
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbene-catalyzed enantioselective construction of a quasi-symmetrical spirocyclic hydroquinone with a minor chiral distinction†
Panlong
Ren
 a,
Qing
Zhao
a,
Yonggui Robin
Chi
*bc and
Tingshun
Zhu
*a
a,
Qing
Zhao
a,
Yonggui Robin
Chi
*bc and
Tingshun
Zhu
*a
aKey Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Chemistry, IGCME, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhutshun@mail.sysu.edu.cn
bState Key Laboratory of Green Pesticide, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Guiyang 550025, China. E-mail: robinchi@ntu.edu.sg
cSchool of Chemistry, Chemical Engineering, and Biotechnology, Nanyang Technological University, Singapore 637371, Singapore
First published on 15th April 2025
Abstract
Constructing a nearly symmetrical chiral center with tiny chiral differences is a challenging task in asymmetric synthesis. The natural antibiotic fredericamycin A (FDM-A), a representative example, has a unique structure with a quasi-symmetrical spirocyclic hydroquinone and remains difficult to chemically synthesize. Herein we developed an N-heterocyclic carbene–catalyzed enantioselective hydroquinone formation reaction with desymmetrization of spirocyclic cyclopentene-1,3-diones to construct these challenging structures. Using our method, the asymmetric synthesis of FDM-A (previously requiring a 26-step or 32-step synthesis) was shortened to 11 steps. Several analogs of FDM-A were also readily made. Moreover, a more challenging all-carbon quaternary chiral center with minimal differences (H vs. D) in a remote position (6 atoms away from the chiral center) was also constructed to investigate the performance of the extremely weakly chiral small molecule.
Introduction
Fredericamycin A (FDM-A) was isolated from Streptomyces griseus (FCRC-48) by Pandey and co-workers in 1981 (Fig. 1a).1a It is a hexacyclic quinone-based natural product with a novel spiro [4.4] nonane ring, not previously observed in any antibiotic connected with naphthoquinone or isoquinoline aromatic moieties.1b The all-carbon quaternary chiral center in FDM-A is a unique symmetrical structure, whose chirality is enabled by a methoxy substituent in a remote position (6 atoms away from the chiral carbon center). The biosynthesis of natural FDM-A probably relies on an enzymatic asymmetric epoxidation of the highly symmetrical benastatin core and the following stereospecific transformations.2 In comparison, the asymmetric chemical synthesis of this symmetrical chiral structure is much more challenging (Fig. 1b). Although several different strategies,3a including the aldol reaction,3b,c radical cyclization,3d–g Diels–Alder reaction,3h,i Tamura annulation3j,k and Hauser–Kraus annulation3l,m have been applied in the total synthesis of FDM-A since the 1990s, few of them can achieve the desired enantioselective control. In 1995, Boger and coworkers obtained both enantiomers by conducting a chiral separation of racemic FDM-A, but with a low separation factor (α = 1.14).3c The only two examples of asymmetric synthesis of FDM-A were reported by the Kita group, involving a 26-step (longest linear sequence, LLS) synthesis with a chiral sulfoxide auxiliary in 1999,3j,k and a 32-step (LLS) synthesis with enzymatic esterification and enantiospecific transformations in 2005.3i In comparison to the racemic synthesis, tedious steps (>15 steps) were required to achieve the enantioselective control. Our research group has continuous interest in building sophisticated aromatic cycles with asymmetric benzannulation.4 Herein we developed a carbene-catalyzed asymmetric hydroquinone formation reaction to synthesize this symmetrical structure with small differences in a remote position by desymmetrization of spirocyclic dienophiles. An 11-step (LLS) synthesis of FDM-A as well as facile synthesis of FDM-A analogs were achieved. Moreover, we obtained a more challenging structure with only H/D differences in the remote position, which can hardly be identified even in the enzyme catalysis or chiral separation. | ||
| Fig. 1 Enantioselective construction of quasi-symmetrical spirocyclic hydroquinone with a minor chiral distinction. | ||
Results and discussion
Our present study commenced with the 6-chloro phthalide 1a and spirocyclic 1,3-cyclopentenedione 2a (ref. 5) (spirocyclic core of FDM-A) used as model substrates (Table 1). With MYTsA (N-methylynetoluenesulfonamide)6 as the coupling reagent, triazolium salt C1 (ref. 4b) as the precatalyst, DABCO as the base and CH3CN as the solvent, the reaction proceeded smoothly at room temperature and furnished the product 3a in 65% yield with a 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 enantiomeric ratio (er) (Table 1, entry 1). Control tests showed that the absence of NHC catalyst or coupling reagent each totally deactivated the reaction (entry 2). Note the considerable difference between catalyst chosen in our reaction and the traditional examples for asymmetric NHC-organocatalysis.7N-Aryl-substituted triazolium salts such as C2 (ref. 4a and 8) or C3,9 which have been widely applied in asymmetric NHC-organocatalysis, were totally inactive in our reaction (entry 3). N-Phenyl triazolium catalyst C4 (ref. 10) showed acceptable enantioselectivity but with only 8% yield (entry 4). N-Benzyl triazolium salts, while easily synthesized from SN2 alkylation of triazole, were normally considered as unsuitable catalysts for asymmetric reactions11 due to the lack of sufficient steric hindrance. Possibly due to the relatively crowded transition state with the phthalide-type Breslow intermediate, the reaction showed remarkable enantioselectivity when we used the N-benzyl triazolium catalyst. Another N-benzyl catalyst, namely C5, also gave the desired product in good yield but with lower er (entry 5). Reactions with other bases such as DBU (entry 6) or Cs2CO3 (entry 7) showed slightly lower yields and enantioselectivities. Other solvents such as CH2Cl2 gave inferior results (entry 8, see the ESI† for details). Performing the reaction at 0 °C instead of 25 °C resulted in a slight enhancement of enantioselectivity (entry 9). Finally, the use of 4 Å molecular sieves to maintain a more anhydrous environment gave the optimized results, affording 3a in a 73% yield with a 97:3 er (entry 10).
:4 enantiomeric ratio (er) (Table 1, entry 1). Control tests showed that the absence of NHC catalyst or coupling reagent each totally deactivated the reaction (entry 2). Note the considerable difference between catalyst chosen in our reaction and the traditional examples for asymmetric NHC-organocatalysis.7N-Aryl-substituted triazolium salts such as C2 (ref. 4a and 8) or C3,9 which have been widely applied in asymmetric NHC-organocatalysis, were totally inactive in our reaction (entry 3). N-Phenyl triazolium catalyst C4 (ref. 10) showed acceptable enantioselectivity but with only 8% yield (entry 4). N-Benzyl triazolium salts, while easily synthesized from SN2 alkylation of triazole, were normally considered as unsuitable catalysts for asymmetric reactions11 due to the lack of sufficient steric hindrance. Possibly due to the relatively crowded transition state with the phthalide-type Breslow intermediate, the reaction showed remarkable enantioselectivity when we used the N-benzyl triazolium catalyst. Another N-benzyl catalyst, namely C5, also gave the desired product in good yield but with lower er (entry 5). Reactions with other bases such as DBU (entry 6) or Cs2CO3 (entry 7) showed slightly lower yields and enantioselectivities. Other solvents such as CH2Cl2 gave inferior results (entry 8, see the ESI† for details). Performing the reaction at 0 °C instead of 25 °C resulted in a slight enhancement of enantioselectivity (entry 9). Finally, the use of 4 Å molecular sieves to maintain a more anhydrous environment gave the optimized results, affording 3a in a 73% yield with a 97:3 er (entry 10).
|
|
|||
|---|---|---|---|
| Entry | Variation of conditions | Yielda,b (%) | e.rc |
| a Conditions: (1) 1a (0.2 mmol), MYTsA (0.24 mmol), CH2Cl2 (1 mL), 25 °C, 0.5 h. (2) 2a (0.1 mmol), C1 (20 mol%), DABCO (50 mol%), CH3CN (2 mL), 25 °C, 24 h. (3) CH3I (0.24 mmol), K2CO3 (0.24 mmol), DMF (1 mL), 25 °C, 2 h. b Yield of isolated product. c Determined using chiral SFC analysis. d Reaction performed at 0 °C. | |||
| 1 | None | 65% | 96:4 |
| 2 | w/o MYTsA or NHC | 0 | — |
| 3 | C2 or C3 instead of C1 | Trace | — |
| 4 | C4 instead of C1 | 8% | 96:4 |
| 5 | C5 instead of C1 | 62% | 75:25 |
| 6 | DBU instead of DABCO | 58% | 93:7 |
| 7 | Cs2CO3 instead of DABCO | 63% | 95:5 |
| 8 | CH2Cl2 instead of CH3CN | 54% | 91:9 |
| 9 | 0 °C instead of 25 °C | 64% | 97:3 |
| 10d | with 100 mg 4 Å MS | 73% | 97:3 |

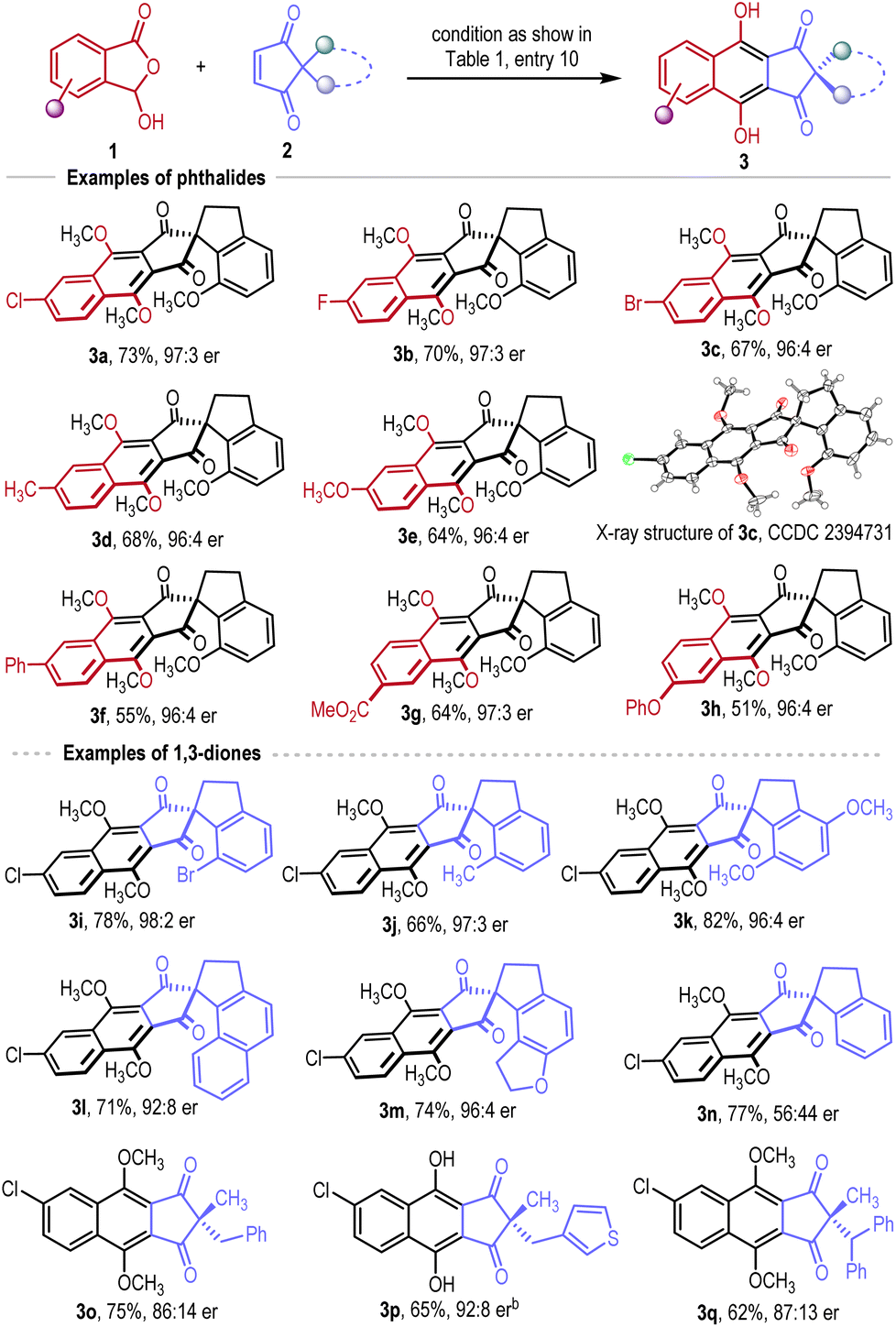
With the optimized conditions in hand, we next turned our attention to examine the scope of the reaction (Scheme 1). First, we evaluated the scope of 3-hydroxyl phthalides by using 2a as a model substrate. Halogenation (products 3a–3c), methylation (product 3d), methoxylation (product 3e), and phenylation (product 3f) in the 6-position of phthalides were all well tolerated, affording the desired products 3a–3f in moderate to good yields (55–73%) with excellent enantioselectivities (96:4–97:3 er). The absolute configurations of 3a and 3c were unambiguously confirmed from the results of single-crystal X-ray diffraction analysis. 5-Substituted phthalides with the electron-withdrawing carbonyl group (product 3g) or electron-donating phenyloxy group (product 3h) were also applicable substrates in our reaction, giving similar good results in both yields and enantioselectivities (51–64% yield, 96:4–97:3 er).
 | ||
| Scheme 1 Substrates scope aAll yields are isolated yields and the er values were determined from the results of chiral SFC analysis. bWithout methylation. | ||
Using 1a as a model phthalide substrate, the scope of the 1,3-cyclopentenediones was also evaluated. Replacing the methoxy group of 2a with a bigger bromo group led to a slightly enhanced enantioselectivity (product 3i, 98:2 er), while replacement with a methyl group gave product 3j in 66% yield with 97:3 er. Introducing a para-methoxy substitution in 2a gave product 3k in 82% yield with 96:4 er. Replacing the phenyl ring of 2a with a naphthyl unit (product 3l) and dihydrobenzofuranyl unit (product 3m) also afforded the desired products with good results (71–74%, 92:8–96:4 er). The rigid spirocyclic structure and substitution maintaining the enantio-facial differences are very important for the enantioselectivity of our reaction. Removing the methoxy group in 2a led to a sharp decline in enantioselectivity (product 3n, 56:44 er). The more flexible 2,2-dialkyl 1,3-cyclopentenediones all gave products with reduced enantioselectivities (products 3o–3q, 86:14–92:8 er).
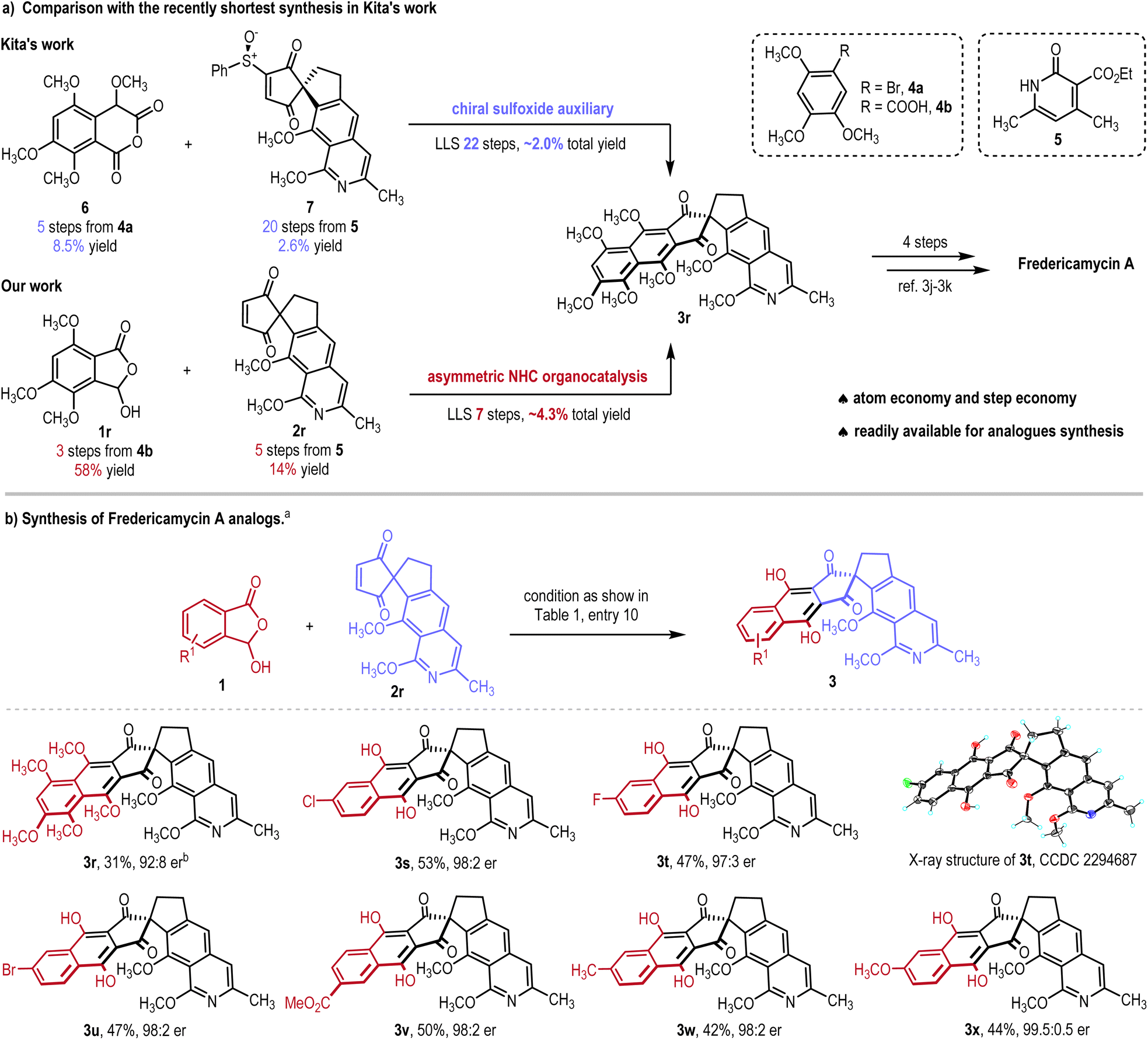
Encouraged by the general good performance of our method as shown in Scheme 1, we turned our attention to the total synthesis of FDM-A. As shown in Scheme 2, the previously shortest synthesis of FDM-A, reported by Kita, relied on a Tamura annulation of the anhydride 6 and the chiral sulfoxide-auxiliary–attached dienophile 7. The anhydride 6 required a 5-step synthesis from 4a (8.5% yield), and the dienophile 7 required a 20-step synthesis from 5 (2.6% total yield).3j,k In our synthesis, phthalide 1r can be obtained with a 3-step synthesis from 4b and the yield was found to be nearly 7 times higher than that of the synthesis of 6 (58% total yield). In comparison to the synthesis of dienophile 7, from the same starting material 5, our synthesis of dienophile 2r only required one-fourth of the synthetic steps (5 steps) and gave more than 5 times the yield (14% yield). Overall, the key intermediate 3r was successfully obtained in 7 LLS steps with 4.3% total yield, that is fewer than one-third of the synthetic steps and more than twice the yield than those in the synthesis by Kita. The key intermediate 3r for the synthesis of FDM-A was also reported in the 4-step synthesis in the work by Kita.3j,k Notably, with dienophile 2r as a model substrate, a variety of FDM-A analogs (3s–3x) were afforded in moderate yields (42–53%) with excellent enantioselectivities (97:3–99.5:0.5 er). The absolute configuration of 3t was unambiguously confirmed from the results of single-crystal X-ray diffraction analysis.
 | ||
| Scheme 2 Asymmetric synthesis of fredericamycin A and its analogs. aUnless otherwise specified, all the reactions were conducted without methylation. bSee the ESI† for details. | ||
Recognizing tiny differences between prochiral substrates for the construction of symmetrical chiral centers is a challenging task in asymmetric organic synthesis.12 To achieve an extremely symmetrical chiral structure with minimal differences, the chloro group of 1a was replaced with a deuterium atom in our reaction, giving the interesting product 3y in 65% yield. As shown in Scheme 3a, the only difference to ensure the all-carbon quaternary chiral center was the difference between hydrogen and deuterium 6 atoms away from the chiral center. Using different enantiomers of the NHC catalyst, both enantiomers of 3y were obtained with similar enantioselectivities. Based on substitutions in compound 1 only slightly influencing the enantioselectivity (Scheme 1, products 3a–3h, 92%∼94% ee), 3y should be obtained in about 92% ee. However, the exact ee of 3y was not confirmed as no chiral-stationary column can separate the two enantiomers. No obvious signal was observed in the ECD spectra of the two enantiomers (Scheme 3b). Only optical rotation provided evidence to recognize the chirality of 3y. The optical rotations of 3y and its enantiomer each at various concentrations were measured, and good linear relationships were found (R2 > 0.99), showing a reliable specific rotation of 0.6 (Scheme 3c). Our method thus can readily provide both enantiomers of the extremely weakly chiral compounds with minimal differences in remote position as challenging examples in chiral separation and chirality measurement.
 | ||
| Scheme 3 Quasi-symmetrical spirocyclic structure with minor chiral distinction. | ||
To better understand the mechanism of the carbene-catalyzed hydroquinone formation reaction, a kinetic isotope effect (KIE) experiment was conducted and the result is shown in Scheme 4a. The parallel KIE experiment revealed a secondary KIE (kH/kD = 1.06), showing the breaking of the C–H bond to not be the rate-determining step (but probably a fast step). The postulated pathway of carbene-catalyzed hydroquinone formation reaction is illustrated in Scheme 4b. Briefly, the reaction starts with the formation of Breslow intermediate Ivia nucleophilic addition of the carbene catalyst to the aldehyde group. The following lactonization step forms the phthalide-type Breslow intermediate II (see ESI† for details of the phthalide-type Breslow intermediate),13 which has a resonance structure II′. The annulation may carry on via a stepwise Michael addition and Dieckmann condensation from intermediate II to intermediate IV (path a, Scheme 4b),14 or via a concerted [4 + 2] annulation from intermediate II′ to IV (path b, Scheme 4b).15 During the annulation process, the chiral indane moiety favors the less steric hindered part and realizes the enantioselective control. The release of carbene catalyst from intermediate IV gives intermediate V and finally a rapid aromatization process with rapid C–H bond breaking gives the hydroquinone product 3a.
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
We have developed an NHC organocatalytic strategy for the enantioselective construction of spirocyclic hydroquinones bearing an all-carbon quaternary chiral center with small differences in a remote position. 3-Hydroxy phthalides were used as easily accessible starting materials and reacted with the prochiral spirocyclic dienophiles to afford the desired hydroquinone products in moderate to good yields with excellent enantioselectivities. Unlike the normal NHC organocatalytic model involving the Breslow intermediate, the annulation described in this manuscript involving the phthalide-type Breslow intermediate was found to favor the N-benzyl triazolium salts rather than the N-aryl ones. The N-benzyl triazolium salts were found to be easier to synthesize via simple alkylation of triazole, facilitating the building of a catalyst library for future investigations. With the help of this powerful method, the facile synthesis of FDM-A was achieved, shortening the synthetic route from 26 steps to 11 steps. Several analogs of FDM-A were readily synthesized as well. A more challenging quasi-symmetrical spirocyclic structure with only differences of hydrogen and deuterium was also successfully synthesized to investigate the special performance of “weak” chirality.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data [for compounds 3a, 3c and 3t] have been deposited at the CCDC [under CCDC 2294687 and 2394730–2394731] and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
P. Ren conducted most of the experiments. Q. Zhao prepared the substrates for the synthesis of fredericamycin A. Additionally, Y. R. Chi and T. Zhu conceptualized and directed the project, and drafted the manuscript with the assistance of all co-authors. All authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (No. 22071269), Pearl River Recruitment Program of Talent (No. 2019QN01L149), Guangdong Provincial Key Laboratory of Construction Foundation (No. 2023B1212060022), Singapore National Research Foundation under its NRF Competitive Research Program (NRF-CRP22-2019-0002), Ministry of Education, Singapore, under its MOE AcRF Tier 1 Award (RG84/22, RG70/21), MOE AcRF Tier 2 (MOE-T2EP10222-0006), and MOE AcRF Tier 3 Award (MOE2018-T3-1-003), a Chair Professorship Grant, Sun Yat-Sen University and Nanyang Technological University.Notes and references
- (a) R. C. Pandey, M. W. Toussaint, R. M. Stroshane, C. C. Kalita, A. A. Aszalos, A. L. Garretson, T. T. Wei, K. M. Byrne, R. F. Geoghegan and R. J. White, J. Antibiot., 1981, 34, 1389–1401 CrossRef CAS PubMed; (b) D. J. Warnick-Pickle, K. M. Byrne, R. C. Pandey and R. J. White, J. Antibiot., 1981, 34, 1402–1407 CrossRef CAS.
- (a) K. M. Byrne, B. D. Hilton, R. J. White, R. Misra and R. C. Pandey, Biochemistry, 1985, 24, 478–486 CrossRef CAS PubMed; (b) E. Wendt-Pienkowski, Y. Huang, J. Zhang, B. Li, H. Jiang, H. Kwon, C. R. Hutchinson and B. Shen, J. Am. Chem. Soc., 2005, 127, 16442–16452 CrossRef CAS PubMed; (c) Y. Chen, Y. Luo, J. Ju, E. Wendt-Pienkowski, E. S. R. Rajski and B. Shen, J. Nat. Prod., 2008, 71, 431–437 CrossRef CAS PubMed; (d) Y. Chen, E. Wendt-Pienkowski and B. Shen, J. Bacteriol., 2008, 190, 5587–5596 CrossRef CAS PubMed; (e) A. Das, P. H. Szu, J. T. Fitzgerald and C. Khosla, J. Am. Chem. Soc., 2010, 132, 8831–8833 CrossRef CAS PubMed; (f) P. H. Szu, S. Govindarajan, M. J. Meehan, A. Das, D. D. Nguyen, P. C. Dorrestein, J. Minshull and C. Khosla, Chem. Biol., 2011, 18, 1021–1031 CrossRef CAS PubMed; (g) B. Tsakem, G. Li and R. B. Teponno, Bioorg. Chem., 2024, 150, 107572 CrossRef CAS PubMed.
- Several different strategies in the total synthesis of FDM-A, see: (a) S. Kotha and A. Fatma, Asian J. Org. Chem., 2021, 10, 129–148 CrossRef CAS; (b) T. R. Kelly, S. H. Bell, N. Ohashi and R. J. Armstrong-Chong, J. Am. Chem. Soc., 1988, 110, 6471–6480 CrossRef CAS; (c) D. L. Boger, O. Hueter, K. Mbiya and M. Zhang, J. Am. Chem. Soc., 1995, 117, 11839–11849 CrossRef CAS; (d) D. L. J. Clive, Y. Tao, A. Khodabocus, Y. Wu, A. G. Angoh, S. M. Bennett, C. N. Boddy, L. Bordeleau, D. Kellner, G. Kleiner, D. S. Middleton, C. J. Nichols, S. R. Richardson and P. G. Vernon, J. Chem. Soc. Chem. Commun., 1992, 1489–1490 RSC; (e) D. L. J. Clive, Y. Tao, N. Khodabocus, Y. Wu, A. G. Angoh, S. M. Bennett, C. N. Boddy, L. Bordeleau, D. Kellner, G. Kleiner, D. S. Middleton, C. J. Nichols, S. R. Richardson and P. G. Vernon, J. Am. Chem. Soc., 1994, 116, 11275–11286 CrossRef CAS; (f) A. V. R. Rao, A. K. Singh, B. V. Rao and K. M. Reddy, Tetrahedron Lett., 1993, 34, 2665–2668 CrossRef CAS; (g) A. V. R. Rao, A. K. Singh, B. V. Rao and K. M. Reddy, Heterocycles, 1994, 37, 1893–1912 CrossRef CAS; (h) Y. Kita, K. Lio, K. Kawaguchi, N. Fukuda, Y. Takeda, H. Ueno, R. Okunaka, K. Higuchi, T. Tsujino, H. Fujioka and S. Akai, Chem.–Eur. J., 2000, 6, 3897–3905 CrossRef CAS PubMed; (i) S. Akai, T. Tsujino, N. Fukuda, K. Iio, Y. Takeda, K. Kawaguchi, T. Naka, K. Higuchi, E. Akiyama, H. Fujioka and Y. Kita, Chem.–Eur. J., 2005, 11, 6286–6297 CrossRef CAS PubMed; (j) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, S. Akai and H. Fujioka, Angew. Chem., Int. Ed., 1999, 38, 683–686 CrossRef CAS; (k) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, K. Ueda, S. Akai and H. Fujioka, J. Am. Chem. Soc., 2001, 123, 3214–3222 CrossRef CAS PubMed; (l) J. A. Wendt, P. J. Gauvreau and R. D. Bach, J. Am. Chem. Soc., 1994, 116, 9921–9926 CrossRef CAS; (m) F. X. Wang, J. L. Yan, Z. Liu, T. Zhu, Y. Liu, S. C. Ren, W. X. Lv, Z. Jin and Y. R. Chi, Chem. Sci., 2021, 12, 10259–10265 RSC.
- (a) K. Xu, W. Li, S. Zhu and T. Zhu, Angew. Chem., Int. Ed., 2019, 58, 17625–17630 CrossRef CAS PubMed; (b) P. Ren, Q. Zhao, K. Xu and T. Zhu, ACS Catal., 2024, 14, 13195–13201 CAS.
- (a) S. M. Bennett and D. L. J. Clive, J. Chem. Soc. Chem. Commun., 1986, 11, 878–880 RSC; (b) C. Wu, Z. Chang, C. Peng, C. Bai, J. Xing and X. Dou, Chem. Sci., 2023, 14, 7980–7987 RSC.
- (a) L. Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, M. Yang, C. Wang and J. Zhao, J. Am. Chem. Soc., 2016, 138, 13135–13138 CrossRef CAS PubMed; (b) S. Xu, D. Jiang, Z. Peng, L. Hu, T. Liu, L. Zhao and J. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202212247 CrossRef CAS PubMed; (c) L. Hu and J. Zhao, Acc. Chem. Res., 2024, 57, 855–869 CrossRef CAS PubMed.
- For selected reviews on NHC catalysis, see: (a) M. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (c) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (d) K. J. R. Murauski, A. A. Jaworskia and K. A. Scheidt, Chem. Soc. Rev., 2018, 47, 1773–1782 RSC; (e) A. T. Biju, in N-Heterocyclic Carbenes in Organocatalysis.,ed. A. T. Biju, Wiley-VCH Verlag GmbH & Co. KGaA, 2019 Search PubMed; (f) X. Chen, H. Wang, Z. Jin and Y. R. Chi, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS; (g) X. Chen, Z. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS PubMed; (h) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem, 2021, 5, 711–725 CrossRef CAS PubMed; (i) R. Song, Y. Xie, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2021, 60, 26026–26037 CrossRef CAS PubMed; (j) A. Ghosh and A. T. Biju, Angew. Chem., Int. Ed., 2021, 60, 13712–13724 CrossRef CAS PubMed; (k) B. Zhang, G. Yang, D. Guo and J. Wang, Org. Chem. Front., 2022, 9, 5016–5040 RSC; (l) Y. Nakano, J. T. Maddigan-Wyatt and D. W. Lupton, Acc. Chem. Res., 2023, 56, 1190–1203 CrossRef CAS PubMed.
- M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS PubMed.
- (a) M. S. Kerr and T. Rovis, J. Am. Chem. Soc., 2004, 126, 8876–8877 CrossRef CAS PubMed; (b) X. Yang, L. Wei, Y. Wu, L. Zhou, X. Zhang and Y. R. Chi, Angew. Chem., Int. Ed., 2022, e202211977 Search PubMed.
- (a) M. S. Kerr, J. R. Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298–10299 CrossRef CAS PubMed; (b) T. Jian, L. He, C. Tang and S. Ye, Angew. Chem., Int. Ed., 2011, 50, 9104–9107 CrossRef CAS PubMed; (c) Q. Wang, S. Wu, J. Zou, X. Liang, C. Mou, P. Zheng and Y. R. Chi, Nat. Commun., 2023, 14, 4878 CrossRef CAS PubMed.
- (a) D. Enders, J. Han and A. Henseler, Chem. Commun., 2008, 3989–3991 RSC; (b) D. Enders and J. Han, Synthesis, 2008, 23, 3864–3868 CrossRef; (c) P. Shao, X. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412–8416 CrossRef CAS PubMed; (d) L. Sun, Z. Liang, W. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 5803–5806 CrossRef CAS PubMed.
- (a) H. Zhou, Y. Zhou, H. Y. Bae, M. Leutzsch, Y. Li, C. K. De, G. Cheng and B. List, Nature, 2022, 605, 84–89 CrossRef CAS PubMed; (b) M. Wang, S. Liu, H. Liu, Y. Wang, Y. Lan and Q. Liu, Nature, 2024, 631, 556–562 CrossRef CAS PubMed.
- M. Sharique and U. K. Tambar, Chem. Sci., 2020, 11, 7239–7243 RSC.
- (a) F. M. Hauser and R. P. Rhee, J. Org. Chem., 1978, 43, 178–180 CrossRef CAS; (b) G. A. Kraus and H. Sugimoto, Tetrahedron Lett., 1978, 19, 2263–2266 CrossRef.
- J. C. Evans, R. C. Klix and R. D. Bach, J. Org. Chem., 1988, 53, 5519–5527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, spectra for all compounds, and crystallographic data. CCDC 2294687 and 2394730–2394731. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01605c |
| This journal is © The Royal Society of Chemistry 2025 |