
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the electrochemical redox-mediated mechanism of oxygen evolution on cobalt sites by hydroxide ion coupling†
Wenjuan
Song‡
a,
Xiaoyue
Duan‡
a,
Poe Ei
Phyu Win
a,
Xiang
Huang
*c and
Jiong
Wang
 *ab
*ab
aInnovation Center for Chemical Science, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215006, P. R. China. E-mail: wangjiong@suda.edu.cn
bJiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, P. R. China
cQuantum Science Center of Guangdong-Hong Kong-Macao Greater Bay Area (Guangdong), Shenzhen 518045, China. E-mail: huangxiang@quantumsc.cn
First published on 8th April 2025
Abstract
Heterogeneous molecular catalysts (HMCs) with cobalt (Co) active sites are potent for the electrochemical oxygen evolution reaction (OER) in energy conversion applications. Such catalysts typically operate through the classical redox-mediated mechanism, where dynamic equilibria of Co2+/3+ and Co3+/4+ redox states are present before and throughout the OER cycle. However, the generation of low-valent Co2+ sites is disadvantageous for catalysis. To address this, sulfate groups embedded in graphene were developed to link a model Co-2,2′-bipyridine complex, resulting in the synthesis of a novel Co-based HMC that generates a specific CoN2O4S1 coordination moiety. These molecular Co sites were induced to oxidize from +2 to +3 oxidation state at open-circuit conditions, due to their proton-coupled electron transfer nature. This process ultimately eliminated the generation of the Co2+ state from its redox equilibrium and efficiently improved the turnover frequencies of Co sites toward OER, showing a two-order dependence on the concentrations of OH− ions. This work provides a novel mechanistic perspective for the rational design of high-performance HMCs.
1 Introduction
The electrochemical conversion of renewable energy into chemical fuels at ambient conditions provides a potential approach for alleviating various environmental issues raised by the combustion of fossil fuels, thereby building a clean and sustainable energy system.1–4 However, the anodic oxygen evolution reaction (OER), commonly involved in electrochemical energy conversion, contains multiple proton-coupled electron transfer (PCET) steps, leading to large barriers that limit the efficiency of energy conversion.5,6 Cobalt (Co)-based materials have been widely identified as potential OER electrocatalysts.7–12 The Co active sites generally exhibit octahedral coordination geometry and follow a redox-mediated mechanism to operate OER. Prior to a catalytic cycle, Co sites exist in a low oxidation state (i.e., +2). As the turnover condition commences, these sites are oxidized to higher oxidation states, coupled with OH−/H+ transfer (i.e., Co3+/4+-oxo species).11,13 As the redox couples of Co2+/3+ and Co3+/4+ are quasireversible, they result in dynamic equilibria among Co2+, Co3+, and Co4+ states. Additionally, the desorption of oxygen species from the high-valent Co sites during the OER could drive reduction. This phenomenon can lead to the occurrence of Co2+ states throughout an OER cycle, which is disadvantageous for oxidizing oxygen species to release O2, interrupting the whole cycle.14,15 Moreover, the redox potentials of Co2+/3+ for generating high-valent Co sites dictate the OER overpotentials, placing an intrinsic limit on catalytic enhancement.16,17 Thus, it is critically important to promote the oxidation of Co2+-to-Co3+ conversion or to stabilize the Co sites at high oxidation states to enhance OER performance.Heterogeneous molecular catalysts (HMCs) possess molecularly well-defined active sites, benefiting the precise optimization of the redox behaviors and related catalytic performance of active sites.18–20 Such catalysts also have solid states, facilitating device assembly for scaled-up applications.21,22 Sulfur (S)-doped graphene has been identified as a potential support for linking molecules with S heteroatom-based groups and developing HMCs.11,23,24 Due to the relatively larger size and greater electronegativity of S atoms compared to those of C atoms, the incorporation of S atoms leads to local geometric distortion and polarization of graphene. The S heteroatoms exist in multiple configurations as sulfides and their oxidized derivatives, accompanied by electron-rich or deficient states.25–27 These properties can significantly influence the electronic migration capabilities, thereby tuning the oxidation states or redox properties of linked molecular catalysts. This, in turn, can optimize the stability of high-valent Co sites and improve the associated OER performance.
In this study, sulfur-doped graphene was synthesized by a hydrothermal method, in which sulfate groups were constructed to link a model molecule of Co-2,2′-bipyridine (CoN2) complexes. This linking strategy forces the linked molecular Co sites to oxidize from +2 to +3 oxidation state at open-circuit conditions due to their intrinsic PCET property. The Co sites interact with OH− ions, inducing electron transfer that eliminates the electrochemical Co2+/3+ redox processes. The Co sites are spontaneously stabilized at +3 oxidation states and higher, ultimately avoiding the occurrence of the Co2+ oxidation state throughout the OER cycle. Accordingly, the turnover frequencies (TOFs) of Co sites for the electrocatalysis of OER are intrinsically improved.
2 Results and discussion
2.1 Structural characterizations
CoN2 was synthesized by reacting Co(NO3)2·6H2O with 2,2′-bipyridine in methanol at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and subsequently isolated from the solvent (Fig. S1†). Graphene oxide (GO) was synthesized using a modified Hummers' method28 and was reduced under hydrothermal conditions with thioacetamide as a modifier to derive S-doped graphene (S1G, Table S1†). CoN2 was then immobilized onto S1G to obtain a heterogeneous cobalt electrocatalyst denoted as S1G-CoN2 (all details are provided in the ESI†). Inductively coupled plasma optical emission spectra (ICP-OES) indicate that the Co content of S1G-CoN2 is 3.0 × 10−6 mol mgS1G−1 (Table S2†). Scanning/transmission electron microscopy (SEM/TEM, Fig. 1a–d) of S1G-CoN2 shows that it formed Co-based clusters over graphene with an average size of 5 nm. Aberration-corrected high-angle annular darkfield scanning transmission electron microscopy (AC HAADF-STEM) images revealed substantial dispersion of molecular Co sites among the clusters (Fig. 1e–g, S3 and S4†). The Co clusters appear to be assemblies of Co complexes, rather than Co oxides or hydroxides, as suggested by Raman spectra and X-ray diffraction (XRD) patterns (Fig. S5†). Ultraviolet-visible (UV-vis, Fig. S6†) spectra demonstrate a slight shift in the π–π* transition from S1G to S1G-CoN2, indicating electron conjugation of S1G with 2,2′-bipyridine. According to X-ray photoelectron spectroscopic (XPS) surveys (Fig. 1h and S7†), the Co 2p electron levels of S1G-CoN2 present two main peaks at 781.2 eV (2p3/2) and 797.4 eV (2p1/2), with simultaneous satellite peaks occurring at 786.4 eV and 803.6 eV, indicating the +2 oxidation state of Co sites. This is consistent with the results of electron paramagnetic resonance measurement (EPR, Fig. S8†). The N 1s electron levels of CoN2 show two peaks at 400.4 eV and 406.9 eV, assigned to the ligated 2,2′-bipyridine, and NO3− ions, respectively (Fig. 1i). However, a peak at 399.8 eV emerges for S1G-CoN2. Combined with the single-crystal structure of CoN2, this variation indicates the replacement of ligated NO3− ions by other ligands. Characteristic peaks of S 2p electron levels occur at 163.4 eV and 167.4 eV for carbonized and oxidized sulfur species, respectively25–27,29 (Fig. 1j), which may be involved in the ligand replacement.
:1 and subsequently isolated from the solvent (Fig. S1†). Graphene oxide (GO) was synthesized using a modified Hummers' method28 and was reduced under hydrothermal conditions with thioacetamide as a modifier to derive S-doped graphene (S1G, Table S1†). CoN2 was then immobilized onto S1G to obtain a heterogeneous cobalt electrocatalyst denoted as S1G-CoN2 (all details are provided in the ESI†). Inductively coupled plasma optical emission spectra (ICP-OES) indicate that the Co content of S1G-CoN2 is 3.0 × 10−6 mol mgS1G−1 (Table S2†). Scanning/transmission electron microscopy (SEM/TEM, Fig. 1a–d) of S1G-CoN2 shows that it formed Co-based clusters over graphene with an average size of 5 nm. Aberration-corrected high-angle annular darkfield scanning transmission electron microscopy (AC HAADF-STEM) images revealed substantial dispersion of molecular Co sites among the clusters (Fig. 1e–g, S3 and S4†). The Co clusters appear to be assemblies of Co complexes, rather than Co oxides or hydroxides, as suggested by Raman spectra and X-ray diffraction (XRD) patterns (Fig. S5†). Ultraviolet-visible (UV-vis, Fig. S6†) spectra demonstrate a slight shift in the π–π* transition from S1G to S1G-CoN2, indicating electron conjugation of S1G with 2,2′-bipyridine. According to X-ray photoelectron spectroscopic (XPS) surveys (Fig. 1h and S7†), the Co 2p electron levels of S1G-CoN2 present two main peaks at 781.2 eV (2p3/2) and 797.4 eV (2p1/2), with simultaneous satellite peaks occurring at 786.4 eV and 803.6 eV, indicating the +2 oxidation state of Co sites. This is consistent with the results of electron paramagnetic resonance measurement (EPR, Fig. S8†). The N 1s electron levels of CoN2 show two peaks at 400.4 eV and 406.9 eV, assigned to the ligated 2,2′-bipyridine, and NO3− ions, respectively (Fig. 1i). However, a peak at 399.8 eV emerges for S1G-CoN2. Combined with the single-crystal structure of CoN2, this variation indicates the replacement of ligated NO3− ions by other ligands. Characteristic peaks of S 2p electron levels occur at 163.4 eV and 167.4 eV for carbonized and oxidized sulfur species, respectively25–27,29 (Fig. 1j), which may be involved in the ligand replacement.
 | ||
| Fig. 1 TEM images of S1G-CoN2 at low (a) and high (b) resolution. (c) A TEM image in a bright field indicates the Co species of S1G-CoN2, and the corresponding elemental mapping images by Energy-dispersive X-ray spectroscopy (EDS) (d). (e) An AC HAADF-STEM image of S1G-CoN2, and the enlarged images of indicate the molecular clusters (f) and dispersed molecules (g) of Co complexes. XPS surveys of Co 2p (h), N 1s (i) and S 2p (j) core electron levels. | ||
X-ray absorption near-edge spectra (XANES) were employed to verify the oxidation states and coordination spheres of the as-fabricated S1G-CoN2. As shown in Fig. 2a, the Co sites of CoN2 and S1G-CoN2 are both approximately in the +2 oxidation state, as indicated by the rising edges. The coordination symmetry of S1G-CoN2 is more distorted compared to that of CoN2, as demonstrated by the decreased intensity of the pre-edge peak at 7709.7 eV. This implies a ligand change of Co sites, as confirmed by the above XPS surveys. Fourier transform (FT) extended X-ray absorption fine structure (EXAFS, Fig. 2b) data of CoN2 before and after the interaction with S1G show that the Co sites maintain one dominant peak at approximately 1.59 Å in the first shell of Co–N/O scattering paths, indicating that CoN2 did not decompose into Co oxides/hydroxides. In the quantitative least-squares, EXAFS fitting analysis of S1G-CoN2 (Fig. 2c, d, S9 and Table S3†), another Co–S bond at 2.2 Å was identified in addition to the Co–N/O scattering paths. Such fitted bonding data are consistent with previous studies.30,31 Accordingly, the coordination sphere of S1G-CoN2 is considered a CoN2O4S1 moiety.
 | ||
| Fig. 2 (a) Normalized Co K-edge XANES data of S1G-CoN2, CoN2 and reference samples. (b) The corresponding FT EXAFS data. Fitted data of Co K-edge EXAFS in R (c) and k (d) spaces of S1G-CoN2. | ||
2.2 Electrochemical OER performance
To evaluate the OER performance of the catalysts, a typical three-electrode system was applied under ambient conditions in 1 M KOH, combined with linear scanning voltammograms (LSVs) and cyclic voltammograms (CVs) (all potentials refer to the reversible hydrogen electrode, RHE, unless specified). During LSV measurements, the working electrode was rotated to counteract bubble accumulation. As control experiments, GO-modified graphene (G) and nitrogen-doped graphene (NG) were synthesized to immobilize CoN2 in the same manner as for S1G-CoN2, resulting in G-CoN2 and NG-CoN2, respectively. Among these samples, S1G-CoN2 exhibited optimal performance with an onset potential of 1.6 V (defined as the potential corresponding to a current density of 10 mA cm−2), which is significantly lower than that of G-CoN2 and NG-CoN2 (Fig. 3a). The Tafel slope of S1G-CoN2 was determined to be 59 mV dec−1, suggesting one electron reversible transfer prior to the rate-limiting step of the OER (Fig. 3b).32 Furthermore, the coordination spheres of Co sites were ligated with 2 and 3 equivalents of 2,2′-bipyridine to achieve CoN4 and CoN6 complexes, respectively, which were immobilized onto different graphene supports (Fig. S10†). S1G-CoN4 remained active and superior to G-CoN4 and NG-CoN4. However, CoN6 exhibited minimal OER activity on any supports, as the Co sites were fully covered by strong ligands, preventing efficient chemisorption of OH− ions. | ||
| Fig. 3 (a) LSVs of G-CoN2, NG-CoN2 and S1G-CoN2, 1 M KOH, 10 mV s−1 scan rate, 1600 rpm. The potentials are compensated by 95% iR loss. (b) The corresponding Tafel slopes derived from LSVs. (c) TOFsurf of Co sites for catalyzing OER. | ||
It was verified that the Co sites served as the active sites for the OER, where OER electrocatalysis was achieved by replacing partial labile ligands with OH− ions. The TOFs of the Co sites were analyzed by normalizing the OER currents with the surface amounts of Co sites on the electrodes, yielding TOFsurf (Fig. 3c and Table S4†). The TOFsurf of S1G-CoN2 reached 0.38 s−1 at an overpotential of 0.35 V, which is higher than those of G-CoN2 and NG-CoN2 and other reported Co-based OER catalysts (Table S5†). The faradaic efficiency (FE) for O2 production on S1G-CoN2 was determined using in-line gas chromatography, which showed values between 98.5 and 99.3% at 10 mA cm−2 (Fig. S11†). A chronoamperometric curve indicates the high durability of S1G-CoN2 for OER catalysis (Fig. S12†). The XRD patterns, Raman spectra, and XPS surveys (Fig. S13–S15†) of S1G-CoN2 did not exhibit significant variations before and after the stability test. Additionally, after the stability test, the electrode was immersed in an ethylenediaminetetraacetic sodium (EDTA) solution for 20 h (Fig. S16†).19 The resultant solution was characterized by UV-vis spectroscopy, showing the characteristic absorbance of the EDTA-Co complex at 540 nm. Under the same conditions, cobalt oxides/hydroxides could not react with EDTA. All these datas suggest that S1G-CoN2 does not decompose into possible oxides or hydroxides during long-term measurements.
To clarify the pH dependence of OER electrocatalysis on the Co sites, CVs of S1G-CoN2 were conducted across an expanded alkaline range (pH 12–14, Fig. S17†). Clear redox features appeared between 0.4 and 0.8 V (vs. Hg/HgO) prior to OER potentials, which are assigned to the Co3+/4+ couple. The Pourbaix slope was fitted to be 59 mV pH−1, representing a typical 1H+–1e− coupled redox process to commence OER (Fig. 4a). The potential dependence of the OER current densities at 0.2 mA cm−2 was fitted to be 135 mV pH−1 (Fig. 4b and S18†). The potential dependence of pH was deconvoluted based on the following eqn (1):
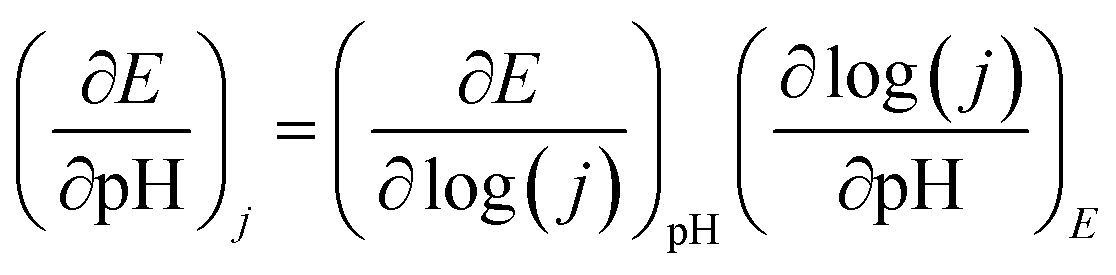 | (1) |
 | ||
| Fig. 4 Redox potentials of Co3+/4+ couple (a), OER potentials (b), and OER current densities (c) as a function of solution pH collected on S1G-CoN2. (d) In situ ATR-FTIR spectra of S1G-CoN2 at different potentials in 1 M KOH. (e) A proposed OER cycle on the Co sites of S1G-CoN2. | ||
The Tafel slope of 59 mV dec−1 on S1G-CoN2 thus establishes a two-order dependence of log(j) on pH. To verify this result, we measured the pH dependence of OER current density using the galvanostatic method, obtaining a linear slope of 2.09 dec pH−1 (Fig. 4c and S19†), reflecting the two-order dependence of OER on the concentration of OH− ions. The OER on S1G-CoN2 was further investigated by in situ attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) measurements (Fig. 4d). At 1.2 V and onward, a wide peak emerged at 840 cm−1, assigned to the Co3+–OH/Co4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species.33 Combining this with the Pourbaix analysis of the Co3+/Co4+ couple, it is inferred that prior to the OER, one Co3+ site of S1G-CoN2, connected with one OH− ion, decouples from one H+ to generate the Co4+O species. Above 1.5 V, another absorption peak appeared at 1167 cm−1, assigned to *OOH species, verifying the O–O bonding for the final evolution of O2 on the Co sites (Fig. 4e).34,35
O species.33 Combining this with the Pourbaix analysis of the Co3+/Co4+ couple, it is inferred that prior to the OER, one Co3+ site of S1G-CoN2, connected with one OH− ion, decouples from one H+ to generate the Co4+O species. Above 1.5 V, another absorption peak appeared at 1167 cm−1, assigned to *OOH species, verifying the O–O bonding for the final evolution of O2 on the Co sites (Fig. 4e).34,35
The correlation of Co oxidation states with OER activity was investigated. To probe the impacts of S doping on the oxidation states of the Co sites and related OER activity, we used various S reagents (i.e., mercaptoacetic acid, benzylthiol, and thioanisole) to incorporate S heteroatoms into graphene, resulting in S2G, S3G, and S4G (Fig. S20†) to immobilize CoN2, leading to S2G/S3G/S4G-CoN2. Their Co contents were all approximately at 10−6 mol mgsupport−1 magnitude. However, S2G/S3G/S4G-CoN2 exhibited inferior OER activities compared to S1G-CoN2, as indicated by the electrocatalytic currents and TOFs (Fig. 5a and b). Notably, S2G/S3G-CoN2 with relatively low OER activity, exhibited significant Co2+/3+ redox at 1.0–1.1 V, while S1G/S4G-CoN2, with relatively high OER activity, showed weak Co2+/3+ redox (Fig. 5c). The Co2+/3+ redox peaks were integrated to calculate the electroactive amounts of Co sites, which was then divided by the surface amounts of Co sites on the electrodes, as obtained by ICP-OES. This derived the Co redox responses for each catalyst, which inversely relate to the TOFsurf (Fig. 5d). This phenomenon can be attributed to: (i) the tethered Co sites having different degrees of electronic coupling with graphene, resulting in continuous electronic bands or finite electron energy levels or (ii) a fraction of Co2+ sites being converted into higher oxidation states due to the PCET nature, stabilized under alkaline conditions. As suggested by Surendranath et al.,36 over-strong electronic coupling could lead to an electron continuum at the interface, eliminating the Co2+/3+ and Co3+/4+ redox peaks of connected complexes. However, this is unlikely as S1G/S4G-CoN2 exhibited clear Co3+/4+ redox at higher potentials, which is even more significant than that of S2G/S3G-CoN2 (Fig. 5e). To verify the latter case, we conducted KOH titrations under a UV-vis spectral monitor.
 | ||
| Fig. 5 (a) LSVs of S1G-CoN2, S2G-CoN2, S3G-CoN2 and S4G-CoN2, 1 M KOH, 10 mV s−1, 1600 rpm. The potentials were compensated by 95% iR loss. (b) TOFsurf of Co sites. (c) CVs of Co2+/3+ redox, 1 M KOH, 50 mV s−1. (d) TOFsurf at an overpotential of 0.4 V correlating with the Co2+/3+ redox responses. (e) CVs of Co2+/3+ and Co3+/4+ redox, 1 M KOH, 50 mV s−1. | ||
The metal-to-ligands charge transfer (MLCT) band of S1G-CoN2 exhibited a blue shift with the addition of KOH solution, suggesting ligand replacement at the Co sites by the nucleophilic attack of OH− ions (Fig. S21†).11,37 In contrast, S3G-CoN2 exhibited an insignificant shift in the MLCT region under the same conditions. Based on the surveys of Co 2p electron levels (Fig. S22†), the two main peaks shifted to 780.3 eV (2p3/2) and 795.1 eV (2p1/2), and the satellites disappeared after immersing S1G-CoN2 into KOH, indicating the increase in the oxidation state of the Co site to +3. Meanwhile, the Co 2p electron levels of S3G-CoN2 did not change upon immersion in KOH, indicating that the oxidation state of the Co sites remained at +2.
Next, we considered different possible structures of the Co sites of S1G-CoN2 based on the fitted EXFAS data, and S-based groups emerging on graphene, as verified by the XPS surveys (Fig. 6a and S23†). These include a CoN2 molecule anchored to an SO3 group (structure A, CoN2O3S1); an SO3 group and an S group (structure B, CoN2O3S2); and an SO3 group and an O group (structure C, CoN2O4S1). Note that S1G contains a small fraction of N dopants. To verify the impacts of N dopants, we intentionally constructed S, N co-doped graphene, on which CoN2 exhibited insignificant OER activity (Fig. S24†). The OER electrocatalysis was theoretically investigated according to the conventional four-electron mechanism using density functional theory (DFT) calculations.38 This mechanism includes four PCET steps, leading to the formation of three intermediates (i.e., *OH, *O, and *OOH). The calculated OER activities of the above Co sites are presented on a two-dimensional volcano plot, which contains four regions corresponding to the four electrochemical steps of OER (Fig. 6b).
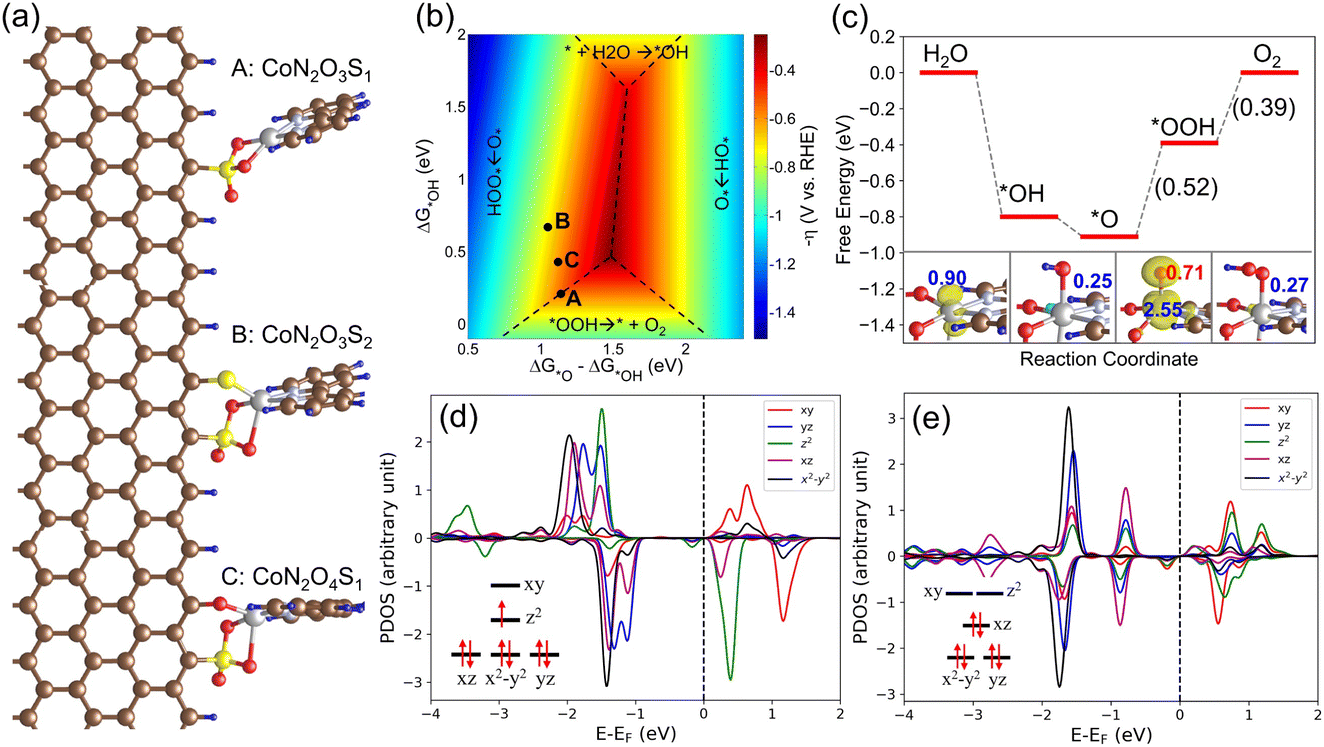 | ||
| Fig. 6 (a) Optimized structures of CoN2 anchoring on S and O terminated graphene edges. (b) A two-dimensional volcano plot for OER on Co sites. (c) The free energy diagram of OER on the Co site of CoN2O4S1 at U = 1.23 V vs. RHE. The reaction free energies of the last two steps are indicated in brackets. Density of states projected onto the d orbitals of the Co site in clean CoN2O4S1 (d) and that with one adsorbed *OH species (e). The insets shown in (c) are the spin densities of clean CoN2O4S1 and those adsorbed with species of *OH, *O, and *OOH. The isosurface value is 0.01e Bohr−3. The values colored in blue are the magnetic moment of Co site and that colored in red is the magnetic moment of adsorbed *O intermediate. The C, O, Co, S, H, and N atoms are represented by brown, red, gray, yellow, blue, and light blue balls, respectively. | ||
Under the equilibrium potential of OER, we found that water readily deprotonated to form *OH and *O species on the Co sites due to the exothermic nature of these reactions and the very low deprotonation barrier, as revealed before.39 However, the formation of *OOH species from *O is highly endothermic, serving as the potential rate-determining step. This is attributed to the strong bonding between the Co site and the OER intermediates. Specifically, the OER intermediates on structure A exhibit much stronger bonding compared to structures B and C, leading to an overpotential of 0.6 V (Fig. S25†). However, when the Co site bonds to one additional S ligand (structure B), the binding energies of the OER intermediates were weakened by 0.3–0.6 eV, increasing the overpotential to 0.76 V (Fig. S26†). Conversely, a suitable weakening in binding energies (0.1–0.2 eV) was observed on structure C, where the Co site was additionally bonded to one O ligand. It exhibited a low overpotential of 0.52 V (Fig. 6c).
To gain insights into the origin of the high activity of structure C, we analyzed the electronic structures of the Co site during the OER process based on the projected density of states (PDOS), spin density, and magnetic moments. In the pristine structure, the PDOS for the d orbitals of the Co site revealed seven occupied electrons at a low spin state (Fig. 6d), resulting in a magnetic moment of 0.9 μB for the Co site. Upon adsorbing one *OH species, the dz2 orbital was depopulated, reducing the magnetic moment of the Co site to 0.25 μB. A similar scenario was observed in the adsorption of *OOH species (Fig. S27†). These results indicate that the bonding between the Co site and the OER intermediates is highly ionic. Given the 3d74s2 configuration of the Co atom, we assigned the oxidation state of +2 to the Co site in structure C, and +3 to the Co site adsorbing *OH and *OOH species (Fig. 6e). Furthermore, we found that this observation aligns with the octet rule as *OH and *OOH species require one electron each to complete their valence electron shells.40 Therefore, it is reasonable to assign the oxidation state of the Co site to +4 when adsorbing one *O species, which was further verified through its magnetic moment of 2.55 μB, corresponding to an intermediate-spin state. Additionally, we observed that the adsorbed *O species has a magnetic moment of 0.71 μB, showing a radical character. This radical nature is reported to reduce the barrier for O–O formation.41,42 Taken together, our theoretical studies identified that the CoN2O4S1 site has excellent OER performance. Moreover, the formation of adsorbed *OH species on a Co site occurs readily at the OER equilibrium potential, indicating that the oxidation state of Co can easily change from +2 to +3 prior to OER, and the oxidation state of the Co sites varies between +3 and +4 during the OER cycle. This aligns closely with our experimental observations.
3 Conclusions
In summary, we demonstrated a novel strategy to enhance OER electrocatalysis by constructing molecular CoN2 linked with sulfur-doped graphene-based support. The linkages of S heteroatoms in a sulfate form resulted in a specific CoN2O4S1 coordination moiety, which exhibited a two-order dependence of the OER on the concentrations of OH− ions while affording a TOF of 0.38 s−1 at an overpotential of 0.35 V in 1 M KOH. While CoN2 loaded on common G and NG is nearly catalytically inert, we found that the as-derived Co sites can convert from +2 to +3 oxidation states through coupling with OH− ions under open-circuit conditions, taking advantage of the PCET nature of CoN2. This property ultimately prevents the emergence of Co2+ states and stabilizes Co sites at a high oxidation state, allowing for efficient progression of the entire OER catalytic cycle. Our results could enable the rational synthesis of precise HMCs, optimizing both reaction kinetics and energetic efficiency.Data availability
All data have been provided in the manuscript and the appendix ESI file.†Author contributions
J. W. conceived and supervised the project. W. S. carried out the synthesis and electrocatalytic measurements. J. W., W. S., and X. D. co-wrote the manuscript. X. H. performed the DFT calculations. P. E. P. W. helped with materials characterization and analysis. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (22002119), the National Natural Science Foundation of Jiangsu, China (BK20200261), and the Programs of Science and Technology of Suzhou, China (ZXL2021448, SYG202137). We sincerely thank Dr Pierre-Yves OLU from John Cockerill Hydrogen for insightful discussions.Notes and references
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS
.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS
- S. Iqbal, B. Safdar, I. Hussain, K. Zhang and C. Chatzichristodoulou, Adv. Energy Mater., 2023, 13, 2203913 CrossRef CAS
- J. Song, C. Wei, Z. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC
- H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS
- J. Yu, F. A. Garcés-Pineda, J. González-Cobos, M. Peña-Díaz, C. Rogero, S. Giménez, M. C. Spadaro, J. Arbiol, S. Barja and J. R. Galán-Mascarós, Nat. Commun., 2022, 13, 4341 CrossRef CAS PubMed
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS
- J. Wang, X. Ge, Z. Liu, L. Thia, Y. Yan, W. Xiao and X. Wang, J. Am. Chem. Soc., 2017, 139, 1878–1884 CrossRef CAS
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, K. Alexander, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, F. István, S. G. A. Mårten and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC
- B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2017, 8, 14839 CrossRef CAS PubMed
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS
- M. N. Jackson, C. J. Kaminsky, S. Oh, J. F. Melville and Y. Surendranath, J. Am. Chem. Soc., 2019, 141, 14160–14167 CrossRef CAS PubMed
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331–341 CrossRef CAS
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, eaap7970 CrossRef PubMed
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- B. Zhang, L. Fan, R. B. Ambre, T. Liu, Q. Meng, B. J. J. Timmer and L. Sun, Joule, 2020, 4, 1408–1444 CrossRef CAS
- J. Wang, X. Huang, S. Xi, J.-M. Lee, C. Wang, Y. Du and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed
- I.-Y. Jeon, S. Zhang, L. Zhang, H.-J. Choi, J.-M. Seo, Z. Xia, L. Dai and J.-B. Baek, Adv. Mater., 2013, 25, 6138–6145 CrossRef CAS PubMed
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a. Chen and S. Huang, ACS Nano, 2011, 6, 205–211 CrossRef
- H. L. Poh, P. Šimek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS
- M. Li, C. Liu, H. Zhao, H. An, H. Cao, Y. Zhang and Z. Fan, Carbon, 2015, 86, 197–206 CrossRef CAS
- S. M. A. M. Bouwens, F. B. M. Vanzon, M. P. Vandijk, A. M. Vanderkraan, V. H. J. Debeer, J. A. R. Vanveen and D. C. Koningsberger, J. Catal., 1994, 146, 375–393 CrossRef CAS
- T. H. M. Lau, X. Lu, J. Kulhavý, S. Wu, L. Lu, T.-S. Wu, R. Kato, J. S. Foord, Y.-L. Soo, K. Suenaga and S. C. E. Tsang, Chem. Sci., 2018, 9, 4769–4776 RSC
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed
- Y. Lin, L. Yu, L. Tang, F. Song, R. Schlögl and S. Heumann, ACS Catal., 2022, 12, 5345–5355 CrossRef CAS
- X. Li and A. A. Gewirth, J. Am. Chem. Soc., 2005, 127, 5252–5260 CrossRef CAS PubMed
- X. Chen, Q. Wang, Y. Cheng, H. Xing, J. Li, X. Zhu, L. Ma, Y. Li and D. Liu, Adv. Funct. Mater., 2022, 32, 2112674 CrossRef CAS
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. S. G. Ahlquist and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
- C. F. Dickens, C. Kirk and J. K. Nørskov, J. Phys. Chem. C, 2019, 123, 18960–18977 CrossRef CAS
- X. Huang, J. Wang, H. B. Tao, H. Tian and H. Xu, Chem. Sci., 2019, 10, 3340–3345 RSC
- Y. Ping, R. J. Nielsen and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 149–155 CrossRef CAS PubMed
- Z. Wang, W. A. Goddard and H. Xiao, Nat. Commun., 2023, 14, 4228 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01674f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |