DOI:
10.1039/D5SC01576F
(Edge Article)
Chem. Sci., 2025,
16, 9406-9412
Post-coordination of Ru(II) controlled regioselective B(4)–H acylmethylation of o-carboranes with sulfoxonium ylides†
Received
27th February 2025
, Accepted 23rd April 2025
First published on 24th April 2025
Abstract
Despite significant progress in the B–H functionalization of carboranes, the development of cost-effective catalytic systems devoid of noble metals, coupled with mechanistic validation of regioselectivity control, remains a formidable challenge. Herein, we disclose an Ag salt-free, redox-neutral, and inexpensive ruthenium(II)-catalyzed protocol that enables exclusive B(4)–H acylmethylation of o-carboranes through a novel post-coordination strategy. By exploiting weakly coordinating carboxylic acid as a traceless directing group, this method achieves excellent mono-site selectivity for B–C(sp3) bond formation using diverse sulfoxonium ylides, demonstrating both functional group tolerance and synthetic scalability. This work not only establishes a practical synthetic platform but also addresses critical mechanistic questions unresolved in prior analogous studies. Through deuterium labeling, in situ high-resolution mass spectrometry (HRMS) tracking, and single-crystal X-ray analysis of critical Ru intermediates, we unequivocally demonstrate that the mono-site selectivity originates from a unique post-coordination mode of Ru(II). The Ru catalyst simultaneously engages both the carboxylic acid and the enolizable acylmethyl moiety in the mono-acylated intermediate, thereby dictating the B(4)–H activation trajectory. Our findings establish a generalizable platform for regiocontrolled carborane functionalization while defining mechanistic paradigms in transition metal-mediated B–H activation chemistry.
Introduction
As a class of three-dimensional (3D) σ-aromatic cousins to benzene, polyhedral boron clusters have triggered a revived interest for decades in building blocks, such as versatile ligands, functional materials, and tuneable pharmacophores.1 In these clusters, icosahedral carboranes, a unique class of valuable bioisosteres of the phenyl group and adamantane, feature sphere-like geometry, 3D delocalization σ aromaticity, inherent robustness, etc.,1a,1b,2 making them considerably attractive scaffolds for applications in materials to medicine (especially applied in boron neutron capture therapy, BNCT) and other numerous areas.3–6 One of the challenges is structural modifications at boron vertexes to enhance the properties and effects. Among carborane derivatives, alkylated-o-carboranes are highly desirable due to their potentially beneficial applications,3b,3c,7 especially in medicinal chemistry.7a,8
The past decades have witnessed the huge development of B–H of carboranes functionalization strategies, including transition-metal-catalysis,9 electrophilic substitution,10 nucleophilic substitution,11 oxidative coupling,12 and others.13 Despite recent advancements in transition metal-catalyzed B–H bond activation of carboranes,1a,9 the development of direct synthetic methods for the preparation of cage B–H alkylated carboranes is still constrained by the challenge of β–H elimination, particularly in the case of mono-alkylated carboranes. Hence, the practical and general mono-alkylation strategy of cage B–H bonds using readily accessible and structurally diverse reagents for potentially beneficial applications is highly desirable. Recently, the regioselective B(4,5)-dialkylation of o-carboranes catalyzed by Rh(III) catalyst was developed through oxidative Heck reactions followed by enol isomerization from o-carborane acids and allylic alcohols with Ag salt as an additive (Scheme 1a).14 Moreover, the Rh(III)-catalyzed B(4)-alkylation of 1-pyridyl-o-carboranes with α-diazodicarboxylates was demonstrated (Scheme 1b).15 On the other hand, sulfoxonium ylides are readily accessible from commercially available reagents. Their safety and stability make them practical surrogates for carbene. Metal-carbene intermediates in which sulfoxonium ylides function as precursors are known to X–H (X = N, O, S, C) bond insertion reactions. However, insertion into the B–H bond was scarcely reported.16 Inspired by the unusual properties of sulfoxonium ylides and the nitrene moiety can insert into the M–B(cage) bond to build a new cage B–N bond,17 we have developed a general an Rh(III)-catalyzed B(4) acylmethylation of 1-pyridyl-o-carboranes with sulfoxonium ylides without any additives (Scheme 1c).18 In that work, the mono-substituted selectivity has been achieved by the post-coordination of the pyridyl, enolizable acylmethyl groups with rhodium. Later, Ir(III)-catalyzed B(4)-acylmethylation or B(3,5)-diacylmethylation of o-carborane acids with sulfoxonium ylides through B(4)–H activation with Ag salt as an additive was also reported (Scheme 1d).19 In pursuing to develop the efficient functionalization of carboranes and broaden the reactivity of sulfoxonium ylide, we wonder whether carbene derived from sulfoxonium ylide could insert into Ru–B(cage) bond to form B–C(sp3) bond and control the mono-vertex selectivity via post-coordination mode. With this in mind, we have explored herein the Ag salt-free, Ru(II)-catalyzed exclusively cage B(4) acylmethylation of o-carborane with α-carbonyl sulfoxonium ylides as the alkylating reagents through direct B–H activation (Scheme 1e).
 |
| | Scheme 1 Transition metal-catalyzed cage B–H alkylation of o-carboranes. | |
Results and discussion
Reaction development
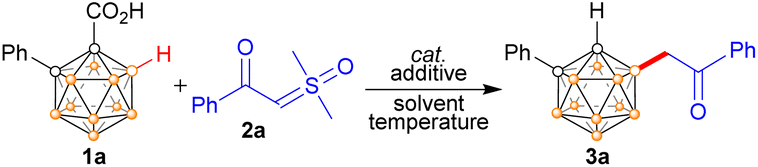
The reaction development is summarized in Table 1. At the outset of the investigation, to evaluate the feasibility, we selected 1-CO2H-2-Ph-o-carborane 1a and α-carbonyl sulfoxonium ylide 2a as the model substrates. In the presence of 5 mol% [Ru(p-cymene)Cl2]2, the treatment of 1a with 1.5 equiv. of 2a in the absence of any additives in HFIP at 100 °C for 12 h afforded the target product 3a in 73% NMR yield (entry 1). However, [Cp*RhCl2]2 does not work in this system, which is an effective catalyst in pyridyl-directed B(4)–H bond alkylation using the same α-carbonyl sulfoxonium ylide as the coupling partner (entry 2).18 In addition, [Cp*IrCl2]2 yielded a 64% yield (entries 3). Attempts to replace the ruthenium catalyst with earth-abundant metal-based catalysts Cp*Co(CO)I2 or Cu(CH3CN)4PF6 were unsuccessful (Table S1†). Other ruthenium catalysts, including Cp*Ru(COD)Cl, [Cp*RuCl2]2, or Ru(COD)Cl2, were also evaluated, but no product was detected (Table S1†). To our delight, [Ru(benzene)Cl2]2 exhibited a significant increase in the yield, reaching 93% (entry 4). Adding Lewis acid PivOH as an additive is detrimental (entry 5). On the contrary, adding acetate (NaOAc or KOAc) improved yields, resulting in a quantitative yield (entry 6 and Table S3†). In addition, no desired product was detected when toluene, CH3CN, DCE, or anisole were used as solvents (entry 7 and Table S2†). On the contrary, TFE and THF provided good yields (89% and 75%, respectively; entry 8 and Table S3†). Furthermore, a quantitative yield was obtained when the reaction was performed at 60 °C for 3 h (Table S4†). A shortened time has little effect on yields (Table S4†). When the reaction was set at 40 °C for 12 h, an 82% yield can be obtained (entry 9). The reduction of 2a to 1.2 equiv. led to the formation of 3a in a slightly lower yield (93%, Table S5†). Notably, when the loading of [Ru(benzene)Cl2]2 was decreased to 2.5 mol%, quantitative yield was also achieved (entry 10). The reaction still afforded a 78% yield when set up under an air atmosphere (entry 11). The results mentioned above indicate that the method is highly robust. No target product was detected in the absence of a catalyst, suggesting that the catalyst plays a critical role in these cross-coupling reactions (Table S5†). Given all the factors, entry 10 was chosen as the optimal reaction condition.
Table 1 Reaction developmenta
|

|
| Entry |
Catalyst |
Additive |
Solvent |
Yieldb [%] |
|
Unless otherwise stated, the reaction in HFIP (1.0 mL) was performed with 1a (0.1 mmol), 2a (0.15 mmol), Cat. (5 mol%), additive (1.0 equiv.) at 100 °C for 12 h under N2.
NMR yield by using CH2Br2 as an internal standard.
The reaction was carried out at 40 °C.
The reaction was carried out at 60 °C for 3 h with Cat. 2.5 mol%.
Isolated yield.
Under air. HFIP: hexafluoroisopropanol; TFE: trifluoroethanol.
|
| 1 |
[Ru(p-cymene)Cl2]2 |
|
HFIP |
73 |
| 2 |
[Cp*RhCl2]2 |
|
HFIP |
0 |
| 3 |
[Cp*IrCl2]2 |
|
HFIP |
64 |
| 4 |
[Ru(benzene)Cl2]2 |
|
HFIP |
93 |
| 5 |
[Ru(benzene)Cl2]2 |
PivOH |
HFIP |
78 |
| 6 |
[Ru(benzene)Cl2]2 |
NaOAc |
HFIP |
Quant |
| 7 |
[Ru(benzene)Cl2]2 |
NaOAc |
Toluene |
0 |
| 8 |
[Ru(benzene)Cl2]2 |
NaOAc |
TFE |
89 |
| 9 |
[Ru(benzene)Cl2]2 |
NaOAc |
HFIP |
82c |
|
10
|
[Ru(benzene)Cl
2
]
2
|
NaOAc
|
HFIP |
Quant.d (99)e |
| 11 |
Ru(benzene)Cl2]2 |
NaOAc |
HFIP |
78d,f |
Scope of sulfoxonium ylides
Subsequently, the scope of (hetero)aryl-substituted sulfoxonium ylides 2 was evaluated with 1a as the model substrate (Scheme 2). Substrates bearing electronically diverse substituents at different positions of phenyl were compatible, giving the corresponding products moderate to excellent yields. Generally, substrates with electron-withdrawing groups in the para position of the phenyl ring showed higher yields than substrates with electron-donating groups. Aryl sulfoxonium ylides bearing electron-donating groups, such as –Me, –OMe, and –OCF3, furnished products (3b–3d) in 80–95% yields. Moreover, aryl sulfoxonium ylides bearing electron-withdrawing groups such as halogen (F, Cl, Br, I), –CF3, –Ph, –CO2Me, and –NO2 furnished 3e–3l in almost quantitative yields. Notably, valuable halogen functional groups and ethers were well tolerated, allowing for late-stage manipulation of the initial products. Ylides with substituents at the ortho- or meta-position were all well tolerated (3m–3p). Substrates bearing two or multiple substituents were also examined, and the yields were comparable to those of the monosubstituted substrates (3q–3s). The lower yield for the substrate bearing –NHCOPh may be attributed to its toxicity to the catalyst or the presence of reactive N–H (3t). Naphthalen-1-yl- and naphthalen-2-yl-substituted sulfoxonium ylides afforded 65% and 99% yields, respectively (3u and 3v). The substrate bearing a furan scaffold was effectively transformed into the corresponding product 3w (94%). Similarly, thiophene and benzothiophene were compatible with the cross-coupling reaction (3x and 3y), which may serve as important frameworks in organic photoelectronic materials. The molecular structures of 3b and 3l were further confirmed by single-crystal X-ray analyses (Schemes S1 and S2†).
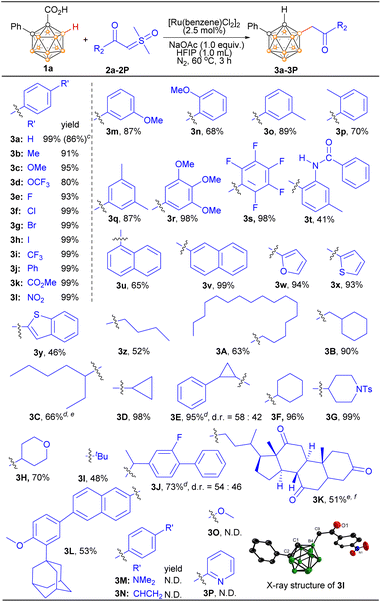 |
| | Scheme 2 Scope of sulfoxonium ylides. aReaction conditions: 1a (0.1 mmol), 2 (1.5 equiv.), [Ru(benzene)Cl2]2 (2.5 mol%), HFIP (1.0 mL), 60 °C, 3 h, N2 atmosphere. bIsolated yield. c1 mmol scale. d2J was used as a racemic mixture. eNo d.r. was determined. fEnantiopure 2K was used. | |
The scope of the alkyl-substituted sulfoxonium ylide coupling partners was also investigated (Scheme 2). With 1a as the model substrate, the coupling reactions with aliphatic sulfoxonium ylides smoothly led to the desired products in good to excellent yields. Both primary and secondary aliphatic acids derived ylides exhibited good to excellent reactivity (3z–3H). Interestingly cyclic secondary alkyl-containing substrates can be converted into respective products in nearly quantitative yields (3D–3F). The piperidine unit, which is a key structural motif in numerous natural products and pharmaceuticals, was well tolerated (3G). Tetrahydropyran was also demonstrated to be a suitable motif (3H). The sterically hindered tertiary alkyl-substituted sulfoxonium ylide can also afford the desired product in a good yield (3I). This state-of-the-art protocol could also be employed to facilitate the direct incorporation of a polyhedral boron cluster unit into pharmaceutical drugs. For instance, flurbiprofen is a non-steroidal anti-inflammatory drug with fever-reducing and pain-relieving properties. Dehydrocholic acid has been utilized to stimulate the secretion of biliary lipids, while adapalene is employed for the topical treatment of acne vulgaris. These drug-derived ylides reacted readily with 1a, affording the mono-alkylated-carboranes 3J, 3K, and 3L in good to high yields. To demonstrate the practicality of the strategy, a scale-up reaction of 1a and 2a at a 1 mmol scale gave 3a in 86% yield. Finally, given that knowing the limits of this reaction might be helpful for the potential user, four unsuccessful substrates were given in Scheme 2 (3M–3P).
Scope of carboranyl acids
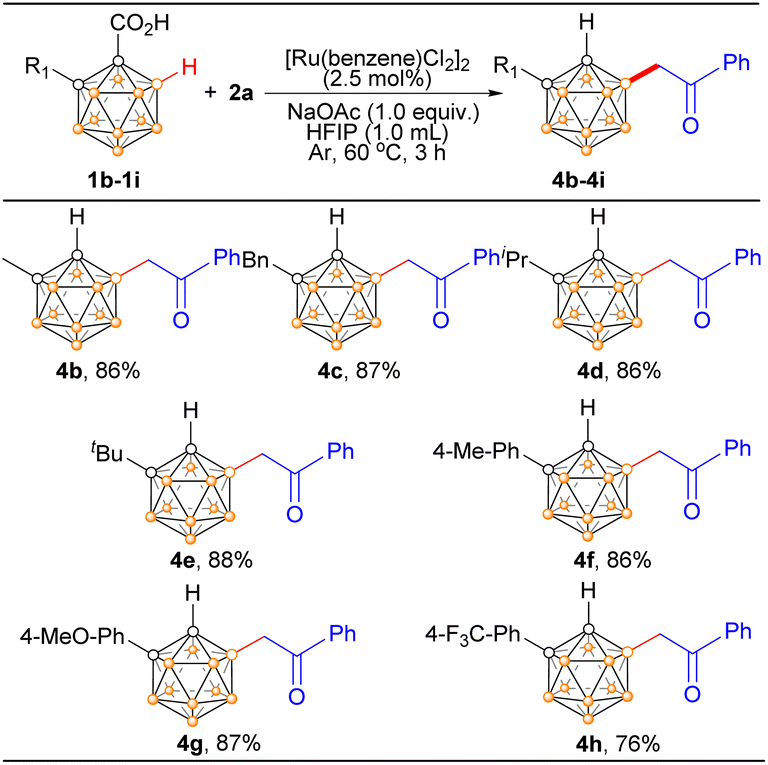
To further expand the substrate scope, we examined the reaction by varying C-substituted o-carboranyl acids using 2a as the model substrate (Scheme 3). The o-carborane acid substrates bearing different substituents at cage C(2) yielded the desired products (4b–4h) in high yields. It is encouraging to note that all the primary, secondary, and tertiary C(2)-alkylated o-carboranyl acids offered the corresponding products in 86–88% isolated yield (4c–4e). Furthermore, C(2)-arylated o-carboranyl acids underwent the B(4) acylmethylation with 2a smoothly and efficiently, producing the corresponding products 4f–4h in 76–87% yields. The substituent (phenyl or alkyl) at the cage C(2) position exhibits negligible impact on B(4) selectivity, which is governed by the synergistic interaction between the –CO2H directing group and the catalytic system.
 |
| | Scheme 3 Scope of carboranyl acids. Reaction conditions: 1 (0.1 mmol), 2a (1.5 equiv.), [Ru(benzene)Cl2]2 (2.5 mol%), HFIP (1.0 mL), 60 °C, 3 h, N2 atmosphere. bIsolated yield. | |
Synthetic applications
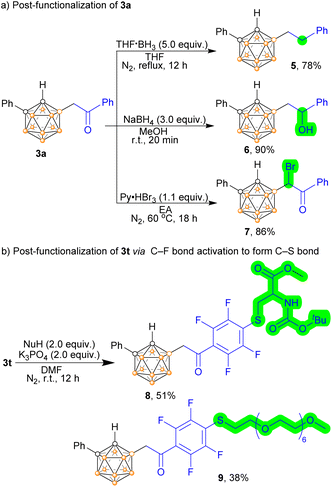
The carbonyl in the acylmethylated compounds allows for additional post-functionalization options, either at the carbonyl site or at its α-carbon position (Scheme 4a). The carbonyl group at 3a can be converted to methylene using THF·BH3 as a reducing reagent, resulting in the mono-alkylated product 5. Additionally, compound 3a can be reduced in the presence of NaBH4 to yield o-carboranyl aliphatic alcohol 6, which serves as an important synthon in the derivation of carboranes and is a crucial structural unit in pharmaceutical compounds. Moreover, the mono-acylmethylated product 3a can undergo mono-bromination at the α-carbon (adjacent to the boron atom) by being treated with Py·HBr3 at 60 °C, as the carbonyl group can activate the neighbouring C–H bond (7). Currently, BNCT drugs and drug candidates often lack either a targeting moiety, a boron-enriched pharmacophore, or a functional group for visualization. Our strategy offers a rational approach for constructing targeted BNCT drug candidates. For instance, the polyhedral boron cluster (carborane), fluorine (a magic element in drug molecules), and the targeting moiety (amino acid) can be merged into a single molecule (Scheme 4b). This is achieved through perfluoroaryl-thiol nucleophilic aromatic substitution chemistry, where penta-fluorobenzyl-containing acylmethylated carborane reacts with nucleophilic thiolated amino acids under mild conditions (8). Polyethylene glycol (PEG) can also be incorporated into the acylmethylated carborane by the same method (9).
 |
| | Scheme 4 Synthetic applications. | |
Mechanism study
Several control experiments were conducted to gain insights into the reaction mechanism, as shown in Scheme 5. The reaction of 1-Ph-carborane 13 and 2a did not proceed as the starting material was fully recovered (Scheme 5a). Therefore, it can be concluded that the directing group carboxyl is essential. Notably, 2-methyl-benzoic acid did not react with sulfoxonium ylide 2a under the optimal reaction conditions (Scheme 5b). This result indicated a significant difference in reactivity between the 3D delocalization σ aromatic carborane and the 2D π aromatic benzene. When employing deuterated 1a-D10 as the substrate, the 1H NMR indicates the deuterium atom was unlikely transferred to the α-carbon of the B(4) (Scheme 5c). In addition, the reaction of 1a and 2a under the optimal reaction conditions in a mixed solution of D2O and HFIP led to the incorporation of deuterium at the α-carbon of the carbonyl and C(1) of the cage with a deuterium level of 53% and 51%, respectively (Scheme 5d). At room temperature, the 2H NMR spectrum of 3a-D3 in dichloromethane displays two signals for the deuterium atoms aforementioned (Fig. S5†). These results indicate that the newly incorporated acylmethyl group attached to boron undergoes facile enolization. In sharp contrast, stirring 3a in D2O and HFIP at 60 °C in the absence of any other reagent or catalyst is insufficient to enable deuterium incorporation at the enolizable positions. This observation demonstrates that the product undergoes reversible enolization and coordination with the ruthenium catalyst.
 |
| | Scheme 5 Control experiments. | |
Based on the aforementioned experimental results, a plausible reaction mechanism has been elucidated by using the reaction of 1a and 2a as an example (Scheme 6a). Firstly, the ruthenium species Ru(benzene)Cl2 reacts with NaOAc, resulting in the formation of Ru(benzene)(OAc)2, which reacts with the carboxyl of 1a to form an active catalytic intermediate A1 or A2, subsequently inducing B–H cleavage to generate a ruthenacyclic complex B containing a Ru–B bond. Subsequently, B undergoes migratory insertion of α-carbonyl sulfoxonium ylide 2avia a metal carbene intermediate C, accompanied by the release of dimethyl sulfoxide (DMSO), to give rise to D. This intermediate then delivers the final product 3avia protodemetallation of D with concomitant regeneration of Ru(benzene)(OAc)2 and decarboxylation of E. Here, HFIP acts as the proton donor in the protodemetallation step.18 Notably, the conversion between D and E should be reversible, as indicated by the deuteration experiment. This post-coordination between the carboxy and enolizable acylmethyl groups of the B(4)-acylmethylated o-carborane and the ruthenium catalyst can prevent the iterative B–H activation pathway caused by metal “cage walking” around the carboranyl surface,20 thereby controlling the selective mono-substitution (Scheme 6b). The molecular structure of A2 was confirmed by single-crystal X-ray determination. Trials to isolate other Ru(II) intermediates were unsuccessful. To our delight, HRMS measurements successfully detected the intermediates A1, B, and D (Fig. S8–S13†). Furthermore, the free DMSO in the catalytic reaction mixture was readily identified by 1H NMR spectroscopy (Fig. S15†). All the results are well-corroborated with the proposed mechanism.
 |
| | Scheme 6 Proposed reaction cycle for Ru(II)-catalyzed B(4)–H alkylation of o-carboranyl acid (a) and illustration of the post-coordination step (b). | |
Conclusions
In conclusion, we have achieved a highly efficient Ru(II)-catalyzed site-selective B(4)–H mono-acylmethylation of o-carborane using sulfoxonium ylides as the alkyl agents. This protocol features a broad substrate scope and directly delivers valuable B(4) alkylated carborane derivatives in moderate to excellent yields. The carborane derivatives contain acidic C–H units, carbonyl, and other newly incorporated organic functional groups, providing new opportunities for post-functionalization. This work establishes a foundation for synthesizing carborane-based functional molecules, which have potential applications in drug discovery (especially for BNCT). Control experiments have enhanced our understanding of the possible reaction mechanism. Notably, the post-coordination of the traceless directing group 1-carboxyl and enol moieties to the ruthenium catalyst prohibits the iterative B–H activation event and results in finely controlled mono-substitution. This post-coordination strategy provides a reliable approach for mono site-selective B–H functionalization of polyhedral boron clusters with similar chemical environments B–H bonds.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
H.-J. C. and H. Y. conceived and supervised the project. H.-J. C., J.-X. L., J.-H. Y. and M.-X. L. performed the experiments and analysed the data. The manuscript was drafted by H.-J. C., Ju. Z. and refined by H.-J. C., Ju. Z., H. Y., Z. Z. and Jie. Z. The ESI† was written by J.-X. L., H.-J.C. and revised by H.-J.C. and Ju. Z. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22201067, 92261202, 22471062) and the Postdoctoral Research Grant in Henan Province (HN2022043).
References
-
(a)
N. S. Hosmane and R. Eagling, Handbook of Boron Science, World Scientific, 2019 Search PubMed;
(b) J. Poater, C. Viñas, M. Solà and F. Teixidor, Nat. Commun., 2022, 13, 3844 CrossRef CAS PubMed;
(c) R. B. King, Chem. Rev., 2001, 101, 1119–1152 CrossRef CAS PubMed.
-
(a)
R. N. Grimes, Carboranes, Academic Press, 2016 Search PubMed;
(b) J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
-
(a) A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem, 2022, 6, 486–504 CrossRef PubMed;
(b) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 Search PubMed;
(c) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed;
(d) R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
-
(a) Y.-F. Han and G.-X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS PubMed;
(b) A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS;
(c) P.-F. Cui, X.-R. Liu and G.-X. Jin, J. Am. Chem. Soc., 2023, 145, 19440–19457 CrossRef CAS PubMed.
-
(a) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed;
(b) J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed;
(c) K. Yuhara and K. Tanaka, Angew. Chem., Int. Ed., 2024, 63, e202319712 Search PubMed;
(d) X. Wei, M.-J. Zhu, Z. Cheng, M. Lee, H. Yan, C. Lu and J.-J. Xu, Angew. Chem., Int. Ed., 2019, 58, 3162–3166 CrossRef CAS PubMed;
(e) K. Tanaka, M. Gon, S. Ito, J. Ochi and Y. Chujo, Coord. Chem. Rev., 2022, 472, 214779 CrossRef CAS;
(f) K. Liu, J. Zhang, Q. Shi, L. Ding, T. Liu and Y. Fang, J. Am. Chem. Soc., 2023, 145, 7408–7415 CrossRef CAS PubMed;
(g) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed;
(h) J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, V. T. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 15758–15765 CrossRef CAS PubMed;
(i) Y. H. Lee, J. Park, J. Lee, S. U. Lee and M. H. Lee, J. Am. Chem. Soc., 2015, 137, 8018–8021 CrossRef CAS PubMed;
(j) W. Ma, J. Zhang, J. Zong, H. Ren, D. Tu, Q. Xu, B. Zhong Tang and H. Yan, Angew. Chem., Int. Ed., 2024, 63, e202410430 CrossRef CAS PubMed;
(k) A. Saha, E. Oleshkevich, C. Vinas and F. Teixidor, Adv. Mater., 2017, 29, 1704238 CrossRef PubMed;
(l) A. M. Cioran, A. D. Musteti, F. Teixidor, Ž. Krpetić, I. A. Prior, Q. He, C. J. Kiely, M. Brust and C. Viñas, J. Am. Chem. Soc., 2012, 134, 212–221 CrossRef CAS PubMed.
-
(a) T. He, H. F. T. Klare and M. Oestreich, Nature, 2023, 623, 538–543 CrossRef CAS PubMed;
(b) Y. Xu, Y. Yang, Y. Liu, Z. H. Li and H. Wang, Nat. Catal., 2023, 6, 16–22 CrossRef CAS;
(c) I. Guerrero, C. Viñas, I. Romero and F. Teixidor, Green Chem., 2021, 23, 10123–10131 RSC;
(d) X. Tan, X. Wang, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2022, 144, 23286–23291 CrossRef CAS PubMed;
(e) C. N. Kona, R. Oku, S. Nakamura, M. Miura, K. Hirano and Y. Nishii, Chem, 2024, 10, 402–413 CrossRef CAS;
(f) O. Tutusaus, S. Delfosse, A. Demonceau, A. F. Noels, C. Viñas and F. Teixidor, Tetrahedron Lett., 2003, 44, 8421–8425 CrossRef CAS.
-
(a) Y. Yin, N. Ochi, T. W. Craven, D. Baker, N. Takigawa and H. Suga, J. Am. Chem. Soc., 2019, 141, 19193–19197 CrossRef CAS PubMed;
(b) H. Xu, J. Liu, R. Li, J. Lin, L. Gui, Y. Wang, Z. Jin, W. Xia, Y. Liu, S. Cheng and Z. Yuan, Coord. Chem. Rev., 2024, 511, 215795 CrossRef CAS;
(c) D. A. Gruzdev, G. L. Levit, V. P. Krasnov and V. N. Charushin, Coord. Chem. Rev., 2021, 433, 213753 CrossRef CAS;
(d) M. Koshino, T. Tanaka, N. Solin, K. Suenaga, H. Isobe and E. Nakamura, Science, 2007, 316, 853 CrossRef CAS PubMed;
(e) L. Kobr, K. Zhao, Y. Shen, A. Comotti, S. Bracco, R. K. Shoemaker, P. Sozzani, N. A. Clark, J. C. Price, C. T. Rogers and J. Michl, J. Am. Chem. Soc., 2012, 134, 10122–10131 CrossRef CAS PubMed.
-
(a) W. Ma, Y. Wang, Y. Xue, M. Wang, C. Lu, W. Guo, Y.-H. Liu, D. Shu, G. Shao, Q. Xu, D. Tu and H. Yan, Chem. Sci., 2024, 15, 4019–4030 RSC;
(b) R. Otero, S. Seoane, R. Sigüeiro, A. Y. Belorusova, M. A. Maestro, R. Pérez-Fernández, N. Rochel and A. Mouriño, Chem. Sci., 2016, 7, 1033–1037 RSC;
(c) M. W. Lee, Jr., Y. V. Sevryugina, A. Khan and S. Q. Ye, J. Med. Chem., 2012, 55, 7290–7294 CrossRef PubMed;
(d) J. Wang, L. Chen, J. Ye, Z. Li, H. Jiang, H. Yan, M. Y. Stogniy, I. B. Sivaev, V. I. Bregadze and X. Wang, Biomacromolecules, 2017, 18, 1466–1472 CrossRef CAS PubMed.
-
(a) Z. Qiu and Z. Xie, Acc. Chem. Res., 2021, 54, 4065–4079 CrossRef CAS PubMed;
(b) X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS;
(c) S. Duttwyler, Pure Appl. Chem., 2018, 90, 733–744 CrossRef CAS;
(d) Y. K. Au and Z. Xie, Bull. Chem. Soc. Jpn., 2021, 94, 879–899 CrossRef CAS;
(e) R. Cheng, J. Zhang, H. Zhang, Z. Qiu and Z. Xie, Nat. Commun., 2021, 12 Search PubMed;
(f) F. Lin, J.-L. Yu, Y. Shen, S.-Q. Zhang, B. Spingler, J. Liu, X. Hong and S. Duttwyler, J. Am. Chem. Soc., 2018, 140, 13798–13807 CrossRef CAS PubMed;
(g) Y.-N. Ma, Y. Gao, Y. Ma, Y. Wang, H. Ren and X. Chen, J. Am. Chem. Soc., 2022, 144, 8371–8378 CrossRef CAS PubMed;
(h) Z. Sun, J. Zong, H. Ren, C. Lu, D. Tu, J. Poater, M. Solà, Z. Shi and H. Yan, Nat. Commun., 2024, 15, 7934 CrossRef CAS PubMed;
(i) F. Sun, S. Tan, H.-J. Cao, C.-s. Lu, D. Tu, J. Poater, M. Solà and H. Yan, J. Am. Chem. Soc., 2023, 145, 3577–3587 CrossRef CAS PubMed;
(j) J. Wu, K. Cao, C.-Y. Zhang, T.-T. Xu, L.-F. Ding, B. Li and J. Yang, Org. Lett., 2019, 21, 5986–5989 CrossRef CAS PubMed;
(k) T.-T. Xu, K. Cao, C.-Y. Zhang, J. Wu, L.-F. Ding and J. Yang, Org. Lett., 2019, 21, 9276–9279 CrossRef CAS PubMed;
(l) Y. Fu, D. Zhang, C. Jin and J. Lu, Chin. J. Org. Chem., 2024, 44, 438–447 CrossRef CAS;
(m) P. Li, J. Yang, J. Zeng, S. Miao, R. Fang, J. Lu and J.-Y. Lu, Org. Lett., 2024, 26, 8134–8138 CrossRef CAS PubMed;
(n) M. J. Zhu, P. Z. Wang, Z. Q. Wu, Y. F. Zhong, L. M. Su, Y. Q. Xin, A. M. Spokoyny, C. Zou and X. Mu, Chem. Sci., 2024, 15, 10392–10401 RSC;
(o) Y. Baek, K. Cheong, G. H. Ko, G. U. Han, S. H. Han, D. Kim, K. Lee and P. H. Lee, J. Am. Chem. Soc., 2020, 142, 9890–9895 CrossRef CAS PubMed;
(p) Y.-F. Liang, L. Yang, B. B. Jei, R. Kuniyil and L. Ackermann, Chem. Sci., 2020, 10764–10769 RSC.
-
(a) Y.-N. Ma, H. Ren, Y. Wu, N. Li, F. Chen and X. Chen, B(9)-OH-o-Carboranes: Synthesis, Mechanism, and Property Exploration, J. Am. Chem. Soc., 2023, 145, 7331–7342 CrossRef CAS PubMed;
(b) Y. Wang, Y. Gao, W. Guo, Q. Zhao, Y.-N. Ma and X. Chen, Org. Chem. Front., 2022, 9, 4975–4980 RSC.
-
(a) Y. Quan, C. Tang and Z. Xie, Dalton Trans., 2019, 48, 7494–7498 RSC;
(b) R. Frank, A. K. Adhikari, H. Auer and E. Hey-Hawkins, Chem.–Eur. J., 2014, 20, 1440–1446 CrossRef CAS PubMed.
-
(a) Z. Yang, W. Zhao, W. Liu, X. Wei, M. Chen, X. Zhang, X. Zhang, Y. Liang, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 11886–11892 CrossRef CAS PubMed;
(b) M. Chen, D. Zhao, J. Xu, C. Li, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 7838–7844 CrossRef CAS PubMed;
(c) S. Xu, H. Zhang, J. Xu, W. Suo, C.-S. Lu, D. Tu, X. Guo, J. Poater, M. Solà and H. Yan, J. Am. Chem. Soc., 2024, 146, 7791–7802 CrossRef CAS PubMed;
(d) D. C. Young, D. V. Howe and M. F. Hawthorne, J. Am. Chem. Soc., 1969, 91, 859–862 CrossRef CAS;
(e) M. Y. Stogniy, S. A. Anufriev and I. B. Sivaev, Inorganics, 2023, 11, 72 CrossRef CAS.
-
(a) H. Ren, P. Zhang, J. Xu, W. Ma, D. Tu, C.-s. Lu and H. Yan, J. Am. Chem. Soc., 2023, 145, 7638–7647 CrossRef CAS PubMed;
(b) D. Olid, R. Nunez, C. Vinas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC;
(c) A. A. Semioshkin, I. B. Sivaev and V. I. Bregadze, Dalton Trans., 2008, 8, 977–992 RSC;
(d) S. Li and Z. Xie, J. Am. Chem. Soc., 2022, 144, 7960–7965 CrossRef CAS PubMed;
(e) H. A. Mills, J. L. Martin, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2020, 142, 4586–4591 CrossRef CAS PubMed.
- Q. Wang, S. Tian, C. Zhang, J. Li, Z. Wang, Y. Du, L. Zhou and J. Lu, Org. Lett., 2019, 21, 8018–8021 CrossRef CAS PubMed.
- G. H. Ko, J. K. Lee, S. H. Han and P. H. Lee, Org. Lett., 2022, 24, 1507–1512 CrossRef CAS PubMed.
-
(a) T. Zhou, P.-F. Qian, J.-Y. Li, Y.-B. Zhou, H.-C. Li, H.-Y. Chen and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 6810–6816 CrossRef CAS PubMed;
(b) Y. W. Xu, X. K. Zhou, G. F. Zheng and X. W. Li, Org. Lett., 2017, 19, 5256–5259 CrossRef CAS PubMed;
(c) S.-S. Zhang, H. Xie, B. Shu, T. Che, X.-T. Wang, D. Peng, F. Yang and L. Zhang, Chem. Commun., 2020, 56, 423–426 RSC;
(d) J. Li, H. He, M. Huang, Y. Chen, Y. Luo, K. Yan, Q. Wang and Y. Wu, Org. Lett., 2019, 21, 9005–9008 CrossRef CAS PubMed.
-
(a) H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2016, 138, 12727–12730 CrossRef CAS PubMed;
(b) H. Li, F. Bai, H. Yan, C. Lu and V. I. Bregadze, Eur. J. Org Chem., 2017, 2017, 1343–1352 CrossRef CAS.
- H.-J. Cao, X. Wei, F. Sun, X. Zhang, C. Lu and H. Yan, Chem. Sci., 2021, 12, 15563–15571 RSC.
-
(a) L.-B. Zhang and Z. Xie, Org. Lett., 2022, 24, 1318–1322 CrossRef CAS PubMed;
(b) G. H. Ko, K. Um, H. C. Noh, J. Y. Kim, H. Jeong, C. Maeng, S. H. Han, G. U. Han and P. H. Lee, Org. Lett., 2022, 24, 1604–1609 CrossRef CAS PubMed.
-
(a) X.-R. Liu, P.-F. Cui, S.-T. Guo, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2023, 145, 8569–8575 CrossRef CAS PubMed;
(b) C. Guo, Z. Qiu and Z. Xie, ACS Catal., 2021, 11, 2134–2140 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jia-Xin
Li
a,
Jia-Hui
Yan
a,
Miao-Xin
Liu
a,
Qianyi
Zhao
*a,
Jia-Xin
Li
a,
Jia-Hui
Yan
a,
Miao-Xin
Liu
a,
Qianyi
Zhao