DOI:
10.1039/D5SC00827A
(Edge Article)
Chem. Sci., 2025,
16, 7838-7846
Minimal catalytic dissipative assemblies via cooperation of an amino acid, a nucleobase precursor and a cofactor†
Received
31st January 2025
, Accepted 20th March 2025
First published on 24th March 2025
Abstract
Functions arising from cooperation between protobiopolymers have fueled the chemical emergence of living matter, which requires a continuous supply of energy to exist in a far-from-equilibrium state. Non-equilibrium conditions imparted by available energy sources have played critical roles in the appearance of complex co-assembled architectures, which exploit the properties of different classes of biopolymers. Such co-assemblies formed from mixtures of nitrogenous heterocycles as protonucleobases and peptide precursors might have acted as early versions of catalytic machinery, capable of sustaining chemical reaction networks. Herein, we show the generation of catalytic non-equilibrium networks from a mixture of a nitrogenous heterocycle, an amino acid and a cofactor driven by an aromatic substrate. The cooperation, a result of supramolecular interactions between different components, rendered the assemblies capable of activating the cofactor towards oxidative degradation of the substrate, which resulted in autonomous disassembly (negative feedback). Furthermore, utilising promiscuous hydrolytic capability, the transient co-assemblies could metabolise a precursor to generate additional amounts of the substrate, enhancing the lifetime (positive feedback) of the assemblies.
Introduction
Living matter requires a continuous supply of energy to sustain it in a far-from-equilibrium state and displays remarkable cooperation between biopolymers, such as nucleic acids and proteins. This collaboration is beautifully manifested in the ribosome, the universal cell organelle.1–6 Ribonucleoprotein, a supramolecular complex composed of3,4 RNA subunits and as many as 70 ribosomal proteins, exploits the properties of both nucleic acids and proteins to exemplify biopolymer collaboration.1a–c,2 Such cooperation, manifested in the form of multicomponent supramolecular assembly, was crucial for the generation of proto-biocatalysts.1c,d,3,4,7 Furthermore, such early catalysts were required for the complexification of reaction networks and protometabolism operating out-of-equilibrium (Fig. 1a).7–15 Under non-equilibrium conditions, early biopolymer cooperation was facilitated as available energy sources drove the polymerization of simple building blocks, leading to supramolecular catalytic entities capable of facilitating biochemical reactions networks.16–25 Designing catalytic reaction networks based on simple precursors of biopolymers, such as protonucleobases26,27a and amino acids,27b along with the prebiotically plausible porphyrin-based co-factors27c that can show functional collaboration, can build on our understanding of the principles leading to complex functions arising from simple molecules during early chemical evolution. However, protocellular forms that preceded the emergence of primitive catalytic machinery possibly did not have the advantage of even short peptide sequences or nucleic acid fragments. From the context of a primitive yet diverse chemical inventory, it is intriguing to conceive that a simple nitrogenous heterocycle (a plausible protonucleobase), peptide precursors and low-molecular-weight cofactors could have coexisted and comixed under non-equilibrium conditions to access supramolecular architectures with catalytic capabilities.24–27 The bottom-up approach of creating catalytic reaction networks under non-equilibrium conditions with co-assembled simple building blocks can contribute towards our understanding of the emergence of living matter like properties.28–33 Herein, we report that a mixture of low-molecular-weight building blocks, i.e., a heterocycle (model protonucleobase), a single amino acid (precursor of proteins) and a cofactor in the presence of an aromatic substrate, can lead to the generation of catalytic non-equilibrium networks (Fig. 1b). The heterocycle (T) and aromatic acid (AR) co-assemble to create the structural framework of the assemblies, which non-covalently bind the amino acid (AA, Fig. 1b) and the cofactor (Cf, hemin) to display emergent peroxidase-like activity. The catalytic co-assembly subsequently degrades the substrate, leading to disassembly and hence, autonomously controls its own lifetime (negative feedback, Fig. 1b). The supramolecular assembly of such simple building blocks related to extant biopolymers, that activate a bound cofactor to demonstrate catalysis and dissipative self-assembly, is an unprecedented observation. Apart from oxidation, the presence of AA in the assembled state helped in the realization of a chemical reaction network by catalyzing a secondary hydrolysis reaction, thereby expanding the catalytic diversity.34 The latent hydrolytic activity benefitted the assemblies by generating more substrate from a precursor. Since the substrate played an important role in inducing co-assembly, regeneration of the substrate thus induced further assembly formation, and increased the lifetimes and mechanical strength of the assemblies and installed positive feedback into the system.
 |
| | Fig. 1 (a) Possible collaboration between peptide fragment and short nucleotides towards primitive catalytic machinery. (b) Generation of dissipative assemblies via co-assembly of a mixture of heterocycle (T, triazine protonucleobase), cofactor (Cf) and single amino acid (AA), driven by AR in an oxidising environment (H2O2). The cooperation between the components leads to peroxidase activity that subsequently degrades AR, leading to autonomous disassembly (negative feedback). | |
Results and discussion
We started with a triazine amine (Fig. 1b, argued to be a possible protonucleobase, T)35,36a and to drive its assembly, an equimolar (50 mM) concentration of an organic acid was added (AR). Hydroxybenzoic acids have been shown to form co-assemblies with heterocycles such as melamine (T) through hydrogen-bonding interactions.36b The methoxy group in AR made the substrate electron rich and subsequently more prone to oxidation. Further, the substrate has been shown to be oxidised by peroxidase enzymes.37 Next, a non-enzymatic yet intrinsic catalytic pathway was installed by adding a single amino acid (methyl acetyl-L-histidinate, AA, 10 mM, where the carboxy group was methylated to impart hydrophobicity) along with a low-molecular-weight cofactor (Cf, 150 μM) (hemin, a prosthetic group of modern peroxidases, Fig. 1b; the intrinsic peroxidase activity of hemin in water is miniscule38). We expected that in the mixture, T and AR would rapidly interact to provide the structural framework of the supramolecular assemblies and would imbibe AA and Cf on the surface of the assemblies.39 In the assembled state, AA would coordinate with Cf to restrict the dimerization to a certain extent and possibly activate it for catalysis (in the presence of H2O2, 30 mM). In an oxidizing environment, this would lead to the catalytic degradation of the electron-rich substrate AR and would help in accessing the (dis)assembly of organized structures via negative feedback. The mixture of T, AR, AA, Cf and H2O2 showed rapid self-assembly, which eventually turned to a light-brown-coloured self-supporting gel in ∼2 min (Fig. 2b, pH ∼ 5.1, 1.5% (v/v) DMF in H2O, Cf is brown in colour). Interestingly, the co-assembled gel showed a slight intensification of the brown colour with time, which suggested ongoing oxidation. In the next ca. 2.5 h, an autonomous transition to a sol was witnessed, suggesting dissipative assembly (Fig. 2a–c). The autonomous (dis)assembly was visualized with time-resolved transmission electron microscopy (TEM), which showed the temporal formation and rupture of fibrillar networks (Fig. 2d–f), yet leaving some kinetically trapped fibres. Atomic force microscopy (AFM) also suggested the presence of fibrillar nanostructures for the assembled state with an average height of ∼2–8 nm (Fig. S1†). Controls done in the absence of either AA or Cf led to kinetically stable co-assemblies with no noticeable visual changes over time (Fig. 2g, S2 and S3†). This suggested the importance of the cofactor–amino acid combination for the emergence of catalysis that led to the realization of the dissipative assembly. As expected, a mixture of T and AR alone led to a kinetically stable gel (Fig. S4†). Further, the omission of T or AR resulted in free-flowing solutions (Fig. 2g, S5a and b†), whereas a mixture of T, AR, AA, and Cf without H2O2 led to a kinetically stable gel (Fig. 2g and S5c†). These observations underline the importance of the substrate-driven generation of catalytically active non-equilibrium networks. Furthermore, variation of the concentration of the four components (T, AR, Cf and AA) and pH led to modulation of the lifetime of the co-assembly, presumably due to different oxidation rates (Fig. S6†). In the present system, the optimum oxidizing activity at pH 5 led to the realization of transient assemblies.38d,40 The generation of a dissipative assembly via cooperation between the building blocks was also evident from time-dependent rheological studies, which revealed an initial rise in the storage modulus (G′) followed by a decline (Fig. 3a and S7†). Controls in the absence of Cf or AA registered a similar increase in G′, but did not show an autonomous decline over a prolonged time, which suggested the role of catalysis in temporally controlling the mechanical properties (Fig. 3b, c, S8 and S9†). A higher maximum G′ was observed in the absence of AA compared to that in the presence of AA, presumably due to an ongoing oxidation process in the latter case. Temporal generation of networks was further probed with the hydrophobic dye rhodamine 110, which showed an increase in fluorescence intensity for up to 1 h upon binding to a hydrophobic environment, followed by a gradual decline, while controls without AA or Cf did not show any temporal changes (Fig. 3d and S10†). To elucidate the interactions leading to the structural framework of the assemblies, Fourier transform infrared spectroscopy (FTIR) of the co-assembled system (T + AR + AA + Cf + H2O2) and the individual components were done. The intermolecular hydrogen bonding between T and AR was evident from the spectra, as peaks at 3000–3500 cm−1 corresponding to N–H vibrations (of T) were significantly broadened in the assembled state (Fig. 3e). A similar effect was observed for kinetically stable assemblies in the presence of T and AR alone, which underpinned the role of both components in the formation of the structural backbone of the assemblies. The dissipative assemblies were next investigated with 1H NMR-based studies. Interestingly, AR showed peak broadening in the gel state due to the reduced transversal relaxation time (T2), which subsequently resulted in signal suppression (Fig. 3f and S11;†T does not have any non-exchangeable protons). As a function of time, the peaks became sharper, which reflected the temporal (dis)assembly process of the supramolecular framework (Fig. 3f and S11†). Next, NMR spectra of the co-assembled system at varying concentrations of AR were recorded. At lower concentration (<10 mM), due to negligible aggregation, the total AR present in the system was equivalent to the amount being observed, whereas beyond 10 mM, the amount observed was significantly lower than the actual amount, which confirmed that AR was incorporated into the co-assemblies (Fig. 3g, h and S12†).36a Powder X-ray diffraction (PXRD) of the co-assembly showed d-spacings of 3.24 Å and 4.45 Å, which suggested an ordered arrangement of the building blocks through π–π interactions (Fig. 4a). PXRD of the stable assemblies consisting of T and AR alone showed peaks at 3.22 Å and 4.35 Å, again indicating that the heterocycle and the aromatic acid were the main
components for the self-assemblies (Fig. 4a). Additionally, density functional theory (DFT) studies at B3LYP/D3BJ/def2-TZVP/CPCM (water) were performed to gain a more detailed understanding of the packing arrangements in assemblies comprising T and AR. Two computational models with two distinct H-bond interaction modes between T and AR were explored: the first one featuring –CO⋯H–N H-bond interaction was preferred (binding energy of −9.9 kcal mol−1) over the second, having -OMe as the H-bond acceptor (binding energy of −8.3 kcal mol−1) (Fig. S13†). Furthermore, parallel stacked-dimer models (Fig. S13c and d†) emphasized a preference for the synergistic –CO⋯H–N H-bond and extended π-stacking interactions at an interplanar distance of 3.28 Å, in agreement with the powder X-ray diffraction (PXRD) studies (d-spacing of 3.22 Å) (Fig. 4b, S13c and d†). We hypothesize that this leads to the extended H-bond network between T and AR forming a cyclic rosette structure (Model A, Fig. 4c and S14†) responsible for catalytic activity. Next, we investigated the role of the assemblies in recruiting highly soluble AA, which can consequently install catalytic potential from the assembled state. For this, the assemblies were centrifuged at different times and the supernatant was separated, with the hope that only the unbound AA would be there in the supernatant, from which the amount of bound AA could be estimated. HPLC of supernatants as a function of time showed that the extent of binding of AA was significantly higher at ca. 10 min (3.9 ± 1.2 mM, ∼39%, Fig. 4d and S15; see ESI† for details) compared to the disassembled states, i.e., at the beginning and at ca. 600 min. The concentrations shown in Fig. 4d are the concentrations of bound AA, which were calculated by subtracting the amount observed from HPLC at each time point from the total amount added. Further, the binding of AA during the formation of the co-assembly was investigated with 1H NMR. Notably, time-dependent 1H NMR of the dissipative gel (T + AR + AA + Cf + H2O2) revealed transient binding of AA to the non-equilibrium assemblies. The concentration of free AA observed from NMR showed a decrease from 10 mM (free AA) to ∼7 mM after 2 min, implying a binding of ∼30%, which is similar to that observed from HPLC studies (Fig. 4e and 3f). The concentration of unbound AA showed a gradual increase to ∼9 mM with time, underpinning the event of disassembly (Fig. 4e, 3f, S11 and S16†). Interestingly, when AA was added to the kinetically stable gel of T and AR, a similar binding tendency of the amino acid (∼30% entrapment) was observed, which did not change as a function of time (Fig. S17 and S18†). Next, the possible interaction of Cf with AA bound to the assemblies was investigated via UV-vis spectroscopy. In contrast to free Cf in water, an 18 nm red shift of the Soret band was observed for the assemblies (Fig. 4f). The results suggested a lower extent of Cf dimerization in the co-assembly and the subsequent formation of an AA–Cf complex (hemin–histidine complex), where AA is co-ordinated to Cf.38 Although we did not observe the sharp bands typically associated with stoichiometric binding, the significantly larger shift compared to AA + Cf alone (a red shift of ∼12 nm) suggested binding of the cofactor to the co-assemblies (Fig. 4f).38 Next, circular dichroism (CD) showed an induced negative signal of achiral Cf at ∼416 nm, which suggested its binding to the chiral microenvironment (due to presence of chiral AA) of the co-assemblies (Fig. S19†). Since the induced CD signal of hemin (Cf) was measured in the presence of assemblies, the concentration of Cf was kept high. Controls done without AA or Cf did not result in the generation of any prominent peak at ∼416 nm (Fig. S19†). In combination, the results confirmed that AA and Cf were non-covalently bound to the structural framework constituted from the T and AR co-assembly. Next, catalytic oxidation by the transient assemblies was investigated. The consumption of AR over time was used to monitor the progress of the reaction, as the complexities of multiple oxidation products prohibited their precise monitoring. Time-resolved HPLC revealed a decrease in peak area of AR, and suggested the consumption of ca. 8.2 mM (∼16%) in 3.5 h (Fig. 4g, S20 and S21;† in the presence of 30 mM H2O2). Interestingly, a control done in the presence of 41.8 mM AR (remaining AR after oxidation; concentrations of T, Cf, AA and H2O2 kept the same) did not result in a self-supporting gel, indicating that the extent of oxidation observed was sufficient for disassembly (Fig. S22†). Controls done in the absence of either T, AA or both (the rest of the components were kept the same) showed ∼6%, ∼2% and ∼6.8% consumption of AR, respectively (Fig. 4g). Furthermore, the consumption of AR was observed to be negligible (∼1%) when Cf was added externally into the pre-formed gel (Fig. 4g; even when high concentrations of Cf were present). This result suggested that the pre-mixing of T and AR before the addition of Cf diminished the catalytic activity, presumably due to the fact that the preformed assemblies were not able to incorporate the cofactor. Expectedly, there was no oxidation in the absence of H2O2 (Fig. 4g). The aromatic organic acid AR along with the heterocycle T form the substrate-driven co-assembly that locally increased the concentrations of AA and Cf. These results strongly confirmed the importance of the co-assemblies, which could recruit AA and Cf for the emergent peroxidase activity, that subsequently installed negative feedback towards its stability. Further, HPLC showed peaks corresponding to the oxidation products for the transient assemblies aged for 3.5 h (Fig. S23; † oxidation in the presence of HRP showed similar product formation,37,41 Fig. S24†) while controls in the absence of T (AR + AA + Cf + H2O2) showed no prominent peak generation (Fig. S23†). HRMS-HPLC investigations confirmed the presence of one of the oxidation products, which matched a report in the literature (Fig. S25†).37 Such acceleration of catalytic ability in the aggregated state is a strategy used in extant living systems to self-regulate non-equilibrium biopolymers to perform work.30a,b,33a,42–45 Importantly, the oxidation percentage of AR and the corresponding lifetime could be tuned by varying the concentrations of different components. Higher concentrations of Cf and AA showed greater extents of oxidation, which resulted in shorter lifetimes (Fig. 4h). To check the recyclability of the system, an additional batch of substrate AR was added when the mechanical strength of the system showed a decrease. Indeed, the mechanical strength registered an increase and subsequent autonomous decline, thus realizing two cycles of (dis)assembly. However, the system could not be recycled further, possibly due to accumulation of oxidation products (Fig. S26†).
 |
| | Fig. 2 Representative images of vials (a–c) and TEM micrographs (d–f) of a system containing T, AA, Cf and H2O2 at different times after the addition of AR. (g) Table showing the final observations of different systems. | |
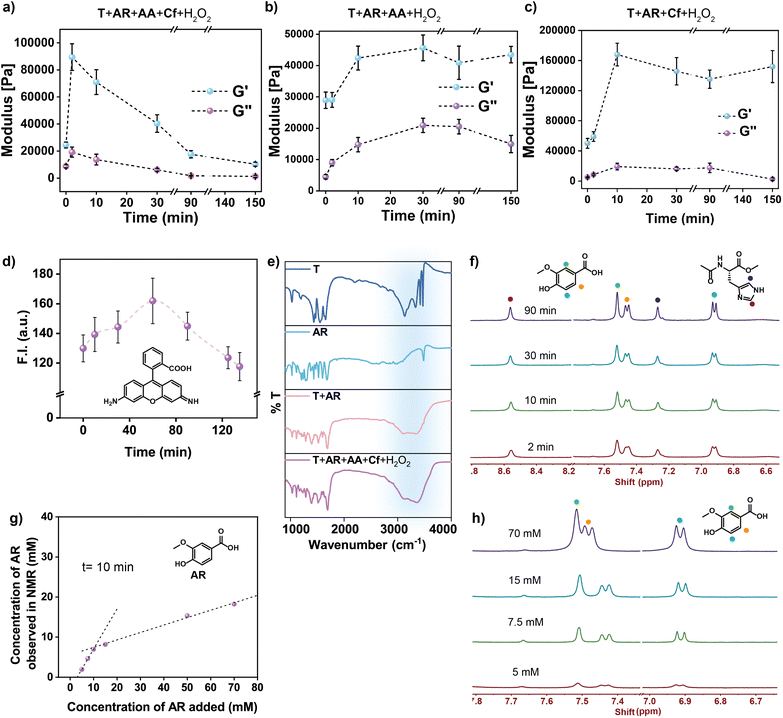 |
| | Fig. 3 (a–c) Time-dependent storage moduli of different systems. (d) Time-dependent fluorescence intensities of Rhodamine 110. (e) FT-IR spectra of different systems. (f) Time-dependent 1H NMR of the dissipative self-assembled system (T + AR + AA + Cf + H2O2). (g) Plot of observed concentration (as determined by NMR) against the total concentration of AR, and (h) selective 1H NMR spectra of the dissipative system (T + AR + AA + Cf + H2O2) using different concentrations of AR. Error bars represent the standard deviation of triplicates. | |
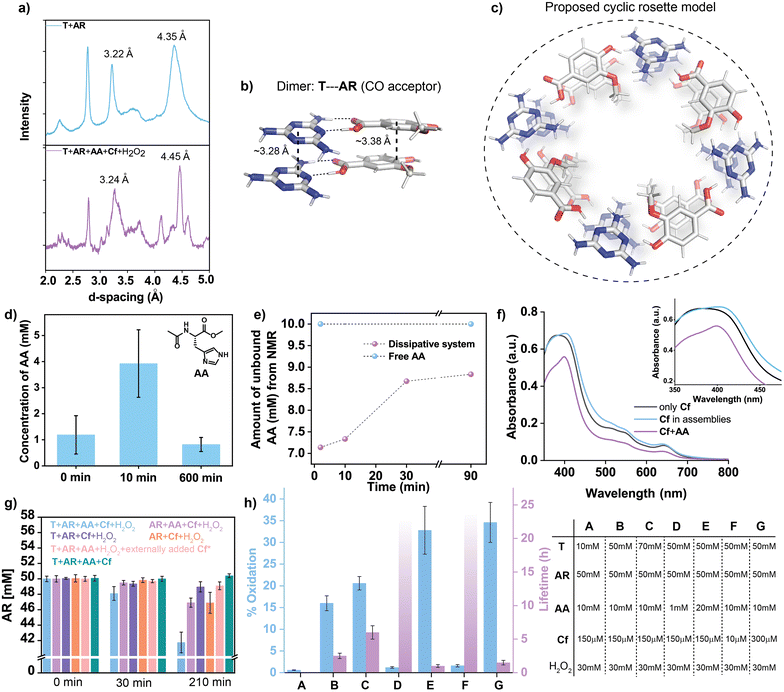 |
| | Fig. 4 (a) PXRD of different systems. (b) Optimized geometries of the computational model of the T and AR dimer and (c) the cyclic rosette model. (d) Concentration of bound AA from HPLC. (e) Change in concentration of AA with time, obtained from time-dependent 1H NMR of the dissipative system (T + AR + AA + Cf + H2O2). (f) Change in absorbance of Cf in the presence of the assemblies and AA alone (enlarged in the inset). (g) Consumption of AR with time in different systems. * represents the systems with 300 μM Cf (the concentrations of the other components were the same). (h) Variation in lifetime and oxidation percentages of the co-assembled system with different compositions. The kinetically stable co-assembly shown with a fading bar implies a lifetime of more than 48 h. Error bars represent the standard deviation of triplicates. | |
As noted, the capability of the co-assembled networks to bind small molecular guests might have been beneficial for the emergence of latent functions: i.e. to promote other biochemical reactions (promiscuity).46 To probe this, the hydrophobic dye Nile red was chosen, and time-resolved confocal microscopy was done. Fluorescent networks were observed at t = 10 min, which gradually dissolved away at t = 2.5 h (Fig. 5a–c and S27†). This suggested the temporal generation of supramolecular fibrillar structures along with the capability to bind to a diverse range of guests. One of the downsides of the reaction cycle realized thus far was the fact that catalysis led to products that were not beneficial for the co-assemblies.46,47 Reaction networks that can exploit recruited guests for feedback loops to self-regulate and, importantly, can utilize the liberated products for its benefit, can demonstrate protometabolism. Since the non-equilibrium assemblies were able to imbibe the histidine-based building block AA,48 we asked whether the assemblies, despite the minimal building blocks, can utilize promiscuous hydrolytic activity (i.e. hydrolytic capabilities due to the presence of AA) to regenerate AR from an ester-based precursor (Pro-AR, AR was coupled with a hydrophobic aromatic group, 7-hydroxycoumarin).32a,48 This would create a hydrolysis–oxidation cascade reaction, where the hydrolysis reaction would create additional amounts of AR, while the methyl ester of AA remains stable throughout the assembly–disassembly cycle and in turn might help in the further generation of the catalysts (dissipative assemblies). Since the substrate played an important role in inducing the co-assembly, the regeneration of more substrate thus installed positive feedback towards the assembly formation (Fig. 5d).49 The generation of an excess amount of AR and subsequently the assemblies, should lead to an increase in the lifetime and the mechanical strength of the assembled state compared to the dissipative assemblies in the absence of Pro-AR. We started with 50 mM T, 50 mM AR and 10 mM Pro-AR along with AA (10 mM), Cf (150 μM) and H2O2 (30 mM) (2.75% (v/v) DMF in H2O; pH was 5.32; the ester was stable at this pH as negligible hydrolysis was observed, Fig. S28†). Interestingly, the lifetime of the gels showed an increase from 150 ± 15 min to 230 ± 30 min (from quadruplicate vials, Fig. 5e). Ensemble measurement, such as the rheological study, also revealed ∼41% higher maximum mechanical strength in the presence of Pro-AR compared to that without the precursor ester (Fig. 5e, S29 and S30†). From HPLC and fluorescence spectroscopy, the generation of 7-hydroxycoumarin from Pro-AR could be observed (Fig. 5f and g; the rate calculated from HPLC was 0.50 ± 0.04 μM min−1, Fig. S31 and S32† shows that AA was stable). Interestingly, in the absence of AR, the storage modulus of the mixture (T + AA + Cf + Pro-AR + H2O2) was almost two orders of magnitude lower compared to samples with AR (Fig. 5e). Samples with more than 10 mM Pro-AR led to precipitation (in the absence of AR). Further, the hydrolysis rate of Pro-AR was found to be dependent on the concentration of AA, which directly translated to the lifetime of the systems (Fig. S33†). To probe the role of the assemblies in catalysing the ester Pro-AR, a control without T (AA + AR + Cf + Pro-AR + H2O2) was done. This free-flowing mixture showed ∼4.5-fold lower hydrolytic activity (0.11 ± 0.03 μM min−1, Fig. S34†). This underlined the importance of the catalytic microenvironment of the dissipative networks to catalyse Pro-AR hydrolysis to generate AR and display positive feedback. In combination, the results suggested that the locally generated AR, despite its modest amount, played a significant role in augmentation of lifetime and mechanical strength (Fig. 5e) and provided positive feedback towards the formation of the co-assemblies.
 |
| | Fig. 5 (a–c) Representative CLSM images of co-assemblies incubated with Nile red. (d) The promiscuous hydrolysis–oxidation-based reaction network. (e) Storage modulus and lifetime of different systems. Statistics were obtained by ANOVA and Bonferroni post hoc tests, *p < 0.05, **p < 0.01, and ***p < 0.001, n = 3. Time-resolved (f) HPLC chromatograms and (g) fluorescence intensities showing the release of 7-hydroxycoumarin. Error bars represent the standard deviation of triplicates. | |
Conclusions
Non-equilibrium conditions imparted by available energy sources played critical roles in the appearance of early versions of catalytic machinery, possibly via the functional collaboration of nitrogenous heterocycles as protonucleobases and peptide precursors. This work demonstrates the non-equilibrium generation of catalytic co-assemblies due to cooperation between minimal precursors of extant biopolymers. An aromatic acid drove the triazine heterocycle (a plausible protonucleobase) towards the formation of dissipative networks that showed emergent peroxidase-like activity by binding a single amino acid and a cofactor, to subsequently degrade the acid (negative feedback). Exclusion of any one of the components led to kinetically stable co-assemblies. Furthermore, through promiscuous hydrolytic activity, the co-assembly could replenish the acid substrate from a precursor, resulting in an increase in its lifetime via positive feedback. The emergence of function due to such cooperation between simple building blocks seen in non-equilibrium assemblies opens up possibilities of recruiting diverse guests required to expand the catalytic repertoire and set the stage for complex reaction networks and protometabolism.
Data availability
We wish to state that all experimental and computational data associated with this work are available in the ESI.†
Author contributions
D. D. conceived and supervised the overall project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
D. D. acknowledges SERB SB/SJF/2020-21/08, MoE-STARS-2/2023-0752. S. P. A., S. R., P. B. acknowledge UGC for fellowship.
Notes and references
-
(a) T. M. Schmeing, R. M. Voorhees, A. C. Kelley, Y. G. Gao, F. V. IV Murphy, J. R. Weir and V. Ramakrishnan, Science, 2009, 326, 688–694 CAS;
(b) A. Harish and G. Caetano-Anollés, PLoS One, 2012, 7, e32776 CAS;
(c) F. Müller, L. Escobar, F. Xu, E. Węgrzyn, M. Nainytė, T. Amatov, C. Chan, A. Pichler and T. Carell, Nature, 2022, 605, 279–284 Search PubMed;
(d) J. T. Goodwin, A. K. Mehta and D. G. Lynn, Acc. Chem. Res., 2012, 45, 2189–2199 CAS.
-
(a) M. Di Giulio, J. Mol. Evol., 1997, 45, 571–578 CAS;
(b) B. M. A. G. Piette and J. G. Heddle, Trends Ecol. Evol., 2020, 35, 397–406 Search PubMed;
(c) A. K. Bandela, H. Sadihov-Hanoch, R. Cohen-Luria, C. Gordon, A. Blake, G. Poppitz, D. G. Lynn and G. Ashkenasy, Isr. J. Chem., 2022, e202200030 Search PubMed.
- K. E. Nelson, M. Levy and S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3868–3871 CAS.
-
(a) M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CAS;
(b) D. Speijer, Life, 2015, 5(1), 230–246 Search PubMed.
- J. C. Bowman, A. S. Petrov, M. Frenkel-Pinter, P. I. Penev and L. D. Williams, Chem. Rev., 2020, 120, 4848–4878 CAS.
- D. M. D. Fialho, T. P. Roche and N. V. Hud, Chem. Rev., 2020, 120, 4806–4830 Search PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Open Biol., 2017, 7, 170050 Search PubMed.
- A. de la Escosura, Life, 2019, 9, 66 CrossRef CAS PubMed.
- Y. Bai, A. Chotera, O. Taran, C. Liang, G. Ashkenasy and D. G. Lynn, Chem. Soc. Rev., 2018, 47, 5444–5456 RSC.
- W. G. Scott, A. Szöke, J. Blaustein, S. M. O'Rourke and M. P. Robertson, Life, 2014, 4(2), 131–141 CrossRef PubMed.
- J. D. Sutherland, Nat. Rev. Chem, 2017, 1, 0012 CrossRef CAS.
- K. B. Muchowska, S. J. Varma and J. Moran, Chem. Rev., 2020, 120, 7708–7744 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CAS.
- D. Deamer and A. L. Weber, Cold Spring Harbor Perspect. Biol., 2010, 2, a004929 Search PubMed.
- J. L. England, Nat. Nanotechnol., 2015, 10, 919–923 CAS.
-
(a) R. D. Astumian, Chem. Commun., 2018, 54, 427–444 CAS;
(b) M. He and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 18560–18569 CAS.
- H. Hess and J. L. Ross, Chem. Soc. Rev., 2017, 46, 5570–5587 CAS.
- R. Pascal, A. Pross and J. D. Sutherland, Open Biol., 2013, 3, 130156 Search PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CAS.
-
(a) H. S. Azevedo, S. L. Perry, P. A. Korevaar and D. Das, Nat. Chem., 2020, 12, 793–794 CAS;
(b) H. Fanlo-Virgós, A. R. Alba, S. Hamieh, M. Colomb-Delsuc and S. Otto, Angew. Chem., Int. Ed., 2014, 53, 11346–11350 CrossRef PubMed.
-
(a) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 CAS;
(b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC;
(c) E. Olivieri, B. Gasch, G. Quintard, J.-V. Naubron and A. Quintard, ACS Appl. Mater. Interfaces, 2022, 14, 24720–24728 CrossRef CAS PubMed.
- S. De and R. Klajn, Adv. Mater., 2018, 282, 1706750 CrossRef PubMed.
-
(a) S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS PubMed;
(b) M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 CrossRef.
- A. Ricardo and J. W. Szostak, Sci. Am., 2009, 301, 54–61 CrossRef CAS PubMed.
- D. C. Wallace, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8947–8953 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
-
(a) B. Patel, C. Percivalle, D. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed;
(b) S. L. Miller, Science, 1953, 117, 528–529 CrossRef CAS PubMed;
(c) G. W. Hodgson and C. Ponnamperuma, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 22–28 CAS.
-
(a) G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Systems chemistry, Chem. Soc. Rev., 2017, 46, 2543–2554 CAS;
(b) N. Singh, A. Lopez-Acosta, G. J. M. Formon and T. M. Hermans, J. Am. Chem. Soc., 2022, 144, 410–415 CAS;
(c) J. Mehak and B. J. Ravoo, Angew. Chem., Int. Ed., 2021, 60, 21062–21068 Search PubMed;
(d) M. K. Ovalle, M. Toyoda, R. C. N. Stindt, S. Crespi and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202214495 CAS;
(e) E. Olivieri, J. M. Gallagher, A. Betts, T. W. Mrad and D. A. Leigh, Nat. Synth., 2024, 3, 707–714 CAS.
-
(a) S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CAS;
(b) M. P. van der Helm, C.-L. Wang, B. Fan, M. Machhione, E. Mendes and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 20604–20611 CAS;
(c) L. Tian, M. Li, J. Liu, A. J. Patil, B. W. Drinkwater and S. Mann, ACS Cent. Sci., 2018, 4, 1551–1558 CAS;
(d) S. M. Morrow, I. Colomer and S. P. A Fletcher, Nat. Commun., 2019, 10, 1011 Search PubMed.
-
(a) P. S. Muñana, G. Ragazzon, J. Dupont, C. Z.-J. Ren, L. J. Prins and J. L.-Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef PubMed;
(b) S. P. Afrose, S. Bal, A. Chatterjee, K. Das and D. Das, Angew. Chem., Int. Ed., 2019, 58, 15783–15787 CrossRef CAS PubMed;
(c) M. P. van der Helm, G. Li, M. Hartono and R. Eelkema, J. Am. Chem. Soc., 2022, 144, 9465–9471 CrossRef CAS PubMed;
(d) C. M. E. Kriebisch, A. M. Bergmann and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 7719–7725 CrossRef CAS PubMed.
-
(a) J. Boekhoven, W. E. Hendriksen, G. J. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed;
(b) M. M. Hossain, J. L. Atkinson and C. S. Hartley, Angew. Chem., Int. Ed., 2020, 59, 13807–13813 CrossRef CAS PubMed;
(c) J. Deng and A. Walther, Adv. Mater., 2020, 32, 2002629 CrossRef CAS PubMed;
(d) J. Rodon Fores, M. Criado-Gonzalez, A. Chaumont, A. Carvalho, C. Blanck, M. Schmutz, F. Boulmedais, P. Schaaf and L. Jierry, Angew. Chem., Int. Ed., 2020, 59, 14558 CrossRef CAS PubMed;
(e) I. Maity, N. Wagner, R. Mukherjee, D. Dev, E. Peacock-Lopez, R. Cohen-Luria and G. Ashkenasy, Nat. Commun., 2019, 10, 4636 Search PubMed.
-
(a) A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 CAS;
(b) P. Adamski, M. Eleveld, A. Sood, A. Kun, A. Szilagyi, T. Czaran, E. Szathmary and S. Otto, Nat. Rev. Chem, 2020, 4, 386–403 CrossRef PubMed;
(c) S. Panja, B. Dietrich and D. J. Adams, ChemSystemsChem, 2020, 2, e1900038 CrossRef CAS;
(d) E. Penocchio and G. Ragazzon, Small, 2023, 19, e2206188 CrossRef PubMed.
-
(a) S. P. Afrose, C. Ghosh and D. Das, Chem. Sci., 2021, 12, 14674–14685 RSC;
(b) T. M. Hermans and N. Singh, Angew. Chem., Int. Ed., 2023, 62, e202301529 CrossRef CAS PubMed;
(c) T. Sangchai, S. A. Shehimy, E. Penocchio and G. Ragazzon, Angew. Chem., Int. Ed., 2023, 62, e202309501 CrossRef CAS PubMed;
(d) K. Dai, M. D. Pol, L. Saile, A. Sharma, B. Liu, R. Thomann, J. L. Trefs, D. Qiu, S. Moser, S. Wiesler, B. N. Balzer, T. Hugel, H. J. Jessen and C. G. Pappas, J. Am. Chem. Soc., 2023, 145, 26086–26094 CrossRef CAS PubMed.
-
(a) M. Fedor and J. Williamson, Nat. Rev. Mol. Cell Biol., 2005, 6, 399–412 CrossRef CAS PubMed;
(b) O. Omosun, M.-C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 Search PubMed;
(c) Z.-G. Wang, Y. Li, H. Wang, K. Wan, Q. Liu, X. Shi and B. Ding, Chem.–Eur. J., 2019, 25, 12576–12582 CrossRef CAS PubMed.
- B. J. Cafferty, D. M. Fialho, J. Khanam, R. Krishnamurthy and N. V. Hud, Nat. Commun., 2016, 7, 11328 CrossRef CAS PubMed.
-
(a) B. J. Cafferty, I. Gallego, M. C. Chen, K. I. Farley, R. Eritja and N. V. Hud, J. Am. Chem. Soc., 2013, 135, 2447–2450 CAS;
(b) B. Roy, P. Bairi, A. Saha and A. K. Nandi, Soft Matter, 2011, 7, 8067–8076 CAS.
- J. A. Buswell, P. Ander, B. Pettersson and K.-E. Eriksson, FEBS Lett., 1979, 103, 98–101 CrossRef CAS PubMed.
-
(a) Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed;
(b) R. Geng, R. Chang, Q. Zou, G. Shen, T. Jiao and X. Yan, Small, 2021, 17, 2008114 CrossRef CAS PubMed;
(c) N. Cvjetan, L. D. Schuler, T. Ishikawa and P. Walde, ACS Omega, 2023, 8, 42878–42899 CrossRef CAS PubMed;
(d) T. Uno, A. Takeda and S. Shimabayashi, Inorg. Chem., 1995, 34, 1599–1607 CAS;
(e) E. Monzani, B. Bonafe, A. Fallarini, C. Redaelli, L. Casella, L. Minchiotti and M. Galliano, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1547, 302–312 CrossRef CAS PubMed.
-
(a) N. K. Wijerathne, M. Kumar and R. V. Ulijn, Chem.–Eur. J., 2019, 25, 11847–11851 CAS;
(b)
N. K. Wijerathne, CUNY Academic Works, 2019, https://academicworks.cuny.edu/gc_etds/3264;
(c) J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., Int. Ed., 2012, 51, 517–520 CAS;
(d) H. Jintoku, T. Sagawa, K. Miyamoto, M. Takafuji and H. Ihara, Chem. Commun., 2010, 46, 7208–7210 CAS.
-
(a) A. Ranieri, F. Bernini, C. Augusto Bortolotti, A. Bonifacio, V. Sergo and E. Castellini, Langmuir, 2011, 27, 10683–10690 CAS;
(b) A. C. Osuji, S. O. O. Eze, E. E. Osayi and F. C. Chilaka, Sci. World J., 2014, 2014, 183163 Search PubMed;
(c) A. J. R. AL-Sa'ady, M. H. A. Al-Bahrani and G. M. Aziz, Int. J. Curr. Microbiol. Appl. Sci., 2018, 7, 328–339 Search PubMed.
- J. Berlin and W. Barz, Z. Naturforsch., C:J. Biosci., 1975, 30c, 650–658 CAS.
- E. Penocchio, R. Rao and M. Esposito, Nat. Commun., 2019, 10, 3865 Search PubMed.
- R. Chen, S. Neri and L. J. Prins, Nat. Nanotechnol., 2020, 15, 868–874 CAS.
- M. Caplow and J. Shanks, J. Biol. Chem., 1990, 265, 8935–8941 CAS.
- R. D. Astumian, Nat. Commun., 2019, 10, 3837 Search PubMed.
- P. Carbonell, G. Lecointres and J. L. Faulon, J. Biol. Chem., 2011, 286, 43994–44004 CAS.
- J. Ottelé, A. S. Hussain, C. Mayer and S. Otto, Nat. Catal., 2020, 3, 547–553 Search PubMed.
-
(a) C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. G. Pappas, V. Narang, T. Wang, C. Maldarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CAS;
(b) S. Pal, A. Reja, S. Bal, B. Tikader and D. Das, Angew. Chem., Int. Ed., 2022, 61, e202111857 CAS;
(c) C. Ghosh, S. Menon, S. Bal, S. Goswami, J. Mondal and D. Das, Nano Lett., 2023, 23, 5828–5835 CAS.
-
(a) A. Y. Mitrophanov and E. A. Groisman, BioEssays, 2008, 30, 542–555 CrossRef CAS PubMed;
(b) B. Novák and J. Tyson, Nat. Rev. Mol. Cell Biol., 2008, 9, 981–991 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Lisa
Roy
b,
Lisa
Roy




