b, and then hyperchromically shifts to yellow (541 nm) producing a mixture of cis/trans-di-fluorinated helicene 74c.165 This indicates that the titration process from 74a to 74b represents a transformation of the B–π–B system into a B–π–D(B) system, where the iCE-CT process results in red-shifted emission. Further transformation to the D(B)–π–D(B) system in 74c leads to blue-shifted emission, resulting from a CQ-CT process (Fig. 24c). Notably, the fluorides in both 74b and 74c exhibit negligible contributions to FMOs due to their weak donor ability.
 | ||
| Fig. 25 (a) Double-fluoride titration of double-boron doped helicene 74a. (b) Titration experiments of 74a in acetone (1.0 × 10−5 M) solution with TBAF, measured using fluorescence spectra (λex = 470 nm). Inset: photographs showing fluorescence of 74a with 0 equiv., 4.00 equiv. and 25 equiv. of TBAF in acetone. Copyright 2023, John Wiley & Sons. | ||
Four-coordinated boron could be dissociable during the photoexcitation process. Yamaguchi and colleagues reported that triarylborane–pyridine complex 75a underwent a photodissociation process in the excited state, resulting in dual fluorescence (Fig. 26).166 The shorter-wavelength fluorescence originates from the LE emission of 75a, while photodissociation in the S1 state generates uncoordinated borane 75b, which is responsible for the longer-wavelength emission. Notably, the HOMO of 75a located at the boron-containing backbone, while the LUMO almost localized on the pyridine group, forming an effective D–π–A(B) system. However, 75a exhibited a blue-shifted emission to 75b due to its relatively weaker CT compared to that of 75b.
 | ||
| Fig. 26 (a) Photoinduced coordination cleavage of triarylborane–pyridine complex 75a. (b) A plausible energy diagram for the photodissociation process. Copyright 2014, American Chemical Society. | ||
Furthermore, we have summarized several representative examples of changes in the CT type and emission wavelength before and after coordination with electron-rich or electron-deficient ligands, and assigned their CT process (Table 1). The CE-CT and CQ-CT mechanisms provide the theoretical foundation both for assigning donor and acceptor roles in tetracoordinate boron molecules and for interpreting FMO analyses, thereby further assisting in understanding the luminescence properties of these molecules.
| Before | CT typea | λ em (nm) | Ligand/type | After | CT typea | λ em (nm) | (i)CE-CT/CQ-CT | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The CT type of these compounds was determined based on the FMO distributions. b The emission wavelengths were measured in solution, annealed film, or powder. c Not available. | ||||||||
| 74a | B–π–B | 522 | F−/D | 74b | D(B)–π–B | 567 | iCE-CT | 165 |
| 74b | D(B)–π–B | 567 | F−/D | 74c | D(B)–π–D(B) | 541 | CQ-CT | 165 |
| 75b | D(π)–B | 550–700 | Pyridine/A | 75a | D(π)–A(B) | 470–530 | —c | 166 |
| 77a | D(π)–B | 540 | –Ph2PO/A |
77b | D(π)–A(B) | 440 | —c | 167 |
| 78b | D–π–B | 511 | –NMe2/D | 78a | D–π–D(B) | 382 | CQ-CT | 168 |
| 79a | D–π–A–B | 475 | –CO/A |
79b | D–π–A(B) | 620 | CE-CT | 169 |
| 80a | D–π–B | 540 | –NHMe/D | 80b | D(B)–π | —c | CQ-CT | 170 |
| 86a | B–D–A–D–B | 485 | F−/D | 86b | D(B)–D–A–D–D(B) | 590 | iCE-CT | 171 |
Since both CE-CT and CQ-CT are caused by the coordination of boron, there could be several prospects and synergies in combining them: (1) by integrating both mechanisms, one can create materials that respond to multiple stimuli. For example, a system that exhibits CE-CT under one set of conditions and CQ-CT under another can be designed to have a more complex and nuanced response profile. This can be particularly useful in creating materials that can adapt to changing environments, such as in adaptive optics or responsive coatings. (2) Combining CE-CT and CQ-CT can lead to materials with tunable emission properties. By carefully designing the coordination environment and the electronic properties of the donor and acceptor moieties, one can achieve a wide range of emission wavelengths and intensities. This can be applied in the development of multi-color emissive materials for displays, lighting, and other applications where a broad color gamut is desired. (3) The ability to switch between CE-CT and CQ-CT can be harnessed to create molecular logic gates. By designing molecules that can respond to multiple inputs (such as different ligands or environmental conditions) and produce distinct outputs (emission or quenched states), one can develop complex molecular computing systems. This can pave the way for the next generation of molecular electronics and nanoscale computing devices.
5.3 Intramolecular dynamic-coordination induced CT
The coordination bond in tetracoordinate boron compounds is usually dynamic, since it can be formed or cleaved in response to external stimuli, such as temperature, solvent and light. OBCTs can be exploited to construct intramolecular dynamic-coordination systems due to their flexible design enabling stimuli-responsive properties in OBCTs by modulating the ICT process upon coordination. 76a with an azobenzene photo-switch can be partially converted to the cis isomer 76b upon irradiation, showing an absorption maximum of 76a at 339 nm while that of 76b is at 460 nm (Fig. 27a).172 Wolf et al. introduced boron–phosphine oxide Lewis pair systems (77a and 77b) (Fig. 27b).167 The open structure 77a is favored in hydrogen-bond-donating (HBD) solvents or at higher temperature, featuring an intense CT fluorescence, while the closed structure 77b exhibits a 100 nm blue-shifted emission compared to 77a, attributed to LE transition of 77b. Introducing mesityl groups on phosphorus impedes the formation of the closed structure due to steric bulk. Subsequently, this system was applied in biological imaging.173 Analogous to the photo-responsive systems 75a and 76a, dibenzoborole derivatives 78a and 78c undergo B–N coordination-bond cleavage to form 78b and 78d upon photoexcitation (Fig. 27c).168 This process is termed bond-cleavage-induced intramolecular charge transfer (BICT) and gives rise to dual emissions, where short-wavelength emissions arise from the LE states of 78a and 78c and long-wavelength emissions originate from the CT states of 78b and 78d. The electron-donating effect of the aryl (Ar) substituent shifts the HOMOs onto dibenzoborole rather than NMe2, thereby enabling the CT transitions in 78b and 78d. In contrast, the unsubstituted molecule exhibits only a single emission, likely because the CT from NMe2 to dibenzoborole is quenched by nonradiative decay. | ||
| Fig. 27 Intramolecular dynamic coordination systems under external stimuli: (a) 76a and 76b; (b) 77a and 77b; (c) 78a and 78b, 78c and 78d; (d) 79a and 79b; (e) 80a and 80b; (f) 81a and 81b. | ||
Weak or bulky donors and boron acceptors are required to ensure intramolecular dynamic coordination properties. Wang and co-workers synthesized 79a featuring bulky BMes2 and an aldehyde group as a weak donor, which can be converted to its closed state 79b in a 92% H2O/THF solution (Fig. 27d).169 H2O molecules could interact with the amino groups in 79a, reducing its donating ability to boron and thus increasing boron's electrophilicity, while the oxygen atom of H2O may also interact with the carbonyl group, enhancing its nucleophilicity. Notably, low temperature and grinding facilitate the formation of molecule 79b, resulting in an emission color shift from blue to red. Subsequently, replacing the aldehyde group with an alkylamino group formed a series of molecules in which the tert-butyl-substituted amino group is responsible for the proton-solvent-responsive absorption.174 The proportion of the closed-state structure increases in proton solvents, reducing the long-wavelength absorption band of the open-state structure. If the ligand is too weak or bulky to form effective coordination bond, there is no dynamic coordination. The stilbene derivative 80a can undergo the B→NH coordination process and form 80b upon light irradiation, which leads to a blue shift in the 388 nm absorption peak and quenched fluorescence (Fig. 27e).170 In contrast, molecules with bulkier nitrogen substituents than those in 80a exhibit enhanced fluorescence upon irradiation due to a transition from trans-ICT to cis-ICT, without the formation of coordination bonds. These cis isomers could revert to their trans forms upon heating. Recently, compounds 81a with a phosphine oxide group ligand showed that the S0–S1 transition of 81a primarily originates from the HOMO on the NMe2-Ph/Mes group to the LUMO + 1 localized at oxygen/phenyl groups of the OPPh2 unit as well as the linker Ph unit (Fig. 27f).17581a can transform into 81b with a red-shifted absorption in polar solvent. Increasing the temperature also promotes this transformation, showing a nearly 15.7-fold enhancement in fluorescence intensity from 137 K to 340 K. In addition, grinding 81a powder induces an 18 nm bathochromic shift, with ΦPL increasing from 4.0% to 13.0%.
In addition to dynamic coordination by switching open/closed states, Wang and co-workers reported 82a with constrained ring systems and an internal dynamic B→N bond (Fig. 28a).176 The B→N bond length in 82a (1.80 Å) is longer than the typical B→N bond length (<1.70 Å), and its boron atom adopts a nearly sp3-hybridized geometry, indicating the formation of a loose B→N bond. Crystals of 82a exhibits weak emission at ambient temperature, while the emission intensity significantly increases as the temperature rises, due to cleavage of the B→N bond to produce 82b. Grinding the crystals of 82a leads to a fluorescence change from blue to green with an increase in emission intensity (Fig. 28b). A similar molecule 83 by changing the substituents on boron and nitrogen exhibited pronounced photochromism upon UV irradiation, switching from short-lived fluorescence to long-lived phosphorescence.177 In addition, a range of extended dimeric B/N-coordinated molecules 84a–84c exhibited different ratios of sp2-and sp3-hybridized boron by varying the number of methyl substituents on the π-linkers.178 The HOMO of 84a localized on the central diaminophenyl linker, while the LUMO delocalized over two BMes2 groups. In contrast, the HOMO of 84b localized on the tetracoordinated B/N Lewis adduct (sp3-B), while the LUMO resided on the other Lewis pair unit (sp2-B) (Fig. 28c). Although the sp3-B/N dimer 84c was non-emissive in solution, grinding the solid-state material resulted in a substantial increase in PLQY from 2% to 60%, attributed to the cleavage of the B→N bond.
 | ||
| Fig. 28 (a) Single BN system and (c) double BN constrained-ring systems with intramolecular dynamic coordination under external stimuli. (b) Photograph showing the temperature increase- and grinding-driven fluorescence change from the single crystal 82a to 82b. Copyright 2018, American Chemical Society. | ||
Similar to the intramolecular dynamic coordination of 79a/79b (Fig. 27d), the open state 85a exists in a crystalline form or in an annealed film and partially transforms into the closed-shell structure 85b upon dissolution or in a cast film, accompanied by red-shifted absorption (Fig. 29).179 The formation of the B→N bond significantly lowers the LUMO level from −1.59 eV in 85a to −2.29 eV in 85b, while the HOMO level increases slightly (+0.18 eV). Additionally, the crystals of 85a change from yellow to dark green upon grinding, likely due to the partial formation of 85b. Coordinating the boron atom in OBCTs with electron-rich ligands could also result in red-shifted absorption and emission, as shown in Fig. 24c. Similarly, 86a featuring 1,4-phenazaborine as a weak electron donor, exhibits red-shifted emission when its boron atoms coordinate with electron-rich fluoride to form molecule 86b.171 This red shift occurs because the weak B–N donor forms a stronger F–B–N donor in 86b, enhancing the CT from the nitrogen fragment to the dibenzo[a,j]phenazine (DBPHZ) unit. Through coordination with anions, 86a-based films achieve tunable photoluminescence ranging from a blue to a deep red–NIR region.
 | ||
| Fig. 29 Molecules with red-shifted emission by changing coordination of boron. | ||
6. Conclusions and perspectives
Organoboron compounds with flexible molecular design are capable of accommodating various CT processes and can be utilized to create innovative CT molecules, in which boron's unique coordination ability forms two distinct systems, three-coordinated and four-coordinated OBCTs. Boron plays a versatile role ranging from an acceptor to a donor–acceptor regulator in these systems. Herein, this perspective has summarized the booming field of three- and four-coordinated OBCTs focusing on several critical aspects: structural design, ICT mechanisms and their impact on optoelectronic properties, as well as the present pros and cons and the future development of OBCTs. Designing desired molecules by exploiting CT mechanisms offers an efficient approach that often leads to remarkable results with less effort. In addition, integrating multiple CT mechanisms or developing novel ones can significantly boost the performance of OBCTs.Among the extensively reported tricoordinate OBCTs, D–A OBCTs with triarylboryl acceptors are the most prevalent, featuring a CT process that is highly dependent on the arrangements and ratios of donors and acceptors. To better understand and refine the structure–property relationships of D–A OBCTs, it's crucial to carry out further research on additional D–A arrangements and spatial configurations, as well as to modify triarylboryl acceptors by introducing electron-donating or electron-withdrawing groups for OBCTs. Most D–A OBCTs undergo a twist excited state process, termed TICT OBCTs. Flexible TICT OBCTs, featuring freely rotatable single bonds between donors and acceptors, often exhibit dual emissions derived from LE and TICT states. The long-wavelength TICT emission is sensitive to solvent polarity and the ratio of TICT to LE can be increased upon cooling. Designing irreversible conversion from the LE state to the TICT state has significant application potential. By incorporating weak intramolecular interactions within the TICT configuration, the TICT state can be stabilized once formed from the LE state, preventing the reverse process. This process enables a gradual shift from dual emissions to single TICT emission over time, making it ideal for “self-destruct after reading” anti-counterfeiting technologies. Conversely, the stabilized TICT states which can revert to the LE states upon external stimuli might possess potential in molecular solar thermal energy storage (MOST).180,181 In addition, TICT OBCTs can achieve white-light emission or exhibit multi-stage stimulus-responsive behavior when capable of undergoing further transformations via proton transfer and other processes. TICT driven intermolecular interactions can also lead to light-controlled supramolecular self-assembly, broadening their potential for macroscopic applications. Rigid TICT OBCTs, featuring restricted rotation around single bonds between donors and acceptors, can achieve TADF with minimal ΔEST. However, forbidden transition in these molecules typically results in a low PLQY. Introduction of LE contribution into rigid TICT OBCTs without altering the rigid twisted structure can be achieved through increasing the π-conjugation of donors or acceptors, which paves the way for high-performance OLED devices.
Incorporating π-bridges between donors and acceptors leads to the formation of D–π–A OBCTs, in which the conjugated bridge facilitates LE transition and amplifies the transition dipole moment, while the functional π-bridge such as tetraphenylethene, macrocyclic and pillar[5]arene, or quinoid conjugated motifs can endow D–π–A OBCTs with AIE, chiroptical properties, and NIR emission, respectively. Correspondingly, introducing a donor–acceptor CT process can enhance the luminescence properties of the functional π-bridge. Exploiting azobenzene, spiropyran, or triphenylethylene as photoresponsive functional π-bridges enables the further construction of highly luminescent D–π–A molecular switches and enhances photoreaction rates. Moreover, introducing additional new functional fragments into D–π–A OBCTs is also worthwhile for presenting additional properties. D–π–A OBCTs with several LE transitions could exhibit an HLCT process, where higher triplet excited states are involved in the RISC process, resulting in the effective utilization of triplet excitons. The boron atom provides an optimal HLCT acceptor candidate as it can easily increase LE contributions by combining with π fragments or heteroatom donors. However, reports on HLCT OBCTs remain scarce, highlighting an untapped area for future exploration.
The boron atom has also been used to design SR-CT molecules. MR-TADF OBCTs utilize the opposing resonance effects of boron and heteroatom donors in the central phenyl rings, to achieve TADF with narrow FWHM emission which can further be red-shifted when combined with B–π–B (LE) or LR-CT strategies. Additionally, TSCT OBCTs with parallel donor–acceptor alignments provide effective coordination, enhance transition dipole moments, and prevent π–π stacking. Meanwhile, rigid TSCT OBCTs also support efficient TADF. To overcome the limitations of existing CT mechanisms and realize innovative CT pathways, it is essential to consider detailed factors, such as resonance effects, the geometry of sp2-hybridized vacant orbitals of boron, and the spatial configuration of molecules during the molecular design. Developing new CT mechanisms for tricoordinate organoboron compounds is challenging, and thus combining multiple CT mechanisms offers an effective strategy to compensate for individual limitations. For instance, designing molecules with medium-range CT mechanisms182 can combine the advantages of LR-CT and SR-CT, while alternating donor–acceptor fragments instead of atoms to emulate MR effects could enable narrow-band emission in D–π–A OBCTs.
CT in tetracoordinate boron compounds has been less developed compared to their tricoordinate boron counterparts, which is due to the filled vacant orbitals in the boron atom, complicating the internal CT mechanism. Therefore, tetracoordinate boron OBCTs are typically exploited to be strong electron-withdrawing substituents as acceptor fragments. An in-depth comprehension of diverse mechanisms of tetracoordinate boron can boost the development of OBCTs. Four-coordinated OBCTs demonstrate unique electronic mechanisms, including resonance effects (e.g., B–N and B→N) and hyperconjugation, while boron substituents and chelating ligands can serve as donor and acceptor components to construct TADF molecules. Our group introduced the CE-CT mechanism, which leverages coordination between boron atoms and electron-deficient substituents, paired with covalent donor linkages, to enhance electron push–pull effects and amplify molecular donor–acceptor strengths. In this perspective, we introduce theoretical models of D–π–B, A–π–B, and B–π–B and explore how coordination affects CT properties based on the CE-CT mechanism. Additionally, dynamic intramolecular coordination can achieve stimuli-responsive molecular designs.
While studying the CT mechanisms of tetracoordinate boron, it is crucial to explore the role of chelation: (1) the resonance effect could switch the role of donors and acceptors, that is, the resonance effect might change a donor to an acceptor, and (2) how hyperconjugation resulting from various chelation of boron (e.g., functional groups and geometry configuration) impacts CT. The unique CE-CT induced by tetracoordinate boron allows precise assignment of donor and acceptor segments in the tetracoordinate OBCTs while the boron atom only acts as a regulatory bridge. However, detailed studies on the impact of ligands in CE-CT are essential, as most current tetracoordinate OBCTs exhibit CQ-CT processes, highlighting the imperative for suitable chelating ligand selection. The CT of pentacoordinate boron systems also deserves exploration, since it can form a three-center four-electron (3c-4e) bond.183 Additionally, the design concept of tricoordinate boron CT can be also used for constructing tetracoordinate OBCTs. For example, constructing D–π–A four-coordinated OBCTs by combining various functional fragments endows molecules with new functions. Alternatively, four-coordinated MR-TADF OBCTs molecules can be designed through CE-CT to achieve highly efficient red and NIR OLEDs. The unique tetrahedral spatial structure of four-coordinated OBCTs is highly suitable for constructing TSCT molecules by changing substituents and constructing TADF molecules with small ΔEST and high PLQYs. By combining three-coordinated and four-coordinated boron, the CT process can proceed from four-coordinated boron as a donor to three-coordinated boron as an acceptor, and the photophysical properties can be further tuned via coordination at the three-coordinated boron.
Not only does the CT mechanism of organoboron compounds and the role played by boron still require further research, but the corresponding application scenarios for OBCTs are also underexplored. Many studies do not explore the corresponding application scenarios or devices after investigating the optoelectronic properties induced by CT. If OBCTs can be used in more practical applications like MR-TADF OLEDs and biological sensing, more attention will be paid to research on OBCTs.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All authors contributed to the writing and revision of the manuscript and approved the final version of the perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (22001211), the Key Research and Development Program of Shaanxi Province (2023YBGY-454) and the Shaanxi Fundamental Science Research Project for Chemistry & Biology (Grant No. 22JHQ015) for financial support.References
- W. Rettig, Angew. Chem., Int. Ed., 1986, 25, 971–988 CrossRef.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
-
R. Misra and S.P. Bhattacharyya, in Intramolecular Charge Transfer, ed. R. Misra and S.P. Bhattacharyya, Wiley-VCH, Weinheim, 2018, ch. 2, pp. 29–69, DOI:10.1002/9783527801916.ch2.
- X. Chen, X. Zhang, X. Xiao, Z. Wang and J. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202216010 CrossRef CAS.
- S. Zeng, X. Liu, Y. S. Kafuti, H. Kim, J. Wang, X. Peng, H. Li and J. Yoon, Chem. Soc. Rev., 2023, 52, 5607–5651 RSC.
- V. G. Pivovarenko and A. S. Klymchenko, Chem. Rec., 2024, 24, e202300321 CrossRef CAS PubMed.
- Z.-L. Che, C.-C. Yan, X.-D. Wang and L.-S. Liao, Chin. J. Chem., 2022, 40, 2468–2481 CrossRef CAS.
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- A. J. T. Lou and T. J. Marks, Acc. Chem. Res., 2019, 52, 1428–1438 CrossRef CAS.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- A. Pal, M. Karmakar, S. R. Bhatta and A. Thakur, Coord. Chem. Rev., 2021, 448, 214167 CrossRef CAS.
- C. Chen, C.-Z. Du and X.-Y. Wang, Adv. Sci., 2022, 9, 2200707 CrossRef CAS.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- X. Chen, D. Tan and D.-T. Yang, J. Mater. Chem. C, 2022, 10, 13499–13532 RSC.
- H. Lv, K. Xiang and D.-T. Yang, Eur. J. Org Chem., 2022, 2022, e202201208 CrossRef CAS.
- X. Liang and Q. Zhang, Sci. China Mater., 2017, 60, 1093–1101 CrossRef.
- L. Wu, M. Holzapfel, A. Schmiedel, F. Peng, M. Moos, P. Mentzel, J. Shi, T. Neubert, R. Bertermann, M. Finze, M. A. Fox, C. Lambert and L. Ji, Nat. Commun., 2024, 15, 3005 CrossRef CAS PubMed.
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS.
- Z. Fan, Y. Liu, T. Zhang, Y. Wang and C. Dou, CCS Chem., 2024, 1–10 Search PubMed.
- L. Yuan, J. Yang, S. Qi, Y. Liu, X. Tian, T. Jia, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202314982 CrossRef CAS PubMed.
- Y. Liu, L. Yuan, J. Guo, W. Sun, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202306911 CrossRef CAS.
- S. S. Kothavale and J. Y. Lee, Adv. Opt. Mater., 2020, 8, 2000922 CrossRef CAS.
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS.
- W. Zhuang, F.-F. Hung, C.-M. Che and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202406497 CAS.
- Y. Xu, Q. Wang, X. Cai, C. Li, S. Jiang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202312451 CrossRef CAS.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- Y. Guo, C. Chen and X.-Y. Wang, Chin. J. Chem., 2023, 41, 1355–1373 CrossRef CAS.
- J. Miao, Y. Wang, J. Liu and L. Wang, Chem. Soc. Rev., 2022, 51, 153–187 RSC.
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC.
- H.-B. Cheng, X. Cao, S. Zhang, K. Zhang, Y. Cheng, J. Wang, J. Zhao, L. Zhou, X.-J. Liang and J. Yoon, Adv. Mater., 2023, 35, 2207546 CrossRef CAS.
- Y. Ma, S.-J. Lou and Z. Hou, Chem. Soc. Rev., 2021, 50, 1945–1967 RSC.
- Q. Xue and G. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- T. Zhang, Y. Xiao, H. Wang, S. Kong, R. Huang, V. Ka-Man Au, T. Yu and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301896 CrossRef CAS.
- S.-Y. Yang, Y.-K. Qu, L.-S. Liao, Z.-Q. Jiang and S.-T. Lee, Adv. Mater., 2022, 34, 2104125 CrossRef CAS.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang, D. Ma, D. Zhang and L. Duan, Nat. Commun., 2023, 14, 2394 CrossRef CAS PubMed.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656–12678 RSC.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jäkle, J. Am. Chem. Soc., 2022, 144, 18908–18917 CrossRef CAS PubMed.
- Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef CAS PubMed.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- S. Oda, B. Kawakami, M. Horiuchi, Y. Yamasaki, R. Kawasumi and T. Hatakeyama, Adv. Sci., 2023, 10, 2205070 CrossRef CAS.
- M. Mamada, A. Aoyama, R. Uchida, J. Ochi, S. Oda, Y. Kondo, M. Kondo and T. Hatakeyama, Adv. Mater., 2024, 36, 2402905 CrossRef CAS.
- X. Cai, Y. Pan, C. Li, L. Li, Y. Pu, Y. Wu and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202408522 CrossRef CAS PubMed.
- Z. Ye, H. Wu, Y. Xu, T. Hua, G. Chen, Z. Chen, X. Yin, M. Huang, K. Xu, X. Song, Z. Huang, X. Lv, J. Miao, X. Cao and C. Yang, Adv. Mater., 2024, 36, 2308314 CrossRef CAS.
- X. Luo, Q. Jin, M. Du, D. Wang, L. Duan and Y. Zhang, Adv. Sci., 2024, 11, 2307675 CrossRef CAS PubMed.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen and L. Duan, Angew. Chem., Int. Ed., 2020, 59, 17499–17503 CrossRef CAS PubMed.
- X. Chen, D. Tan, J. Dong, T. Ma, Y. Duan and D.-T. Yang, J. Phys. Chem. Lett., 2022, 13, 10085–10091 CrossRef CAS.
- J. Dong, L. Zhang, D. Tan, J. Wu, N. Wang, S. K. Mellerup, S. Wang and D.-T. Yang, Chem. Commun., 2021, 57, 9882–9885 RSC.
- M. Zhao and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 21289–21294 CrossRef CAS PubMed.
- C. Chen, J. Lu, Y. Lv, Y. Yan, Q. Sun, A. Narita, K. Müllen and X.-Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212594 CrossRef CAS.
- W. Li, C.-Z. Du, X.-Y. Chen, L. Fu, R.-R. Gao, Z.-F. Yao, J.-Y. Wang, W. Hu, J. Pei and X.-Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201464 CrossRef CAS PubMed.
- S. Zhou, Y. Liu, W. Jin, T. Qin, X. Liu, C. Zhao, Z. Liu and X. Yu, Org. Lett., 2023, 25, 1573–1577 CrossRef CAS.
- Y. Zhang, W. Li, R. Jiang, L. Zhang, Y. Li, X. Xu and X. Liu, J. Org. Chem., 2022, 87, 12986–12996 CrossRef CAS.
- Y. Yu, L. Wang, D. Lin, S. Rana, K. S. Mali, H. Ling, L. Xie, S. De Feyter and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202303335 CrossRef CAS.
- L. Chen, J. Dong, D. Tan, J. Wu and D.-T. Yang, Chin. J. Chem., 2024, 42, 3069–3074 CrossRef CAS.
- T. Ma, J. Dong and D.-T. Yang, Chem. Commun., 2023, 59, 13679–13689 RSC.
- Y. Yu, C. Wang, F.-F. Hung, C. Chen, D. Pan, C.-M. Che and J. Liu, J. Am. Chem. Soc., 2024, 146, 22600–22611 CrossRef CAS.
- T. Kushida, S. Shirai, N. Ando, T. Okamoto, H. Ishii, H. Matsui, M. Yamagishi, T. Uemura, J. Tsurumi, S. Watanabe, J. Takeya and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 14336–14339 CrossRef CAS PubMed.
- M. Ito, S. Shirai, Y. Xie, T. Kushida, N. Ando, H. Soutome, K. J. Fujimoto, T. Yanai, K. Tabata, Y. Miyata, H. Kita and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, 61, e202201965 CrossRef CAS.
- Y. H. Lee, W. Lee, T. Lee, D. Lee, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 45778–45788 CrossRef CAS.
- A. Michaelis, A. Michaelis and H. v. Soden, Liebigs Ann. Chem., 1885, 229, 295–334 CrossRef.
- P. J. Grisdale, J. L. R. Williams, M. E. Glogowski and B. E. Babb, J. Org. Chem., 1971, 36, 544–549 CrossRef CAS.
- J. C. Doty, B. Babb, P. J. Grisdale, M. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1972, 38, 229–236 CrossRef CAS.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Chem.–Eur. J., 2006, 12, 2358–2370 CrossRef CAS PubMed.
- A. Proń, M. Baumgarten and K. Müllen, Org. Lett., 2010, 12, 4236–4239 CrossRef.
- C.-C. Tsai, W.-C. Huang, H.-Y. Chih, Y.-C. Hsh, C.-W. Liao, C.-H. Lin, Y.-X. Kang, C.-H. Chang, Y. J. Chang and C.-W. Lu, Org. Electron., 2018, 63, 166–174 CrossRef CAS.
- Y. Liu, G. Xie, K. Wu, Z. Luo, T. Zhou, X. Zeng, J. Yu, S. Gong and C. Yang, J. Mater. Chem. C, 2016, 4, 4402–4407 RSC.
- Y. Liu, H. Huang, T. Zhou, K. Wu, M. Zhu, J. Yu, G. Xie and C. Yang, J. Mater. Chem. C, 2019, 7, 4778–4783 RSC.
- C. Qu, G. Xia, Y. Xu, Y. Zhu, J. Liang, H. Zhang, J. Wang, Z. Zhang and Y. Wang, J. Mater. Chem. C, 2020, 8, 3846–3854 RSC.
- D. Zhong, S. Liu, L. Yue, Z. Feng, H. Wang, P. Yang, B. Su, X. Yang, Y. Sun and G. Zhou, Chem. Sci., 2024, 15, 9112–9119 RSC.
- E. Lippert, W. LÜDer and H. Boos, in Proceedings of the IVth International Meeting on Molecular Spectroscopy, ed. A. Mangini, Pergamon, 1962, vol. 1, DOI:10.1016/B978-1-4832-1332-3.50070-6.
- K. Rotkiewicz, K. H. Grellmann and Z. R. Grabowski, Chem. Phys. Lett., 1973, 19, 315–318 CrossRef CAS.
- J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2011, 50, 8072–8076 CrossRef CAS PubMed.
- X. Liu, S. Li, J. Feng, Y. Li and G. Yang, Chem. Commun., 2014, 50, 2778–2780 RSC.
- T. Taniguchi, J. Wang, S. Irle and S. Yamaguchi, Dalton Trans., 2013, 42, 620–624 RSC.
- C. Arivazhagan, A. Maity, K. Bakthavachalam, A. Jana, S. K. Panigrahi, E. Suresh, A. Das and S. Ghosh, Chem.–Eur. J., 2017, 23, 7046–7051 CrossRef CAS PubMed.
- Y.-J. Lien, T.-C. Lin, C.-C. Yang, Y.-C. Chiang, C.-H. Chang, S.-H. Liu, Y.-T. Chen, G.-H. Lee, P.-T. Chou, C.-W. Lu and Y. Chi, ACS Appl. Mater. Interfaces, 2017, 9, 27090–27101 CrossRef CAS.
- P. Ganesan, D.-G. Chen, W.-C. Chen, P. Gnanasekaran, J.-A. Lin, C.-Y. Huang, M.-C. Chen, C.-S. Lee, P.-T. Chou and Y. Chi, J. Mater. Chem. C, 2020, 8, 4780–4788 RSC.
- D. Pecorari, A. Mazzanti, S. Gianvittorio, S. Foschi, S. Stagni, V. Fiorini and M. Mancinelli, Org. Chem. Front., 2021, 8, 4496–4507 RSC.
- D. Pecorari, E. Giuliani, A. Mazzanti, S. Stagni, V. Fiorini, G. Vigarani, F. Zinna, G. Pescitelli and M. Mancinelli, J. Org. Chem., 2023, 88, 871–881 CrossRef CAS.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- Y. Kitamoto, T. Namikawa, D. Ikemizu, Y. Miyata, T. Suzuki, H. Kita, T. Sato and S. Oi, J. Mater. Chem. C, 2015, 3, 9122–9130 RSC.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS PubMed.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- K. Matsuo and T. Yasuda, Chem. Sci., 2019, 10, 10687–10697 RSC.
- D. H. Ahn, H. Lee, S. W. Kim, D. Karthik, J. Lee, H. Jeong, J. Y. Lee and J. H. Kwon, ACS Appl. Mater. Interfaces, 2019, 11, 14909–14916 CrossRef CAS PubMed.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- T. Agou, K. Matsuo, R. Kawano, I. S. Park, T. Hosoya, H. Fukumoto, T. Kubota, Y. Mizuhata, N. Tokitoh and T. Yasuda, ACS Mater. Lett., 2019, 2, 28–34 CrossRef.
- Q. Chen, Y. Xiang, X. Yin, K. Hu, Y. Li, X. Cheng, Y. Liu, G. Xie and C. Yang, Dyes Pigm., 2021, 188, 109157 CrossRef CAS.
- J. Wang, N. Li, Q. Chen, Y. Xiang, X. Zeng, S. Gong, Y. Zou and Y. Liu, Chem. Eng. J., 2022, 450, 137805 CrossRef CAS.
- Y. H. Lee, Y.-S. Shin, T. Lee, J. Jung, J.-H. Lee and M. H. Lee, Chem. Eng. J., 2021, 423, 130224 CrossRef CAS.
- G. Xia, C. Qu, Y. Zhu, J. Ye, K. Ye, Z. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 9598–9603 CrossRef CAS.
- X. Gong, W. Yang, H. Zhang, W. Ning, S. Gong, X. Gao and C. Yang, Sci. China Mater., 2024, 67, 3537–3542 CrossRef CAS.
- Y.-W. Chen, C.-C. Tsai, H.-Y. Chih, H.-Y. Tsai, W.-Y. Wang, G.-Y. Liu, M.-Y. Wu, C.-H. Chang and C.-W. Lu, Dyes Pigm., 2022, 197, 109892 CrossRef CAS.
- Y. Shirota, M. Kinoshita, T. Noda, K. Okumoto and T. Ohara, J. Am. Chem. Soc., 2000, 122, 11021–11022 CrossRef CAS.
- C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- M. Ito, E. Ito, M. Hirai and S. Yamaguchi, J. Org. Chem., 2018, 83, 8449–8456 CrossRef CAS.
- H. Shi, D. Xin, S.-D. Bai, L. Fang, X.-E. Duan, J. Roose, H. Peng, S. Chen and B. Z. Tang, Org. Electron., 2016, 33, 78–87 CrossRef CAS.
- G. Turkoglu and T. Ozturk, Dalton Trans., 2022, 51, 2715–2725 RSC.
- J. Jin, Y. Tao, H. Jiang, R. Chen, G. Xie, Q. Xue, C. Tao, L. Jin, C. Zheng and W. Huang, Adv. Sci., 2018, 5, 1800292 CrossRef PubMed.
- O. S. Ipek, S. Topal and T. Ozturk, Dyes Pigm., 2021, 192, 109458 CrossRef CAS.
- R. Oshimizu, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, 61, e202209394 CrossRef CAS PubMed.
- P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994–7998 CrossRef CAS PubMed.
- P. Chen, X. Yin, N. Baser-Kirazli and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 10768–10772 CrossRef CAS.
- A. Ito, M. Uebe, R. Kurata, S. Yano, H. Fueno and T. Matsumoto, Chem.–Asian J., 2018, 13, 754–760 CrossRef CAS.
- J.-F. Chen, X. Yin, B. Wang, K. Zhang, G. Meng, S. Zhang, Y. Shi, N. Wang, S. Wang and P. Chen, Angew. Chem., Int. Ed., 2020, 59, 11267–11272 CrossRef CAS.
- P. Li, D. Shimoyama, N. Zhang, Y. Jia, G. Hu, C. Li, X. Yin, N. Wang, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200612 CrossRef CAS.
- Y. Jia, P. Li, K. Liu, C. Li, M. Liu, J. Di, N. Wang, X. Yin, N. Zhang and P. Chen, Chem. Sci., 2022, 13, 11672–11679 RSC.
- F. Zhao, J. Zhao, H. Liu, Y. Wang, J. Duan, C. Li, J. Di, N. Zhang, X. Zheng and P. Chen, J. Am. Chem. Soc., 2023, 145, 10092–10103 CrossRef CAS PubMed.
- R. Kurata, A. Ito, M. Gon, K. Tanaka and Y. Chujo, J. Org. Chem., 2017, 82, 5111–5121 CrossRef CAS.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos, M. Holzapfel, H. Braunschweig, C. Lambert, A. Steffen, L. Ji and T. B. Marder, Chem.–Eur. J., 2017, 23, 13164–13180 CrossRef CAS PubMed.
- Z.-B. Sun, J.-K. Liu, D.-F. Yuan, Z.-H. Zhao, X.-Z. Zhu, D.-H. Liu, Q. Peng and C.-H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 4840–4846 CrossRef CAS PubMed.
- M. Uebe, D. Sakamaki and A. Ito, ChemPlusChem, 2019, 84, 1305–1313 CrossRef CAS.
- M. Ito, M. Sakai, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2021, 60, 21853–21859 CrossRef CAS PubMed.
- T. Jairam and W. P. Hong, J. Mater. Chem. C, 2022, 10, 16173–16217 RSC.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- P. Jin, Y. Han, F. Tian, L. Wang, X. Zhao, C. Zhang and J. Xiao, Chem.–Eur. J., 2020, 26, 3113–3118 CrossRef CAS.
- Z. Lu, D. Hu, S.-W. Chen, R. Wang, L. Xing, Y. Zhu, J. Lin, Y. Huo and S. Ji, J. Mater. Chem. C, 2024, 12, 9929–9938 RSC.
- J. Lv, J. Li, S. Wang, H. Shen, L. Xia, Y. Liu, S. Xue, D. Ma, S. Ying and S. Yan, J. Mater. Chem. C, 2024, 12, 17475–17481 RSC.
- X.-Q. Gan, Z.-M. Ding, D.-H. Liu, W.-Q. Zheng, B. Ma, H. Zhang, X. Chang, L. Wang, Y. Liu, X. Wu, S.-J. Su and W. Zhu, Adv. Opt. Mater., 2023, 11, 2300195 CrossRef CAS.
- R. Wu, K. Sun, G. Shi, Y. Han, T. Gong, Y. Xu, S.-T. Zhang and B. Yang, Adv. Funct. Mater., 2024, 34, 2403501 CrossRef CAS.
- G. Li, K. Xu, J. Zheng, X. Fang, W. Lou, F. Zhan, C. Deng, Y.-F. Yang, Q. Zhang and Y. She, J. Am. Chem. Soc., 2024, 146, 1667–1680 CrossRef CAS.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 CrossRef CAS.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 CrossRef CAS.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 CrossRef CAS.
- C. Lv, X. Wang, Q. Zhang and Y. Zhang, Mater. Chem. Front., 2023, 7, 2809–2827 RSC.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- P. Keerthika and R. K. Konidena, Adv. Opt. Mater., 2023, 11, 2301732 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- M. Hayakawa, X. Tang, Y. Ueda, H. Eguchi, M. Kondo, S. Oda, X.-C. Fan, G. N. Iswara Lestanto, C. Adachi and T. Hatakeyama, J. Am. Chem. Soc., 2024, 146, 18331–18340 CrossRef CAS PubMed.
- X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475–5478 CrossRef CAS PubMed.
- D.-R. Bai, X.-Y. Liu and S. Wang, Chem.–Eur. J., 2007, 13, 5713–5723 CrossRef CAS PubMed.
- Z. M. Hudson, X.-Y. Liu and S. Wang, Org. Lett., 2011, 13, 300–303 CrossRef CAS PubMed.
- H. Pan, G.-L. Fu, Y.-H. Zhao and C.-H. Zhao, Org. Lett., 2011, 13, 4830–4833 CrossRef CAS PubMed.
- M.-Y. Zhang, Z.-Y. Li, B. Lu, Y. Wang, Y.-D. Ma and C.-H. Zhao, Org. Lett., 2018, 20, 6868–6871 CrossRef CAS PubMed.
- Y. H. Lee, S. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 24035–24042 CrossRef CAS.
- Y. H. Lee, S. Park, J. Oh, S.-J. Woo, A. Kumar, J.-J. Kim, J. Jung, S. Yoo and M. H. Lee, Adv. Opt. Mater., 2018, 6, 1800385 CrossRef.
- A. Kumar, W. Lee, T. Lee, J. Jung, S. Yoo and M. H. Lee, J. Mater. Chem. C, 2020, 8, 4253–4263 RSC.
- X.-L. Chen, J.-H. Jia, R. Yu, J.-Z. Liao, M.-X. Yang and C.-Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS.
- M. Ouyang, L. Xing, Q. Chen, H. Huang, M. Zhu, K. Hu, Y. Liu, W.-C. Chen, Y. Huo and C. Yang, J. Mater. Chem. C, 2021, 9, 1678–1684 RSC.
- C. Wu, W. Liu, K. Li, G. Cheng, J. Xiong, T. Teng, C.-M. Che and C. Yang, Angew. Chem., Int. Ed., 2021, 60, 3994–3998 CrossRef CAS.
- B. Du, X. Wang, F. Chen, Q. Yang, S. Shao, L. Wang, X. Jing and F. Wang, Chem. Commun., 2021, 57, 7144–7147 RSC.
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S.-J. Su, S. Yan and Z. Ren, Angew. Chem., Int. Ed., 2022, 61, e202210864 CrossRef CAS.
- P. Zuo, Y.-J. Yang, F.-M. Liu, J.-R. Wu, Q. Zheng, H.-T. Yuan, L.-S. Liao, D.-Y. Zhou and Z.-Q. Jiang, Adv. Opt. Mater., 2024, 12, 2400860 CrossRef CAS.
- S. Luo, J. Wang, N. Li, X.-F. Song, X. Wan, K. Li and C. Yang, Angew. Chem., Int. Ed., 2023, 62, e202310943 CrossRef CAS.
- X.-F. Luo, L. Shen, J.-Y. Wang and X. Xiao, Chem. Commun., 2024, 60, 574–577 RSC.
- A. Kumar, H. Y. Shin, T. Lee, J. Jung, B. J. Jung and M. H. Lee, Chem.–Eur. J., 2020, 26, 16793–16801 CrossRef CAS.
- H. Narita, H. Min, N. Kubo, I. Hattori, T. Yasuda and S. Yamaguchi, Angew. Chem., Int. Ed., 2024, 63, e202405412 CrossRef CAS PubMed.
- Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS.
- A. C. Murali, P. Nayak and K. Venkatasubbaiah, Dalton Trans., 2022, 51, 5751–5771 RSC.
- X. Shao, M. Liu, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205893 CrossRef CAS PubMed.
- C. Zhu, X. Ji, D. You, T. L. Chen, A. U. Mu, K. P. Barker, L. M. Klivansky, Y. Liu and L. Fang, J. Am. Chem. Soc., 2018, 140, 18173–18182 CrossRef CAS PubMed.
- Y.-J. Shiu, Y.-C. Cheng, W.-L. Tsai, C.-C. Wu, C.-T. Chao, C.-W. Lu, Y. Chi, Y.-T. Chen, S.-H. Liu and P.-T. Chou, Angew. Chem., Int. Ed., 2016, 55, 3017–3021 CrossRef CAS PubMed.
- L. Zhou, F. Ni, N. Li, K. Wang, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202203844 CrossRef CAS PubMed.
- L. Jiang, Y. Wang, D. Tan, X. Chen, T. Ma, B. Zhang and D.-T. Yang, Chem. Sci., 2022, 13, 5597–5605 RSC.
- D.-T. Yang, T. Nakamura, Z. He, X. Wang, A. Wakamiya, T. Peng and S. Wang, Org. Lett., 2018, 20, 6741–6745 CrossRef CAS.
- J. Dong, L. Chen, Q. Feng and D. Yang, Angew. Chem., Int. Ed., 2025, 64, e202417200 CrossRef CAS PubMed.
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D.-T. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS.
- K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 CrossRef CAS PubMed.
- Y. Cao, J. K. Nagle, M. O. Wolf and B. O. Patrick, J. Am. Chem. Soc., 2015, 137, 4888–4891 CrossRef CAS PubMed.
- T. Matsumoto, K. Tanaka, K. Tanaka and Y. Chujo, Dalton Trans., 2015, 44, 8697–8707 RSC.
- Y.-g. Shi, J.-w. Wang, H. Li, G.-f. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC.
- G. Ji, N. Wang, X. Yin and P. Chen, Org. Lett., 2020, 22, 5758–5762 CrossRef CAS PubMed.
- N. Aota, R. Nakagawa, L. E. de Sousa, N. Tohnai, S. Minakata, P. de Silva and Y. Takeda, Angew. Chem., Int. Ed., 2024, 63, e202405158 CrossRef CAS PubMed.
- N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909–3911 CrossRef CAS.
- Y. Cao, X. Wang, X. Shi, S. M. Clee, P. L. McGeer, M. O. Wolf and C. Orvig, Angew. Chem., Int. Ed., 2017, 56, 15603–15606 CrossRef CAS.
- H.-J. Li, S. K. Mellerup, X. Wang and S. Wang, Org. Lett., 2019, 21, 2838–2842 CrossRef CAS.
- Y. Wang, K. Liu, P. Chen, X. Yin, T. Peng, J. Iqbal and N. Wang, J. Mater. Chem. C, 2022, 10, 10981–10987 RSC.
- Q. Hou, L. Liu, S. K. Mellerup, N. Wang, T. Peng, P. Chen and S. Wang, Org. Lett., 2018, 20, 6467–6470 CrossRef CAS PubMed.
- Y. Shi, Y. Zeng, P. Kucheryavy, X. Yin, K. Zhang, G. Meng, J. Chen, Q. Zhu, N. Wang, X. Zheng, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202213615 CrossRef CAS PubMed.
- Y. Shi, C. Li, H. Ma, Z. Cao, K. Liu, X. Yin, N. Wang and P. Chen, Org. Lett., 2022, 24, 5497–5502 CrossRef CAS PubMed.
- H. Shimogawa, O. Yoshikawa, Y. Aramaki, M. Murata, A. Wakamiya and Y. Murata, Chem.–Eur. J., 2017, 23, 3784–3791 CrossRef CAS.
- Z. Wang, H. Hölzel and K. Moth-Poulsen, Chem. Soc. Rev., 2022, 51, 7313–7326 RSC.
- R. C. Richter, S. M. Biebl, R. Einholz, J. Walz, C. Maichle-Mössmer, M. Ströbele, H. F. Bettinger and I. Fleischer, Angew. Chem., Int. Ed., 2024, 63, e202405818 CrossRef CAS.
- X. He, J. Lou, B. Li, X. Dong, F. Zhong, W. Liu, X. Feng, D. Yang, D. Ma, Z. Zhao, Z. Wang and B. Z. Tang, Adv. Mater., 2024, 36, 2310417 CrossRef CAS.
- C. Dou, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 9346–9349 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |