
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of the tetrahydroxynaphthalene-derived meroterpenoid furaquinocin via reductive deamination and intramolecular hydroalkoxylation of an alkene†
Tomohiro
Noguchi
 a,
Fan
Zhao
a,
Yoshitaka
Moriwaki
ab,
Hideaki
Yamamoto
a,
Kei
Kudo
a,
Ryuhei
Nagata
a,
Takeo
Tomita
ab,
Tohru
Terada
ab,
Kentaro
Shimizu
ab,
Makoto
Nishiyama
ab and
Tomohisa
Kuzuyama
*ab
a,
Fan
Zhao
a,
Yoshitaka
Moriwaki
ab,
Hideaki
Yamamoto
a,
Kei
Kudo
a,
Ryuhei
Nagata
a,
Takeo
Tomita
ab,
Tohru
Terada
ab,
Kentaro
Shimizu
ab,
Makoto
Nishiyama
ab and
Tomohisa
Kuzuyama
*ab
aGraduate School of Agricultural and Life Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan. E-mail: utkuz@g.ecc.u-tokyo.ac.jp
bCollaborative Research Institute for Innovative Microbiology, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan
First published on 31st March 2025
Abstract
Hybrid isoprenoid-polyketides, known as meroterpenoids, are a family of natural products that exhibit various bioactivities and are promising drug scaffolds. Despite the structural diversity of 1,3,6,8-tetrahydroxynaphthalene (THN)-derived meroterpenoids, such as furaquinocin, naphterpin, and furanonaphthoquinone, several biosynthetic genes for these compounds are conserved, suggesting a shared biosynthetic mechanism. However, the common biosynthetic mechanism and pathway-specific structural diversification mechanisms of these meroterpenoids are not yet fully understood. This study reveals the biosynthetic pathway for furaquinocin, demonstrating that it involves reductive deamination to form a key hydroquinone intermediate essential for subsequent reactions, including a unique cyclization step. We identified the mechanism of reductive deamination of the biosynthetic intermediate 8-amino-flaviolin through transient diazotization, leading to the formation of the hydroquinone intermediate 1,2,4,5,7-pentahydroxynaphthalene (PHN). Structural and computational studies confirmed that PHN is a key substrate for the subsequent methylation. We also showed that the hydroquinone intermediates are prerequisites for the subsequent pathway-specific reactions, including prenylation and novel intramolecular hydroalkoxylation of an alkene. This hydroalkoxylation reaction is notable in that a methyltransferase homolog catalyzes it in an S-adenosylmethionine-independent manner. Our findings provide a new model for furaquinocin biosynthesis, offering insights into the biosynthetic strategies for THN-derived meroterpenoids.
Introduction
Meroterpenoids are hybrid natural products harboring a terpenoid skeleton fused with other molecules, such as polyketides. Meroterpenoids exhibit various biological activities, such as antitumor, antibacterial, and antioxidant activities.1–3 The 1,3,6,8-tetrahydroxynaphthalene (THN)-derived meroterpenoids produced by actinomycetes include furaquinocin (1),4,5 an antitumor active compound produced by Streptomyces sp. KO-3988 and Streptomyces reveromyceticus SN-593; naphterpin (2),6 produced by Streptomyces sp. CL190; furanonaphthoquinone,7 produced by Streptomyces cinnamonensis DSM 1042; and napyradiomycin,8 produced by Streptomyces sp. CNQ525 and Streptomyces aculeolatus (Fig. 1). Despite the structural diversity of these final products, several of the biosynthetic genes are conserved; for example, the gene clusters for the biosynthesis of THN-derived meroterpenoids contain a common gene cassette (Fig. S1†). Given the gene conservation, we inferred that these meroterpenoids share a common biosynthetic mechanism and showed that 8-amino-flaviolin (8-AF, 3), as a common biosynthetic intermediate, is biosynthesized via C2v symmetric intermediates, THN and mompain, by three conserved enzymes (e.g., Fur1, 2, and 3 for furaquinocin biosynthesis) (Fig. 1).9,10 In contrast to 3, none of the final products contained an amino group at the C8 position. In the conversion of 3 to the final products, the amino group is replaced by a hydrogen atom, indicating that a reductive deamination step is indispensable during the biosynthesis of THN-derived meroterpenoids. However, the mechanism by which the amino group is removed during the biosynthesis of these meroterpenoids has not been elucidated to date.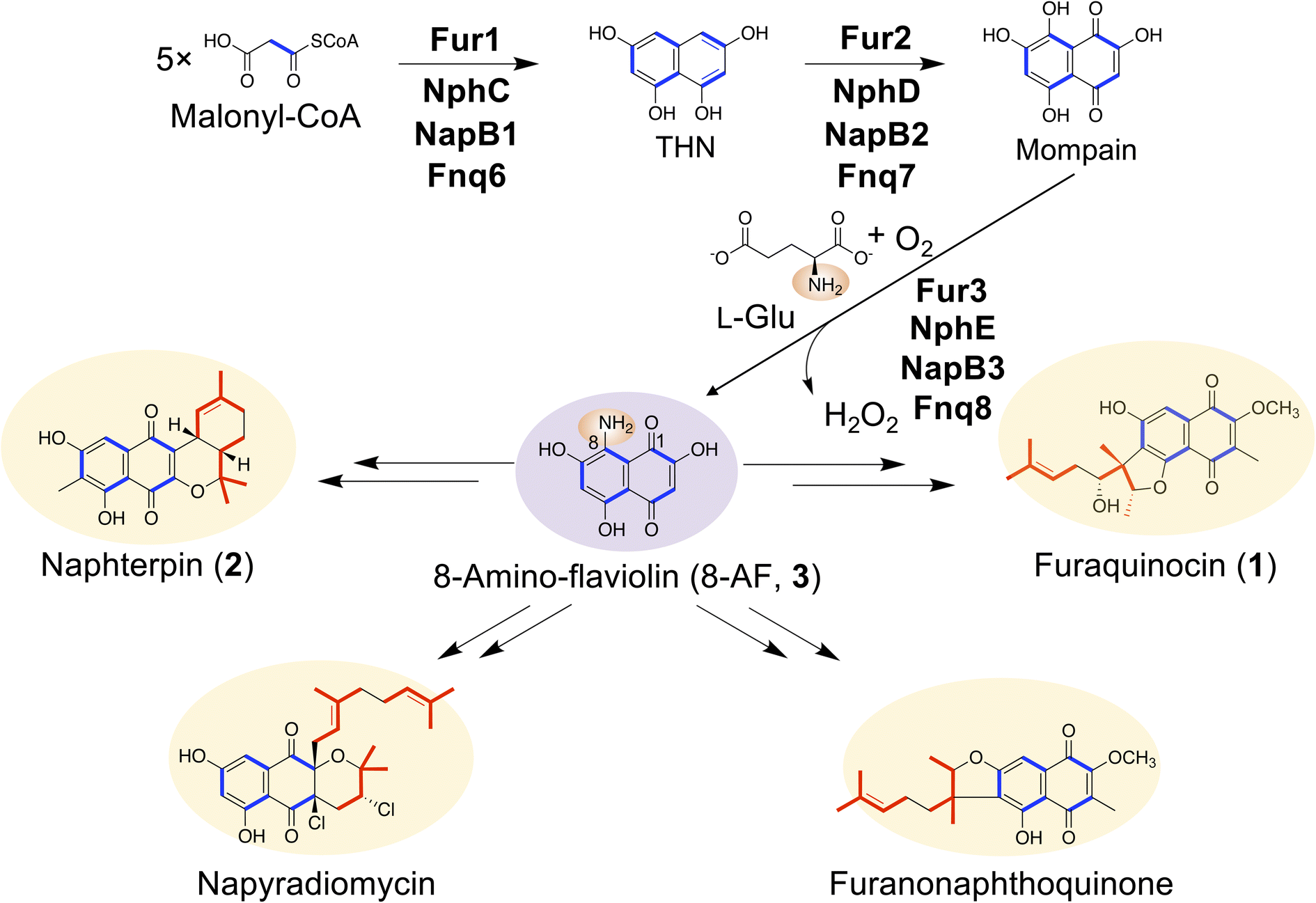 | ||
| Fig. 1 Common biosynthetic pathway for meroterpenoids produced by Streptomyces. 8-AF (3) is a common biosynthetic intermediate for THN-derived meroterpenoids. However, all the meroterpenoids shown here do not contain an amino group. Blue, polyketide moiety; red, terpenoid moiety. | ||
Questions also remain regarding the pathway-specific cyclization mechanisms underlying the structural diversity of THN-derived meroterpenoids. As pathway-specific cyclases, vanadium-dependent haloperoxidases (VHPOs) have been identified as being involved in the biosynthesis of napyradiomycin and the merochlorin class of THN-derived meroterpenoids (Fig. S2†).11,12 In the biosynthesis of non-THN-derived bacterial meroterpenoids including xiamycin, hapalindoles, and atolypene A, unique cyclization mechanisms have been reported (Fig. S3A–C†).13–19 However, the cyclization patterns of THN-derived meroterpenoids, including furaquinocin, naphterpin, and furanonaphthoquinone, differ from those observed in other bacterial meroterpenoids. In addition, the gene clusters responsible for the biosynthesis of these THN-derived meroterpenoids do not contain genes encoding homologs of the known bacterial meroterpenoid cyclases, such as VHPOs. Consequently, the mechanisms driving the cyclization and structural diversification of these THN-derived meroterpenoids remain unclear, limiting the understanding of the source of their structural diversity.
The complete biosynthetic pathway of furaquinocin remains to be elucidated.20 In addition to intermediate 3, 2-methoxy-3-methylflaviolin (MMF, 4) has been identified as the substrate for the prenyltransferase Fur7.21 However, the enzymes responsible for the conversion of 3 to 4 and the subsequent cyclization reactions after prenylation remain unclear.
In this study, a comprehensive analysis of furaquinocin biosynthesis reveals a complete biosynthetic pathway involving the hydroquinone intermediates (Fig. 2). This pathway involves the transient introduction of a diazo group, leading to the formation of a key hydroquinone-based intermediate. This reduced hydroquinone intermediate plays a crucial role in two subsequent methylation steps. The introduction of two methyl groups determines the position of the carbon atom for prenylation. Following prenylation, the cyclic structure of the prenyl moiety of furaquinocin is achieved by cyclization via intramolecular hydroalkoxylation of an alkene catalyzed by an unprecedented type of cyclase as a methyltransferase homolog.
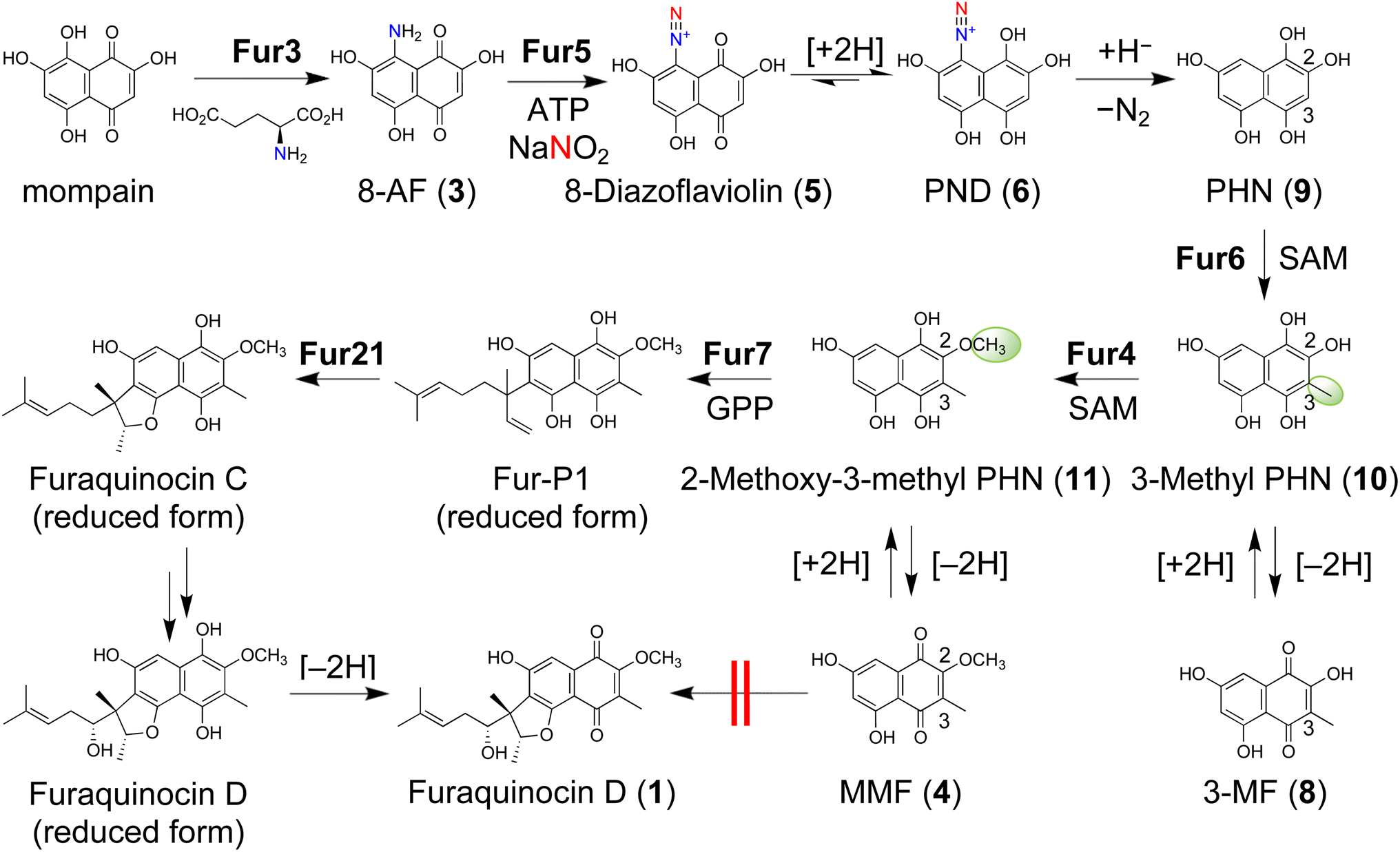 | ||
| Fig. 2 Proposed biosynthetic pathway for furaquinocin. With initiation by the diazotization of 3 through Fur5, furaquinocin is biosynthesized in the reduced hydroquinone form via N2 gas emission and subsequent modification reactions. The equilibrium between 5 and 6 is biased toward 6 because of the introduction of an electron-withdrawing diazo group into 3, thereby allowing the subsequent nonenzymatic diazo group elimination in 6 to proceed. The diazo group in 6 is removed as nitrogen gas by the action of NADPH to facilitate the reaction. See the text for details on the other reaction steps. | ||
Results and discussion
Nitrite formation by Fur16 and Fur17
We identified the furaquinocin biosynthetic gene (fur) cluster (fur1-21, Table S1†) and achieved the heterologous production of furaquinocin in Streptomyces albus.20 We showed that Fur7 catalyzes the geranylation of MMF (4) to form 6-prenyl-2-methoxy-3-methylflaviolin (Fur-P1) (Fig. S4†).21 As two methylation reactions and reductive deamination are necessary to convert 8-AF (3) to 4, we hypothesized that the two methylation reactions involving 3 are catalyzed by the putative methyltransferases Fur4 and Fur6. In addition, after closely investigating the fur cluster, we found that Fur16 and Fur17, homologs of the nitrite-producing enzymes CreD (59% identity) and CreE (59% identity), respectively, in the cremeomycin biosynthetic gene cluster are also present in the fur cluster. The CreD and CreE pair, known as ANS pathway enzymes, generate nitrite from aspartate.22 In addition, the nitrite-forming enzyme pair NphK and NphL and the pair Fnq20 and Fnq21 are also conserved in the biosynthetic gene clusters of naphterpin (nph) and furanonaphthoquinone (fnq), respectively (Fig. S1†).To investigate whether Fur16 and Fur17 produce nitrite, N-terminally His-tagged fur16 and fur17 were overexpressed in Escherichia coli, and the recombinant proteins were purified to homogeneity (Fig. S5A†). When Fur16 and Fur17 were incubated with L-aspartate (Asp), nitrite was produced (Fig. S5B†). Unexpectedly, the fur cluster contains a nitrite biosynthetic system involving the ANS pathway, even though the structure of furaquinocin does not possess nitrogen atoms. In organic synthesis, nitrite is used as a reagent for the diazotization of amino groups.23 Thus, we inferred that nitrite might be involved in the diazotization of the amino group of 3. We identified Fur5, which is a homolog of the diazo-forming enzyme AvaA6 (29% identity) involved in avenalumic acid biosynthesis, in the fur cluster.24 The AvaA6 homologs NphH (28% identity) and Fnq11 (28% identity) are also conserved in the nph and fnq clusters, respectively (Fig. S1†). In the biosynthesis of avenalumic acid, reductive deamination is achieved through diazotization by AvaA6 and subsequent hydride reduction by AvaA7. Although the fur cluster does not contain an AvaA7 homolog, we hypothesized that furaquinocin may be biosynthesized by reductive deamination via diazotization of 3, which could involve Fur5.
Biochemical analysis of Fur5 and NphH
To identify the function of Fur5, we deleted the fur5 gene from the furaquinocin heterologous expression plasmid pWHM-fura2 (hereafter referred to as pWFQ) through λ red recombination-mediated in-frame deletion. The resulting construct pWFQΔfur5 was subsequently transformed into S. albus G153. The resulting strain S. albus G153/pWFQΔfur5 exhibited significant accumulation of the previously reported biosynthetic intermediate 3 but exhibited loss of 1 production (Fig. S6†). This result indicated that 3 is a substrate for Fur5. To confirm Fur5 activity, C-terminally His-tagged fur5 was overexpressed in S. albus G153, and a small amount of recombinant protein was purified to homogeneity (Fig. S7A†). When 3 was incubated with Fur5 in the presence of ATP and nitrite, the purple color derived from 3 disappeared within a few minutes, indicating that 3 was converted to an unknown product (Fig. S7B†). To identify the reaction product of Fur5, we performed LC-MS/MS analysis on the Fur5 reaction mixture; we detected two compounds, with m/z values of 233.02 and 235.03, that were produced in a Fur5-dependent manner (Fig. 3). When the Fur5 reaction was performed with 15N-labeled nitrite, both products were detected with the m/z increasing by 1 (m/z 234.02 and 236.03, Fig.s S8 and S9†). This result indicated that both products contained nitrogen atoms derived from nitrite. MS/MS fragment analysis suggested that the product corresponding to m/z 233.02 was 8-diazoflaviolin (5), in which the amino group of 3 was converted to a diazo group (Fig. S8†). The structure of 5 was also supported by the 1H NMR spectrum obtained after derivatization with ethyl 2-methylacetoacetate (Fig. S10†). Owing to the very low yield and low stability of Fur5, it was difficult to purify enough 5 for 13C NMR analysis. Similarly, the product corresponding to m/z 235.03 was identified as 2,4,5,7,8-pentahydroxynaphthalene-1-diazonium (PND, 6), a reduced form of 5 (Fig. S9†). During the investigation of the Fur5 reaction, flaviolin (7) was detected in addition to 5 and 6 (Fig. 3). Compounds 5 and 6 are unstable and therefore readily converted to 7, as further discussed in the next section. In addition, the spontaneous conversion of 5 and 6 to 7 also suggests that 5 and 6 contain unstable diazo groups. Next, we investigated how ATP is utilized in the Fur5 reaction. As ATP was converted to AMP during the diazotization process, we hypothesize that the adenylation reaction toward nitrite activates nitrite to facilitate the nucleophilic attack of 3 (Fig. S11†).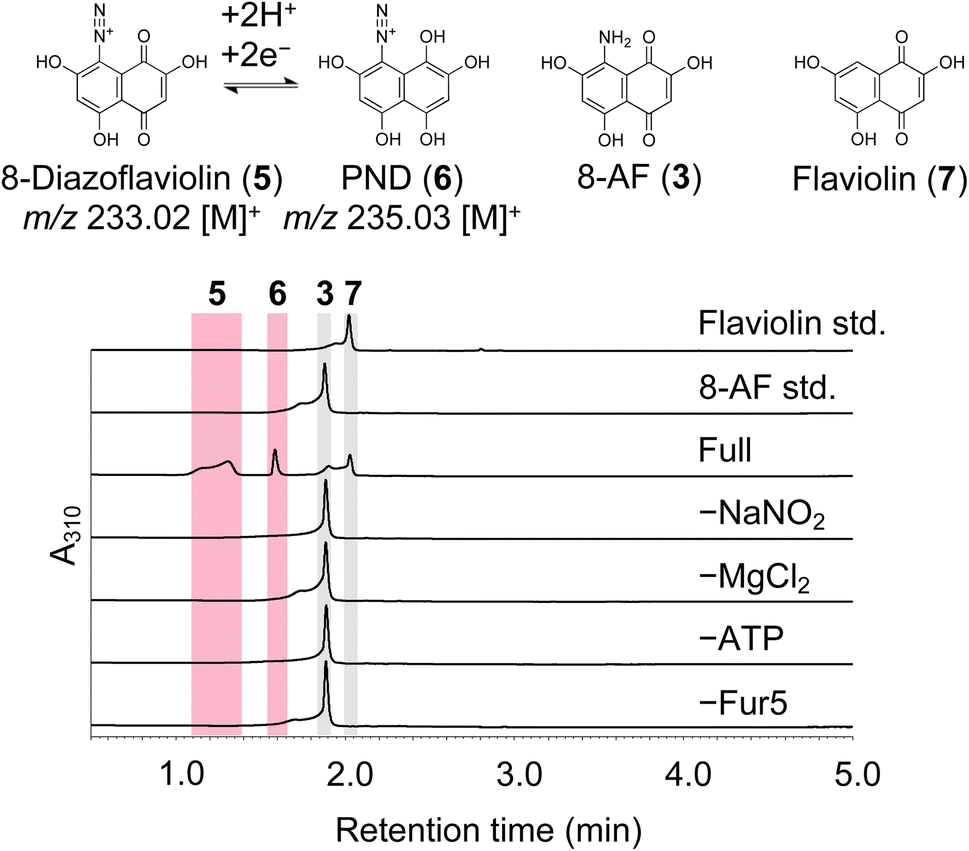 | ||
| Fig. 3 In vitro assays of the enzymatic activity of Fur5. HPLC-UV/VIS analysis of the Fur5 reaction mixture. The assay conditions are described in the ESI.†A310, absorbance at 310 nm. | ||
The Fur5 homolog NphH (77% identity) is found in the biosynthetic gene cluster of the naphterpin-producing Streptomyces sp. CL190. Therefore, we hypothesized that NphH is also involved in the diazotization of the amino group of 3. Inactivation of the nphH gene in CL190 resulted in the complete loss of naphterpin production and the accumulation of 3 (Fig. S12B and C†). Complementation of CL190ΔnphH with the nphH gene restored naphterpin production (Fig. S12D†), indicating that nphH is essential for naphterpin biosynthesis. Purified NphH was unstable and aggregated rapidly during the in vitro reaction but converted 3 to 5 and 6 as Fur5 catalyzed (Fig. S13†). These results indicate that the biosynthesis of the THN-derived meroterpenoids naphterpin and furaquinocin involves a biosynthetic system involving the diazotization of the amino group of 3. Furthermore, the conservation of genes encoding 8-AF (3)-forming enzymes, nitrite-forming enzymes, and diazotization enzymes in the fur, nph, and fnq clusters (Fig. S1†) suggests the existence of a shared diazotization system in the biosynthesis of THN-derived meroterpenoids such as furaquinocin, naphterpin, and furanonaphtoquinone.
Fur6 methyltransferase-catalyzed reactions
As mentioned above, when the Fur5-catalyzed reaction mixture was left at room temperature for one day and then analyzed by LC-MS again, 5 and 6 were found to have been fully converted to 7 (Fig. S14†). We expected that 5, 6, or 7 would be the authentic substrate for the subsequent methylation reaction and examined various reaction conditions. Surprisingly, Fur6 exhibited methylation activity when 7 was used as a substrate in the presence of the strong reducing agent dithionite, whereas methylation activity was not detected under reaction conditions without dithionite (Fig. 4A). Large-scale reactions with Fur6 and NMR spectroscopy analysis revealed that the methylated product was 3-methylflaviolin (3-MF (8), Fig. 4A and S15†). This result indicates that the quinone moiety of 7 is reduced by dithionite, the resultant 1,2,4,5,7-pentahydroxynaphthalene (PHN, 9) serves as a substrate for Fur6 methyltransferase, and subsequent aerobic oxidation yields 8 (Fig. 4A). On the other hand, when NADPH was added instead of dithionite, 8 was not formed, suggesting that NADPH cannot reduce 7 directly (Fig. 4A). | ||
| Fig. 4 Characterization of Fur6. (A) HPLC-UV/vis analysis of the Fur6 reaction mixture. The assay conditions are described in the ESI.†A310, absorbance at 310 nm. (B) Ligand interactions in the active site of Fur6. Carbon atoms in PHN, SAM, and Fur6 residues are shown in yellow, orange, and green, respectively. Oxygen, nitrogen, sulfur, and hydrogen atoms are shown in red, blue, yellow-orange, and white, respectively. The distance between the C3 atom of PHN and the carbon atom of the transferable methyl group of SAM is shown as a black dashed line. Potential hydrogen bonds are shown as yellow dashed lines. | ||
Crystal structure of Fur6 methyltransferase
To gain more insight into the Fur6-catalyzed reaction, we prepared crystals of Fur6 and determined its three-dimensional structure at 2.12 Å resolution via X-ray crystallography (PDB ID 8HAR; Fig. S16†). Only the S-adenosylmethionine (SAM) analog S-adenosylhomocysteine (SAH) was bound to the active site. Since 9 is readily oxidized in air and therefore obtaining the 9-bound crystal structure would be difficult, we decided to perform docking simulations to reveal the binding pocket of 9.The initial coordinates of Fur6 were obtained from the crystal structure mentioned above. The position of the transferable methyl group of SAM was modeled by superimposing an optimized SAM structure over that of SAH observed in the crystal structure. The model of 9 was generated in the monoanionic form. The pKa values of 9 calculated using the software program Epik2021-1 for pKa prediction suggested that the hydroxy group at C5 was deprotonated (Fig. S17†).25,26 The following assumptions were applied when 9 was manually docked into the Fur6 structure. First, the overall structure of 9 was assumed to be near the position of the methyl group of SAM, which is necessary for initiating the SN2-like reaction.27 Second, the C3 atom of 9 was assumed to be located within 4 Å from the carbon atom of the transferable methyl group of SAM to facilitate the methylation reaction. Third, 9 was assumed to be surrounded by several residues that stabilize the ligand in the active site through hydrogen bonds or π–π stacking. On the basis of these assumptions, we constructed 12 complex models. MD simulation was then performed to evaluate the stability of each model. The simulation revealed a stable binding mode of 9 in Fur6 (Fig. 4B). The C3 atom of 9 in the complex was located at an average distance of 3.41 Å from the carbon atom of the methyl group of SAM (Fig. S18†), which is sufficiently close for the methylation reaction. Trp150 and Asp252 form hydrogen bonds with 9, and the aromatic moieties of Tyr98, Tyr137, His249, Phe297, Phe298, Phe301, and Phe302 appear to stabilize the ligand in the active site. When residues potentially involved in hydrogen bond formation with 9 were mutated, the methylation activity of Fur6 disappeared because of mutations at Tyr137, Trp150 and Asp252. (Fig. S19 and S20†), suggesting that Tyr137, Trp150, and Asp252 recognize 9 in the Fur6 active site. These results also indicate that 9 is the authentic substrate of Fur6.
The hydroxy group at C5 exhibited the lowest pKa (Fig. S17†). This group is first deprotonated to phenolate under physiological conditions. Deprotonated 9 binds to the active site of Fur6 and is correctly oriented by Tyr137, Trp150, and Asp252. In contrast, the hydroxy group at C5 of 7 forms a hydrogen bond with the ketone at C4, which becomes difficult to deprotonate, partially explaining why the methylation reaction does not proceed against 7 (Fig. S21†). Although 9 is easily oxidized in air and cannot be directly detected, 9 may also be an intermediate in the biosynthesis of (R)-5-hydroxyscytalone and an important intermediate in secondary metabolism.28
Nonenzymatic reduction of compound 6
While it was established that compound 9 is the substrate for Fur6, the mechanism by which 9 is generated from compound 3 during the formation of 8 remained unknown. To elucidate this mechanism, we attempted to generate 8 by using 3 as a substrate and conducting a simultaneous reaction catalyzed by Fur5 and Fur6; however, 8 was not detected after this reaction (Fig. 5). In contrast, when NADPH was simultaneously added to the reaction mixture, 8 was detected. Compound 8 was also generated through sequential reactions catalyzed by Fur5 and Fur6, wherein Fur5 was removed from the reaction mixture by ultrafiltration before NADPH was added (see the Methods section, Fig. S22†). These findings suggest that 9, which could be generated by nonenzymatic reduction of 6 by the action of NADPH, serves as a substrate for Fur6, resulting in methylation at C3 and the subsequent formation of 8 by aerobic oxidation (Fig. 5). The addition of a wide range of other bioreducing agents, such as GSH and cysteine, also enabled the Fur5- and Fur6-catalyzed formation of 8, as did the addition of NADPH. These observations imply that 6 might also undergo nonenzymatic reduction in vivo (Fig. S23†). | ||
| Fig. 5 In vitro assays for the simultaneous Fur5 and Fur6 reaction mixture. HPLC-UV/VIS analysis of the simultaneous Fur5 and Fur6 reaction mixture. The assay conditions are described in the ESI.†A310, absorbance at 310 nm. | ||
The conversion of 9 from 3 proceeds via reductive deamination, which is accomplished by diazotization with Fur5 and nonenzymatic reduction with reducing agents, such as NADPH. In our subsequent investigation, we postulated that the diazo group in compound 6 would be liberated as nitrogen gas during the reductive deamination process. To test this hypothesis, we used 15N-labeled 3 and 15N-labeled nitrous acid in the Fur5 and Fur6 reaction mixture. 15N-labeled 3 was synthesized with 15N-labeled L-Glu via the oxidative transamination of mompain with Fur3 (Fig. S24†).9 Fully labeled N2 gas (15N15N) was produced in a labeled substrate- and Fur5-dependent manner, as detected by GC-MS analysis of the gas phase in the reaction vial (Fig. S25†). This result indicates that during the reductive deamination process, the nitrogen atoms in the amino groups that are converted to diazo groups are released as nitrogen gas.
Deamination reactions in vivo, such as those catalyzed by PLP-dependent aminotransferases, glutamate dehydrogenase, and flavoenzymes, typically replace the amino group with an oxygen atom29,30 and differ from reductive deamination (Fig. S26†). Reductive deamination is poorly understood, with GMP reductase being the only known single enzyme to catalyze this process, releasing ammonia and forming inosine monophosphate (IMP).31 Recently, a novel reductive deamination pathway involving the diazo-forming enzyme AvaA6 and the NAD(P)-dependent oxidoreductase AvaA7 was identified as being involved in avenalumic acid biosynthesis.24 The reductive deamination via diazotization catalyzed by Fur5 is also one of the few examples highlighting the structural diversity achieved by rare diazo-forming enzymes, such as AvaA6, CreM, Aha11, and AzpL (Fig. S27†).32–34
Naphthoquinone is reduced nonenzymatically by NADH,35 but many of the intermediates of furaquinocin, such as 3, cannot be reduced nonenzymatically by NADH or NADPH. Electron-donating groups lower the redox potential of quinones36,37 to accelerate the autoxidation of hydroquinones.38 Therefore, we suspect that the electron-donating hydroxy groups at C2, C5, and C7 and an amino group at C8 lower the redox potential of the naphthoquinone intermediates and prevent them from being reduced nonenzymatically by NADPH. On the other hand, the introduction of an electron-withdrawing diazo group by Fur5 increases the redox potential of the naphthoquinone intermediate. Thus, the equilibrium between 5 and 6 is biased toward 6, presumably allowing the subsequent elimination of the nonenzymatic diazo group in 6 to proceed. Further biochemical analyses of Fur5 are needed to clarify its characteristic function in reductive deamination.
Fur4 methyltransferase-catalyzed reaction
Compound 8, detected in the sequential reaction of Fur5 and Fur6, also accumulated in the culture medium of S. albus G153/pWFQΔfur4, which was prepared by deleting the fur4 gene from pWFQ (Fig. S28†). On the basis of this result, we hypothesized that the substrate of Fur4, the SAM-dependent O-methyltransferase homolog among the Fur enzymes necessary for conversion to 4, is also a reduced form of 8. To test this hypothesis, we incubated Fur4 with 8 in the presence of the reducing agent dithionite, as previously performed with Fur6. HPLC analysis revealed that significant quantities of 4 were generated in the reaction mixture, indicating that 3-methyl PHN (10), a reduced form of 8, is the authentic substrate for Fur4 (Fig. S29†). This result demonstrates that 4 is produced by aerobic oxidation of 2-methoxy-3-methyl PHN (11), a reaction product catalyzed by Fur4.Fur7 prenyltransferase-catalyzed reaction
Similar to the methylation reactions catalyzed by Fur6 and Fur4, prenylation catalyzed by Fur7 was greatly enhanced when the substrate was in the reducing state (Fig. S30†), with 4 reduced to 2-methoxy-3-methyl PHN (11) by the action of dithionite. Although the quinone form intermediate 4 has previously been proposed as a substrate for Fur7, this result reveals that the authentic substrate of Fur7 is 11, the reduced form of 4, and the reaction product of Fur7 is 6-prenyl-2-methoxy-3-methyl PHN, which is subsequently oxidized to Fur-P1.21 We suspect that in previous studies, a large amount of Fur7 was incubated with 4 over an extended reaction period; therefore, prenylation of 4, which is a side reaction, was observed.21Fur21-catalyzed reaction
We determined that Fur4 and Fur6 are responsible for the two methylation reactions in furaquinocin biosynthesis, but, curiously, another methyltransferase homolog, Fur21, is present in the fur cluster. Therefore, we hypothesized that Fur21 has activities other than methylation. Finally, experiments with our reductive in vitro reaction system revealed that Fur21 catalyzes the cyclization of a reduced form of Fur-P1 to produce a reduced form of furaquinocin C (Fig. 6). Fur21 is a homolog of SAM-dependent methyltransferases but does not require SAM for this cyclization reaction (Fig. 6). Several homologs of SAM-dependent methyltransferases catalyze cyclization reactions, but the reaction properties of Fur21 differ from those of the other homologs.39,40 The cyclization reaction catalyzed by TleD requires methylation by SAM, whereas cyclization by Fur21 does not require such methylation.28,41 SlnM is similar to Fur21 in that it does not require methylation but differs from Fur21 in that SAM is required for its activity. SlnM activity is preserved by the SAM analog sinefungin but inhibited by SAH, indicating that the positive charge of SAM is essential for SlnM activity.42 Another example of an enzyme in which the positive charge of SAM is important for activity is LepI. LepI is a SAM-dependent multifunctional enzyme that catalyzes the ambimodal Diels–Alder/hetero-Diels–Alder reaction and retro-Claisen rearrangement.43,44 SpnF is also a SAM-dependent methyltransferase-like enzyme that catalyzes cyclization, but its cyclization pattern is a [4 + 2] cycloaddition reaction (Fig. S31†).45 The reaction catalyzed by Fur21 can be regarded as an intramolecular hydroalkoxylation of an alkene, which is a challenge in stereoselective organic chemical synthesis.46 PhnH from Penicillium herquei NRRL 1040 is the only enzyme reported to catalyze intramolecular hydroalkoxylation, similar to Fur21.47,48 PhnH is a small protein composed of approximately 150 amino acid residues belonging to the DUF3237 superfamily and does not show sequence similarity with Fur21 (Fig. S32A†). The cyclization mechanism catalyzed by Fur21 is also intriguing from the perspective of a novel noncanonical terpenoid cyclization reaction.3,19,49,50 Although further biochemical and structural analyses of Fur21 are needed, Fur21-catalyzed stereoselective intramolecular hydroalkoxylation of an alkene may be mediated by an acid–base catalyzed mechanism proposed in the PhnH-catalyzed reaction (Fig. S32B†).47,48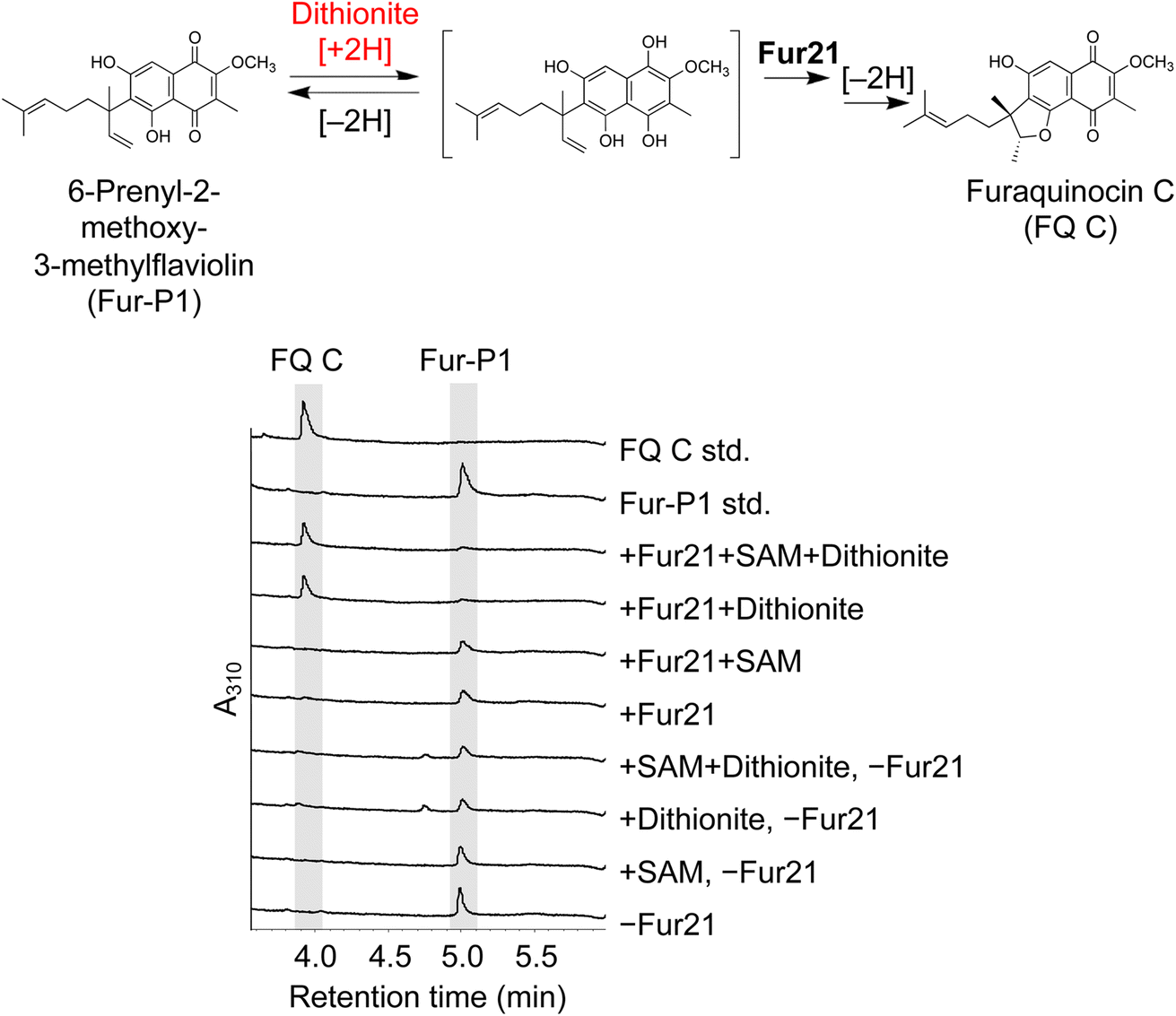 | ||
| Fig. 6 Cyclization by Fur21. LC-UV/VIS-MS spectra of Fur21. Cyclization by Fur21 proceeded under reduced conditions. The assay conditions are described in the ESI.†A310, absorbance at 310 nm. Under the +Fur21 + dithionite condition, the substrate Fur-P1 was completely consumed and converted to Furaquinocin C. The cyclization occurred in a SAM-independent manner. | ||
Conclusion
We have shown that the biosynthetic pathway of the THN-derived meroterpenoid furaquinocin involves the reductive deamination of 8-AF (3) and intramolecular hydroalkoxylation-mediated cyclization. This study highlights the importance of the amino group of 3 in the formation of a hydroquinone intermediate via diazotization common to the biosynthesis of THN-derived meroterpenoids such as furaquinocin and naphterpin. The amino group of 3 is a prerequisite for the formation of 8-diazoflaviolin (5) via diazotization. The resulting diazo group elevates the redox potential of the quinone moiety of 5 to facilitate the formation of the key hydroquinone intermediate, PHN (9) via PND (6). Compound 9 is critical for the formation of the subsequent hydroxyquinone intermediates of furaquinocin biosynthesis. In the later stage of the biosynthetic pathway after 9, two methylation events regulate the position of geranylation, which is a prerequisite for cyclization in furaquinocin biosynthesis. Our discovery regarding furaquinocin biosynthesis through the reduced hydroquinone intermediates led to the revision of the previously proposed model for THN-derived furaquinocin biosynthesis through the oxidized quinone intermediates.20,21Data availability
The data supporting this article have been included as part of the ESI.† This study was carried out using publicly available data from DDBJ nucleotide sequence database at https://www.ddbj.nig.ac.jp/index-e.html with AB212624.Author contributions
TN – conceptualization, investigation, formal analysis, methodology, visualization, writing-original draft, funding acquisition. FZ – conceptualization, formal analysis, software, writing – original draft. YM – conceptualization, formal analysis, supervision. HY – investigation. KK – investigation. RN – writing-original draft, writing – review & editing, supervision. TTo – investigation, formal analysis, writing – original draft, writing – review & editing. TTe – conceptualization, writing – review & editing, supervision, funding acquisition. MS – supervision. MN – supervision. TK – conceptualization, writing – review & editing, supervision, project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported in part by the grants from JSPS KAKENHI (16H06453 and 22H05120 to T. K. and 22H05126 to T. Te). This research was partially supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Numbers JP21am0101070 and JP23ama121027. T. N. was supported by a Grant-in-Aid for JSPS fellows (19J22569).References
- T. Kuzuyama, J. Antibiot., 2017, 70, 811–818 CrossRef CAS PubMed.
- L. A. M. Murray, S. M. K. McKinnie, B. S. Moore and J. H. George, Nat. Prod. Rep., 2020, 37, 1334–1366 RSC.
- J. D. Rudolf, T. A. Alsup, B. Xu and Z. Li, Nat. Prod. Rep., 2021, 38, 905–980 RSC.
- K. Komiyama, S. Funayama, Y. Anraku, M. Ishibashi, Y. Takahashi and S. Omura, J. Antibiot., 1990, 43, 247–252 CrossRef CAS PubMed.
- M. Ishibashi, S. Funayama, Y. Anraku, K. Komiyama and S. Omura, J. Antibiot., 1991, 44, 390–395 CrossRef CAS PubMed.
- K. Shin-ya, S. Imai, K. Furihata, Y. Hayakawa, Y. Kato, G. D. Vanduyne, J. Clardy and H. Seto, J. Antibiot., 1990, 43, 444–447 CrossRef CAS PubMed.
- P. Sedmera, S. Pospíšil and J. Novák, J. Nat. Prod., 1991, 54, 870–872 CrossRef CAS.
- K. Shiomi, H. Nakamura, H. Naganawa, K. Isshiki, T. Takeuchi and H. Umezawa, J. Antibiot., 1986, 39, 494–501 CrossRef CAS PubMed.
- S. Isogai, M. Nishiyama and T. Kuzuyama, Bioorg. Med. Chem. Lett., 2012, 22, 5823–5826 CrossRef CAS PubMed.
- T. Noguchi, S. Isogai, T. Terada, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2022, 144, 5435–5440 CrossRef CAS PubMed.
- Z. D. Miles, S. Diethelm, H. P. Pepper, D. M. Huang, J. H. George and B. S. Moore, Nat. Chem., 2017, 9, 1235–1242 CrossRef CAS PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 CrossRef CAS PubMed.
- H. Li, Y. Sun, Q. Zhang, Y. Zhu, S.-M. Li, A. Li and C. Zhang, Org. Lett., 2015, 17, 306–309 CrossRef CAS PubMed.
- H. Li, Q. Zhang, S. Li, Y. Zhu, G. Zhang, H. Zhang, X. Tian, S. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2012, 134, 8996–9005 CAS.
- Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CAS.
- S. Li, A. N. Lowell, F. Yu, A. Raveh, S. A. Newmister, N. Bair, J. M. Schaub, R. M. Williams and D. H. Sherman, J. Am. Chem. Soc., 2015, 137, 15366–15369 CAS.
- Q. Zhu and X. Liu, Angew. Chem., Int. Ed., 2017, 56, 9062–9066 CAS.
- R. M. Hohlman and D. H. Sherman, Nat. Prod. Rep., 2021, 38, 1567–1588 RSC.
- Z. Wang, T. A. Alsup, X. Pan, L.-L. Li, J. Tian, Z. Yang, X. Lin, H.-M. Xu, J. D. Rudolf and L.-B. Dong, Chem. Sci., 2025, 16, 310–317 CAS.
- T. Kawasaki, Y. Hayashi, T. Kuzuyama, K. Furihata, N. Itoh, H. Seto and T. Dairi, J. Bacteriol., 2006, 188, 1236–1244 CrossRef CAS PubMed.
- T. Kumano, T. Tomita, M. Nishiyama and T. Kuzuyama, J. Biol. Chem., 2010, 285, 39663–39671 CrossRef CAS PubMed.
- Y. Sugai, Y. Katsuyama and Y. Ohnishi, Nat. Chem. Biol., 2016, 12, 73–75 CrossRef CAS PubMed.
- J. Chen, X. Xie, J. Liu, Z. Yu and W. Su, React. Chem. Eng., 2022, 7, 1247–1275 RSC.
- S. Kawai, R. Hagihara, K. Shin-Ya, Y. Katsuyama and Y. Ohnishi, Angew. Chem., Int. Ed., 2022, 61, e202211728 CrossRef CAS PubMed.
- J. R. Greenwood, D. Calkins, A. P. Sullivan and J. C. Shelley, J. Comput.-Aided Mol. Des., 2010, 24, 591–604 CrossRef CAS PubMed.
- J. C. Shelley, A. Cholleti, L. L. Frye, J. R. Greenwood, M. R. Timlin and M. Uchimaya, J. Comput.-Aided Mol. Des., 2007, 21, 681–691 CrossRef CAS PubMed.
- M. F. Hegazi, R. T. Borchardt and R. L. Schowen, J. Am. Chem. Soc., 1976, 98, 3048–3049 CrossRef CAS PubMed.
- S. M. Husain, M. A. Schätzle, S. Lüdeke and M. Müller, Angew. Chem., Int. Ed., 2014, 53, 9806–9811 CrossRef CAS PubMed.
- N. Singh, S. J. Maniscalco and H. F. Fisher, J. Biol. Chem., 1993, 268, 21–28 CrossRef CAS.
- M. Ozeir, L. Pelosi, A. Ismail, C. Mellot-Draznieks, M. Fontecave and F. Pierrel, J. Biol. Chem., 2015, 290, 24140–24151 CrossRef CAS PubMed.
- L. K. B. Martinelli, R. G. Ducati, L. A. Rosado, A. Breda, B. P. Selbach, D. S. Santos and L. A. Basso, Mol. Biosyst., 2011, 7, 1289–1305 RSC.
- A. J. Waldman and E. P. Balskus, J. Org. Chem., 2018, 83, 7539–7546 CrossRef CAS.
- G.-L. Ma, H. Candra, L. M. Pang, J. Xiong, Y. Ding, H. T. Tran, Z. J. Low, H. Ye, M. Liu, J. Zheng, M. Fang, B. Cao and Z.-X. Liang, J. Am. Chem. Soc., 2022, 2022, 1622–1633 CrossRef.
- S. Kawai, Y. Sugaya, R. Hagihara, H. Tomita, Y. Katsuyama and Y. Ohnishi, Angew. Chem., Int. Ed., 2021, 60, 10319–10325 CrossRef CAS PubMed.
- N. Scherbak, Å. Strid and L. A. Eriksson, Chem. Phys. Lett., 2005, 414, 243–247 CrossRef CAS.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed.
- K. Naiki, J. Fiber Sci. Technol., 1960, 16, 251–264 Search PubMed.
- R. Munday, in Methods in Enzymology, Academic Press, 2004, vol. 382, pp. 364–380 Search PubMed.
- Q. Sun, M. Huang and Y. Wei, Yao Xue Xue Bao, 2021, 11, 632–650 CAS.
- Y.-H. Lee, D. Ren, B. Jeon and H.-W. Liu, Nat. Prod. Rep., 2023, 40, 1521–1549 RSC.
- T. Awakawa, J. Nat. Med., 2021, 75, 467–474 CrossRef CAS PubMed.
- C. Jiang, Z. Qi, Q. Kang, J. Liu, M. Jiang and L. Bai, Angew. Chem., Int. Ed., 2015, 127, 9225–9228 CrossRef.
- M. Ohashi, F. Liu, Y. Hai, M. Chen, M. C. Tang, Z. Yang, M. Sato, K. Watanabe, K. N. Houk and Y. Tang, Nature, 2017, 549, 502–506 CrossRef PubMed.
- Y. Cai, Y. Hai, M. Ohashi, C. S. Jamieson, M. Garcia-Borras, K. N. Houk, J. Zhou and Y. Tang, Nat. Chem., 2019, 11, 812–820 CrossRef CAS PubMed.
- C. D. Fage, E. A. Isiorho, Y. Liu, D. T. Wagner, H. W. Liu and A. T. Keatinge-Clay, Nat. Chem. Biol., 2015, 11, 256–258 CrossRef CAS PubMed.
- J. L. Kennemur, R. Maji, M. J. Scharf and B. List, Chem. Rev., 2021, 121, 14649–14681 CrossRef CAS PubMed.
- S. S. Gao, M. Garcia-Borràs, J. S. Barber, Y. Hai, A. Duan, N. K. Garg, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2017, 139, 3639–3642 CrossRef CAS PubMed.
- Y. Feng, X. Yu, J. W. Huang, W. Liu, Q. Li, Y. Hu, Y. Yang, Y. Chen, J. Jin, H. Li, C. C. Chen and R. T. Guo, Biochem. Biophys. Res. Commun., 2019, 516, 801–805 CrossRef CAS PubMed.
- J. D. Rudolf and C.-Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08319a |
| This journal is © The Royal Society of Chemistry 2025 |