
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intrinsic self-healable supramolecular dynamic covalent elastomer for sustainable high-performance tactile sensing†
Ding
Yang
,
Jiahui
Zhao
,
Fang-Yu
Liu
,
Meng
Chen
* and
Da-Hui
Qu
 *
*
Key Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: ac0105@ecust.edu.cn; dahui_qu@ecust.edu.cn
First published on 23rd April 2025
Abstract
Supramolecular chemistry empowers polymeric materials with versatile beneficial features encompassing stimulus adaptation, e.g. self-healing, to truly function in a biomimetic manner. To seek an effective self-healing mechanism for current polymers with no trade-offs in other property perspectives still remains a challenge. Herein, we present a sustainable alternative to the conventional covalent elastomers, a dynamic covalent disulfide polymer highly crosslinked by bio-catechol hydrogen bonds and coordinative metallic dopants. The polymeric elastomer exhibits mechanical tailorability, ambient intrinsic self-healing with an efficiency reaching 90%, and closed-loop recycling capability with no property deterioration. The assembled microstructured capacitive pressure sensor possesses a sensitivity up to 1.58 kPa−1, an effective working range up to 35 kPa and an exceptional response time of a few milliseconds, which makes it particularly promising for contemporary wearable devices for a spectrum of applications like physiological monitoring and voice-cancelling communication.
Introduction
Ever since the concept of supramolecular chemistry, ‘contemporary chemistry beyond individual molecules’, was coined by Lehn, this field has been fertilised with a rapidly growing number of publications in a multidisciplinary, interdisciplinary and futuristic manner.1–3 Supramolecular chemistry, with the inclusion of non-covalent/dynamic covalent interactions,4 specifically projects designed polymeric systems with various advances, e.g. manipulatable mechanics, stimulus-responsiveness5 and sustainability.6 Stimulus-responsiveness, e.g. self-healing, is of particular interest, not only endowing the established polymer with a biomimetic character, but also prolonging the fabricated material's optimum usage in an elegant, smart and eco-friendly way. The recent prime focus has shifted from extrinsic to intrinsic self-healing, commonly achieved via incorporating non-covalent interchain crosslinking, e.g. H-bonds,7–9 metal coordination,10,11 host–guest recognition12 and π–π interactions,13 into conventional networks, where the H-bond, a thermoreversible weak bond with a binding energy of 5 to 100 kJ mol−1, has sparked the major interest: the famous quadruple H bond motif 2-ureido-4[1H]-pyrimidinone UPy could endow networks with reconstruction ability upon gentle heating;14,15 other motifs, e.g. multi-urea/carboxylate acids, catechols and polysaccharides also appear to deliver efficient healing performance.16,17 Meanwhile, metal–organic coordination has its own charm: the broad spectrum of metal and ligand choices endow this sacrificial interaction with a wide range of possible binding energies, 10 to 400 kJ mol−1, and coordination structures, thus making it capable of establishing networks with various mechanics;18 in particular, ion-catecholate (carboxylic acid, polydopamine) and zinc–histidine complexes, initially discovered in biological systems, e.g. mussel byssus and spider fang, have repeatedly unveiled their role in stiffing, toughening and endowing the material with self-healing and shape memory ability.19 Although reports have been scattered throughout the recent literature, most cases do require the facilitation of external stimuli (pH, heat, moisture), and decent ambient self-healing initiated by singular/dual supramolecular interactions still remains rare.20Polymeric materials for wearable electronics like pressure sensors have been incentively explored with breadth and depth in the recent century, mapping their appearances from human activity recognition to non-invasive on-skin/injectable biosensing and on to human–robot interaction.21–23 Classic polymeric skeletons, especially polydimethylsiloxane (PDMS) and polyurethane (PU),24 have been deeply involved, with an enormous amount of effort put into endowing them with self-healing. Versatile healing mechanisms have been intensively evolved, including the application of multi H bond (acylhydrazone, dis/tri-carboxylic acid, MXenes),25,26 metal coordination(cobalt-triazole, zinc-diamidepyridine),27,28 electrostatic (ionic liquid)29 and dynamic covalent interactions (imine, disulfide).30,31 Feng et al. utilised the dynamicity of boron ester and UPy, allowing PDMS to be self-healed with a peak efficiency of 80% at 40 °C.32 Huang et al. reported that a PDMS elastomer decorated with disulfide and silver–thiolate complexes could exhibit decent ambient self-reconstruction within 6 hours.33 Thermal-induced self-healable PU networks were also reported by Bao and Xia,34,35 where lanthanide metals, e.g. europium and terbium, were introduced to form the sacrificial coordinative interchain crosslinking. As for most polymers that possess self-healing, increased structural complexities were inevitable, detrimental to their recycling capabilities. Zha et al.'s work was one of the few to circumvent this by incorporating the urea-structured quadruple H bond into the polymer.36 However, it is still challenging to install an effective self-healing mechanism into a suitable polymer network, with satisfactory properties, maximum healing efficiency, and no hindrance towards recycling, for deliberate implications like advanced pressure sensors.
Herein, we have reported a prospective sustainable alternative to the classic elastomers like PDMS with the potential to be utilised in contemporary capacitive wearable sensors: a dynamic covalent disulfide skeleton has been continuously reported by our group,37–40 covered with H bond binding functionalities, straightforwardly mixed and blended with bioderived catechol-structured H bond dopants, bio-based caffeic acid and coordinative metallic dopants to construct a sustainable elastomer with fascinating ambient self-healing and close-looped recycling capabilities (Fig. 1). The elastomer was contextualised in the aspects of supramolecular interaction-directed mechanical tailorability, dynamic viscoelasticity, ambient intrinsic self-healing with an 80% healing efficiency in 2 hours, and dynamic disulfide dithiolane-initiated closed-loop recycling; later the utilisation of this particular elastomer was demonstrated as the dielectric active layer of a capacitive pressure sensor, whereby the assembled device exhibited a sensitivity up to 1.58 kPa−1, an effective working range up to 35 kPa, and an exceptional response time of 7–9 ms with outstanding performance durability.
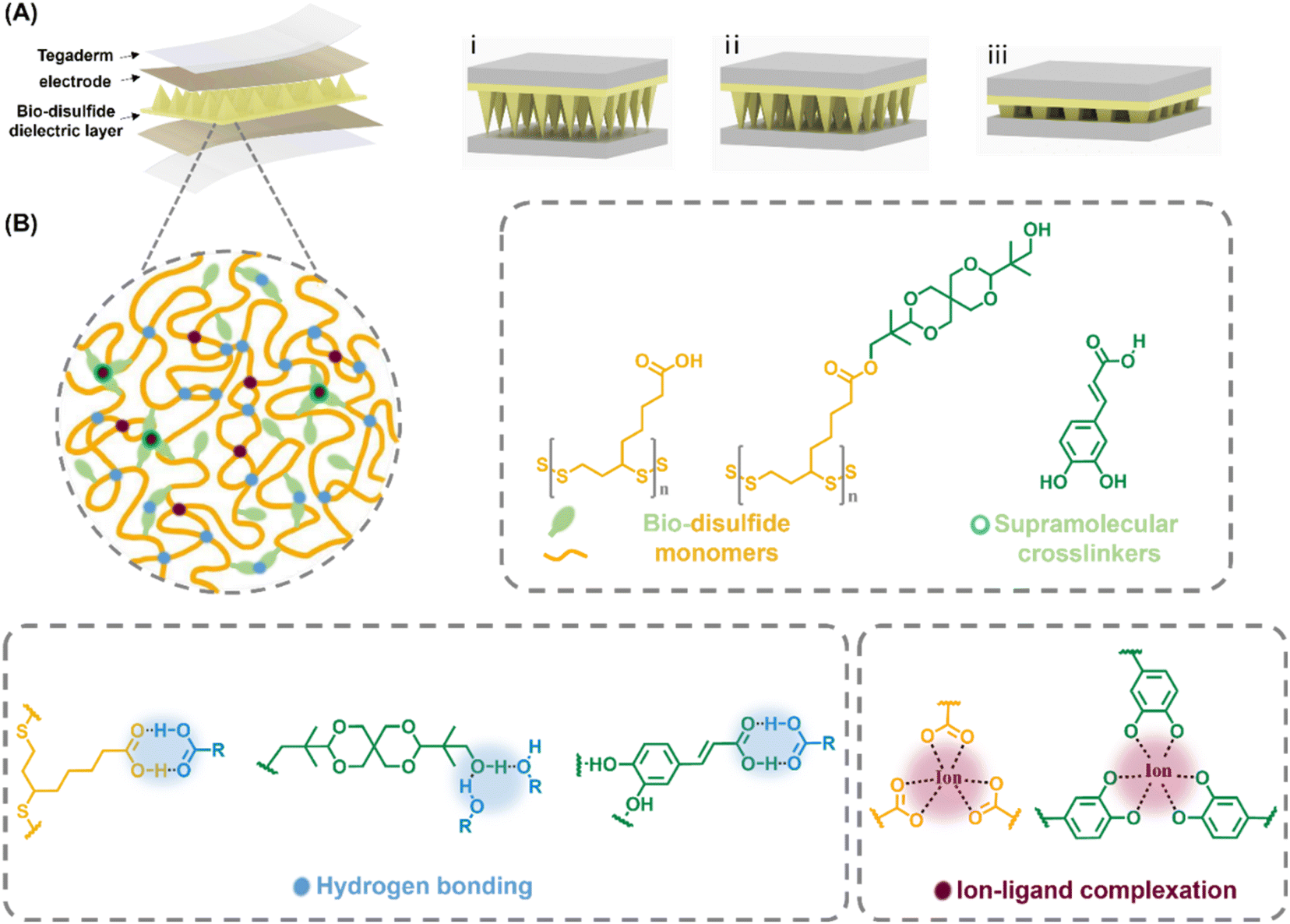 | ||
| Fig. 1 Graphical illustrations of the micropyramid-structured dynamic poly(disulfide)-based capacitive pressure sensor. (A) Conceptual illustrations of the dynamic disulfide polymer-based wearable capacitive pressure sensor and its physical appearance distortion upon different external pressures. (B) Graphical illustrations of the chemical composition of the micropyramid dielectric dynamic polymer. | ||
Results and discussion
We deliberately designed a highly supramolecular-crosslinked dynamic disulfide polymer and later evaluated its capability as the active dielectric layer for recyclable high-functional wearable sensor devices. The designed poly(disulfide) consisted of naturally occurring lipoic acid (TA), bio-spiroglycol decorated lipoic acid derivate (SPG), bio-caffeic acid and metal ions, where the SPG was obtained from selective esterification of TA and spiroglycol (where the spiroglycol molecule was only attached with one TA molecule). The fabricated micropatterned poly(disulfide) was generated through hot-melting followed by curing in a microstructured mould, referred to as TA–SPGx–y, where x stands for the molar ratio of TA and SPG (of the polymer), e.g. x = 1, 5 and 10 represent TA-to-SPG molar ratios of 1 to 1, 5 to 1, 10 to 1; as Fe3+ ions, introduced as a solution of FeCl3, could form tri-coordinated complexes with both the carboxyl groups of TA and caffeic acid, the molar amount of Fe3+ was always adjusted relative to TA's carboxyl groups, expressed by y as the molar ratio of Fe3+versus TA's carboxyl groups in percentage; caffeic acid was subsequently introduced to maintain its molar concentration at a level 3 times higher than that of Fe3+.Characterisation of the microstructured dielectric elastomer
This series of highly supramolecular-crosslinked elastomers, TA–SPGs, was subjected to Raman spectroscopy (Fig. 2A and S5†), where the conversion of the characteristic singlet peak of monomeric TA (TA mono, 510 cm−1) into doublet peaks of TA–SPGs (TA–SPG5, 506 cm−1 and 524 cm−1; TA–SPG10, 509 cm−1 and 527 cm−1) was realised. It is also in line with the literature that the polymeric dithiolane ring-opening process of both TA and SPG was evidenced by a peak's appearance at 3000 cm−1 denoted as high degree-polymerised poly(disulfide)s, commonly seen for curved ones.41 For monomeric SPG, a characteristic diffraction peak at 17.4° was recorded for the distinguishing twisted bicyclic ring structure of spiroglycol.42 This diffraction peak was significantly weakened, close to negligible, for all TA–SPGs, suggesting the studied disulfide copolymers were fully amorphous regardless of the exact polymer composition (Fig. 2B and S6–S8†). | ||
| Fig. 2 TA–SPG dielectric polymer characterisation. (A) Processed Raman spectra. (B) Processed X-Ray Diffraction (XRD). (C) Processed Thermal Gravimetric Analyzer (TGA) curves. (D and E) Processed Differential Scanning Calorimetry (DSC) curves. The tensile stress–strain curves of TA–SPG polymers (F and G) with different TA/SPG compositions and different caffeic acid/metal ion(III) concentrations. (H) Cyclic loading curves of TA–SPG10–0.1%, loaded (100% strain), unloaded, and immediately reloaded 10 times. (I) The compression-rebound cycles of TA–SPG10–0.1%. All tensile and compression tests were carried out with a tensile speed of 20 mm min−1. | ||
Thermogravimetric analysis (TGA) (Fig. 2C) indicated that monomeric SPG started to decompose at 299 °C, 40 °C higher than monomeric TA (250 °C). For TA–SPGs (Fig. S9†), they reached their 5 wt% weight loss at approximately 255 °C, similar to that of the TA homopolymer, but reached their 80 wt% weight loss at 459 °C, 100 °C higher than that of the TA homopolymer; the dual supramolecular crosslinker (caffeic acid and Fe3+) had a negligible effect on these copolymers' thermal stability, suggesting this high thermal stability was solely owing to the presence of rigid and steric spiroglycol, often responsible for the high Tg of conventional bio-polymers.43–45 Differential scanning calorimetry (DSC) first recorded the melting points (Fig. 2D) of the monomeric SPG and TA, 125 °C and 59 °C, where Tm of TA is in line with the literature, and the higher Tm of SPG, close to that (140 °C) of the monomer for the reported supramolecular H-bond network, was attributed to spiroglycol's thermal stabilising effect.46,47 Recorded Tg values (Fig. 2E) for TA–SPG5, TA–SPG10, and TA–SPG10–0.1% were 16.2, 11.4, and 15.5 °C, while they were 14.0, 12.4, and 19.6 °C for the spiroglycol-TA dimer, which indicated that: (a) Tg increased with the spiroglycol content; (b) Tg rose with the addition of the dual supramolecular crosslinker; (c) compared with the dimer counterparts, TA–SPGs had a lower content of rigid covalent ester-structured crosslinking, suggesting the increased degree of crosslinking with the rise of the supramolecular dopant content.44 To understand the role of Fe3+ ions in this supramolecular network, the control of TA–SPG10–0.1% was also fabricated, TA–SPG10–0.1%CA, where the polymer had identical compositions of TA, SPG and caffeic acid as TA–SPG10–0.1%, but was Fe3+ ion-free. Compared with TA–SPG10–0.1%, ion-free TA–SPG10–0.1%CA has a lower Tg, 12.9 °C, implying the formation of the ion–ligand complexation noticeably increased the elastic network's thermal stability upon ion inclusion as the metal–ligand coordination interactions usually possessed a higher interaction energy than H bonding.48–50
Mechanical tensile tests revealed that TA–SPGs possessed a composition-depended property behaviour (Fig. 2F): (a) by increasing the TA content from TA–SPG5 to TA–SPG10, the corresponding copolymers experienced a strength reduction from 1.5 MPa to 50.9 KPa, owing to the significant content drop of the rigid spiroglycol, which was not only responsible for high thermal stability but also for decent mechanical strength,51 suggesting spiroglycol could surely bring rigidity into the polymer skeleton regardless of the way it was incorporated into the network (it is often involved via diester linkages in conventional polymers), including this rare case, where only one of spiroglycol's diol hydroxyl groups was covalently bonded to the disulfide skeleton, while the other, untouched, was deliberately designed for H-bond binding. TA–SPG10 was picked to further incorporate with the dual supramolecular crosslinkers, caffeic acid and Fe3+, as TA–SPG5 was too brittle to exhibit a rubbery character to fulfil the requirement of the later sensor study. By adjusting the supramolecular crosslinker content, the mechanical properties peaked at TA–SPG10–0.1%, with a 1.26 MPa strength, a 195% strain and a 2.15 MJ m−3 toughness, which were 25-fold, 5-fold, and 325-fold higher than these of TA–SPG10 (Table S1†), confirming the role of caffeic acid and Fe3+ together in strengthening the polymeric network via forming versatile H-bonds and ion–ligand complexation crosslinking motifs. This is also in agreement with the earlier thermal study, in which TA–SPG10–0.1% had a higher degree of crosslinking including covalent ester, supramolecular H-bond and ion–ligand complexation bonding, compared to TA–SPG10 mainly crosslinked by covalent esters, and it exhibited a higher thermal stability, higher Tg, better mechanical strength and resilience. To further investigate the effect of this dual supramolecular crosslinker, the mechanical properties of aforementioned control polymer TA–SPG10–0.1%CA (Fig. 2G) were also recorded and the introduction of caffeic acid in TA–SPG10 raised the polymer's flexibility by nearly 4-fold with a slight drop in strength, implying the bulk caffeic acid squeezed into the flexible disulfide network and formed new H bonds by partially disrupting the initial H bond layout generated by the TA's carboxylic group and SPG's hydroxyl group; in this way the overall network gained some extra flexibility but with a small strength compensation. The much lower strength and strain of TA–SPG10–0.1%CA, compared with TA–SPG10–0.1%, well illustrated that the ion more profoundly integrated the TA–SPG copolymer's strength and toughness, relative to caffeic acid, supported by the fact that the ion–ligand complexation is stronger than the H bond in strength according to simulations.48 The tensile test rate had a positive effect on the tested polymers' Young's modulus, but did not alter their rubbery character (Fig. S10†).
A moderate elastic performance of TA–SPG10–0.1% was illustrated by repeated cyclic tests. TA–SPG10–0.1% was stretched to 1× its original length and unloaded for 10 cycles (Fig. 2H). The stress-softening effect had not been profound since cycle 1. A noticeable residual strain of 30% was observed in all cycles. If we set the first cycle's energy dissipation, 0.036 MJ m−3, as 100%, 24% of energy was dissipated as heat in Cycle 2, and 49% in Cycle 10. TA–SPG10–0.1%'s elasticity was not ideal compared with PDMS, but certainly more advanced than other TA-based elastomers.44,52 The repeated compression loading–unloading test (Fig. 2I) suggested that similar to PDMS, the polymer exhibited (a) a linear stress region as the strain increased from 0 to 35%, (b) a plateau between 35% and 45%, and (c) an exponentially increasing region between 45% and 67%.53 This medium compressibility was attributed to TA–SPG10–0.1%'s high content of reversible supramolecular crosslinking: these sacrificial linkages would undergo instant bond dissociation to achieve decent energy dissipation upon force-induced rupture, followed by a rapid bond re-association to restore the dynamic elastic network. The repeated compression Cycle 2 and 3 moderately overlapped, indicating that TA–SPG10–0.1%'s compressibility was fully reversible. The swelling test suggested this supramolecular crosslinked elastic material exhibited a decent anti-swelling behaviour towards water, petroleum ether and triethylamine, and a moderate anti-swelling behaviour towards ethyl ether and aqueous hydrochloric acid (Fig. S11†).
To investigate the dynamic viscoelasticity of TA–SPG10–0.1% constructed by supramolecular crosslinking, a cyclic temperature ramp test was performed from 20 °C to 130 °C (Fig. 3A). Both G′ and G′′ exhibited a downward trend with ramped-up temperature, while an opposite transition was spotted for ramped-down temperature, G′ superposed G′′ throughout the process, implying the TA–SPG10–0.1% network exhibited a thermoelastic character; the ramp curves coincided nicely at high temperature (>80 °C) but experienced a hysteresis at low temperature (<80 °C), attributed to the thermo-sensitive disulfide skeleton being activated at high temperature and undergoing instant dynamic exchange along with other thermo-activated sacrificial supramolecular crosslinking, resulting in an excellent TA–SPG10–0.1% thermoreversibility. Meanwhile, at lower temperatures, the sluggish dissociation–reassociation behavior of both the disulfide and non-covalent crosslinks led to a delayed restoration of the sample material’s moduli. No distinct peak was observed for tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ, indicating the absence of a phase transition during the tested temperature range, consistent with the findings in DSC.
δ, indicating the absence of a phase transition during the tested temperature range, consistent with the findings in DSC.
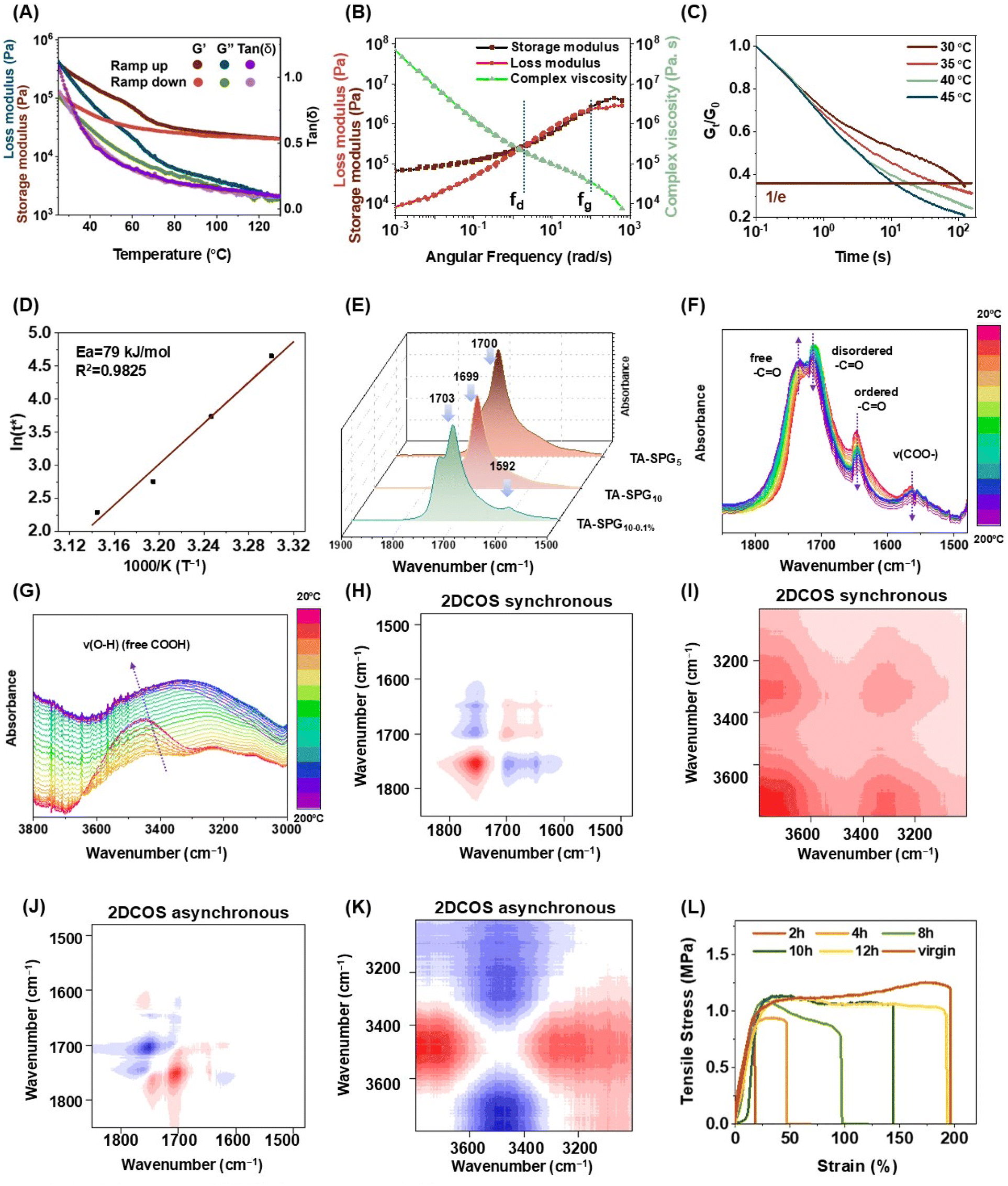 | ||
| Fig. 3 TA–SPG10–0.1% polymer characterisation and self-healing analysis. (A) Cyclic temperature ramp curves. (B) Time–temperature superposition rheological master curves. (C) Stress relaxation of TA–SPG10–0.1% at different temperatures over a period of under 112 s. (D) Processed Arrhenius plot of the characteristic ln(τ*) versus 1000/T of TA–SPG10–0.1%. (E) Fourier Transform Infrared Spectrometer spectra (FTIR) of TA–SPG polymers, temperature-variable FTIR spectra of TA–SPG10–0.1%. (F) From 1850 to 1480 cm−1. (G) From 3800 to 3000 cm−1. (H and I) Two-Dimensional Correlation Spectroscopy (2D-COS) synchronous spectra generated from (F and G). (J and K) 2D-COS asynchronous spectra generated from (F and G), red represents positive spectra intensities, while blue represents negative ones. (L) The tensile stress–strain curves of virgin/self-healed TA–SPG10–0.1% under different healing durations. All tensile tests were carried out with a tensile speed of 20 mm min−1. | ||
Time–temperature superposition (TTS) through small amplitude oscillatory shear experiments of TA–SPG10–0.1% were also conducted to disclose its dynamic behaviours at different time scales (Fig. 3B). The master curves were recorded at a frequency range of 103 to 10−3 rad s−1 with a reference temperature of 20 °C, divided into three regions: (a) when f > fg (102 rad s−1), G′ > G′′ was defined as the glassy regime in which the polymer material displayed solid-state properties and the crosslinked dynamic network was in the frozen state with minimal chain movements; when fd (100 rad s−1) < f < fg (102 rad s−1), G′ < G′′ was described as the dissipative regime dominated by Tg where the superimposed master curves may be attributed to the weakened supramolecular crosslinking, including H bond and ion–ligand complexation, as well as the instant dynamic exchange of the disulfide units; when f < fd (100 rad s−1), a classic rubbery plateau was observed, similar to other supramolecular crosslinked networks, and following that, G′ superposed G′′ with the lowered frequency, imparting a well-intact dynamic polymer network with firmly implanted non-covalent crosslinking.54,55
To unveil the correlation between TA–SPG10–0.1%'s viscoelastic behaviour and temperature, the stress relaxation experiment was conducted (Fig. 3C). The polymer exhibited a quasi-uniform speed stress relaxation under all conditions but with a positive temperature association of the relaxation rate. Specifically, for the same level of stress relaxation, it took 100 s at 30 °C while at 45 °C only 10 s was needed. It could be speculated that at the lower temperature, the polymer network was predominantly frozen and the stress relaxation took place at a fairly slow pace; the reason behind the faster relaxation at higher temperatures should encompass many polymer structural features, including the network becoming less intact and all supramolecular crosslinking being loosely bound. The aforementioned observations are in line with other previously reported supramolecularly crosslinked polymers.39,56 TA–SPG10–0.1%'s temperature-dependent viscosity behaviour nicely adhered to the Arrhenius equation based on the Maxwell model, where the minimal energy required for the polymer system to lose its chain entanglement, referred to as the activation energy, Ea derived from the Arrhenius relation plot, was estimated to be 79 kJ mol−1 (Fig. 3D). The high supramolecular crosslinking content of TA–SPG10–0.1% has led it to have a lower Ea, compared with a transesterification vitrimer, 88 kJ mol−1, and dynamically crosslinked polyrotaxane, 89 kJ mol−1.57,58
Attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) was able to unveil the correlation between the significant functionalities and polymer compositions (Fig. 3E, S12 and S13†): (a) TA content tied in with the H bond-directed low frequency shift of the peaks denoted as a carboxylate motif including carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, sp2 C–O, sp3 C–O and O–H, to be exact, 1699, 1253, 1083, and 2850 cm−1 for TA–SPG10 and 1700, 1261, 1084, and 2854 cm−1 for TA–SPG5, where the correlation was seemingly weak but certainly noticeable; (b) compared with the solely H bond crosslinked polymer, the dual ion/H-bond crosslinked one exhibited a high frequency shift of the carbonyl CO upon the addition of caffeic acid and Fe3+, 1703 cm−1 for TA–SPG10–0.1% and 1699 cm−1 for TA–SPG10; this was also accompanied by an observed peak of ion-carboxylate complexation, 1592 cm−1 only for TA–SPG10–0.1%, implying the formation of the ion complexation crosslinking was in contrast to the existing H bonding within the supramolecular dynamic network.59 To further elucidate the variations of the significant functionalities within TA–SPG10–0.1%, this well-established network, upon the heating effect, was further subjected to temperature-variable FTIR spectroscopic analysis operating at a temperature range of 20 to 200 °C: (a) on the basis of spectra detailed in the region of 1500–1800 cm−1 (Fig. 3F), apart from free (unboned) CO, 1733 cm−1, experiencing a rising intensity, other characteristic peaks of disorderly H-bonded CO, 1711 cm−1, orderly H-bonded CO, 1645 cm−1, and metal coordinated carboxylate COO−, 1565 cm−1, were all found to have their intensities attenuated; as for 3000–3800 cm−1 (Fig. 3G), the O–H signal originating from the carboxylate group was intensified alongside a higher frequency shift from 3335 to 3460 cm−1; both regions coherently unlocked the highly supramolecular crosslinked nature of TA–SPG10–0.1%, with direct pieces of evidence for the appearance of the thermal-sensitive sacrificial H bonding, accompanied by metal coordination. Two-dimensional correlation spectra (2DCOS), including synchronous (Fig. 3H and I) and asynchronous ones (Fig. 3J and K), were interpreted regarding Noda's rule, the appearing sequence of the observed chemical motifs was in the order of orderly H-bonded ν(CO) 1645 → free ν(CO) 1733 → disorderly H-bonded ν(CO) 1711 cm−1 in the lower frequency region, and H-bonded ν(O–H) 3335 → free ν(O–H) 3460 cm−1 in the higher region (→ means prior appearance; see determination details in Table S8†).60,61
O, sp2 C–O, sp3 C–O and O–H, to be exact, 1699, 1253, 1083, and 2850 cm−1 for TA–SPG10 and 1700, 1261, 1084, and 2854 cm−1 for TA–SPG5, where the correlation was seemingly weak but certainly noticeable; (b) compared with the solely H bond crosslinked polymer, the dual ion/H-bond crosslinked one exhibited a high frequency shift of the carbonyl CO upon the addition of caffeic acid and Fe3+, 1703 cm−1 for TA–SPG10–0.1% and 1699 cm−1 for TA–SPG10; this was also accompanied by an observed peak of ion-carboxylate complexation, 1592 cm−1 only for TA–SPG10–0.1%, implying the formation of the ion complexation crosslinking was in contrast to the existing H bonding within the supramolecular dynamic network.59 To further elucidate the variations of the significant functionalities within TA–SPG10–0.1%, this well-established network, upon the heating effect, was further subjected to temperature-variable FTIR spectroscopic analysis operating at a temperature range of 20 to 200 °C: (a) on the basis of spectra detailed in the region of 1500–1800 cm−1 (Fig. 3F), apart from free (unboned) CO, 1733 cm−1, experiencing a rising intensity, other characteristic peaks of disorderly H-bonded CO, 1711 cm−1, orderly H-bonded CO, 1645 cm−1, and metal coordinated carboxylate COO−, 1565 cm−1, were all found to have their intensities attenuated; as for 3000–3800 cm−1 (Fig. 3G), the O–H signal originating from the carboxylate group was intensified alongside a higher frequency shift from 3335 to 3460 cm−1; both regions coherently unlocked the highly supramolecular crosslinked nature of TA–SPG10–0.1%, with direct pieces of evidence for the appearance of the thermal-sensitive sacrificial H bonding, accompanied by metal coordination. Two-dimensional correlation spectra (2DCOS), including synchronous (Fig. 3H and I) and asynchronous ones (Fig. 3J and K), were interpreted regarding Noda's rule, the appearing sequence of the observed chemical motifs was in the order of orderly H-bonded ν(CO) 1645 → free ν(CO) 1733 → disorderly H-bonded ν(CO) 1711 cm−1 in the lower frequency region, and H-bonded ν(O–H) 3335 → free ν(O–H) 3460 cm−1 in the higher region (→ means prior appearance; see determination details in Table S8†).60,61
Self-healing and reprocessing
By mimicking the human skin, self-healing allows wearable sensor devices to tolerate certain abrasions upon usage without a major dysfunction. TA–SPG10–0.1% was picked and subjected to a cut-and-heal analysis, where the sample specimen was cut into two pieces which were brought into gentle contact for ambient temperature self-healing. The process was evaluated by the self-healed sample's mechanical behaviour and was monitored for up to 12 hours (Fig. 3I, S15 and S16†). The self-healed TA–SPG10–0.1% exhibited the same rubbery character as the pristine one. The polymer strength was recovered to 80% within 2 hours and could be restored to 88% at the max. The strain was retrieved at a lower rate but achieved a quantitative recovery within 12 hours. The toughness also achieved its maximum recovery, 90%, at 12 hours. This autonomous self-healing was primarily driven by the intrinsic rich H bonding and ion–ligand complexation within the studied dynamic disulfide network, and the fast reversible dissociation–reassociation nature of these supramolecular interactions allowed TA–SPG10–0.1%'s repair to occur rapidly without the facilitation of any external stimuli, beneficial for constructing it into smart durable wearable sensors. The effect of laboratory humidity on TA–SPG10–0.1%'s self-healing performance was also investigated: three different humidity environments, 0%, 50% and 70%, were deliberately designed and realised to mimic the laboratory environments across four seasons; the polymer demonstrated a similar mechanical property recovery after healing for 12 hours regardless of the humidity levels, strength recovery of 86–96% and quantitative strain recovery, implying a negligible moisture effect on the self-healing process. TA–SPG10–0.1% self-healing performance was comparable with H-bond crosslinked urea-PDMS,62 dynamic imine crosslinked PDMS vitrimer,63 dynamic disulfide crosslinked PDMS-graphene oxide nanocomposites,64 ahead of imine crosslinked MXene-PDMS65 and coordinative cobalt(II) crosslinked PDMS and less profound than imine crosslinked PDMS.28,66With the fast-growing scale of e-waste, sustainability is inevitable in designing current electronic devices including e-skin. To unveil the reprocessing capability of TA–SPG10–0.1%, the polymer was subjected to three consecutive closed-loop recycling cycles, accompanied by a structural identification of the recycled streams and a quantitative analysis of the recycled polymers, including the recycling yields and recovered mechanical properties. For each recycling cycle, TA–SPG10–0.1% was subjected to a triflic acid-promoted fragmentation in chloroform at 70 °C, converted into small soluble fragments and underwent complete dissolution owing to the reversibility of the dynamic disulfide network and facile dissociation of the supramolecular crosslinking embedded in the polymer skeleton. The fragmentation product was directly subjected to a second polymerisation without any further component separation, regaining the recycled TA–SPG10–0.1% chip (Fig. 4A). The recycling yield was elucidated as the percentage ratio of the fragmentation product mass to the fragmented polymer mass, 82% for Cycle 1, 80% for Cycle 2, and 75% for Cycle 3, where the slightly reduced yield would likely be attributed to the experimental loss. For all cycles, the fragmentation products exhibited nearly identical chemical shift profiles in 1H Nuclear magnetic resonance (NMR) spectra, where the chemical shifts implied that they all contained monomeric SPG (Fig. 4B, S19 and S20†), as the spiroglycol monoester structure was recorded, assigned as 4.25, 3.91, 3.55, 3.44, 3.33, 0.95, and 0.93 ppm. The appearance of TA could not be fully confirmed due to the complete signal overlap of the five-membered dithiolane ring of TA and SPG. However, mass spectroscopy confirmed the occurrence of TA in the fragmentation product, a correlated peak with a mass-charge ratio of 206, while SPG and caffeic acid were also observed, assigned peaks with mass-charge ratios of 492 and 219, respectively (Fig. S21–S23†). The average molecular weight recorded by gel permeation chromatography GPC was 904 Da (Table S7†), which was equivalent to two SPG or four TA molecules; the completion of the fragmentation was reasonably high relative to the literature, and the fragmentation product was believed to appear as dimerised/trimerised fragments rather than large oligomers.41 Compared with the pristine polymer, the recycled TA–SPG10–0.1% exhibited a similar degree of polymerisation of the dynamic disulfide skeleton according to Raman analysis (Fig. 4C), with the same characteristic structural motifs, including H bond-induced low frequency-shifted carbonyl CO and the ion–ligand coordinative complexation, recorded by ATR-FTIR (Fig. 4D). When TA–SPG10–0.1% was recycled for the first time, the reobtained polymer chip possessed close-to-identical rubbery mechanical properties to the pristine one, with a tensile stress of 1.13 MPa, a strain of 188% and a toughness of 2.1 MJ m−3 (Fig. 4E), which implied that: (a) the closed-loop chemical recycling had no degradation effect on the polymer properties, (b) the triflic acid did not promote the carbonate hydrolysis and left the spiroglycol monoester motif untouched, different to the literature.41 However, with the repeated recycling cycles, the TA–SPG10–0.1%'s rubbery character was slightly disrupted, as evidenced by a gradual reduction in toughness, from 2.0 MJ m−3 to 1.7 MJ m−3 from Cycle 2 to 3. With the recent development of the sensing elastomeric material, ones with complex compositions, including inorganic dopants like carbon nanotubes, have been widely studied, but they commonly suffer from non-ideal recycling due to the uneven distribution of the inorganic dopants during the polymer refabrication, potentially resulting in a dysfunction of the sensing material.67,68 Herein, TA–SPG10–0.1%, a dynamic elastomer containing organic/inorganic dopants, presented an interesting closed-loop recycling capability, in which the material could be depolymerised through a simple chemical process without dopant separation and refabricated in a one-pot manner to yield a recycled polymer to have close-to-identical properties to the pristine polymer, which is of great significance for these dielectric elastomers in sensing devices.
 | ||
| Fig. 4 TA–SPG10–0.1% closed-loop recycling assessment. (A) Graphic illustrations of the closed-loop solvolytic recycling. The comparisons of (B) 1H Nuclear Magnetic Resonance (1H NMR) spectra of the TA–SPG monomeric precursor. (C) Raman spectra. (D) FTIR spectra and (E) tensile stress–strain curves of virgin/recycled TA–SPG10–0.1%. | ||
Sensing performance evaluation and physiological signal monitor application
The state-of-the-art of designing current high-performance pressure sensors is to boost their working parameters in all aspects, e.g. sensitivity, working range, and response time. The integration of these parameters might be in inverse variations; it is therefore crucial to evolve sensor devices and their constituents in a well-balanced manner as well as based on the speculation of the working circumstance demands. A electrode–dielectric polymer–electrode laminated sensing device was hence fabricated, where the dielectric polymer was TA–SPG10–0.1%, pre-moulded into a micro-sized pyramid array well known for Zhennan Bao's on-skin electronics.69,70 With the facilitation of the micropyramid array, the dielectric layer's configuration variations in terms of surface area and total volume upon minimal pressure were notably amplified, thus generating consistent detectable signals.The key sensor performance, sensitivity, of this micropyramid-structured TA–SPG10–0.1% was deduced from the trilinear curve of the normalised capacitance versus functional pressure, with three calculated sensitivities of 1.58, 0.41, and 0.02 kPa−1 in the range of 0–1.5, 1.5–5.6, and >5.6 kPa−1, with an observed detection limit of 40 Pa (Fig. 5A), which was profoundly superior to the performance delivered by its solid plain counterparts, 0.02 kPa−1 and 0.001 kPa−1 in the ranges of 0–5 kPa and >5 kPa (Fig. S24†). Its sensitivity has outperformed many other reported sensing systems, e.g. MXene nanosheets (≤0.55 kPa−1), wrinkled polypyrrole (≤0.51 kPa−1), and tissue paper coated with silver nanowires (≤1.5 kPa−1), but surpassed by others, e.g. carbon nanotube doped polydimethylsiloxane.71–76 It is known that the real-time sensor capacitance is proportional to the dielectric constant of the dielectric layer ε and surface area A, and inversely proportional to the electrode distance d. It is also worth noting that the dielectric layer for this assembled sensor was composed of air and dynamic disulfide elastomer TA–SPG10–0.1%. At the low-pressure range, the pyramid apex was extremely sensitive to pressure, resulting in a drastic apex deformation as well as the sharp decrease of d, mainly responsible for the high sensitivity; at the medium-pressure range, the pyramid array was less prone to higher pressures, and much higher external stress/pressure would be required for the sensor to achieve a similar level of dielectric layer configuration variation as the low-pressure range; at high pressure, the sensor behaved in the same manner as the device with a solid plain dielectric layer as almost all the air gaps had been squeezed out. The assembled device was able to fully function up to 35 kPa. The response and recovery durations were deduced from the assembled sensor capacitive behaviour of a complete loading–loading cycle of varied pressure, 0.51 to 9.32 kPa (Fig. 5B). It is suggested the sensor was able to instantly respond, in 7 to 9 ms, to varied external pressure, and to restore its initial status, 7 to 8 ms, once the pressure was released, on account of the combinatory effect of the disulfide dielectric elastomer's elasticity and the dielectric layer's microstructured layout. This sensor has exhibited a better performance than many other systems, e.g. multi-walled carbon nanotube-coated silicone rubber (45 ms), palladium ion-coated polyacrylonitrile (48 ms), and silver nanowire-coated polyvinylidene fluoride (166.9 ms).74,77–86 The hysteresis curves were demonstrated via performing a complete pressure loading–unloading cycle (Fig. 5C), illustrated that the instant relative capacitance curve of the increasing pressure was superimposed with that of the decreasing pressure, owing to the intrinsic elasticity of the disulfide dielectric elastomer capable of simultaneous structure reconfiguration without integrity deterioration. To demonstrate the electromechanical stability of the assembled sensor, the repeated loading–unloading cyclic test was performed with a constant loading pressure of 0.25 kPa at a frequency of 2 Hz (Fig. 5D); the device was able to generate fast steady responses for 400 consecutive cycles with no sign of performance failure; the first 20 consecutive capacitance signal outputs (Fig. 5E) suggested the signal amplitude fluctuated at a neglectable level as the cyclic test was conducted manually. The loading–unloading cyclic tests were also conducted at 0.6 kPa with incremental frequency ranging from 0.06 Hz to 0.5 Hz (Fig. 5F). For all frequencies, the device was able to generate consistent responses with constant amplitudes, and the signal output could also instantly return to the original value. These cyclic tests indicated the assembled TA–SPG10–0.1% sensor not only exhibited remarkable stability towards different performance environments, but generated steady instantaneous signals. In general, the assembled capacitive sensor possessed many performance advantages compared to many reported sensors (Fig. 5G and H).
 | ||
| Fig. 5 TA–SPG10–0.1%-based micropyramid capacitive sensor performance evaluation. (A) Sensitivities, described by the capacitance change as a function of applied pressure. (B) Device response/recovery time. (C) Pressure response curve for instrument hysteresis analysis measured from a single loading–unloading cycle. (D) Long-term stability evaluation under 0.24 kPa for 400 consecutive response cycles with its first 20 response cycles detailed in (E and F). Dynamic capacitance measurements under 0.6 kPa with different frequencies, 0.5, 0.13, 0.1, and 0.06 Hz. Sensor performance comparisons with previously reported systems. (G) Sensitivity versus working range. (H) Response time versus working range. | ||
To put the fabricated sensor device in real practice, the device was firstly affixed to a forefinger to monitor the bending motions in real-time: the sensor was able to precisely track the bending motions, generating reliable digital signal outputs, as the finger was bent to various angles stepwisely increasing from 10° to 110° with a 10° interval between two consecutive bending angles (Fig. 6A); the capacitive signal outputs were discovered to be linearly correlated with the bending angles (Fig. 6B), reflecting the elegance of the fabricated sensor, a combination of precision and decent sensitivity, beneficial for assembling smart devices capable of predicting undisclosed motion with a pressure calibration curve. The dielectric layer of the device studied was subjected to the cut-and-heal as well as closed-loop recycling processes, and it was depicted that the sensor could function in the same manner as the pristine one even after being ambiently self-healed and recycled multiple times, implying the robustness of the fabricated device (Fig. 6C). The device could be allocated at various body joints to detect versatile physiological motions (Fig. 6D), including back-flatting, knee-flexion/extension, squatting, walking, running and cycling. Complex motions could also be monitored by simply employing multiple devices in the sensing area. The device retained its full function regardless of whether the monitored joints were covered with/without clothing. Not only large-scale body motions but also fine finger configurations could be monitored (Fig. 6E): the capacitive digital signals of 6 different hand configurations, representing the numbers 1 to 6 were successfully demonstrated, similar to the other reported glove hands, unveiling the device's potential in remote control in robotics as well as virtual reality.86 The device's potential utilisation in voice-free communication was also projected (Fig. 6F): by simply varying the pressing duration, the classic dashes and dots, comprising all Morse code signals, could be easily generated by altering the sensor capacitance for signal recognition; the Morse code of ECUST, initials of East China University of Science and Technology, was used as an example, confirming the device effectiveness as a special-purpose communication tool.
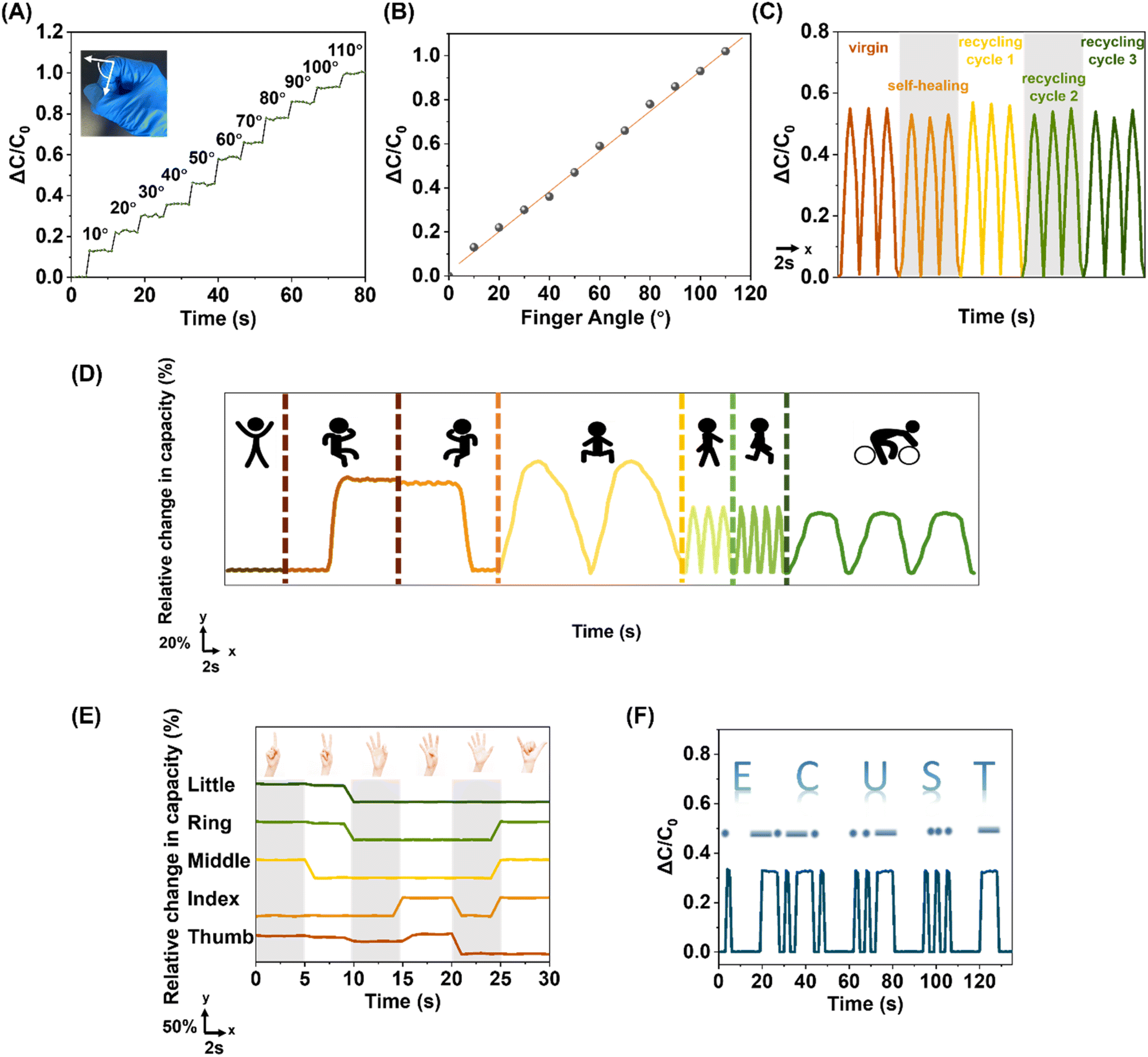 | ||
| Fig. 6 Demonstration of the versatile implications of the TA–SPG10–0.1% capacitive sensor. (A) Stepwise angle-dependent capacitance response of a finger joint. (B) A linear signal plot of the finger joint with different bending angles. (C) Dynamic capacitance measurements of the virgin, self-healed, and recycled sensor devices. (D) Capacitive signal detection of various physiological movements, where the signal responses arose from lying, sitting, standing, squatting, walking, running, and riding. (E) Relative changes in capacitance versus time with different finger configurations. (F) Generation demonstrations of Morse code ‘E’, ‘C’, ‘U’, ‘S’, and ‘T’. | ||
Conclusions
We have reported here a sustainable alternative to the conventional covalent elastomers like PDMS, a dynamic covalent disulfide polymer supramolecularly crosslinked by bio-catechol hydrogen bonding and coordinative metallic dopants. The polymeric elastomer exhibited decent thermal stability and supramolecular interaction-directed mechanical tailorability. Its competent mechanical elasticity was together demonstrated by the cyclic loading and the cyclic compression-rebound analyses. Temperature/frequency-dependent rheology confirmed the elastomer's dynamic thermoelasticity; both the reversible disulfide polymer skeleton and the dual supramolecular crosslinking immersed in the network were prone to high temperature/frequencies; this dynamicity was further confirmed by temperature-variable FTIR spectroscopy. Its intrinsic self-healing analysis was conducted, monitored under ambient conditions up to 12 hours, and the elastomer could restore 80% of its strength within merely 2 hours without any external stimuli facilitation, which was not often seen. The elastomer's closed-loop recycling cycles were all achieved via a mild temperature solvolysis followed by a same one-pot polymerisation to yield the recycled elastomers with no significant property deterioration relative to the pristine one, where the full process was traced by 1H NMR, mass spectroscopy, GPC, Raman and FTIR analyses. The subsequently assembled micropyramid-structured capacitive pressure sensor possessed a sensitivity up to 1.58 kPa−1, an effective working range up to 35 kPa, an exceptional response time of 7–9 milliseconds and outstanding performance durability, which is particularly promising for contemporary wearable devices for a spectrum of applications like physiological monitoring, robotic remote control and voice-canceling communication.Ethical statement
All experiments were performed in accordance with the Guidelines of the Measures for Ethical Review of Life Science and Medical Research Involving Human Beings Article 32, and approved by the ethics committee at The East China University of Science and Technology. Informed consent was obtained from the human participants of this study.Data availability
Additional experimental details and data are provided in the ESI.†Author contributions
All authors discussed the results and commented on the manuscript. Design of the study: M. C. and D.-H. Q.; acquisition of funding: M. C. and D.-H. Q.; project supervision: D. Y., J. Z., F.-Y. L., and M. C.; design and chemical synthesis: D. Y. and J. Z.; writing original draft: M. C.; writing – review and editing: D. Y., J. Z., F.-Y. L., and D.-H. Q. with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22025503, 22220102004), Science and Technology Commission of Shanghai Municipality (24DX1400200), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD40), the Programme of Introducing Talents of Discipline to Universities (grant no. B16017) and the Fundamental Research Funds for the Central Universities. The authors thank the Research Center of Analysis and Test of East China University of Science and Technology for help with material characterisation.Notes and references
- J.-M. Lehn, Pure Appl. Chem., 1977, 49, 857–870 CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- J.-M. Lehn, Angew Chem. Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- Z. Zhang, G. N. T. Le, Y. Ge, X. Tang, X. Chen, L. Ejim, E. Bordeleau, G. D. Wright, D. C. Burns, S. Tran, P. Axerio-Cilies, Y. T. Wang, M. Dong and G. A. Woolley, Nat. Chem., 2023, 15, 1285–1295 CrossRef CAS PubMed.
- C.-Y. Shi, Q. Zhang, H. Tian and D.-H. Qu, Smart Mat., 2020, 1, e1012 Search PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042 RSC.
- H. Qiao, B. Wu, S. Sun and P. Wu, J. Am. Chem. Soc., 2024, 146, 7533–7542 CrossRef CAS PubMed.
- H. Ye, B. Wu, S. Sun and P. Wu, Nat. Commun., 2024, 15, 885 CrossRef CAS.
- X. Wang, T. Liu, F. Sun, J. Zhang, B. Yao, J. Xu and J. Fu, Smart Mol., 2024, 2, e20240008 CrossRef.
- Z. Zhao, S. Chen, P. Zhao, W. Luo, Y. Luo, J. Zuo and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202400758 CrossRef CAS.
- J.-C. Lai, L. Li, D.-P. Wang, M.-H. Zhang, S.-R. Mo, X. Wang, K.-Y. Zeng, C.-H. Li, Q. Jiang, X.-Z. You and J.-L. Zuo, Nat. Commun., 2018, 9, 2725 CrossRef.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS.
- Y. Pan, J. Hu, Z. Yang and L. Tan, ACS Appl. Polym. Mater., 2019, 1, 425–436 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- G. Yan, G. Chen, Z. Peng, Z. Shen, X. Tang, Y. Sun, X. Zeng and L. Lin, Adv. Mater. Interfaces, 2021, 8, 2100239 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- A. Campanella, D. Döhler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700739 CrossRef.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- K. Buaksuntear, P. Limarun, S. Suethao and W. Smitthipong, Int. J. Mol. Sci., 2022, 23, 6902 CrossRef CAS PubMed.
- C. Zhao, J. Park, S. E. Root and Z. Bao, Nat. Rev. Bioeng., 2024, 2, 671–690 CrossRef CAS.
- H. C. Ates, P. Q. Nguyen, L. Gonzalez-Macia, E. Morales-Narváez, F. Güder, J. J. Collins and C. Dincer, Nat. Rev. Mater., 2022, 7, 887–907 CrossRef PubMed.
- S.-D. Ma, Y.-T. Wu, J. Tang, Y.-M. Zhang, T. Yan and Z.-J. Pan, Chin. J. Polym. Sci., 2023, 41, 1238–1249 CrossRef CAS.
- C.-C. Lu, W.-C. Gao, P. Li, W. Wu, R. K. Y. Li and H. Zhao, Chin. J. Polym. Sci., 2023, 41, 1037–1050 CrossRef CAS.
- K. Zhang, J. Sun, J. Song, C. Gao, Z. Wang, C. Song, Y. Wu and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 45306–45314 CrossRef CAS PubMed.
- N. Roy, Ž. Tomović, E. Buhler and J. Lehn, Chem.–Eur. J., 2016, 22, 13513–13520 CrossRef CAS.
- Z. Wang, C. Xie, C. Yu, G. Fei, Z. Wang and H. Xia, Macromol. Rapid Commun., 2018, 39, 1700678 CrossRef.
- X.-Y. Jia, J.-F. Mei, J.-C. Lai, C.-H. Li and X.-Z. You, Chem. Commun., 2015, 51, 8928–8930 RSC.
- Z. Zhang, L. Qian, B. Zhang, C. Ma and G. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202410335 CrossRef CAS PubMed.
- S. Kim, H. Jeon, S. Shin, S. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- X.-Y. Chen, Y.-B. Fu, X.-L. Yan and L. Liu, Chin. J. Polym. Sci., 2024, 42, 1610–1618 CrossRef CAS.
- H. Yu, Y. Feng, L. Gao, C. Chen, Z. Zhang and W. Feng, Macromolecules, 2020, 53, 7161–7170 CrossRef CAS.
- K. Zhang, X. Shi, J. Chen, T. Xiong, B. Jiang and Y. Huang, Chem. Eng. J., 2021, 412, 128734 CrossRef CAS.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J. Lai, L. Liu, C. Li, X. Yan, C. Liu, J. B. -H. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, 30, 1801435 CrossRef.
- Z. Wang, C. Xie, C. Yu, G. Fei, Z. Wang and H. Xia, Macromol. Rapid Commun., 2018, 39, 1700678 CrossRef.
- J.-H. Gao, B. Wan, M.-S. Zheng, L. Luo, H. Zhang, Q.-L. Zhao, G. Chen and J.-W. Zha, Mater. Horiz., 2024, 11, 1305–1314 RSC.
- Y. Deng, Z. Huang, B. L. Feringa, H. Tian, Q. Zhang and D.-H. Qu, Nat. Commun., 2024, 15, 3855 CrossRef CAS PubMed.
- S.-W. Zhou, C. Yu, M. Chen, C.-Y. Shi, R. Gu and D.-H. Qu, Smart Mol., 2023, 1, e20220009 CrossRef.
- C.-Y. Shi, D.-D. He, Q. Zhang, F. Tong, Z.-T. Shi, H. Tian and D.-H. Qu, Natl. Sci. Rev., 2023, 10, nwac139 CrossRef CAS.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, D.-D. He, H. Tian and D.-H. Qu, CCS Chem., 2023, 5, 1422–1432 CrossRef CAS.
- M. Chen, R. Yang, H. Wu, Q. Wang, C. Shi, S.-W. Zhou, D. Yang, F.-Y. Liu, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2024, 63, e202409200 CrossRef CAS PubMed.
- M. Yin, X. Zhang, H. Wang, Z. Yuan, H. Deng, X. Jiang, H. Hu, J. Zhu and J. Wang, ACS Sustainable Chem. Eng., 2024, 12, 6343–6354 CrossRef CAS.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- D. Yang, K. Zhao, R. Yang, S.-W. Zhou, M. Chen, H. Tian and D.-H. Qu, Adv. Mater., 2024, 36, 2403880 CrossRef CAS PubMed.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- C.-Y. Shi, D.-D. He, B.-S. Wang, Q. Zhang, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2023, 135, e202214422 CrossRef.
- Q. Zhang, Y. Deng, C.-Y. Shi, B. L. Feringa, H. Tian and D.-H. Qu, Matter, 2021, 4, 1352–1364 CrossRef CAS.
- S. Zhang, Z. Chen, Y. Lu, Z. Xu, W. Wu, W. Zhu, C. Peng and H. Liu, RSC Adv., 2015, 5, 74284–74294 RSC.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- D. G. Sycks, D. L. Safranski, N. B. Reddy, E. Sun and K. Gall, Macromolecules, 2017, 50, 4281–4291 CrossRef CAS.
- S. Zhang, C. Ge and R. Liu, Sens. Actuators, A, 2022, 341, 113580 CrossRef CAS.
- G. Zhao, F. Qian, X. Li, Y. Tang, Y. Sheng, H. Li, J. Rao, M. V. Singh, H. Algadi, M. Niu, W. Zhang, Z. Guo, X. Peng and T. Chen, Adv. Compos. Hybrid Mater., 2023, 6, 184 CrossRef CAS.
- X. Yang, L. Cheng, Z. Zhang, J. Zhao, R. Bai, Z. Guo, W. Yu and X. Yan, Nat. Commun., 2022, 13, 6654 CrossRef CAS.
- J. Qin, Y. Wang, T. Wang, N. Wang, W. Xu, L. Cheng, W. Yu, X. Yan, L. Gao, B. Zheng and B. Wu, Angew. Chem., Int. Ed., 2024, 63, e202400989 CrossRef CAS PubMed.
- Z. Zhang, L. Cheng, J. Zhao, H. Zhang, X. Zhao, Y. Liu, R. Bai, H. Pan, W. Yu and X. Yan, J. Am. Chem. Soc., 2021, 143, 902–911 CrossRef CAS PubMed.
- L. Wang, L. Cheng, G. Li, K. Liu, Z. Zhang, P. Li, S. Dong, W. Yu, F. Huang and X. Yan, J. Am. Chem. Soc., 2020, 142, 2051–2058 CrossRef CAS PubMed.
- L. Yang, Y. Wang, G. Liu, J. Zhao, L. Cheng, Z. Zhang, R. Bai, Y. Liu, M. Yang, W. Yu and X. Yan, Angew. Chem., Int. Ed., 2024, 63, e202410834 CAS.
- M. H. Lan, X. Guan, D. Y. Zhu, Z. P. Chen, T. Liu and Z. Tang, ACS Appl. Mater. Interfaces, 2023, 15, 19447–19458 CrossRef CAS.
- H. Wan, B. Wu, L. Hou and P. Wu, Adv. Mater., 2024, 36, 2307290 CrossRef CAS PubMed.
- H. Qiao, B. Wu, S. Sun and P. Wu, J. Am. Chem. Soc., 2024, 146, 7533–7542 CrossRef CAS PubMed.
- D. Döhler, J. Kang, C. B. Cooper, J. B.-H. Tok, H. Rupp, W. H. Binder and Z. Bao, ACS Appl. Polym. Mater., 2020, 2, 4127–4139 CrossRef.
- Z. Feng, B. Yu, J. Hu, H. Zuo, J. Li, H. Sun, N. Ning, M. Tian and L. Zhang, Ind. Eng. Chem. Res., 2019, 58, 1212–1221 CrossRef CAS.
- B. Krishnakumar, M. Singh, V. Parthasarthy, C. Park, N. G. Sahoo, G. J. Yun and S. Rana, Nanoscale Adv., 2020, 2, 2726–2730 RSC.
- K. Zhang, J. Sun, J. Song, C. Gao, Z. Wang, C. Song, Y. Wu and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 45306–45314 CrossRef CAS PubMed.
- D.-P. Wang, Z.-H. Zhao, C.-H. Li and J.-L. Zuo, Mater. Chem. Front., 2019, 3, 1411–1421 RSC.
- M. Pruvost, W. J. Smit, C. Monteux, P. Poulin and A. Colin, npj Flexible Electron., 2019, 3, 7 CrossRef.
- K. Ha, W. Zhang, H. Jang, S. Kang, L. Wang, P. Tan, H. Hwang and N. Lu, Adv. Mater., 2021, 33, 2103320 CrossRef CAS PubMed.
- D. Zhong, C. Wu, Y. Jiang, Y. Yuan, M. Kim, Y. Nishio, C.-C. Shih, W. Wang, J.-C. Lai, X. Ji, T. Z. Gao, Y.-X. Wang, C. Xu, Y. Zheng, Z. Yu, H. Gong, N. Matsuhisa, C. Zhao, Y. Lei, D. Liu, S. Zhang, Y. Ochiai, S. Liu, S. Wei, J. B.-H. Tok and Z. Bao, Nature, 2024, 627, 313–320 CrossRef CAS PubMed.
- Y. Luo, M. R. Abidian, J.-H. Ahn, D. Akinwande, A. M. Andrews, M. Antonietti, Z. Bao, M. Berggren, C. A. Berkey, C. J. Bettinger, J. Chen, P. Chen, W. Cheng, X. Cheng, S.-J. Choi, A. Chortos, C. Dagdeviren, R. H. Dauskardt, C. Di, M. D. Dickey, X. Duan, A. Facchetti, Z. Fan, Y. Fang, J. Feng, X. Feng, H. Gao, W. Gao, X. Gong, C. F. Guo, X. Guo, M. C. Hartel, Z. He, J. S. Ho, Y. Hu, Q. Huang, Y. Huang, F. Huo, M. M. Hussain, A. Javey, U. Jeong, C. Jiang, X. Jiang, J. Kang, D. Karnaushenko, A. Khademhosseini, D.-H. Kim, I.-D. Kim, D. Kireev, L. Kong, C. Lee, N.-E. Lee, P. S. Lee, T.-W. Lee, F. Li, J. Li, C. Liang, C. T. Lim, Y. Lin, D. J. Lipomi, J. Liu, K. Liu, N. Liu, R. Liu, Y. Liu, Y. Liu, Z. Liu, Z. Liu, X. J. Loh, N. Lu, Z. Lv, S. Magdassi, G. G. Malliaras, N. Matsuhisa, A. Nathan, S. Niu, J. Pan, C. Pang, Q. Pei, H. Peng, D. Qi, H. Ren, J. A. Rogers, A. Rowe, O. G. Schmidt, T. Sekitani, D.-G. Seo, G. Shen, X. Sheng, Q. Shi, T. Someya, Y. Song, E. Stavrinidou, M. Su, X. Sun, K. Takei, X.-M. Tao, B. C. K. Tee, A. V.-Y. Thean, T. Q. Trung, C. Wan, H. Wang, J. Wang, M. Wang, S. Wang, T. Wang, Z. L. Wang, P. S. Weiss, H. Wen, S. Xu, T. Xu, H. Yan, X. Yan, H. Yang, L. Yang, S. Yang, L. Yin, C. Yu, G. Yu, J. Yu, S.-H. Yu, X. Yu, E. Zamburg, H. Zhang, X. Zhang, X. Zhang, X. Zhang, Y. Zhang, Y. Zhang, S. Zhao, X. Zhao, Y. Zheng, Y.-Q. Zheng, Z. Zheng, T. Zhou, B. Zhu, M. Zhu, R. Zhu, Y. Zhu, Y. Zhu, G. Zou and X. Chen, ACS Nano, 2023, 17, 5211–5295 CrossRef CAS PubMed.
- G. Cheng, H. Xu, N. Gao, M. Zhang, H. Gao, B. Sun, M. Gu, L. Yu, Y. Lin, X. Liu, G. He and D. Wei, Carbon, 2023, 204, 456–464 CrossRef CAS.
- Y. Guo, M. Zhong, Z. Fang, P. Wan and G. Yu, Nano Lett., 2019, 19, 1143–1150 CrossRef CAS PubMed.
- C. Yang, L. Li, J. Zhao, J. Wang, J. Xie, Y. Cao, M. Xue and C. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 25811–25818 CrossRef CAS PubMed.
- L. Gao, C. Zhu, L. Li, C. Zhang, J. Liu, H.-D. Yu and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 25034–25042 CrossRef CAS PubMed.
- S. Chen, L. Sun, X. Zhou, Y. Guo, J. Song, S. Qian, Z. Liu, Q. Guan, E. Meade Jeffries, W. Liu, Y. Wang, C. He and Z. You, Nat. Commun., 2020, 11, 1107 CrossRef CAS PubMed.
- Z. He, W. Chen, B. Liang, C. Liu, L. Yang, D. Lu, Z. Mo, H. Zhu, Z. Tang and X. Gui, ACS Appl. Mater. Interfaces, 2018, 10, 12816–12823 CrossRef CAS PubMed.
- S. Yin, H. Li, W. Qian, M. A. M. Hasan and Y. Yang, Int. J. Extreme Manuf., 2024, 6, 055502 CrossRef CAS.
- J. Qiu, X. Guo, R. Chu, S. Wang, W. Zeng, L. Qu, Y. Zhao, F. Yan and G. Xing, ACS Appl. Mater. Interfaces, 2019, 11, 40716–40725 CrossRef CAS PubMed.
- Y. Wan, Z. Qiu, Y. Hong, Y. Wang, J. Zhang, Q. Liu, Z. Wu and C. F. Guo, Adv. Electron. Mater., 2018, 4, 1700586 CrossRef.
- Y.-Q. Liu, J.-R. Zhang, D.-D. Han, Y.-L. Zhang and H.-B. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 38084–38091 CrossRef CAS PubMed.
- S. C. B. Mannsfeld, B. C.-K. Tee, R. M. Stoltenberg, C. V. H.-H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed.
- A. Tewari, S. Gandla, S. Bohm, C. R. McNeill and D. Gupta, ACS Appl. Mater. Interfaces, 2018, 10, 5185–5195 CrossRef CAS PubMed.
- C. M. Boutry, Y. Kaizawa, B. C. Schroeder, A. Chortos, A. Legrand, Z. Wang, J. Chang, P. Fox and Z. Bao, Nat. Electron., 2018, 1, 314–321 CrossRef.
- M. Xu, Y. Gao, G. Yu, C. Lu, J. Tan and F. Xuan, Sens. Actuators, A, 2018, 284, 260–265 CrossRef CAS.
- R. Han, Y. Liu, Y. Mo, H. Xu, Z. Yang, R. Bao and C. Pan, Adv. Funct. Mater., 2023, 33, 2305531 CrossRef CAS.
- R. Zhang, L. Qi, X. Chao, H. Lian, J. Luo and S. Chen, Chem. Eng. J., 2024, 485, 149729 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01404b |
| This journal is © The Royal Society of Chemistry 2025 |