
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization-induced self-assembly enables access to diverse highly ordered structures through kinetic and thermodynamic pathways†
Ibuki
Shibata
a,
Ayae
Sugawara-Narutaki
 ab and
Rintaro
Takahashi
*c
ab and
Rintaro
Takahashi
*c
aDepartment of Energy Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan
bLaboratory for Biomaterials and Bioengineering, Institute of Integrated Research, Institute of Science Tokyo, 2-3-10, Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan
cDepartment of Macromolecular Science, Graduate School of Science, The University of Osaka, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: takahashi@chem.sci.osaka-u.ac.jp
First published on 31st March 2025
Abstract
Polymerization-induced self-assembly (PISA) has emerged as a powerful technique for generating microphase-separated structures, but research has primarily focused on systems exhibiting “disordered” structures. Here, we demonstrate the facile construction of various highly ordered microphase-separated structures via PISA, with and without kinetic control through manipulation of the glass transition temperature (Tg) of the core-forming blocks. We synthesized diblock copolymers in an ionic liquid (40 wt% solute) by polymerizing styrene or 2-hydroxyethyl acrylate from one end of poly(ethylene glycol). When using polystyrene as the core-forming block, its high Tg relative to the polymerization temperature resulted in the formation of kinetically trapped structures, including pure hexagonal close-packed (HCP) spheres exhibiting X-ray diffraction peaks up to the 17th-order. Conversely, lower-Tg core-forming block [poly(2-hydroxyethyl acrylate)] led to thermodynamically stable, highly ordered structures, including a double-gyroid morphology. These results highlight the efficacy of PISA for generating diverse, highly ordered microphase-separated structures from simple diblock copolymers and demonstrate its potential to access structures unattainable through conventional ex situ polymerization.
Introduction
Self-assembly and microphase separation are fundamental processes with significant implications across diverse fields. These intricate processes often yield kinetically controlled structures, highlighting their importance in producing unique and functional architectures.1–3 In this context, polymerization-induced self-assembly (PISA) has garnered considerable interest due to its ability to efficiently prepare micelles and vesicles of controlled structure and size. During PISA, polymerization and self-assembly proceed concurrently; the kinetics of both processes interactively influence the resulting structures.3–13When in situ polymerization of copolymers is conducted in concentrated solution or bulk, microphase separation occurs. This phenomenon, pioneered by the group of Hillmyer,14–16 is termed polymerization-induced microphase separation (PIMS), a subset of PISA. PIMS has garnered increasing attention as a facile route to microphase-separated (nanoporous) structures and for its potential applications in electroconductive materials, gas adsorbents, and membranes.17–27 Hereinafter, we will use ‘PISA’ to include ‘PIMS’.
However, most PISA studies have focused on disordered microphase separation, characterized by broad diffraction peaks in small-angle X-ray scattering (SAXS) profiles. In contrast, microphase separation prepared through conventional ex situ polymerization or solution casting methods is well-established to exhibit various long-range ordered structures, such as lamellae (LAM), hexagonally packed cylinders (HEX), face-centered cubic (FCC), and body-centered cubic (BCC).18,28,29 As a notable exception, the group of Armes30 reported long-range ordered microphase-separation with sharp SAXS diffraction peaks via PISA synthesis of hydrogenated polybutadiene and benzyl methacrylate (BzMA) monomer in n-dodecane. Their work focused on a BCC-to-HEX transition process, rather than a broader exploration of ordered structures.
Despite these advancements, creating highly ordered microphase-separated structures through PISA remains challenging, which is crucial for their use as electroconductive and gaseous adsorptive materials. In particular, not all structures observed in ex situ-prepared diblock copolymers have been replicated using PISA; for example, double-gyroid (GYR) structures have not yet been reported via PISA. Moreover, PISA processes in dilute or semidilute solutions can become kinetically trapped, leading to unusual behaviors of micelles/vesicles.3,31–36 Similarly, PISA syntheses in concentrated solution or bulk conducted kinetically controlled conditions are expected to yield microphase-separated structures that are inaccessible through ex situ polymerization.
In this study, we have comprehensively explored PISA synthesis in concentrated solutions with various degrees of polymerization for the core-forming blocks to prepare well-defined, ordered microphase-separated structures. We examined two types of core-forming blocks with glass transition temperatures (Tg) both above and below the polymerization temperature. Specifically, styrene or 2-hydroxyethyl acrylate (HEA) was polymerized from one end of poly(ethylene glycol) [PEG; macromolecular chain-transfer agent (macro-CTA)] via reversible addition–fragmentation chain-transfer (RAFT) polymerization at 60 °C in an ionic liquid, 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]) (Scheme 1). While styrene and HEA are soluble in [BMIM][PF6], their corresponding polymers are insoluble, causing the resulting block copolymers (PEG45–PSn or PEG45–PHEAn) to self-assemble and undergo microphase separation as polymerization progresses. Polystyrene (PS) is known to have Tg of approximately 100 °C,37 while Tg of poly(2-hydroxyethyl acrylate) (PHEA) is around 15 °C.38,39 Hence, the self-assembled PS blocks become glassy, whereas the PHEA blocks remain rubbery. The glassy core domains may become kinetically trapped, potentially preventing structural transformation. Consequently, we demonstrate that PISA synthesis can generate a range of highly ordered microphase-separated structures, influenced by the Tg of the core-forming block. Furthermore, we show that kinetically controlled PISA can lead to unusual structures not observed in conventional microphase separation of diblock copolymers.
 | ||
| Scheme 1 PISA synthesis used in this study: RAFT polymerization to synthesize PEG45–PSn (high Tg core-forming block) and PEG45–PHEAn (low Tg core-forming block), where the subscripts indicate the target number-average degree of polymerization. V-65 (2,2′-azobis(2,4-dimethylvaleronitrile)) is an azo-type initiator with a 10-hours half-life temperature of 51 °C. | ||
Results and discussion
PISA synthesis
The macro-CTA was synthesized via Steglich esterification of hydroxy-terminated PEG with a carboxylic acid-functionalized CTA (Scheme S1†).26,27,30,34 Complete functionalization of PEG by CTA was confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy and UV spectroscopy (Fig. S1 and S2†). The RAFT polymerization (Scheme 1) exhibited good control, as indicated by the monomer conversions exceeding 92% and the dispersity (Ð) below 1.22 for all samples prepared in this study (Fig. S3, S4, 1a, b, and Table S1†). Ð is defined as Mw/Mn, where Mw and Mn denote the weight- and number-average molecular weights.The Tg values of the PS and PHEA blocks, in the presence of [BMIM][PF6], were investigated by differential scanning calorimetry (DSC) (Fig. 1c and d). The results confirm Tg of approximately 102 °C for PS (the exothermic shift after the glass transition may be caused by the relaxation of internal stress). In contrast, Tg of PEG45–PHEA136 was −13 °C, and no distinct Tg signal was observed for lower n samples of PEG45–PHEAn, despite the reported Tg value of bulk PHEA being around 15 °C.38,39 Additionally, PEG45–PHEA136 was prepared in ethyl acetate (without [BMIM][PF6]), followed by evaporation of the ethyl acetate and DSC analysis. This samples exhibited slightly higher Tg (−10 °C). These data indicate that PS domains are almost solvent-free and exist in a glassy state at the polymerization temperature, whereas PHEA domains likely contain solvent molecules and are in a rubbery state.
 | ||
| Fig. 1 SEC traces (a and b) and DSC thermograms (c and d) of PEG45–PSn (a and c) and PEG–PHEAn (b and d). DSC was performed with the non-volatile solvent [BMIM][PF6] present, at the polymer concentration of 40 wt%, except for the dotted curve in panel (d). Arrows in panels (c) and (d) indicate Tg. In panels (a) and (b), “molecular weight” represents the relative molecular weight obtained with poly(methyl methacrylate) standards. | ||
Ordered spheres with and without kinetic control
Regarding the microphase-separated structures, it is remarkable that the SAXS profiles for these samples exhibited sharp, high-order diffraction peaks (Fig. 2). PEG45–PS23 displayed diffraction peaks whose positions align with a BCC lattice (the ratio to the first-order peak was 1, √2, √3, √4,…). This indicates that the PS block formed spherical core domains arranged on a BCC lattice. The domain spacing (center-to-center distance) between the nearest-neighbor spheres was , where q* stands for the q value of the primary peak. BCC structures are commonly observed in the microphase separation of diblock copolymer solutions prepared by ex situ polymerization when the core-forming blocks are short.
, where q* stands for the q value of the primary peak. BCC structures are commonly observed in the microphase separation of diblock copolymer solutions prepared by ex situ polymerization when the core-forming blocks are short.
 | ||
| Fig. 2 (a) SAXS profiles for PEG45–PSn in [BMIM][PF6] with varied degrees of polymerization of PS at 25 °C, prepared by PISA at the solute concentration of 40 wt%. Short vertical lines for n = 75 indicate the allowed reflections of GYR. (b) Close-up of the SAXS profile of PEG45–PS45 and schematic illustration of HCP structure. Allowed reflections for HCP (n = 45) correspond to the following ratio of the peak positions: √32, √36, √41, √68, √96, √113, √128, √132, √137, √144, √164, √176, √209, √224, √233, √240, and √260. (c) Two-dimensional SAXS pattern of PEG45–PS45. | ||
Intriguingly, more than 10 peaks were observed in PEG45–PS45, all of which matched the allowed reflections from a hexagonal close-packed (HCP) structure with a nearest-neighbor domain spacing of  (Fig. 2a–c). While the (013), (020), and (014) reflections were not detected, likely due to the form factor of spherical PS domains, a 17th-order peak (122) was observed, despite the use of non-microbeam SAXS (with an X-ray spot size of 0.2 mm in diameter). It is worth noting that no peaks attributable to other structures were observed. The presence of an FCC structure, for example, would have resulted in a shoulder peak around q = 0.45 nm−1;40 however, no such feature is evident in the SAXS profile. These results confirm the formation of a highly ordered, single-phase HCP structure. This scattering profile with the high-order peaks does not result from an exceptionally high X-ray contrast (difference in electron density between polystyrene and the ionic liquid; Table S2†).
(Fig. 2a–c). While the (013), (020), and (014) reflections were not detected, likely due to the form factor of spherical PS domains, a 17th-order peak (122) was observed, despite the use of non-microbeam SAXS (with an X-ray spot size of 0.2 mm in diameter). It is worth noting that no peaks attributable to other structures were observed. The presence of an FCC structure, for example, would have resulted in a shoulder peak around q = 0.45 nm−1;40 however, no such feature is evident in the SAXS profile. These results confirm the formation of a highly ordered, single-phase HCP structure. This scattering profile with the high-order peaks does not result from an exceptionally high X-ray contrast (difference in electron density between polystyrene and the ionic liquid; Table S2†).
Nanoparticles typically order into either FCC or BCC structures, depending on the interparticle interaction potential, but rarely into HCP structures.41–43 Hard spherical colloids and spherical micelles formed from block copolymers with short corona chains tend to order into FCC structure, while block copolymers with sufficiently long corona chains favor BCC structure due to long-range repulsive interactions. The resulting structure is determined by the interfacial area between the core and corona regions, as well as the uniformity of chain stretching.44,45 Due to the packing frustration (i.e., the difficulty in achieving uniform chain stretching), spherical interfaces formed from low-dispersity linear block copolymers (prepared by ex situ polymerization) typically adopt a BCC structure.44,45 Close-packed structures can be stabilized by using star block copolymers27,46–49 or by blending a homopolymer with a diblock copolymer.50,51 As far as the authors know, pure HCP structures have not been reported in linear diblock copolymers, with the exception of specific polymers synthesized via ex situ polymerization by Whittaker, Hawker, Bates, and coworkers.40 Their polymers were ABA triblock copolymers with A = poly(2,2,2-trifluoroethyl acrylate) and B = poly(2-dodecyl acrylate) or poly(4-dodecyl acrylate). The stability of HCP structure in these systems was dependent on the side alkyl length of B, suggesting a specific effect arising from the local structure. In the context of PISA, Armes and coworkers30 reported the observation of HCP structures coexisting with BCC or HEX structures during the polymerization of BzMA from hydrogenated polybutadiene in concentrated n-dodecane solution at 90 °C. Although Tg of PBzMA block (ca. 56 °C52) is lower than the polymerization temperature (90 °C), the HCP structure appeared to be in kinetically trapped states and did not transform to HEX. We infer that the HCP formation is favored in PISA unless Tg of the core-forming block is significantly lower than the polymerization temperature. It is emphasized that in contrast to previous studies,30,40 our current work demonstrates the formation of a pure HCP structure without any coexisting structures in the conventional diblock copolymer, PEG45–PS45.
To investigate the kinetic influence of the PISA process on HCP formation, we prepared a control sample using a solution casting method: PEG45–PS45 (including [BMIM][PF6]) was dissolved in ethyl acetate, a solvent in which PEG, PS, and [BMIM][PF6] are all soluble. The ethyl acetate was then evaporated under vacuum at 60 °C (given the non-volatility of [BMIM][PF6]). This solution-cast sample exhibited disordered spheres (DIS; Fig. S5†), suggesting that the HCP structure is a consequence of the kinetic pathway inherent to the PISA process and cannot be obtained by ex situ polymerization.
Furthermore, no HCP structure was observed for PEG45–PHEAn series. PEG45–PHEA29 formed DIS, as evidenced by the halo in its SAXS profile, with a domain spacing of 2π/q* = 12.2 nm (Fig. 3a). This disordered structure is attributed to the weaker segregation of PEG45–PHEAn compared to PEG45–PSn. The SAXS profiles of PEG45–PHEA34 and PEG45–PHEA45 exhibited diffraction peaks at positions corresponding to HEX structures (with ratios of 1, √3, √4, √7, √9, and √11). The domain spacing of the nearest-neighbor cylinders  was 15.4 nm for PEG45–PHEA34 and 16.8 nm for PEG45–PHEA45. The 16.8 nm spacing observed for PEG45–PHEA45 (HEX) is smaller than that the 19.0 nm spacing for PEG45–PS45 (HCP). The dimensional relationship is consistent with previous observations, suggesting that the kinetically trapped HCP structure cannot readily transform into HEX.30,53 These results indicate that the formation of the pure HCP structure in PISA of PEG45–PS45 can be attributed to the higher Tg of the PS block, which more effectively hinders the rearrangement of spherical core domains and prevents morphological transformation to cylinders.
was 15.4 nm for PEG45–PHEA34 and 16.8 nm for PEG45–PHEA45. The 16.8 nm spacing observed for PEG45–PHEA45 (HEX) is smaller than that the 19.0 nm spacing for PEG45–PS45 (HCP). The dimensional relationship is consistent with previous observations, suggesting that the kinetically trapped HCP structure cannot readily transform into HEX.30,53 These results indicate that the formation of the pure HCP structure in PISA of PEG45–PS45 can be attributed to the higher Tg of the PS block, which more effectively hinders the rearrangement of spherical core domains and prevents morphological transformation to cylinders.
 | ||
| Fig. 3 (a) SAXS profiles for PEG45–PHEAn with varied degrees of polymerization of PHEA at 25 °C, prepared through PISA at the solute concentration of 40 wt%. (b) Close-up of the SAXS profile of PEG45–PHEA75 and schematic illustration of GYR structure. Allowed reflections for GYR (n = 75) correspond to the following ratio of the peak positions: √6, √8, √14, √16, √20, √22, √24, √26, √30, √32, √38, √40, √42, √46, √48, √50, and √52. (c) Two-dimensional SAXS pattern of PEG45–PHEA75. | ||
Highly ordered gyroid structure
A notable finding within PEG45–PHEAn series is the formation of a well-defined double-gyroid (GYR) structure in PEG45–PHEA75. GYR formation was confirmed by the intensity of the primary peak being approximately 10 times higher than that of the secondary peak, with the peak positions accurately matching the allowed reflections of GYR (Fig. 3). Of particular interest, a 16th-order peak was observed, despite the invisibility of the (431), (440), and (541) reflections. A GYR structure was also observed in PEG45–PS75, although the diffraction peaks were less distinct (Fig. 2) than those of PEG45–PHEA75. The lattice parameters were for PEG45–PHEA75 and 41.7 nm for PEG45–PS75. The smaller lattice parameter observed in PEG45–PS75 may be attributed to the glassy nature of the PS domains, which could hinder the expansion of the GYR structure during the PISA process. GYR is a known thermodynamically stable structure in the microphase separation of block copolymers, but it typically exists within a narrow compositional window (or is sometimes absent) for linear diblock copolymers.29,54–56 To the best of our knowledge, such high-order diffraction peaks for a GYR structure have not been previously reported for diblock copolymers prepared via ex situ polymerization. Additionally, this is the first observation of GYR structures through PISA. These results suggest that PISA, with optimized core-forming and stabilizing block length, is an effective method for preparing well-defined GYR structures, and that low-Tg core-forming blocks may be particularly suitable for their formation.
The lattice parameters were for PEG45–PHEA75 and 41.7 nm for PEG45–PS75. The smaller lattice parameter observed in PEG45–PS75 may be attributed to the glassy nature of the PS domains, which could hinder the expansion of the GYR structure during the PISA process. GYR is a known thermodynamically stable structure in the microphase separation of block copolymers, but it typically exists within a narrow compositional window (or is sometimes absent) for linear diblock copolymers.29,54–56 To the best of our knowledge, such high-order diffraction peaks for a GYR structure have not been previously reported for diblock copolymers prepared via ex situ polymerization. Additionally, this is the first observation of GYR structures through PISA. These results suggest that PISA, with optimized core-forming and stabilizing block length, is an effective method for preparing well-defined GYR structures, and that low-Tg core-forming blocks may be particularly suitable for their formation.
Kinetically controlled competition between lamella and vesicle
Another unexpected observation is the broadening of diffraction peaks as the degree of polymerization of the PS block exceeds 75. PEG45–PS91 exhibited broad peaks with positions of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (Fig. 2a), and the volume fraction of the PS domain is adequately high. This observation indicates lamella formation, but the structure is not well-defined. Moreover, PEG45–PS136 exhibited much broader peaks, without higher-order peaks beyond the second order, indicating a disordered structure. In contrast, PEG45–PHEA136 exhibited relatively distinct peaks consistent with a lamella structure (Fig. 3). This transition behavior of PEG–PHEA (BCC → HEX → GYR → LAM) is similar to that observed in conventional ex situ-prepared block copolymers. To further consider the disordered structure of PEG45–PS136, we saw the SAXS profile as a double-logarithmic plot, which shows that the scattering intensity is close to the scaling relationship of “intensity ∼ q−2” at the low-q region57 (Fig. 4a). This scaling suggests the presence of vesicles. To confirm the vesicle formation in PEG45–PS136, we performed transmission electron microscopy (TEM). Consequently, vesicles (VES) with the coexistence of LAM were observed (Fig. 4b and c). We note that the solvent, [BMIM][PF6], is non-volatile and electron-rich, and thus, the dark-colored inside of the vesicles implies that the vesicles contain [BMIM][PF6]. The broad SAXS peaks can therefore be attributed to the presence of vesicles, as the periodic structure becomes disordered. The domain spacing of the lamella determined by SAXS was 2π/q* = 27.1 nm, which agrees well with the TEM result of 25 ± 6 nm.
:2 ratio (Fig. 2a), and the volume fraction of the PS domain is adequately high. This observation indicates lamella formation, but the structure is not well-defined. Moreover, PEG45–PS136 exhibited much broader peaks, without higher-order peaks beyond the second order, indicating a disordered structure. In contrast, PEG45–PHEA136 exhibited relatively distinct peaks consistent with a lamella structure (Fig. 3). This transition behavior of PEG–PHEA (BCC → HEX → GYR → LAM) is similar to that observed in conventional ex situ-prepared block copolymers. To further consider the disordered structure of PEG45–PS136, we saw the SAXS profile as a double-logarithmic plot, which shows that the scattering intensity is close to the scaling relationship of “intensity ∼ q−2” at the low-q region57 (Fig. 4a). This scaling suggests the presence of vesicles. To confirm the vesicle formation in PEG45–PS136, we performed transmission electron microscopy (TEM). Consequently, vesicles (VES) with the coexistence of LAM were observed (Fig. 4b and c). We note that the solvent, [BMIM][PF6], is non-volatile and electron-rich, and thus, the dark-colored inside of the vesicles implies that the vesicles contain [BMIM][PF6]. The broad SAXS peaks can therefore be attributed to the presence of vesicles, as the periodic structure becomes disordered. The domain spacing of the lamella determined by SAXS was 2π/q* = 27.1 nm, which agrees well with the TEM result of 25 ± 6 nm.
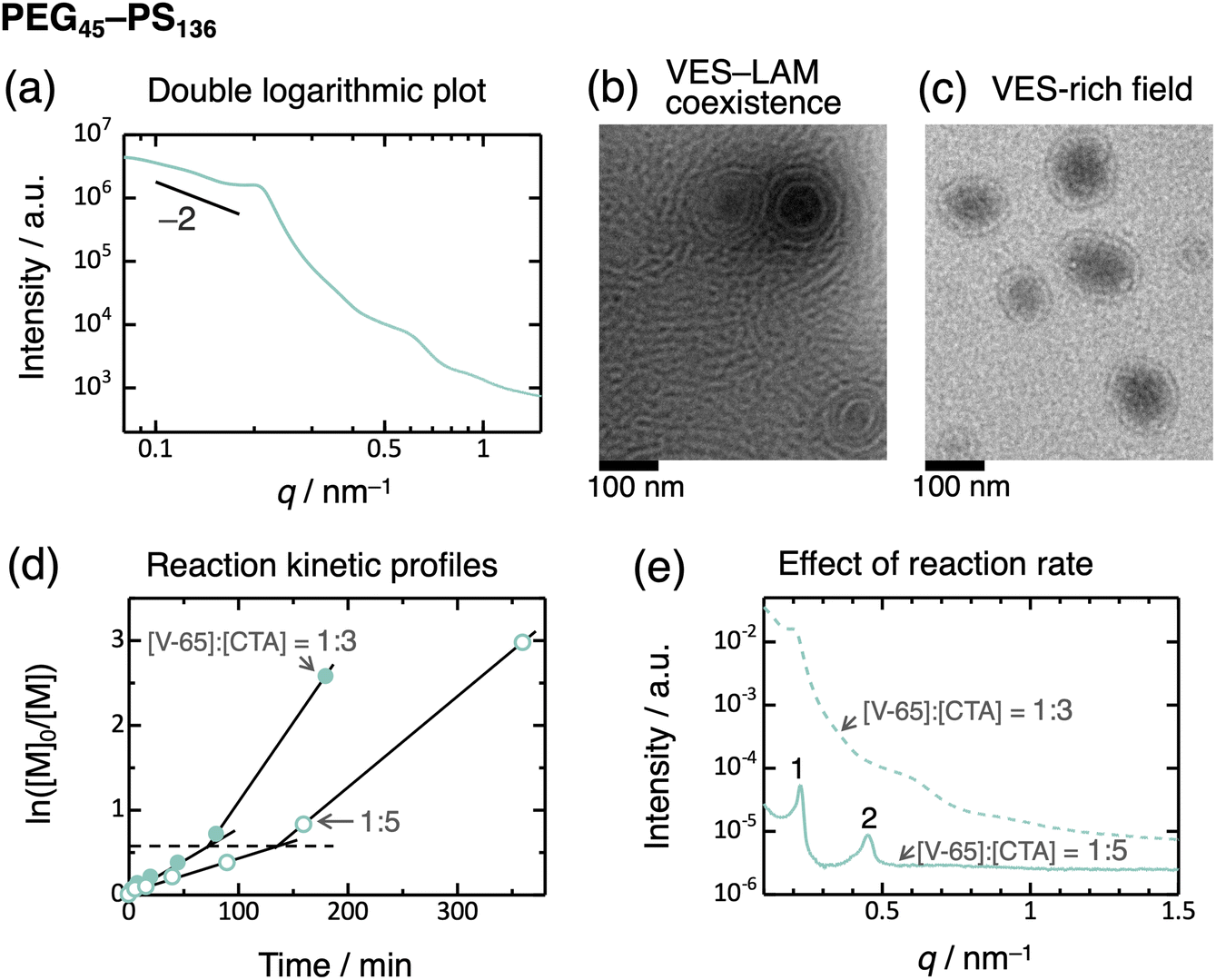 | ||
| Fig. 4 (a) Double logarithmic plot of the SAXS profile for PEG45–PS136 in [BMIM][PF6] (the same data as in Fig. 2a). (b and c) TEM images of PEG45–PS136 in [BMIM][PF6]. Panels (b and c) show different fields of view of the same sample; panel b displays vesicle–lamella coexistence, and panel (c) shows a vesicle-rich field. (d) Reaction kinetic profiles with [V-65]:[CTA] ratios of 1:5 (unfilled circle) and 1:3 (filled circle) at 60 °C. The broken line represents the ln([M]0/[M]) value at the inflection points. (e) Effect of the reaction rate on the SAXS profiles of PEG45–PS136 in [BMIM][PF6] at 25 °C. The PISA synthesis was performed at 60 °C for 6 hours with a [V-65]:[CTA] ratio of 1:5 (solid curve) and for 3 hours with a [V-65]:[CTA] ratio of 1:3 (broken curve; same data as in Fig. 2a). | ||
Vesicles typically grow through fusion and/or molecular exchange, and sedimentation of these enlarged vesicles results in LAM formation. This process reduces the bilayer curvature and minimizes the bending energy.58–62 However, if the growth mechanisms are inhibited, the vesicle structure may become trapped. In a dilute solution, the low collision probability allows vesicles to remain metastable for extended periods. In contrast, LAM is typically observed in more concentrated solutions rather than VES,29 as the higher collision probability should lead to LAM formation. In this study, the glassy PS core domains of VES hinder both fusion and molecular exchange. As a result, the VES structure becomes kinetically trapped in spite of the high solute concentration (40 wt%).
To validate this hypothesis, we performed PISA synthesis at a reduced reaction rate of the polymerization by decreasing the molar ratio of initiator (V-65) to CTA ([V-65]:[CTA]) to 1:5.63 As expected, the reaction rate decreased, as evidenced by the shallower slope in the plot of ln[M]0/[M] versus time (Fig. 4d), where [M] stands for the molar concentration of monomer, and [M]0 is the initial [M] value. An inflection point, indicative of acceleration of reaction rate, was observed in this plot. This phenomenon is often seen during PISA syntheses and is attributed to the onset of self-assembly11 (monomer encapsulation within the micelle core, leading to an increased local monomer concentration). In this study, the inflection occurred at the same ln[M]0/[M] values (i.e., similar conversions) for both [V-65]:[CTA] ratios. That is, the self-assembly initiates at a degree of polymerization independent of reaction rates investigated. Nonetheless, the SAXS profile of the PEG45–PS136 prepared using the lower initiator concentration exhibited relatively sharp diffraction peaks (up to the second order) of a LAM structure (Fig. 4e). This suggests that rapid polymerization of the high-Tg core-forming block during PISA leads to rapid vitrification of the membrane, trapping the VES structure and preventing its transformation to LAM. Conversely, slow polymerization allows the growth of VES and the transformation to LAM before becoming glassy. In other words, the ordered/disordered states can be controlled by the reaction rate.
On the other hand, the low Tg of the PHEA blocks facilitates fusion and molecular exchange, leading to LAM formation with far fewer coexisting vesicles, as indicated by the sharper diffraction peaks, despite the lack of the TEM images due to the unfortunately poor contrast between the PEG and PHEA blocks in the presence of [BMIM][PF6]. This result is consistent with the competition between VES and LAM as discussed above.
Relationship between structure and viscoelastic/electrical properties
The observed structures are summarized in Fig. 5a as a quasi-phase diagram. We conducted oscillatory rheometry and impedance spectroscopy to characterize these structures (Fig. S6–S9†). The plateau moduli, determined from the storage modulus at the minimum of the loss tangent, varied by more than an order of magnitude depending on the structure; the ordered structures tended to exhibit higher plateau moduli than the disordered structures (Fig. 5b and Table S1†). In contrast, the electrical conductivity remained relatively constant across the different structures due to the high ionic liquid content (60 wt%) and was primarily influenced by the choice of core-forming block (PS or PHEA). Thus, structural control can enhance the modulus while maintaining the conductivity. | ||
| Fig. 5 (a) Quasi-phase diagram of PEG45–PSn (high-Tg core-forming block) and PEG45–PHEAn (low-Tg core-forming block) prepared via PISA in [BMIM][PF6] in 40 wt% solute. (b) Plateau modulus obtained from oscillatory rheometry plotted against electrical conductivity for PEG45–PSn (turquoise symbols) and PEG45–PHEAn (pink symbols) in [BMIM][PF6] with 40 wt% solute at 25 °C. | ||
Conclusions
We explored ordered structures through PISA during the syntheses of diblock copolymers with various degrees of polymerization and two distinct core-forming blocks with differing Tg. With the high-Tg core-forming blocks (PEG45–PSn), we observed structural transitions of BCC → HCP → GYR → DIS (VES + LAM) as the degree of polymerization of the PS block increased (Fig. 5a). Notably, a highly ordered, pure HCP structure was observed. The formation of the HCP structure is unusual in conventional diblock copolymers prepared via ex situ polymerization and is attributed to kinetic effects inherent to the PISA process. Similarly, the presence of the VES structure is also likely kinetically driven, as the competition between the VES (disordered) and LAM (ordered) states is influenced by the polymerization rate. In contrast, the low-Tg system (PEG45–PHEAn) did not exhibit HCP and VES + LAM structures. Instead, DIS → HEX → GYR → LAM transitions were observed with increasing the PHEA block length (Fig. 5a). This transition behavior qualitatively aligns with conventional microphase separation observed in ex situ-synthesized diblock copolymers, although ordered spherical phases were not formed due to the weak segregation of PEG45–PHEAn. However, we found a highly ordered GYR structure compared to previously reported GYR structures of ex situ-synthesized diblock copolymers.Moreover, the plateau modulus was tunable by more than one order of magnitude through structural variation while maintaining electrical conductivity. While the absence of core-crosslinking in this study limits the achievable moduli compared to chemically crosslinked solid polymer electrolytes,18,22,23 these structure–property relationships are crucial for future design of high-modulus solid polymer electrolytes by combining precise structural control with crosslinking strategies. Additionally, despite the electrical conductivity being independent of the structure due to the high ionic liquid content (60 wt%), the gyroid (GYR) structure is known to enhance ion transport.56,64 Thus, insights into the fabrication of well-defined GYR structures are valuable for the applications. Therefore, PISA synthesis provides a facile route to a wide range of highly ordered microphase-separated structures from simple, conventional diblock copolymers, offering significant potential for applications. Furthermore, the kinetic effect afforded by the PISA process with high-Tg core-forming blocks enables the formation of structures inaccessible through conventional ex situ polymerization.
Data availability
The data are available from the authors upon reasonable request.Author contributions
I. S.: data curation, formal analysis, investigation, validation, writing – review & editing. A. S.-N.: resources, supervision, writing – review & editing. R. T.: conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Noboru Ohta (JASRI) for the setting up of the SAXS experiments. JASRI is acknowledged for the provision of the synchrotron beam time in SPring-8 (proposal no. 2023A1244, 2023B1174, 2024A1165, and 2024B1209). TEM observations were conducted in the High Voltage Electron Microscope Laboratory at Nagoya University. This work was partially funded by JSPS KAKENHI (Grant No. 24K17722), The Hibi Science Foundation, The Naito Science & Engineering Foundation, and The Asahi Glass Foundation.References
- A. Pross, J. Phys. Org. Chem., 2008, 21, 724 CrossRef CAS.
- R. Pascal, A. Pross and D. Sutherland, Open Biol., 2013, 3, 130156 CrossRef PubMed.
- S. D. P. Fielden, J. Am. Chem. Soc., 2024, 146, 18781 CrossRef CAS PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2018, 40, 1800279 CrossRef PubMed.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368 CrossRef PubMed.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192 RSC.
- C. Györy and S. P. Armes, Angew. Chem., Int. Ed., 2023, 62, e202308372 CrossRef PubMed.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493 CrossRef CAS PubMed.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581 CrossRef CAS.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755 CrossRef CAS.
- G. L. Maitland, M. Liu, T. J. Neal, J. Hammerton, Y. Han, S. D. Worrall, P. D. Topham and M. J. Derry, Chem. Sci., 2014, 15, 4416 RSC.
- M. Seo and M. A. Hillmyer, Science, 2012, 336, 1422 CrossRef CAS.
- S. Saba, M. P. S. Mousavi, P. Bühlmann and M. A. Hillmyer, J. Am. Chem. Soc., 2015, 137, 8896 CrossRef CAS PubMed.
- M. Seo, S. Kim, J. Oh and S.-J. Kim, J. Am. Chem. Soc., 2015, 137, 600 CrossRef CAS PubMed.
- Z. Zhao, S. Lei, M. Zeng and M. Huo, Aggregate, 2024, 5, e418 CrossRef CAS.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2023, 62, e202307329 CrossRef CAS PubMed.
- T. Oh, S. Cho, C. Yoo, W. Yeo, J. Oh and M. Seo, Prog. Polym. Sci., 2023, 145, 101738 CrossRef CAS.
- X. Shi, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Small, 2024, 20, 2305268 CrossRef CAS.
- V. A. Bobrin, Y. Yao, Y. Xiu, J. Zhang, N. Corrigan and C. Boyer, Nat. Commun., 2022, 13, 3577 CrossRef CAS PubMed.
- K. Lee, Y. Shang, V. A. Bobrin, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2204816 CrossRef CAS.
- D. Melodia, A. Bhadra, K. Lee, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, Small, 2023, 19, 2206639 CrossRef CAS.
- K. Yamauchi, H. Hasegawa, T. Hashimoto, H. Tanaka, R. Motokawa and S. Koizumi, Macromolecules, 2006, 39, 4531 CrossRef CAS.
- J. Lee and M. Seo, ACS Nano, 2021, 15, 9154 CrossRef CAS PubMed.
- R. Yamanaka, A. Sugawara-Narutaki and R. Takahashi, Macromolecules, 2023, 56, 4354 CAS.
- R. Yamanaka, A. Sugawara-Narutaki and R. Takahashi, ACS Macro Lett., 2024, 3, 1050 CrossRef PubMed.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3 CrossRef CAS.
- T. P. Lodge, B. Pudil and K. J. Hanley, Macromolecules, 2002, 35, 4707 CrossRef CAS.
- M. J. Rymaruk, C. T. O'Brien, C. György, B. Darmau, J. Jennings, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2021, 60, 12955 CrossRef CAS PubMed.
- W. Hou, X. Yin, Y. Zhou, Z. Zhou, Z. Liu, J. Du, Y. Shi and Y. Chen, J. Am. Chem. Soc., 2024, 146, 24094 CrossRef CAS.
- P. Yang, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Macromolecules, 2016, 49, 6731 CrossRef CAS PubMed.
- E. R. Jones, O. O. Mykhaylyk, M. Semsarilar, M. Boerakker, P. Wyman and S. P. Armes, Macromolecules, 2016, 49, 172 CrossRef CAS PubMed.
- H. Zeng, X. Liang, D. A. Roberts, E. R. Gillies and M. Müllner, Angew. Chem., Int. Ed., 2024, 63, e202318881 CrossRef CAS.
- Y. Shi, W. Zhu, D. Yao, M. Long, B. Peng, K. Zhang and Y. Chen, ACS Macro Lett., 2014, 3, 70 CrossRef CAS PubMed.
- S. Guan, Z. Deng, T. Huang, W. Wen, Y. Zhao and A. Chen, ACS Macro Lett., 2019, 8, 460 CrossRef CAS PubMed.
- J. Rieger, J. Therm. Anal. Calorim., 1996, 46, 965 CrossRef CAS.
- J. A. G. Tejedor, J. C. R. Hernández, J. L. G. Ribelles and M. M. Pradas, J. Macromol. Sci., Part B:Phys., 2007, 46, 43 CrossRef CAS.
- G. Damonte, L. Maddalena, A. Fina, D. Gavallo, A. J. Müller, M. R. Gaputo, A. Mariani and O. Monticelli, Eur. Polym. J., 2022, 171, 111226 CrossRef CAS.
- C. Zhang, D. L. Vigil, D. Sun, M. W. Bates, T. Loman, E. A. Murphy, S. M. Barbon, J.-A. Song, B. Yu, G. H. Frederickson, A. K. Whittaker, C. J. Hawker and C. M. Bates, J. Am. Chem. Soc., 2021, 143, 14106 CrossRef CAS PubMed.
- G. A. McConnell, A. P. Gast, J. S. Huang and D. Smith, Phys. Rev. Lett., 1993, 71, 2102 CrossRef CAS PubMed.
- A. P. Gast, Langmuir, 1996, 12, 4060 CrossRef CAS.
- I. W. Hamley, C. Daniel, W. Mingvanish, S.-M. Mai and C. Booth, Langmuir, 2000, 16, 2508 CrossRef CAS.
- M. W. Matsen, Phys. Rev. Lett., 2007, 99, 148304 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 7641 CrossRef CAS.
- L. Chen, Y. Qiang and W. Li, Macromolecules, 2018, 51, 9890 CrossRef CAS.
- Y. Qiang, W. Li and A.-C. Shi, ACS Macro Lett., 2020, 9, 668 CrossRef CAS.
- W. Jiang, Y. Qiang, W. Li, F. Qiu and A.-C. Shi, Macromolecules, 2018, 51, 1529 CrossRef CAS.
- Q. Xie, Y. Qiang, L. Chen, Y. Xia and W. Li, ACS Macro Lett., 2020, 9, 980 CrossRef CAS PubMed.
- Y.-Y. Huang, H.-L. Chen and T. Hashimoto, Macromolecules, 2003, 36, 764 CrossRef CAS.
- L.-T. Chen, C.-Y. Chen and H.-L. Chen, Polymer, 2019, 169, 131 CrossRef CAS.
- T. K. Mandal and E. M. Woo, Polym. J., 1999, 31, 226 CrossRef CAS.
- M. J. Park, J. Bang, T. Harada, K. Char and T. P. Lodge, Macromolecules, 2004, 37, 9064 CrossRef CAS.
- A. K. Khandpur, S. Forster, F. S. Bates, I. W. Hamley, A. J. Ryan, W. Bras, K. Almdal and K. Mortensen, Macromolecules, 1995, 28, 8796 CrossRef CAS.
- M. W. Matsen, Macromolecules, 2012, 45, 2161 CrossRef CAS.
- J. Park and K. I. Winey, JACS Au, 2022, 2, 1769 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128 CrossRef CAS.
- R. G. Laughlin, Colloids Surf., A, 1997, 128, 27 CrossRef CAS.
- R. Takahashi, T. Narayanan, S. Yusa and T. Sato, Macromolecules, 2022, 55, 684 CrossRef CAS.
- R. Bleul, R. Thiermann and M. Maskos, Macromolecules, 2015, 48, 7396 CrossRef CAS.
- S. J. Marrink and A. E. Mark, J. Am. Chem. Soc., 2003, 125, 11144 CAS.
- J. E. Nielsen and R. Lund, Langmuir, 2022, 38, 374 CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433 CAS.
- L. Yan, C. Rank, S. Mecking and K. I. Winey, J. Am. Chem. Soc., 2020, 142, 857 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra for macro-CTA as well as PEG45–PSn and PEG45–PHEMAn, UV spectra for macro-CTA, complex moduli, and impedance spectra. See DOI: https://doi.org/10.1039/d5sc01703c |
| This journal is © The Royal Society of Chemistry 2025 |