
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiversifying fluoroalkanes: light-driven fluoroalkyl transfer via vinylboronate esters†
Kaushik
Chakrabarti‡
 a,
Chandana
Sunil‡
a,
Benjamin M.
Farris
a,
Simon
Berritt
b,
Kyle
Cassaidy
c,
Jisun
Lee
b and
Nathaniel K.
Szymczak
*a
a,
Chandana
Sunil‡
a,
Benjamin M.
Farris
a,
Simon
Berritt
b,
Kyle
Cassaidy
c,
Jisun
Lee
b and
Nathaniel K.
Szymczak
*a
aDepartment of Chemistry, University of Michigan, 930N. University, Ann Arbor, Michigan 48109, USA. E-mail: nszym@umich.edu
bMedicine Design, Pfizer Inc., Eastern Point Rd., Groton, CT 06340, USA
cChemical Research and Development, Pfizer Inc., Eastern Point Rd., Groton, CT 06340, USA
First published on 24th March 2025
Abstract
We outline a new synthetic strategy to prepare tertiary difluoromethylene-containing molecules from fluoroalkane precursors and vinyl-pinacol boronic ester (vinyl-BPin) reagents. Under irradiation, fluoroalkyl(vinyl)pinacol boronate esters [vinyl-BPin-CF2R]− undergo a conjugate radical addition process to form new C–C bonds, which does not require air-free conditions and tolerates oxygen and nitrogen-containing heterocycles as well as many classical functional groups. We demonstrate the versatility of this method through a one-pot synthetic protocol using RCF2H precursors and vinyl-BPin reagents in the presence of a Brønsted base. Widely available fluoroalkanes (HFC-23 and HFC-32) and difluoromethyl heteroarenes are used in this protocol, representing distinct strategies to generate tertiary –CF2H, –CF3 and –CF2-heteroarene molecules. Experimental and theoretical mechanistic investigations reveal a reaction sequence involving radical initiation followed by an ionic 1,2-boronate rearrangement.
Introduction
Targeted fluorination of organic compounds can often favorably modulate their physical and chemical properties,1–3 and this strategy is prominently used in the development of medicinal chemistry, agrochemistry, and material sciences.4–6 Importantly, ∼30% of marketed pharmaceuticals contain one or more fluorine atoms.4,6 While trifluoromethyl (–CF3) architectures are the most widely occurring fluoroalkyl group in marketed drugs,7–9 other fluoroalkyl units, such as difluoromethylenes, are attractive candidates for pharmacophore development because they are bioisosteres of carbonyl and ether groups (Fig. 1a).10–12 | ||
| Fig. 1 (a) Pharmaceutically relevant compounds featuring tertiary fluoroalkyl groups (b) prior examples of α-trifluoromethyl boronic ester synthesis, and (c) current approach: synthesis of α,-difluoroalkylated boronic esters. | ||
Fluorinated boronic esters (such as α-trifluoromethyl and α-difluoroalkyl boronic esters) represent an attractive entry point for the construction of more complex fluoroalkylated compounds because they can be diversified through cross-coupling and homologation chemistry.13–20 Current strategies to prepare α-trifluoromethyl boronic esters include the addition of either 2,2,2-trifluorodiazoethane or 2-trifluoromethyl oxirane derivatives to an organoboron precursor (Fig. 1b); however, these methods are limited to –CF3 units21–23 and synthetic routes to prepare α-difluoroalkylated boronic esters are not known.
1,2-Boronate rearrangements are a class of reactions used for the construction of new C–CF2R bonds and proceed with retention of the boronic ester.24–27 Although the addition of electrophiles to a boronate ester is the most common method to induce 1,2-boronate rearrangements,28–39 recent reports by Studer,40–42 Aggarwal,43,44 and Renaud45 showed an alternative approach: radical addition.40,41,43–46 These reactions enable the construction of tertiary boronic esters through the addition of a C–X bond to a vinyl BPin. However, these prior examples are limited to non-fluorinated nucleophiles, likely due to challenges in forming the key RCF2− vinyl boronate.47
Our group previously established that fluoroalkyl vinyl-BPin can serve as an entry point to α,α-difluoroalkylated olefin products,47 and can be prepared from the corresponding B3N3Me6 adducts [RCF2B3N3Me6]−, ultimately derived from deprotonation of the difluoroalkane (i.e. RCF2H). Key precedent using non-fluorinated alkyl groups has established that radical-induced conjugate addition triggers 1,2-boronate rearrangements,40–45,47 and in this manuscript, we develop this strategy to prepare tertiary carbon centers that are uniquely substituted with fluoroalkyl groups (RCF2H; R = H, F, Ph, heteroarene) and -BPin units (Fig. 1c).
We previously found that the fluoroalkyl binding affinity of borane Lewis acids can be used to describe and predict the stability of fluoroalkyl borane adducts.48–50 The free energy of CF3− binding (ΔG) to boron-based Lewis acids is a useful metric to correlate with the stability of a given Lewis acid-CF3 adduct.49 Large −ΔG values indicate irreversible binding of CF3− to the Lewis acid while adducts with small −ΔG values are prone to decomposition. To determine the fluoroalkyl affinity of BPin Lewis acids, we calculated the ΔG values using DFT at the M06-2X-D3/6-311++G(d,p)/SMD(1,2-dimethoxyethane) level of theory (Fig. 2, ESI S28–S32†). Based on the DFT calculations, vinyl-BPin species have a higher affinity for fluoroalkyl anions than B3N3Me6. Additionally, CF3− has the lowest affinity for vinyl-BPin species while CF2H− has the highest.
 | ||
| Fig. 2 Calculated ΔG (kcal mol−1) values for fluoroalkyl binding. M06-2X-D3/6-311++G(d,p)/SMD(1,2-dimethoxyethane), T = 25 °C. | ||
Results and discussion
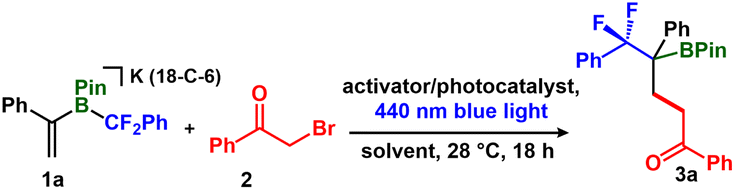
To establish the feasibility of conjugate addition, we allowed [K(18-crown-6)(vinyl-BPin-CF2Ph)] (1a) and 2-bromoacetophenone (2) to react in tetrahydrofuran-dimethyl sulfoxide (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 18 h at 28 °C under 440 nm irradiation (Kessil lamp). In the presence of 1 mol% Ru(bpy)3Cl2·6H2O as a photocatalyst, the reaction afforded the conjugate addition product 3a with 38% conversion, as assessed by 19F NMR spectroscopy (Table 1, entry 1). We found that even in the absence of Ru(bpy)3Cl2·6H2O, the product formed in 35% yield, suggesting that no photocatalyst is required (Table 1, entry 2). No reaction occurred in the absence of blue light, even when heated to 80 °C (Table 1, entry 3, ESI S11†), consistent with a photo- rather than a thermal-induced reaction. We evaluated a series of solvents and activators and found that a combination of 50 mol% NaI/15-C-5 in tetrahydrofuran-dimethoxyethane (2:1) furnished the final product with up to 70% conversion, as assessed by 19F NMR spectroscopy (Table 1, entries 4–7, ESI S11–S13†). In the absence of 15-C-5, we observed <10% NaF by 19F NMR spectroscopy (−121.3 ppm), which we attribute to Na-promoted fluoride elimination.51 To clarify the role of NaI as a reaction additive, we performed two control experiments: (i) a reaction using PhCOCH2I, which afforded 3a in a similar yield (62%) (Table 1, entry 8), and (ii) using tetrabutyl ammonium iodide ([nBu4N]I) as an alternative iodide source, which yielded 3a in 68%. (Table 1, entry 9). These results are consistent with an iodide (I−) promoted Finkelstein reaction that generates a weaker C–I (vs. C–Br) bond, thus facilitating homolysis.43,52,53
:1) for 18 h at 28 °C under 440 nm irradiation (Kessil lamp). In the presence of 1 mol% Ru(bpy)3Cl2·6H2O as a photocatalyst, the reaction afforded the conjugate addition product 3a with 38% conversion, as assessed by 19F NMR spectroscopy (Table 1, entry 1). We found that even in the absence of Ru(bpy)3Cl2·6H2O, the product formed in 35% yield, suggesting that no photocatalyst is required (Table 1, entry 2). No reaction occurred in the absence of blue light, even when heated to 80 °C (Table 1, entry 3, ESI S11†), consistent with a photo- rather than a thermal-induced reaction. We evaluated a series of solvents and activators and found that a combination of 50 mol% NaI/15-C-5 in tetrahydrofuran-dimethoxyethane (2:1) furnished the final product with up to 70% conversion, as assessed by 19F NMR spectroscopy (Table 1, entries 4–7, ESI S11–S13†). In the absence of 15-C-5, we observed <10% NaF by 19F NMR spectroscopy (−121.3 ppm), which we attribute to Na-promoted fluoride elimination.51 To clarify the role of NaI as a reaction additive, we performed two control experiments: (i) a reaction using PhCOCH2I, which afforded 3a in a similar yield (62%) (Table 1, entry 8), and (ii) using tetrabutyl ammonium iodide ([nBu4N]I) as an alternative iodide source, which yielded 3a in 68%. (Table 1, entry 9). These results are consistent with an iodide (I−) promoted Finkelstein reaction that generates a weaker C–I (vs. C–Br) bond, thus facilitating homolysis.43,52,53
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst/activator | Solvent | Yield 3a |
| a 1a (0.023 mmol), PhCOCH2Br (0.035 mmol), THF/DMSO or DME (1.0 mL), 18 h, 28 °C, 440 nm blue light. 19F NMR yields are reported (PhOCF3 used as internal standard). b Performed in the absence of blue light. c 50 mol% 15-C-5 was used. d PhCOCH2I used instead of PhCOCH2Br. | |||
| 1 | Ru(bpy)3Cl2·6H2O (1 mol%) | THF:DMSO (2:1) |
38% |
| 2 | — | THF:DMSO (2:1) |
35% |
| 3 | Ru(bpy)3Cl2·6H2O (1 mol%) | THF:DMSO (2:1) |
0%b |
| 4 | NaI (50 mol%) | THF:DMSO (2:1) |
53% |
| 5 | NaI (150 mol%) | THF:DMSO (2:1) |
36% |
| 6 | NaI (50 mol%) | THF:DME (2:1) |
63% |
| 7 | NaI (50 mol%) | THF:DME (2:1) |
70%c |
| 8 | — | THF:DME (2:1) |
62%d |
| 9 | [nBu4N]I (50 mol%) | THF:DME (2:1) |
68% |
After achieving 70% conversion of 3a, we assessed the reaction scope by varying the vinyl-BPin derivatives (Fig. 3, entries 3a–3d). Aliphatic vinyl-BPin reagents responded moderately, with 28–65% isolated yields (Fig. 3, entries 3b–3d). We explored the scope of radical coupling partners by investigating p-substituted 2-bromoacetophenone derivatives (Fig. 3) and found both electron-rich and deficient substrates afforded similar yields (3e (53%), 3f (62%) and 3g (60%)). Pyridine and benzofuran units are common structural motifs in pharmaceutical and medicinal chemistry,54–58 and substrates with these motifs afforded 3i (59%) and 3j (55%) in good isolated yields. We examined the viability of a series of radical precursors with this methodology (Fig. 3). 2-Iodoacetonitrile afforded the difluoromethyl-containing conjugate addition product 3k in 58% yield. Alternatively, 3k was formed from acetonitrile in 48% yield when 2 equivalents of redox-active ester (1,3-dioxoisoindolin-2-yl adamantane-1-carboxylate) was used as a radical initiator. Ethyl haloacetates furnished 3l in 51% and 28% yield for X = I and X = Br, respectively. 2-Iodo-N,N-dimethylacetamide afforded 3m in 75% yield. Finally, 2-bromo-1-cyclopropylethan-1-one furnished 3n in 42% yield. These results demonstrate compatibility of the method with radical precursors across a wide range of electronic environments.
 | ||
| Fig. 3 Scope of vinyl-BPin, 2-bromo acetophenone derivatives and other radical precursors. Fluoroalkyl vinyl-BPin (0.1 mmol), R2COCH2Br (0.15 mmol), NaI (0.05 mmol), 15-C-5 (0.05 mmol), THF/DME (2.4 mL), 18 h, 28 °C, 440 nm light [Me6B3N3CF2R]K(18-C-6) (0.15 mmol), vinyl-BPin (0.15 mmol), ArCOCH2Br (0.23 mmol), NaI (0.075 mmol), 15-C-5 (0.075 mmol), THF/DME (3.0 mL), 18 h, 28 °C, 440 nm light. 19F NMR yields (PhOCF3 used as internal standard). Isolated yields are in parentheses. a19F NMR yield only (unstable to column chromatography). b Iodide radical precursor was used instead of bromide (No NaI or 15-C-5 added). c2.0 equiv. redox-active ester (1,3-dioxoisoindolin-2-yl adamantane-1-carboxylate) was used. | ||
To improve the broad accessibility of the protocol, we evaluated the requirement of B3N3Me6 by examining the direct deprotonation of ArCF2H by 1.5 equiv. KN(iPr)2 in the presence of vinyl-BPin (condition III).47 We found that deprotonation and subsequent conjugate radical addition were achievable in a one-pot sequence where both the fluoroalkyl and 2-bromoacetophenone were modified (Fig. 4). 2-Bromoacetophenones with p-H and p-CH3 afforded moderate isolated yields (3a (62%) and 3e (50%)) comparable to the borazine protocol (cf.Fig. 3), and 1-(difluoromethyl)-4-fluorobenzene furnished 4a with 23% yield.
 | ||
| Fig. 4 Scope of one-pot reactions (top) and scope in fluoroalkanes (bottom). Vinyl-BPin (0.15 mmol), fluoroalkane (RCF2H) (0.15 mmol), R2–Br (0.23 mmol), NaI (0.075 mmol), 15-C-5 (0.075 mmol), THF/DME (3.0 mL), 18 h, 28 °C, 440 nm light. 19F NMR yields (PhOCF3 used as internal standard). Isolated yields in parentheses. a19F NMR yield. bUnstable to column chromatography. c No NaI or 15-C-5 was added as iodide radical precursor was used instead of bromide. | ||
We also investigated the scope of the fluoroalkyl units derived from hydrofluorocarbons (HFCs) and difluoromethyl heteroarenes. Our group previously established a strategy to repurpose refrigerants or HFCs as chemical synthons for –CF3 and –CF2H sources.48,49,59 We evaluated the feasibility of a one-pot difluoromethylation and trifluoromethylation reaction using [K(18-C-6)(B3N3Me6-CF2H)] and [K(18-C-6)(B3N3Me6-CF3)] with a vinyl-BPin derivative (Fig. 4) and obtained 4b and 4c with 54% and 12% conversion, respectively. We found that 3-(difluoromethyl) pyridine and N-benzyl-2-difluoromethyl-benzimidazole both were also viable fluoroalkyl precursors and afforded 4d (51%) and 4e (46%) when using iodoacetonitrile as the radical source (see details in ESI†).
To elucidate the operative mechanistic pathway, we performed a series of experiments. Without irradiation, no product formed, which suggests that photochemical activation is necessary (Table 1, entry 3). To examine whether radical propagation is operative in this system, we found that, although 42% conversion of 3a occurred within 1.5 h of constant irradiation, no additional product formed after stirring in the absence of light for another 16.5 h (Fig. 5a).60 Additionally, during quantum yield measurements, we found that Φ = 0.21 for the formation of 3k (details in ESI†). Unfortunately, neither of these results (no reactivity post-irradiation and Φ < 1) can confirm or refute a radical propagation mechanism.60 To investigate the presence of radical intermediates, we introduced the radical quencher, TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl), which completely shut down the reaction (i.e. 0% conversion of product 3a), with the formation of the TEMPO-CF2Ph adduct (5b-P; Fig. 5b). These results implicate the intermediacy of fluoroalkyl radicals that may undergo intra- or intermolecular coupling. To examine the feasibility of the latter pathway, we performed a crossover experiment using vinylboronate esters containing a CF2Ph group (1b) and a CF2H group (1c). When subjected to a conjugate addition reaction in the presence of PhCOCH2Br (Fig. 5c), we only observed 3b and 4b, with no crossover products (3b′ and 4b′). These results suggest that intermolecular fluoroalkyl transfer does not occur, and the intramolecular pathway was further investigated by DFT calculations.
 | ||
| Fig. 5 (a) Light/dark reactivity. (b) TEMPO as a radical trap. (c) Crossover experiment. | ||
DFT analysis of the reaction mechanism was performed using M06-2X-D3/6-311++G(d,p) + LANL2DZ(I)/SMD(1,2-dimethoxyethane) and modeled using iodoacetonitrile. Initiation of the reaction occurs via homolysis of the C–I bond in iodoacetonitrile (ΔG = 42.3 kcal mol−1) (Fig. 6 and ESI S28–S30†). Addition of the ˙CH2CN radical to the fluoroalkylated vinyl-Bpin (ΔG‡ = 10.8 kcal mol−1) generates an anionic radical species that can undergo single electron transfer (SET) to I˙ (termination) or iodoacetonitrile (propagation). Importantly, the termination step is thermodynamically favorable (ΔG = −34.8 kcal mol−1, E = 1.51 V) while the propagation step is not (ΔG = 7.5 kcal mol−1, E = −0.33 V) (ESI S28 and S29†), suggesting that radical propagation is not a significant contributor in the reaction mechanism. After SET, the boronate ester undergoes ionic rearrangement (ΔG‡ = 6.4 kcal mol−1) to afford the final product, and these calculated results are consistent with mechanistic proposals on related non-fluoroalkylated vinyl boronate esters.45 Alternate mechanisms were also investigated using DFT analysis (Fig. 6 (left), also see full computational details in ESI†). A non-radical mechanism can proceed via a 1,2-boronate rearrangement as the initial step to produce a carbanion intermediate; however, the barrier (ΔG‡ > 23.5 kcal mol−1) is significantly higher than that when radical addition is the first step. A third pathway beginning with single-electron transfer leads to radical release (trifluoromethyl or 2-propenyl); however, this pathway is inconsistent with the absence of radical crossover products.
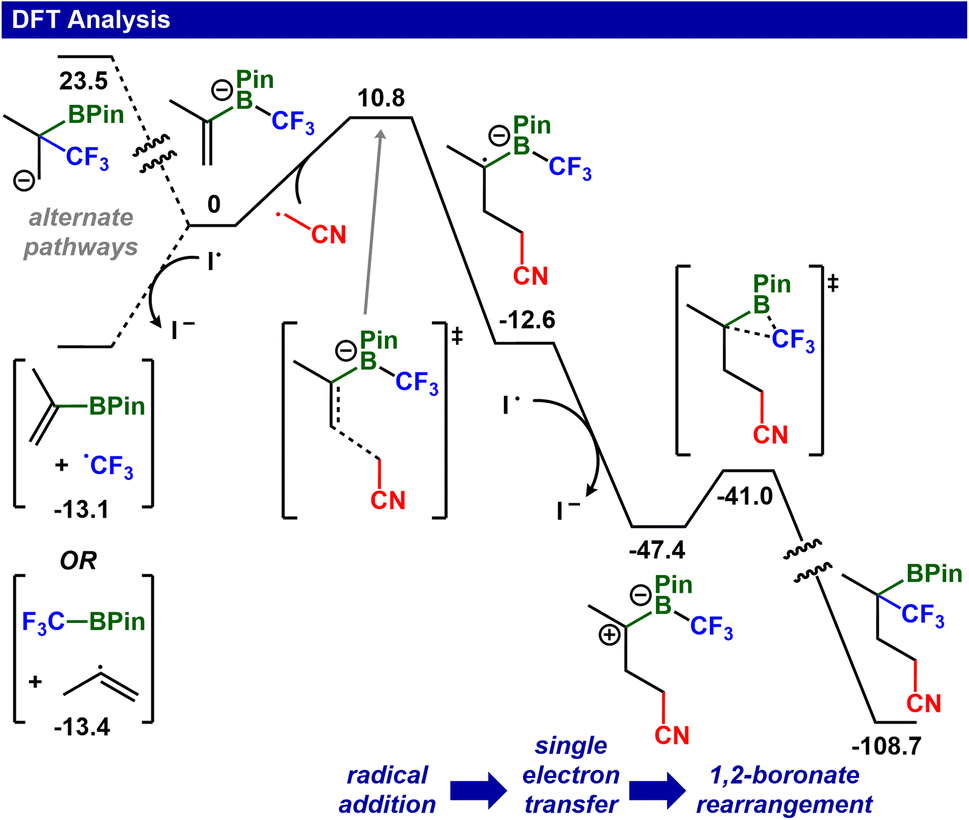 | ||
| Fig. 6 Calculated Gibbs free energies (in kcal mol−1) of the proposed mechanism. M06-2X-D3/6-311++G(d,p) + LANL2DZ(I)/SMD(1,2-dimethoxyethane), T = 25 °C. The analysis starts with ˙CH2CN, which is formed from homolysis of ICH2CN with light. | ||
The developed reaction sequence provides a unique protocol for accessing α-difluoromethylene-containing molecules featuring both BPin and carbonyl units. To evaluate the tolerance of the developed method to common functional groups, we assessed how the yield of a representative reaction (forming 3a) responds to a variety of exogenous additives.61 We found that the reaction was highly robust, with minimal changes to the yield in the presence of aldehydes, ketones, amides, acyl chlorides, amines, alcohols, water, and air (Table 2). These results implicate a high probability of reaction compatibility with many functional groups used in pharmaceutical development.
|
|
|
|---|---|
| Additive | Yield (%) |
| a 1a (0.015 mmol), PhCOCH2Br (0.023 mmol), additive (0.023 mmol) THF/DME (1.2 mL), 18 h, 28 °C, 440 nm blue light. 19F NMR yields reported (PhOCF3 used as internal standard). | |
| — | 70 |
| PhCHO | 68 |
| PhCOPh | 69 |
| PhCOCl | 68 |
| PhCONMe2 | 74 |
| PhCO2Et | 67 |
| PhOH | 58 |
| PhCH2NH2 | 56 |
| Et3N | 77 |
| i-Pr2NEt | 78 |
| H2O | 73 |
| PhCH2I | 70 |
| Undistilled DME | 68 |

To examine compatibility with more complex substrates, we evaluated the viability of the methodology with an estrone derivative 7a (47%), which furnished 7b in 50% isolated yield. Importantly, this substrate highlights high selectivity and reaction compatibility, properties that are needed when pursuing late-stage fluorination of biologically-relevant steroid cores (Fig. 7 and ESI S19†). Finally, we found that the Bpin unit in 3a can be induced to undergo an elimination reaction when treated with KHF2, forming monofluoroalkene 7c in 21% yield (Fig. 7). We propose the monofluoroalkene forms by defluorination of an intermediate –BF3K intermediate (BF4− noted in the 19F NMR spectrum). Importantly, such monofluoroalkenes are a sought-after class of molecules that are bioisosteres of amides, exhibiting enhanced stability and bioactivity,10,62 highlighting another potential application of the developed methodology.
 | ||
| Fig. 7 Reactivity of estrone derivative (top) and derivatization reaction to form a monofluoroalkene (bottom). | ||
Conclusion
In conclusion, we developed a metal-free strategy to generate quaternary carbon centers containing both a fluoroalkyl and a BPin unit by coupling a conjugate radical addition to vinyl-pinacol boronate esters with 1,2-boronate rearrangements. This synthetic strategy provides a unique application of directly using fluoroalkyl nucleophiles derived from fluoroalkanes for the synthesis of complex fluorinated molecules. We demonstrated the versatility of this method through one-pot syntheses of tertiary difluoromethylene-containing molecules directly from RCF2H precursors with vinyl-BPin reagents in the presence of a Brønsted base. This reaction sequence can enable the use of widely available CH2F2 (HFC-32) and HCF3 (HFC-23) as chemical synthons, which are currently underutilized precursors. The method is versatile and compatible with undistilled solvents, competitive functional groups, and biologically active scaffolds, providing access to unique fluoroalkylated compounds that may be subjected to additional derivatization reactions to generate medicinally relevant fluoroalkylated compounds.Data availability
All relevant experimental data and characterization details are provided in the ESI.†Author contributions
The manuscript was written through the contributions of all authors. The project was designed by K. Chakrabarti and N. K. Szymczak with input during execution from all authors. All optimizations, syntheses and characterizations were performed by K. Chakrabarti and C. Sunil. The theoretical calculations and actinometry were performed by B. M. Farris. S. Berritt, K. Cassaidy and J. Lee provided insights related to the project development.Conflicts of interest
NKS holds a patent relating to difluoroalkyl transfer reagents.Acknowledgements
This work was supported by the NSF (CHE 1955284) and by the Advanced Research Computing at the University of Michigan for computational resources. We thank Dr Russell Bornschein for Mass Spectrometry assistance.Notes and references
- Y. Liu, L. Jiang, H. Wang, H. Wang, W. Jiao, G. Chen, P. Zhang, D. Hui and X. Jian, Nanotechnol. Rev., 2019, 8, 573–586 CAS.
- W. D. G. Brittain, C. M. Lloyd and S. L. Cobb, J. Fluorine Chem., 2020, 239, 109630 CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 Search PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 CAS.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CAS.
- F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS.
- J. Zhang, L. Li, Q. Lv, L. Yan, Y. Wang and Y. Jiang, Front. Microbiol., 2019, 10, 1–17 CrossRef PubMed.
- K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817–829 Search PubMed.
- H. C. Brown, G.-M. Chen, M. P. Jennings and P. V. Ramachandran, Angew. Chem., Int. Ed., 1999, 38, 2052–2054 CrossRef CAS PubMed.
- P. V. Ramachandran and M. P. Jennings, Org. Lett., 2001, 3, 3789–3790 CrossRef CAS PubMed.
- T. Braun, M. Ahijado Salomon, K. Altenhöner, M. Teltewskoi and S. Hinze, Angew. Chem., Int. Ed., 2009, 48, 1818–1822 CrossRef CAS PubMed.
- V. V. Levin, P. K. Elkin, M. I. Struchkova and A. D. Dilman, J. Fluorine Chem., 2013, 154, 43–46 CrossRef CAS.
- X. Huang, M. Garcia-Borràs, K. Miao, S. B. J. Kan, A. Zutshi, K. N. Houk and F. H. Arnold, ACS Cent. Sci., 2019, 5, 270–276 CrossRef CAS PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- W.-X. Lv, Q. Li, J.-L. Li, Z. Li, E. Lin, D.-H. Tan, Y.-H. Cai, W.-X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544–16548 CrossRef CAS PubMed.
- O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed.
- G. A. Molander and D. Ryu, Angew. Chem., Int. Ed., 2014, 53, 14181–14185 CrossRef CAS PubMed.
- M. Nandakumar, B. Rubial, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 1187–1191 CrossRef CAS PubMed.
- M. E. D. Hillman, J. Am. Chem. Soc., 1962, 84, 4715–4720 CrossRef CAS.
- D. S. Matteson and R. W. H. Mah, J. Am. Chem. Soc., 1963, 85, 2599–2603 CrossRef CAS.
- R. Blieck and A. de la Torre, Eur. J. Org Chem., 2022, 2022, e202200920 CrossRef CAS.
- S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24–39 CrossRef CAS PubMed.
- G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS.
- D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS.
- R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 49, 3323–3336 CrossRef CAS.
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed.
- G. J. Lovinger, M. D. Aparece and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 3153–3160 CrossRef CAS PubMed.
- E. K. Edelstein, S. Namirembe and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 5027–5030 CrossRef CAS PubMed.
- J. A. Myhill, L. Zhang, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2018, 57, 12799–12803 CrossRef CAS PubMed.
- S. Namirembe and J. P. Morken, Chem. Soc. Rev., 2019, 48, 3464–3474 RSC.
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- D. S. Matteson and K. M. Sadhu, J. Am. Chem. Soc., 1983, 105, 2077–2078 CrossRef CAS.
- D. S. Matteson and R. Ray, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS.
- K. M. Sadhu and D. S. Matteson, Organometallics, 1985, 4, 1687–1689 CrossRef CAS.
- F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503–8518 RSC.
- D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2019, 141, 14126–14130 Search PubMed.
- M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Science, 2017, 355, 936–938 Search PubMed.
- M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736–5739 CrossRef CAS PubMed.
- M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS PubMed.
- N. D. C. Tappin, M. Gnägi-Lux and P. Renaud, Chem.–Eur. J., 2018, 24, 11498–11502 CrossRef CAS PubMed.
- M. Kischkewitz, F. W. Friese and A. Studer, Adv. Synth. Catal., 2020, 362, 2077–2087 Search PubMed.
- K. Chakrabarti, M. M. Wade Wolfe, S. Guo, J. W. Tucker, J. Lee and N. K. Szymczak, Chem. Sci., 2024, 15, 1752–1757 CAS.
- J. B. Geri, E. Y. Aguilera and N. K. Szymczak, Chem. Commun., 2019, 55, 5119–5122 CAS.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 9811–9814 CAS.
- J. B. Geri, M. M. Wade Wolfe and N. K. Szymczak, J. Am. Chem. Soc., 2018, 140, 9404–9408 CAS.
- C. B. Chan, N. H. Tioh and G. T. Hefter, Polyhedron, 1984, 3, 845–851 CAS.
- G. Evano, A. Nitelet, P. Thilmany and D. F. Dewez, Front. Chem., 2018, 6, 1–18 Search PubMed.
- Y. Takahashi and M. Seki, Org. Process Res. Dev., 2021, 25, 1974–1978 CAS.
- S. De, A. Kumar S K, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, RSC Adv., 2022, 12, 15385–15406 Search PubMed.
- E. Khan, ChemistrySelect, 2021, 6, 3041–3064 Search PubMed.
- G. Khodarahmi, P. Asadi, F. Hassanzadeh and E. Khodarahmi, J. Res. Med. Sci., 2015, 20, 1094–1104 CAS.
- Y.-h. Miao, Y.-h. Hu, J. Yang, T. Liu, J. Sun and X.-j. Wang, RSC Adv., 2019, 9, 27510–27540 Search PubMed.
- H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483–504 CAS.
- J. B. Geri, M. M. Wade Wolfe and N. K. Szymczak, Angew. Chem., Int. Ed., 2018, 57, 1381–1385 CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CAS.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: https://doi.org/10.1039/d5sc01776a |
| ‡ These authors are contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |