
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriggering anti-Kasha organic room temperature phosphorescence of clusteroluminescent materials†
Jingyu
Zhang
a,
Yishan
Jin
a,
Xinchi
Lu
a,
Chengxi
Sun
a,
Wei
Ma
a,
Yuhang
Li
a,
Longyan
Zhang
a and
Runfeng
Chen
 *ab
*ab
aState Key Laboratory of Flexible Electronics (LoFE), Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications, 9 Wenyuan Road, Nanjing 210023, China. E-mail: iamrfchen@njupt.edu.cn
bSchool of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China
First published on 24th March 2025
Abstract
Clusterization-triggered emission (CTE) from organic materials without π-conjugated structures for room temperature phosphorescence (RTP) is fascinating with extraordinary photophysical properties and diversified applications, but rather challenging in material design owing to the limited mechanism understanding. Here, we demonstrate a facile strategy to construct CTE polymers with stimuli-responsive emission, anti-Kasha RTP and organic ultralong RTP (OURTP) by introducing ions into the hydrolyzed nonconjugated maleic anhydride and acrylamide copolymers. Thanks to the synergistic effects of hydrogen and ionic bonding with the ion-triggered electrostatic and coordinate interactions to suppress non-radiative decays and promote intersystem crossing, the amorphous copolymers show efficient photoluminescence with quantum efficiencies up to 13.5%, anti-Kasha RTP blue-shift of 29 nm, and OURTP lifetime up to 420 ms. Moreover, the temperature-dependent and water-sensitive anti-Kasha RTP and OURTP are also observed due to the formation of highly emissive CTE structure regulated by ionization. With the excellent processability and flexibility of the copolymer, lifetime-, temperature- and color-encrypted information anti-counterfeiting is designed and explored. The anti-Kasha RTP in CTE materials realized for the first time demonstrates impressive potential for multi-level encryption/anti-counterfeiting applications and more importantly, providing fundamental mechanism understanding for the rational modulation and design of CTE materials with extraordinary photophysical properties.
Introduction
Room-temperature phosphorescence (RTP) materials with unique photophysical properties, rich excited-state features, high flexibility, and excellent structural designability play important roles in anticounterfeiting, bioimaging, photodynamic therapy, and organic light emitting diodes.1–5 To promote RTP, efficient intersystem crossing (ISC) to populate the triplet excited states and suppressed nonradiative decays to support the radiative transition from the lowest triplet excited state (T1) to the ground state (S0) are essential.6,7 Therefore, various strategies have been proposed to facilitate ISC and restrain nonradiative decays, including heteroatom incorporation,8 crystallization/aggregation engineering,9 polymerization approach,10 host–guest doping,11etc. However, most of these strategies inevitably lead to red-shifted RTP in comparison to fluorescence, owing to the large Stokes shift as well as the solid state and triplet excited state emitting nature of the RTP emitters.12,13According to Kasha's rule,14 photons can only be emitted from the lowest excited state of a given multiplicity, resulting in the fact that most of the current RTP emissions originate from the radiative transitions at the T1 energy level for the unavoidable redshift of the RTP spectrum. Kasha's rule generally holds true when the rates of internal conversion (IC) and vibrational relaxation are sufficiently higher than the luminescence rate; but, when the luminescence rate of the high-lying excited state is significantly enhanced, the emission will not obey Kasha's rule and anti-Kasha emissions from the radiative decays of the high-lying singlet (Sn) and triplet (Tn) excited states can be observed. There are three distinct mechanisms of anti-Kasha emission: (1) direct fluorescence radiative decay from high-lying Sn states due to the Sn–S0 transitions,15,16 (2) time-delayed fluorescence originating from the reverse ISC between upper Tn and Sn states for the Tn–Sn–S0 emission,17 and (3) the spin-forbidden RTP emission from high-energy Tn states occurs after efficient ISC following Sn–Tn–S0 transitions.18 Notably, the anti-Kasha phenomenon will lead to the blue-shift of the RTP when the oscillator strength (f) of the Tn–S0 transition for emission is large. And, there are two ways to realize the anti-Kasha RTP: (1) the energy gap between T1 and T2 is large enough to reduce the rate of IC and the phosphorescence from T2 becomes competitive; (2) the T2–T1 energy gap and f of the S0–T1 transition are very small, at which the thermal population of T2 from T1 is facilitated for the appearance of T2 emission (Fig. 1a). Therefore, there are only a limited number of reports on anti-Kasha RTP due to the challenges in satisfying the critical requirements of both anti-Kasha and RTP emission simultaneously, although as early as in the 1970s, Chu et al. first observed the anti-Kasha phosphorescence emission of aromatic carbonyl compounds in a rigid medium at 77 K.19 In 2017, Tang et al. reported the dual RTP in dibenzoketone derivatives, arising from the radiative decays of the low-lying T1 and high T2 states, respectively.20 In 2022, Pan et al. reported a novel organic ultralong RTP (OURTP) material with dynamic phosphorescence from multiple triplet excited states of T2, T1, and stabilized T1 (T*) for blue, green and red OURTP, respectively.21 These studies demonstrate that it is possible to achieve anti-Kasha phosphorescence for blue-shifted RTP,15,22 but the blue-shift is generally low and the emission wavelength of the anti-Kasha RTP is still longer than that of the fluorescence in most cases (Table S1†).
 | ||
| Fig. 1 Material design of anti-Kasha RTP. (a) Kasha and anti-Kasha rules for RTP emission. (b) Molecular design and preparation procedure of the ionic CL polymers based on AM and MA. | ||
Non-traditional cluster luminescent (CL) materials without π-conjugated structures or even π–electrons but with strong emissions have recently attracted considerable attention due to their excellent processability, structural diversity, and fascinating photophysical properties.23 Generally, the cluster refers to the aggregates of materials with entirely non-conjugated structures based on saturated C–C, C–O, or C–N bonds that exhibit bright visible emission in aggregated or solid-state forms. The formation of a cluster can be identified by the following features: (1) the chemical structure of the luminescent material is non-conjugated and functional groups containing non-bonding or π–electrons are separated by a saturated σ-bonded backbone; (2) when the material is dissolved in solution at low concentrations, it is non-luminescent, but strong luminescence appears at high concentrations, especially in the solid state; (3) the emission wavelengths are excitation-dependent and the excitation at longer wavelengths results in red-shifted emissions.23,24 To stimulate cluster-triggered emission (CTE) from π-conjugation-free structures, the electron-rich atoms (N, O, S, etc.) should be aggregated in the cluster to generate the through-space conjugation (TSC) by intermolecular weak interactions including hydrogen bonding, orbital overlap, and electrostatic interactions.25,26 In this way, CTE has been modulated to achieve fluorescence,27,28 thermally activated delayed fluorescence (TADF),29 RTP/OURTP,30 hybridized local and charge transfer,31 and circularly polarized luminescence.32 Specifically, CL materials with TSC aided by heteroatom incorporation and strong intermolecular interactions are promising for RTP with facilitated ISC and suppressed nonradiative decays; the photoluminescent quantum yield (PLQY) and lifetime of their RTP can reach up to 87.5% and 1.7 s, respectively.33 Compared to the RTP materials based on π-conjugated structures, the non-conjugated CL materials with highly tunable and versatile TSC that are sensitive to many means have rich photophysical properties, but the lack of clarity in their structure–property relationship leads to difficulties in finely modulating the energy levels, let alone the high-lying excited states for anti-Kasha emission.16 Therefore, it remains a formidable challenge to realize anti-Kasha RTP in non-conjugated chromophores and an anti-Kasha RTP CL material is yet to be developed.
Here, we propose a new strategy to design anti-Kasha CL materials with efficient RTP by copolymerizing the widely used CL RTP monomer of acrylamide (AM) with maleic anhydride (MA), which can be hydrolyzed and can interact with metal ions.34 The thus-obtained ionic copolymers have abundant ionic and hydrogen bonding as well as electrostatic and coordinate interactions, which are important for the formation of TSC with promoted ISC and suppressed nonradiative decays and thus efficient RTP and even OURTP.35 Impressively, the anti-Kasha RTP from radiative decay of T2 was observed in the non-conjugated copolymers (Fig. 1), which is due to the effectively elevated T2 energy levels in the aggregated structure to minimize the IC of T2 → T1 after ionization. More importantly, a large blueshift of up to 29 nm compared to the fluorescence peak was observed, representing the largest blue-shift to date for anti-Kasha RTP materials (Table S1†). This blue-shifted anti-Kasha RTP (∼399 nm) with a lifetime of 36 ms shows temperature-dependent but excitation wavelength-independent emission properties due to the formation of a highly emissive RTP cluster with efficient T2 emission through copolymerization and ionization. Moreover, the ionic CL copolymer also shows OURTP behavior with a green-colored emission and a lifetime of 420 ms. This is the first time that anti-Kasha RTP has been realized in CL materials. Based on these extraordinary anti-Kasha RTP and OURTP with different emission colors and lifetimes, a heart/lock pattern with temperature-/water-responsive multilevel encryption was realized. This work not only demonstrates an efficient method in modulating the TSC and energy levels of CL materials for anti-Kasha RTP emission but also offers important clues in understanding the CTE mechanism for the stimuli-responsive RTP/OURTP emission, paving the way for the design of nonconventional luminophores with extraordinary photophysical properties and applications.
Results and discussion
Design and preparation of CL copolymers
The polymeric and oligomeric MA is a typical non-conjugated emitter with relatively abundant carbonyl groups that can promote ISC and the formation of hydrogen bonding networks.36,37 In addition, the MA unit can be easily hydrolyzed in alkaline solutions to form ionic bonds with metal ions, which can further suppress the nonradiative decays and enhance the emission of the polymers.34 On the other hand, polyacrylamide (PAM), which is rich in carbonyl groups and hydrogen bonding for a rigid network, is a well-known RTP polymer with excellent CTE characteristics and RTP performance.38 Therefore, highly pure MA and AM were chosen as comonomers to prepare their copolymers (PAMMA) with 2 mol% MA by free radical copolymerization at 75 °C for 6 h in 1,4-dioxane (Scheme S1 and Fig. S1†). After stirring the obtained copolymer in tetrahydrofuran/H2O at room temperature for 1 h, the MA unit is hydrolyzed to afford the hydrolyzed copolymer (PAMMA-h).37 Then, the copolymer was reacted in NaOH aqueous solution at 60 °C for 12 h to obtain the ionic copolymer (PAMMA-Na, Scheme S2†). The ionic copolymer can be turned back to the hydrolyzed copolymer (PAMMA-H) by treating its aqueous solution with hydrochloric acid to break the ionic bonds (Fig. 1b).34 All these copolymers were carefully purified by Soxhlet extraction using acetone, petroleum ether, and dichloromethane each for 12 h. The copolymers are highly soluble in water and can be facilely prepared in film state by drying their aqueous solution in an oven for 12 h. Gel permeation chromatography data suggest that the number average molecular weight of PAMMA is about 8200 g mol−1 with a polydispersity of 3.2. From the proton areas of MA to AM revealed by the nuclear magnetic resonance hydrogen spectra (Fig. S2†), the ratio of MA to AM in the copolymer was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48.7, which is rather close to the feed ratio (1:50). For comparison studies, the homopolymer of PAM and copolymers with different contents of MA in the feed were also prepared; similar molecular weights and copolymer composition close to the feeding content were observed (Table S2†).
:48.7, which is rather close to the feed ratio (1:50). For comparison studies, the homopolymer of PAM and copolymers with different contents of MA in the feed were also prepared; similar molecular weights and copolymer composition close to the feeding content were observed (Table S2†).
Compared to PAM, PAMMA shows a similar Fourier transform infrared spectrum since the actual content of MA is only 2 mol% (Fig. S3†). After the introduction of Na+, PAMMA-Na exhibits reduced stretching vibration of the carbonyl groups at 1664 cm−1 owing to the formation of electrostatic interaction between the metal ions and lone-pair electrons of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, while the strengthened and broadened stretching vibration band of the N–H bond near 3346 cm−1 suggests the presence of stronger hydrogen bonding in the ionic copolymer with the aid of a large number of carbonyl and amide groups.39 The strengthened intermolecular interaction with both hydrogen bonds and ionic bonds for the rigid copolymer was further confirmed by the X-ray diffraction (XRD) spectra and wide-angle X-ray scattering (WAXS) patterns (Fig. S4 and 5†). The broad XRD bands and scattering bands around 1.5 Å from WAXS suggest the amorphous nature and disordered molecular stacking structure of the polymers; the increased diffuse scattering peaks after ionization indicate the reduced packing distances for the compact molecular structures of the ionic polymers.34 Moreover, the ionic PAMMA-Na with a decomposition temperature (Td) of 167 °C and a glass transition temperature (Tg) of 125 °C shows close thermal stability to PAMMA (Td = 177 °C; Tg = 145 °C) as revealed by thermogravimetric analyses (Fig. S6†). These good physical properties of the ionic CL copolymers are important to stimulate efficient RTP and fabricate diversified devices for applications.
O, while the strengthened and broadened stretching vibration band of the N–H bond near 3346 cm−1 suggests the presence of stronger hydrogen bonding in the ionic copolymer with the aid of a large number of carbonyl and amide groups.39 The strengthened intermolecular interaction with both hydrogen bonds and ionic bonds for the rigid copolymer was further confirmed by the X-ray diffraction (XRD) spectra and wide-angle X-ray scattering (WAXS) patterns (Fig. S4 and 5†). The broad XRD bands and scattering bands around 1.5 Å from WAXS suggest the amorphous nature and disordered molecular stacking structure of the polymers; the increased diffuse scattering peaks after ionization indicate the reduced packing distances for the compact molecular structures of the ionic polymers.34 Moreover, the ionic PAMMA-Na with a decomposition temperature (Td) of 167 °C and a glass transition temperature (Tg) of 125 °C shows close thermal stability to PAMMA (Td = 177 °C; Tg = 145 °C) as revealed by thermogravimetric analyses (Fig. S6†). These good physical properties of the ionic CL copolymers are important to stimulate efficient RTP and fabricate diversified devices for applications.
Photophysical properties of the ionic copolymers
To explore the effect of copolymerization, hydrolyzation, ionization, and deionization on the luminescence properties of the MA and AM copolymers, the steady state photoluminescence (SSPL) and phosphorescence spectra were investigated (Fig. 2a). Due to the absence of conjugated groups, AM and MA exhibit very weak emissions and even at low temperatures, very weak fluorescence and phosphorescence emissions were observed (Fig. S7†). The copolymerization of MA leads to both red-shifted SSPL (428 nm) and RTP (496 nm) emission bands along with an elongated RTP lifetime (Fig. 2b) and enhanced PLQY and RTP efficiency (PhQY) over 1.2 fold (Fig. 2c) in comparison to those of the homopolymer of PAM. Interestingly, different from the excitation-dependent emission of PAM due to its CTE nature,40 the copolymer shows almost identical RTP peaks at different excitation wavelengths (Fig. S8†); this should be because the incorporation of MA significantly red-shifts and enhances the UV-vis absorption and emission properties for the much increased PLQY and PhQY (Fig. 2c and S9†) and the triplet energy levels of different clusters are almost the same, although both MA and AM monomers are poor in emission either in solution or in the solid state.41 Hydrolyzation has limited influence on the SSPL and phosphorescence spectra, but effectively increases the RTP lifetime, PLQY, and PhQY, because of the enhanced hydrogen bonding network after the turning of the MA unit to MA acid. In sharp contrast to the neglectable effects on SSPL, ionization shows significant impacts on the phosphorescence spectrum, resulting in the extraordinary blue-shifted RTP bands located at 399 and 488 nm. The first RTP band is even shorter than the SSPL band at 428 nm. Moreover, the OURTP lifetime of PAMMA-Na is significantly improved to 420 ms and the PLQY and PhQY also reach the maximums of 13.5% and 8.9%, respectively. Upon ionization of the copolymer, the ionic bonding will be generated to create a rigid environment that can stabilize the triplet excitons and suppress the non-radiative leaps, resulting in the longest RTP lifetime and the highest PLQY and PhQY of the copolymer. Upon the addition of an acid, the carboxylate anions are converted back into the neutral carboxylic groups and the ionic interactions are destroyed. Therefore, the deionized PAMMA-H exhibits similar emission spectra, RTP lifetime, PLQY, and PhQY to PAMMA-h, since they have identical polymer structure and substituent groups. Considering the non-conjugated molecular structure of the copolymers, the excitation wavelength-dependent SSPL emission, and enhanced emission at high concentrations (Fig. S10 and 11†), these copolymers should be CL materials with the CTE feature. | ||
| Fig. 2 Photophysical properties of the anti-Kasha RTP. (a–c) SSPL spectra (blue line) and phosphorescence spectra (red line, delayed by 10 ms) excited at 365 nm (a), the 488 nm RTP decay curves (b) and total PLQY and PhQY (c) of the polymers in film state. (d) The 399 nm RTP decay curve with photographs (insets) taken after ceasing irradiation for different times of the PAMMA-Na film. (e) Phosphorescence spectra at different excitation wavelengths of the PAMMA-Na film with the intensity changes of the 399 and 488 nm emission peaks (inset). (f and g) Excitation-phosphorescence mapping (f) and transient photoluminescence decay image (g) of the PAMMA-Na film; the inset shows the phosphorescence spectra excited at 365 nm with different delay times. | ||
To investigate the significantly blue-shifted phosphorescence spectrum, the lifetimes and emission intensities of the two RTP bands of PAMMA-Na at different excitation wavelengths were measured (Fig. 2d and e). The 399 nm RTP band has a lifetime of 36 ms, which is much shorter than that of the 488 nm RTP (420 ms); they should have different photophysical processes.42 Also, the photoluminescence excitation spectra of the 428 nm SSPL and 488 nm RTP emission peaks show similar excitation spectra (Fig. S12†), whereas the 399 nm emission peak has a significant difference, suggesting that the 428 and 488 nm emissions are from the same species but the 399 nm emission is different. Given the long lifetime of 36 ms and shorter emission wavelength, it should be related to a stable high-lying triplet exciton. With the rather different lifetimes and origins of the RTP bands, dynamically changed RTP color from bluish to green results after switching off the excitation source (Fig. 2d). Again, excitation wavelength independent RTP peaks were observed in PAMMA-Na, but when the excitation wavelength is increased, the intensity of the 399 nm RTP band decreases first and then enhances while the 488 nm RTP band has the opposite behavior, suggesting there is a competition between them (Fig. 2e). This extraordinary relation between the two RTP bands and the excitation wavelength of the ionic copolymer has also been recorded in the phosphorescence-excitation mapping (Fig. 2f), revealing that PAMMA-Na is an atypical CTE polymer. Compared to typical CTE materials with both excitation wavelength-dependent SSPL and RTP spectra with shifting emission peaks, the non-shifted RTP peaks of PAMMA-Na reveal that the luminophores for SSPL and RTP should be different; the phosphors with the spin-forbidden triplet excitons may have similar triplet conjugation length in these ionic CTE materials.41 The instantaneous photoluminescence decay image and time-resolved spectroscopy spectra (Fig. 2g) confirm that the 399 nm emission diminishes as the delay time increases from 10 to 80 ms (inset of Fig. 2g) and the T1 emission at 488 nm dominates the long-lived RTP, revealing that T1 has a longer lifetime than T2.
To identify the optimal copolymerization composition for the highest RTP performance, copolymers with different MA feed contents, from 0.1 to 4 mol%, were tested (Fig. S13†). When the MA content is too high, the molecular weight of the copolymer (Table S2†) decreases with reduced RTP performance (Fig. S14†). And the 2 mol% MA has the longest RTP lifetime and the highest PLQY and PhQY (Table S3†). Therefore, the optimal MA content in the feed is 2 mol%, and at this point the ionic copolymer has the best condition to form highly emissive triplet emitters for RTP emission, dense ionic and hydrogen bonding networks to suppress non-radiative decays, excellent oxygen barrier properties for stable RTP in air (Fig. S15†), and heteroatom effects to enhance the spin–orbit coupling (SOC) and ISC. It should be noted that the addition of water can disrupt the ionic and hydrogen bonding networks, leading to the water-sensitive RTP behavior of the water-soluble copolymers.43 For instance, when PAMMA-Na was placed under 80% humidity for 12 h, the SSPL and phosphorescence intensities decreased by 25% and 33%, respectively (Fig. S16†). Besides Na+ for constructing the ionic polymer to trigger the anti-Kasha RTP, we further tested Ca2+ and Ba2+ using Ca(OH)2 and Ba(OH)2 as alternative ion sources. Similar anti-Kasha phenomena were observed, where the T2 state emission around 400 nm is significantly blue shifted compared to the S1 state emission at 425–430 nm and the T1 state at ∼487 nm (Fig. S17†).
Mechanism of the blue-shifted RTP emission
To probe the mechanism of the blue-shifted RTP peak of the ionic copolymer which shifts to even shorter wavelengths than that of SSPL, the photoluminescence behaviors at different temperatures, in different media and under different ionization levels were studied. The PAMMA-Na film at 78 K has a slightly red-shifted SSPL spectrum (10 nm) compared to that at room temperature, but its phosphorescence spectrum becomes quite different, where the first RTP peak (399 nm) disappears leaving only the second RTP peak (488 nm) (Fig. 3a). Further measurements show that when the temperature increases, the 399 nm RTP peak appears and gradually enhances along with the decreased 488 nm emission band (Fig. 3b). The decay curves reveal that the 488 nm emission has decreasing lifetimes when temperature increases, while the 399 nm emission reaches the longest lifetime of 189 ms at 228 K (Fig. 3c), suggesting that this emission band can be thermally activated.44 Due to the temperature-dependent anti-Kasha RTP with varying lifetimes and peak intensities, blue and green phosphorescence is observed at 249 K and 298 K, respectively. Monitoring the phosphorescence spectra of PAMMA and PAMMA-Na in different polar solvents at low temperatures reveals that there was no solvation effect and charge transfer was low in shifting the emission spectra (Fig. S18†).45 When the ionization level is reduced from 100% to 50%, both the SSPL and 488 nm emission bands remain unchanged but the 399 nm emission is significantly reduced, revealing the close relationship between this emission band and the Na+ ionized structure (Fig. S19†). With these photophysical measurements, the 428 nm SSPL band should be the fluorescence due to the radiative decay of S1 with short lifetimes of 5.2 and 4.7 ns at 78 K and room temperature, respectively (Fig. S20†). The 488 nm band should be the phosphorescence related to the radiative decay of T1 with rather long lifetime at lower temperatures (Fig. S21†). As to the 399 nm emission, the shorter emission wavelength than the fluorescence (428 nm) rules out the possibility of TADF emission, and its millisecond lifetime suggests its phosphorescence nature,44i.e. anti-Kasha phosphorescence from the radiative decay of a high-lying triplet excited state.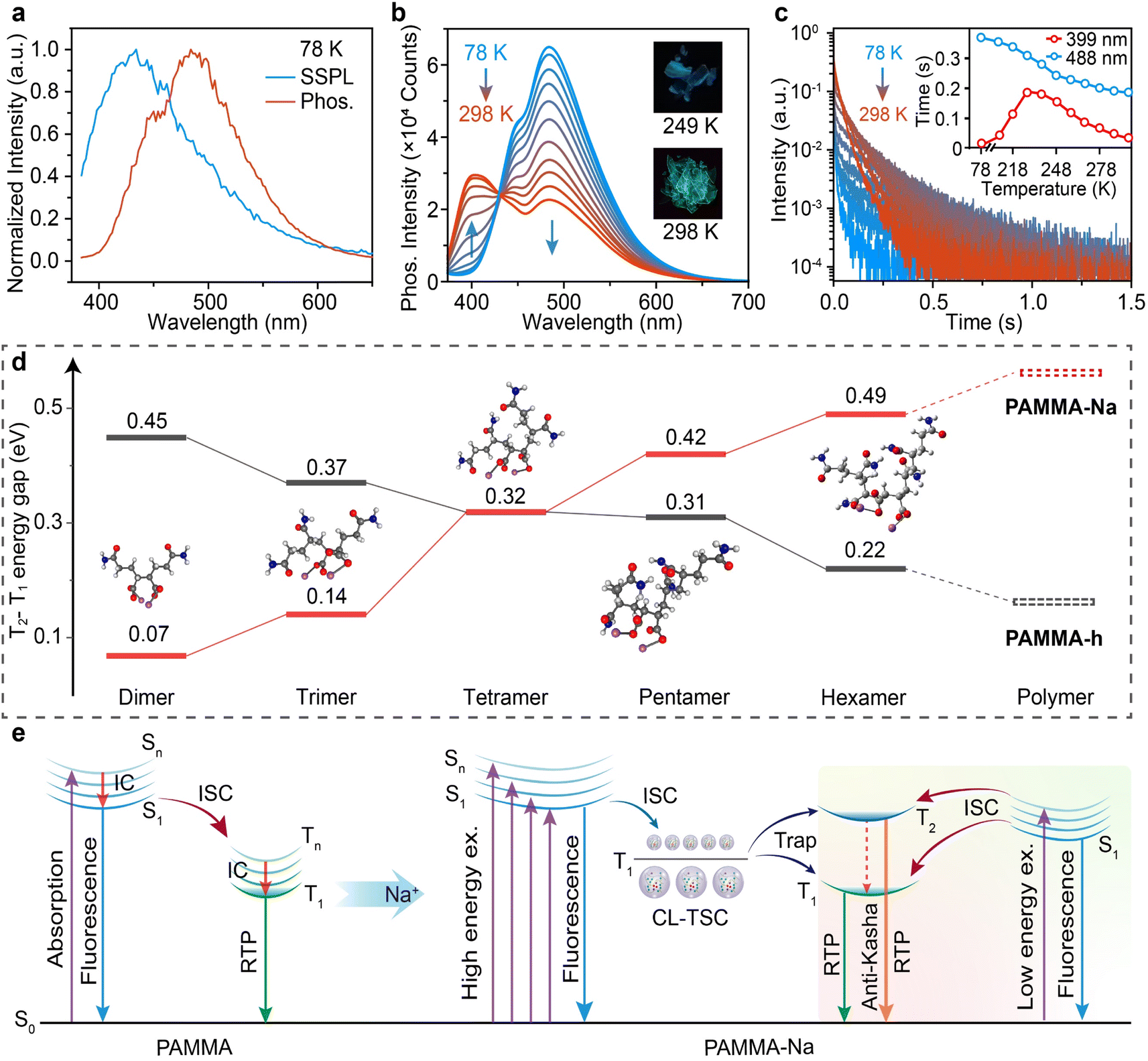 | ||
| Fig. 3 Mechanism of the anti-Kasha RTP. (a) SSPL (blue line) and phosphorescence spectra (red line, delay 10 ms) of the PAMMA-Na film at 78 K. (b and c) Phosphorescence spectra (b) and decay curves (c) of the PAMMA-Na film at different temperatures from 78 to 298 K. Insets show photographs taken after turning off the UV light (365 nm) at 249 and 298 K (b) and the lifetime changes (c) of the two RTP bands (399 and 488 nm) at different temperatures. (d) Theoretical predictions of the T2–T1 energy gaps of PAMMA-h and PAMMA-Na from their dimer, trimer, tetramer, pentamer, and hexamer. (e) Proposed mechanism for the anti-Kasha RTP. | ||
To confirm the anti-Kasha RTP nature of the 399 nm emission, time-dependent density functional theory (TD-DFT) calculations were performed to predict the excited state structure of the CL copolymers.18,46 From the MA and AM oligomers of dimer, trimer, tetramer, pentamer to hexamer with decreasing MA ratios that are close to PAMMA (Fig. S22†), the energy gaps between T1 and T2 of the oligomers decrease consistently and the simulative value of PAMMA-h is only 0.15 eV (Fig. S23†), while the ionized oligomers show continuously enlarged T2–T1 energy gaps that reach 0.61 and 0.56 eV in PAMMA-Na as predicted by linear extrapolation and experimentally measured through RTP peaks, respectively (Fig. 3d); such high values can block the IC for efficiently populating the T1 state of PAMMA-Na. This extraordinary triplet excited state structure of the ionic copolymer provides important evidence for the anti-Kasha RTP emission. Moreover, intramolecular interactions were also theoretically verified by the reduced density gradient isosurface calculations based on the hexamer of AM and MA before and after ionization (Fig. S24†).47 Strong and abundant intramolecular attractive interactions with denser interaction regions were uncovered in the ionized oligomer, which are helpful to suppress the non-radiative decay and promote the ISC in improving the RTP performance by ionization of the CL materials. More importantly, the ionized oligomers show small singlet–triplet splitting energy (ΔEST) and large SOC to efficiently promote ISC for populating the triplet excited states (Table S4†), and the triplet states become highly emissive from the significantly enlarged oscillation strength (Table S5†). It can be expected that after ionization of the copolymer, a highly emissive cluster will be formed and will dominate the RTP and anti-Kasha RTP emissions. Notably, although the TD-DFT calculations are rather preliminary without considering the solid state structures and interpolymeric interactions of the amorphous clusters, these theoretical revelations still suggest the anti-Kasha RTP behavior of the ionic copolymer.
With these experimental and theoretical understandings, a possible mechanism can be proposed for the firstly observed anti-Kasha RTP in CL materials (Fig. 3e). Through the ionization of the CL copolymer, the abundant hydrogen bonding and ionic interactions not only effectively suppress non-radiative transitions in forming a rigid environment, but also generate different clusters with different sizes for the excitation-dependent SSPL emissions and the highly emissive RTP emitting cluster with a wide energy gap between T1 and T2 for the anti-Kasha RTP emission. When the ionic CL copolymer is irradiated by a high energy photon with short excitation wavelength, the small size cluster will be excited and the high-lying T1 forms after ISC. The resulting T1 will be trapped by either T1 or T2 of the highly emissive RTP cluster for the normal and anti-Kasha RTP, respectively, but the high-lying T1 tends to be trapped by T2 of the emissive RTP clusters according to their smaller energy differences, leading to the anti-Kasha dominated RTP spectrum. With the reduction of the excitation energy, the larger cluster will be excited to generate the low-lying T1 excitons, which will be mostly trapped by T1 of the emissive RTP cluster since its energy moves close to the low-lying T1 state; therefore, the normal RTP will dominate the RTP spectrum. When the excitation energy further reduces, the highly emissive RTP cluster itself will be directly excited to Sn and subsequently undergo effective ISC to populate T2 efficiently for the anti-Kasha dominated RTP spectrum again. Therefore, the T2 emission (399 nm) first decreases and then increases as the excitation wavelength increases, while the T1 emission (488 nm) exhibits the opposite trend, reflecting the competition among these multiple photophysical processes (Fig. 2e). This mechanism explains the extraordinary RTP emission behavior of the ionic CL copolymer.
Anti-counterfeiting and encryption application
Considering the extraordinary excitation-wavelength dependent SSPL and temperature- and water-sensitive RTP/OURTP emissions of the ionic CL copolymer, PAMMA and PAMMA-Na were used in the pattern anti-counterfeiting study.48 PAMMA and PAMMA-Na were dissolved in water at a high concentration of 1 g mL−1 for screen printing on a common A4 paper to get a pattern in a “heart/lock” shape by a two-step screen printing method (Fig. 4a). After drying in an oven at 70 °C for 12 h, a transparent pattern was obtained. When it was excited at 365 nm, the blue fluorescence was the same as the background of the paper and no pattern could be recognized. After turning off the UV excitation, a greenish “heart” pattern can be observed and lasts for 4 s at room temperature (298 K); at low temperature (249 K), a green half-circle with blue “lock” pattern appears first then reverts to a green “heart” pattern after 1 s (Fig. 4b). Since the AM-based polymers have excellent water solubility and water can quench the emission of CL polymers, only the dry patterns can show these temperature- and color-resolved pattern encryption (Fig. 4c). On the other hand, PAM, PAMMA-h, and PAMMA-Na can be dissolved in water to make RTP inks for writing the word “studious” (Fig. 4d). Because the CL polymers are transparent, the word can be hardly observed under daylight and on excitation by a 365 nm UV lamp. But, after switching off the 365 nm UV lamp, the word emits a green OURTP that can be clearly read with the information “studious” at room temperature (298 K); after 2 s, the pattern shows another information “studio” due to the short lifetime of PAM. When the pattern was excited with the UV lamp at 249 K, the letters written with PAMMA-Na ink show blue anti-Kasha RTP, so that the hidden message “COO” appears within 1 s, and then transforms to “studio” after 2 s. Therefore, the full message “studious COO studio” can be read, which demonstrates the potential for application in the field of multilevel message encryption (Fig. 4e). Also, considering the excellent processing properties of PAMMA-Na, a large variety of 3D RTP models can be facilely fabricated by pouring the high-concentrated water solution into various molds and drying in a 70 °C oven for 12 h (Fig. 4f). Importantly, the material still has good luminescence properties with slightly reduced SSPL and RTP intensities and lifetimes after several solution and molding cycles, exhibiting its excellent recyclability (Fig. S25†). | ||
| Fig. 4 Anti-counterfeiting and encryption application. (a) Schematic diagram of the screen printing of the pattern anti-counterfeiting; (b and c) temperature (b) and water (c) encrypted RTP pattern after excitation with a 365 nm UV lamp at 298 K and 249 K; (d and e) schematic diagram (d) and photographs (e) of letter encryption using CL polymers with different RTP colors and lifetimes at different temperatures after turning off the UV excitation (365 nm). (f) Photographs of various mold-casted 3D models under 365 nm UV light (UV on) after the light is turned off (UV off). | ||
Conclusions
In summary, we have successfully achieved anti-Kasha RTP in CL materials by introducing ions into the hydrolyzed nonconjugated MA and AM copolymers. The copolymerization and ionization lead to significantly promoted triplet excited state emission for RTP, enlarged T2–T1 energy gap for blocking IC, and enhanced intra/inter-molecular interactions by abundant hydrogen and ionic bonding networks for suppressing nonradiative decays. Thus, the ionic CTE copolymers exhibit anti-Kasha RTP with a significantly blue-shifted emission peak up to 29 nm compared to the fluorescent peak and a lifetime of 36 ms; and this blue-shift is the largest ever reported. Moreover, an efficient OURTP was also observed, showing a lifetime of 420 ms and a PhQY up to 8.9%. Taking advantage of the lifetime/temperature-dependent RTP and excellent processability of the ionic copolymers, we achieved their applications in multi-level information encryption and anti-counterfeiting. This unusual photophysical behavior that violates the Kasha rule in CTE materials offers a deeper understanding on the population, equilibrium, and radiative processes of the higher triplet excited states for RTP, providing important mechanism understanding and unprecedented opportunities for the material design to stimulate efficient TSC, anti-Kasha emission and intrinsic stimuli-responsive RTP and OURTP behaviors for advanced applications.Data availability
The data that support the findings of this study have been included in the main text and ESI.† All other information can be obtained from the corresponding author upon request. Source data are provided with this paper.Author contributions
J. Z., Y. J., L. Z., and R. C. conceived and designed the experiments. J. Z., Y. J., X. L., and W. M. were primarily responsible for the material preparation. J. Z., Y. J., and R. C. measured and analyzed the photophysical properties. C. S. performed the computational calculations. J. Z., Y. J., and Y. L. fabricated the applications. J. Z., Y. J., and R. C. wrote the manuscript and all authors contributed to the data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (22275097, 62374093, and 62288102), the Huali Talents Program of Nanjing University of Posts and Telecommunications, the Open Research Fund of State Key Laboratory of Organic Electronics and Information Displays, State Key Laboratory of Advanced Optical Communication Systems and Networks Shanghai Jiao Tong University (2024GZKF001), Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX22_0250), Jiangsu Government Scholarship for Study Abroad.Notes and references
- Y. Miao, F. Lin, D. Guo, J. Chen, K. Zhang, T. Wu, H. Huang, Z. Chi and Z. Yang, Sci. Adv., 2024, 10, adk3354 CrossRef PubMed.
- M. Cao, Y. Ren, Y. Wu, J. Shen, S. Li, Z.-Q. Yu, S. Liu, J. Li, O. J. Rojas and Z. Chen, Nat. Commun., 2024, 15, 2375 CrossRef CAS PubMed.
- M. Zeng, W. Wang, S. Zhang, Z. Gao, Y. Yan, Y. Liu, Y. Qi, X. Yan, W. Zhao, X. Zhang, N. Guo, H. Li, H. Li, G. Xie, Y. Tao, R. Chen and W. Huang, Nat. Commun., 2024, 15, 3053 CrossRef CAS PubMed.
- S. Shen, Q. Xie, S. R. Sahoo, J. Jin, G. V. Baryshnikov, H. Sun, H. Wu, H. Ågren, Q. Liu and L. Zhu, Adv. Mater., 2024, 36, 2404888 CrossRef CAS.
- Y. Zhao, J. Yang, C. Liang, Z. Wang, Y. Zhang, G. Li, J. Qu, X. Wang, Y. Zhang, P. Sun, J. Shi, B. Tong, H. Y. Xie, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2023, 63, e202317431 Search PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CAS.
- J. Zhang, S. Xu, L. Zhang, X. Wang, Y. Bian, S. Tang, R. Zhang, Y. Tao, W. Huang and R. Chen, Adv. Mater., 2022, 34, 2206712 CAS.
- G. Lu, J. Tan, H. Wang, Y. Man, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Nat. Commun., 2024, 15, 3705 CAS.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CAS.
- H. Peng, G. Xie, Y. Cao, L. Zhang, X. Yan, X. Zhang, S. Miao, Y. Tao, H. Li, C. Zheng, W. Huang and R. Chen, Sci. Adv., 2022, 8, eabk2925 CAS.
- S. Xu, W. Wang, H. Li, J. Zhang, R. Chen, S. Wang, C. Zheng, G. Xing, C. Song and W. Huang, Nat. Commun., 2020, 11, 4802 CAS.
- X.-Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem., 2023, 7, 854–874 CAS.
- X. Luo, B. Tian, Y. Zhai, H. Guo, S. Liu, J. Li, S. Li, T. D. James and Z. Chen, Nat. Rev. Chem., 2023, 7, 800–812 Search PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 Search PubMed.
- L.-L. Zhu, Y.-E. Huang, L.-K. Gong, X.-Y. Huang, X.-H. Qi, X.-H. Wu and K.-Z. Du, Chem. Mater., 2020, 32, 1454–1460 CAS.
- D. Malpicci, E. Lucenti, C. Giannini, A. Forni, C. Botta and E. Cariati, Molecules, 2021, 26, 6999 CrossRef CAS PubMed.
- B. H. Jhun, D. Y. Jeong, S. Nah, S. Y. Park and Y. You, J. Mater. Chem. C, 2021, 9, 7083–7093 RSC.
- J. Li, X. Li, G. Wang, X. Wang, M. Wu, J. Liu and K. Zhang, Nat. Commun., 2023, 14, 1987 CAS.
- S.-Y. Chu, Chem. Phys. Lett., 1975, 32, 24–27 CAS.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 Search PubMed.
- Q. S. Zhang, S. C. Wang, X. H. Xiong, P. Y. Fu, X. D. Zhang, Y. N. Fan and M. Pan, Angew. Chem., Int. Ed., 2022, 61, e202205556 CAS.
- W. Xie, W. Huang, J. Li, Z. He, G. Huang, B. S. Li and B. Z. Tang, Nat. Commun., 2023, 14, 8098 CAS.
- T. Yang, Y. Li, Z. Zhao and W. Z. Yuan, Sci. China: Chem., 2023, 66, 367–387 CAS.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- T. Zhu, T. Yang, Q. Zhang and W. Z. Yuan, Nat. Commun., 2022, 13, 2658 CrossRef CAS PubMed.
- X. Zhang, L. Du, W. Zhao, Z. Zhao, Y. Xiong, X. He, P. F. Gao, P. Alam, C. Wang, Z. Li, J. Leng, J. Liu, C. Zhou, J. W. Y. Lam, D. L. Phillips, G. Zhang and B. Z. Tang, Nat. Commun., 2019, 10, 5161 CrossRef PubMed.
- B. Chu, H. Zhang, L. Hu, B. Liu, C. Zhang, X. Zhang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 61, e202114117 CrossRef PubMed.
- X. Ji, W. Tian, K. Jin, H. Diao, X. Huang, G. Song and J. Zhang, Nat. Commun., 2022, 13, 3717 CrossRef CAS PubMed.
- T. Yang, Y. Wang, J. Duan, S. Wei, S. Tang and W. Z. Yuan, Research, 2021, 2021, 9757460 CAS.
- Z. Dai, G. Wang, F. Xiao, D. Lei and X. Dou, Adv. Mater., 2023, 35, 2300526 CrossRef CAS.
- H. Gong, H. Yu, Y. Zhang, L. Feng, Y. Tian, G. Cui and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202219085 CrossRef CAS.
- H. Li, J. Gu, Z. Wang, J. Wang, F. He, P. Li, Y. Tao, H. Li, G. Xie, W. Huang, C. Zheng and R. Chen, Nat. Commun., 2022, 13, 429 CrossRef CAS PubMed.
- Y. Wang, S. Tang, Y. Wen, S. Zheng, B. Yang and W. Z. Yuan, Mater. Horiz., 2020, 7, 2105–2112 RSC.
- B. Zhao, S. Yang, X. Yong and J. Deng, ACS Appl. Mater. Interfaces, 2021, 13, 59320–59328 CAS.
- J. Zhang, P. Alam, S. Zhang, H. Shen, L. Hu, H. H. Y. Sung, I. D. Williams, J. Sun, J. W. Y. Lam, H. Zhang and B. Z. Tang, Nat. Commun., 2022, 13, 3492 CAS.
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- K. Chen, Y. Wang, B. Chu, Z. Yan, H. Li, H. Zhang, S. Hu, Y. Yang, B. Liu and X.-H. Zhang, J. Mater. Chem. C, 2022, 10, 16420–16429 CAS.
- X. Zhang, M. Zeng, Y. Zhang, C. Zhang, Z. Gao, F. He, X. Xue, H. Li, P. Li, G. Xie, H. Li, X. Zhang, N. Guo, H. Cheng, A. Luo, W. Zhao, Y. Zhang, Y. Tao, R. Chen and W. Huang, Nat. Commun., 2023, 14, 475 CAS.
- H. Wang, H. Shi, W. Ye, X. Yao, Q. Wang, C. Dong, W. Jia, H. Ma, S. Cai, K. Huang, L. Fu, Y. Zhang, J. Zhi, L. Gu, Y. Zhao, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 18776–18782 CAS.
- S. Wang, D. Wu, S. Yang, Z. Lin and Q. Ling, Mater. Chem. Front., 2020, 4, 1198–1205 CAS.
- P. Liao, T. Wu, C. Ma, J. Huang and Y. Yan, Adv. Opt. Mater., 2022, 11, 2202482 Search PubMed.
- J. Zhang, S. Xu, Z. Wang, P. Xue, W. Wang, L. Zhang, Y. Shi, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2021, 60, 17094–17101 CAS.
- L. Zhang, H. Peng, J. Zhang, Z. Guo, Y. Jin, S. Zhang, Y. Tao, C. Zheng and R. Chen, J. Mater. Chem. C, 2022, 10, 16893–16902 CAS.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CAS.
- G. Li, D. Jiang, G. Shan, W. Song, J. Tong, D. Kang, B. Hou, Y. Mu, K. Shao, Y. Geng, X. L. Wang and Z. Su, Angew. Chem., Int. Ed., 2021, 61, e202113425 Search PubMed.
- Q. Mu, K. Zhang, H. Zou, H. Liu, Y. Song, C.-K. Wang, L. Lin and J. Fan, Dyes Pigm., 2022, 205, 110560 CrossRef CAS.
- T. Wang, A. K. Gupta, S. Wu, A. M. Z. Slawin and E. Zysman-Colman, J. Am. Chem. Soc., 2023, 145, 1945–1954 CrossRef CAS.
- H. Li, H. Li, J. Gu, F. He, H. Peng, Y. Tao, D. Tian, Q. Yang, P. Li, C. Zheng, W. Huang and R. Chen, Chem. Sci., 2021, 12, 3580–3586 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01471a |
| This journal is © The Royal Society of Chemistry 2025 |