
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general copper catalytic system for cross-coupling of aryl iodides with chlorosilanes under reductive conditions†
Liping
Qiu
a,
Yiqi
Liu
a,
Han
Chen
b,
Lijuan
Song
 b and
Weilong
Xie
*a
b and
Weilong
Xie
*a
aCollege of Chemistry and Chemical Engineering, Donghua University, Shanghai 201620, China. E-mail: weilongxie@dhu.edu.cn
bSchool of Science, Harbin Institute of Technology, Shenzhen 518055, China
First published on 16th April 2025
Abstract
Directly forged linkages between commercially available electrophiles are powerful synthetic tools for chemical bond construction. This strategy could eliminate the pre-synthesis of reactive organometallic reagents in couplings with electrophiles, thus providing efficient, easily-handled and step-economical routes in organic synthesis. Reported approaches are mainly utilized in carbon–carbon bond formations, whereas carbon–silicon bond construction employing halosilanes with carbon electrophiles is still underexplored. Copper-catalysis has made significant achievements in the coupling reactions of carbon halides in the past decades, yet silyl electrophiles are seldom involved in these systems. Herein, we establish a practical, efficient, and economical copper system catalyzing the construction of Csp2–Si bonds by directly using aryl/vinyl iodides with various chlorosilanes under ligand-free and reductive conditions, thus providing a general platform for organosilane synthesis with broad scope, high functionality tolerance, scalability and operational simplicity. An unprecedented mechanistic motif was obtained to suggest that the copper catalyst was likely to lower the energy barrier in the reaction of the in situ generated arylzinc with halosilanes, rather than proceed via the traditional metal–aryl species.
Introduction
Practical and convenient methods to forge linkages between chemical feedstocks are of paramount importance in organic synthesis, and the last few decades have witnessed the rapid development of transition-metal catalysis for the assembly of diverse molecules from a plethora of organic motifs, thus bringing remarkable achievements in synthetic chemistry, materials science, and pharmaceutical industries.1 In this context, organosilanes attract immense interest and enjoy widespread application in numerous fields, and extensive studies have disclosed that introducing silyl groups to the carbon skeletons or replacing a carbon atom with the silicon element, namely “silicon switch”, in molecule design would modulate the physicochemical and biological properties.2Traditional synthetic routes for organosilanes are heavily dependent on the couplings of organometallic reagents and silyl electrophiles with or without transition-metal catalysts,3 whereas the preparation and utilization of highly reactive compounds such as organolithium and Grignard reagents undermined their efficiency, generality and safety issues in synthesis (Scheme 1a). Thus, further efforts are required to provide efficient and practical approaches for organosilane synthesis catering to the increasing demand of functional organosilanes.
 | ||
| Scheme 1 Representative approaches for the transformations of halosilanes to arylsilanes. | ||
Chlorosilanes are readily available feedstocks, and thus the development of facile and practical catalytic systems with such reagents would provide appealing yet underexplored approaches to the access of organosilanes. Oshima and Yorimitsu et al. reported silver-catalyzed couplings of aryl and alkenyl Grignard reagents with chlorosilanes to furnish tetraorganosilanes.4 Recently, Watson and colleagues also demonstrated coupling of halosilanes with pre-generated alkylzinc or Grignard reagents forming the C–Si bonds with the precious Pd-catalysis.5 Other silane coupling reagents derived from halosilanes, such as metallic silyl reagents,6,7 hydrosilanes,8 disilanes9 and silylborons,10 have also been reported in the sheer variety of silylation systems. However, the instability or difficult accessibility of such reagents inhibits their applications in synthesis.
Although considerable progress has been achieved in coupling with halosilanes, the precedent systems often require the pre-synthesis of carbon- or silicon-derived nucleophiles.4–7 Therefore, a practical, safe and step-economical strategy that eliminates the preparation of those reactive reagents would provide a most straightforward, efficient and operationally simple approach, which would obviously be of great value in the synthesis of organosilane chemicals.11
Recently, Shu et al. disclosed that bipyridine ligands enabled nickel-catalyzing reductive couplings of aryl and alkenyl triflates with vinyl chlorosilanes (Scheme 1b(i)).12 They were later able to employ chlorohydrosilanes containing Si–H bonds in silylation catalyzed by Ni(cod)2 and phenanthroline-type ligands (Scheme 1b(ii)).13 Latterly, chlorosilacyclobutanes were also successful in coupling with aryl electrophiles catalyzed by a diphosphine-nickel system (Scheme 1b(iii)).14 Although nickel-catalysis accelerated by diverse ligands granted access to the couplings of aryl electrophiles with certain halosilanes and attained significant achievements recently. There is room to improve this chemistry based on the status quo.12–14 The case-by-case optimization with different halosilanes and ligands, and limited silyl scope demand us to develop a general and efficient protocol. Additionally, other nickel-catalyzed silyl-reductive couplings were also explored with activated alkyl halides.15 Chromium and cobalt catalyses have also been developed in couplings of chlorosilanes with other carbon electrophiles.16
Copper, as one of the representative non-precious metals,17 has emerged as a powerful system for new chemical bond construction, and the exploration of such economically attractive and environmentally benign catalysis attracted broad interest. For instance, copper-catalyzed Ullmann reactions of aryl halides with various nucleophiles forming biaryls, amines, alcohols, and thiols are well-recognized catalytic transformations in organic synthesis.17 Notably, the majority of the reported copper systems are mainly employed for the coupling reactions of carbon-based organic halides,17,18 whereas the utilization of silicon-based halosilane reagents still remains scarce. In continuation of our research interest with copper-catalysis,19 we herein first present a ligand-free and operational simple copper-catalyzed cross-coupling of aryl and vinyl iodides with various chlorosilanes under reductive conditions, featuring high efficiency and functional group tolerance (Scheme 1c). The current system shows excellent catalytic competence by the aid of a simple, commercial catalyst, and the broad substrate applicability, scalability, and operational simplicity render the system to serve as the general platform for organosilane synthesis. Additionally, mechanistic investigation indicated that the reaction of the present system occurs via unprecedent pathways, where the copper catalyst is likely to lower the energy barrier in the σ-bond metathesis of the in situ generated arylzinc species with halosilanes, thus enabling the Csp2–Si bond formation. Compared with the reported nickel systems,12–14 our system shows different patterns in both reaction pathways and substrate scope.
Results and discussion
Reaction optimization
At the outset, we commenced to identify the optimal catalytic conditions for the proposed cross-couplings of aryl iodides with chlorosilanes. Thus, the investigation of the reactions with iodobenzene (1) and trimethyl chlorosilane (2) as the model substrates in the presence of copper catalysts and the zinc reductant was performed (Table 1, and also see Tables S1–S5 in the ESI†). The desired silylation product (3) was obtained only in trace quantities with 10 mol% of CuBr·SMe2 (entry 1, 6%). Significantly, the yields were dramatically increased with the increment of the reductant (entries 2 and 3, 28% and 52%, respectively). The choice of reductant was observed to be crucial, and the use of other reducing agents such as magnesium, manganese and indium was unsuccessful (entry 4). Unfortunately, the addition of auxiliary ligands such as N-heterocyclic carbenes (NHCs), phosphines, phenanthrolines and bipyridines did not show the enhancement effect on the reaction efficiency (see Table S3 in the ESI for details†).|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Reductant | Solvent | Yieldb (%) |
| a Reaction conditions: iodobenzene (0.40 mmol), trimethyl chlorosilane (0.80 mmol), catalyst (10 mol%), and the reductant in the indicated solvent (0.50 mL) at 100 °C for 12 h under nitrogen. b Yields were determined by GC analyses against a calibrated internal standard of n-hexadecane. c X = Br. d X = OTf, homocoupling product biphenyl was observed in 46% yield. e CoBr2, Co(acac)2 or Fe(acac)2 was used as the catalyst. f The reaction was performed at 80 °C. g The reaction was performed at 120 °C. h The reaction was loaded in air and then sealed. | ||||
| 1 | CuBr·SMe2 | Zn (1.0 equiv.) | PhH (∼0.80 M) | 6 |
| 2 | CuBr·SMe2 | Zn (2.0 equiv.) | PhH | 28 |
| 3 | CuBr·SMe2 | Zn (3.0 equiv.) | PhH | 52 |
| 4 | CuBr·SMe2 | Mg, Mn, In (3.0 equiv.) | PhH | <1–3 |
| 5 | CuBr | Zn (3.0 equiv.) | PhH | 3 |
| 6 | CuBr2 | Zn (3.0 equiv.) | PhH | 18 |
| 7 | CuBr + n-Bu2S | Zn (3.0 equiv.) | PhH | 56 |
| 8 | CuTc | Zn (3.0 equiv.) | PhH | 23 |
| 9 | Cu(OAc)2 | Zn (3.0 equiv.) | PhH | 50 |
| 10 | — | Zn (3.0 equiv.) | PhH | <1 |
| 11 | Pd(OAc)2 | Zn (3.0 equiv.) | PhH | 20 |
| 12 | AgNO3 | Zn (3.0 equiv.) | PhH | <1 |
| 13 | NiX2 | Zn (3.0 equiv.) | PhH | <1c or 10d |
| 14e | Co, Fe | Zn (3.0 equiv.) | PhH | <1–9 |
| 15 | CuBr·SMe2 | Zn (3.0 equiv.) | 1,4-Dioxane | 45 |
| 16 | CuBr·SMe2 | Zn (3.0 equiv.) | DMF | <1 |
| 17 | CuBr·SMe2 | Zn (3.0 equiv.) | PhF | 58 |
| 18 | CuBr·SMe2 | Zn (3.0 equiv.) | PhF (∼2.0 M) | 68 |
| 19 | CuBr·SMe2 | Zn (3.0 equiv.) | PhF (∼0.67 M) | 33 |
| 20f | CuBr·SMe2 | Zn (3.0 equiv.) | PhF (∼0.67 M) | 31 |
| 21 | CuBr·SMe 2 | Zn (3.0 equiv.) | PhF (∼2.0 M) | 95 |
| 22h | CuBr·SMe2 | Zn (3.0 equiv.) | PhF (∼2.0 M) | 37 |
Using copper bromide as the catalyst promoted the reaction in a low yield (entry 5, 3%), which was probably ascribed to its low solubility. Divalent copper salt, CuBr2, promoted the silylation process in a yield of 18% (entry 6). The addition of a catalytic amount of sulfide to the CuBr-catalyzed reaction would increase the productive yield from 3% to 56%, indicating that the solubility of the copper catalyst was probably accounting for the efficiency (entries 5 and 7). Next, other copper salts such as CuTc and Cu(OAc)2 gave the product in moderate to good yields (entries 8 and 9, 23% and 50%, respectively). The reaction could not occur in the absence of copper, indicating that copper was essential for the current coupling system (entry 10). Other transition-metal catalysts were also examined in the current system (entries 11–14). For instance, precious palladium only gave 20% yield of the silylation product (entry 11). Silver nitrate (AgNO3), an effective catalyst for the coupling of aryl Grignard reagents with halosilanes in Oshima and Yorimitsu's protocol,4 was tested to be inapplicable in the current system (entry 12, <1% yield). Nickel bromide (NiBr2) could not promote the current coupling reaction, while nickel triflate [Ni(OTf)2] mainly gave the homocoupling biphenyl side product in 46% yield along with a 10% yield of the silylation product (entry 13). Cobalt and iron catalysts were examined to be almost ineffective (entry 14, <1–9%).
Solvents have a pronounced effect on the reaction efficiency, and polar solvents of 1,4-dioxane and DMF gave diminished yields (entries 15 and 16). Nonpolar solvent fluorobenzene generated the product in a good yield (entry 17, 58%). Intriguingly, the yields were observed to be highly associated with the concentration of the system, and a high concentration gave an increased chemical yield, while decreasing the concentration would cause a significant drop in the efficiency (entries 18 and 19, 68% and 33%, respectively).
Further experimental data disclosed that the reaction temperature showed a remarkable influence on the coupling efficiency, and the reaction performing at 80 °C afforded the product in a mediocre yield (entry 20, 31%), and an excellent efficiency was secured at an elevated temperature (entry 21, 95% at 120 °C). Unfortunately, a significant decrease in productive yield was observed when the reaction was loaded in air (entry 22, 37%), indicating that the current system was quite sensitive to the air atmosphere. It deserves particular mention that the majority of the reported copper systems require complex ligands to accelerate the efficiency,18 and it was highly fascinating to see the current system working effectively with the aid of a simple and commercial copper catalyst, thus providing a practical, efficient and step-economical method for arylsilane synthesis.
Reaction scope
With the optimal conditions in hand, we proceeded to test the feasibility and the scope of aryl iodides (Table 2). Iodobenzene reacted with trimethyl chlorosilane forming the product 3 in 81% isolated yield, and the lower isolation yield than the GC yield was probably due to its volatility property. Variants of aryl iodides with carbon-substituents such as methyl, tert-butyl, and phenyl groups worked equally well (4–7, 74–94%). Aryl coupling partners with electronically distinct functional groups were further examined. Compounds with electron-rich silyloxyl and methmercapto groups furnished the silylation products smoothly (8 and 9, 87% and 75%, respectively). Reagents bearing electron-deficient halides, including fluorine, chlorine and bromine, all generated the products in fair to good yields (10–12, 59–73%), and the remaining halides would allow the downstream transformations for complex molecules. Two isomeric diiodobenzenes formed the bis-silylation products efficiently (13 and 14).| a Reaction conditions: aryl iodide (0.40 mmol), chlorosilane (0.80 mmol), CuBr·SMe2 (20 mol%), and Zn (1.20 mmol) in anhydrous fluorobenzene (0.20 mL) at 120 °C for 24 h under nitrogen, isolated yields. b CuBr·SMe2 (10 mol%), 12 h. c Chlorosilane (0.40 mmol), aryl iodide (1.60 mmol), Zn (6.0 equiv.). d Chlorosilane (0.40 mmol), aryl iodide (2.40 mmol), Zn (9.0 equiv.). |
|---|

|
A highly electron-deficient perfluoroiodobenzene was further tested, and the product 15 was obtained in a good yield (62%). It is worth mentioning that the lithiation of aryl halides containing ortho-fluorine substituents would cause an erosion of aryl metal species due to the rapid fluoride elimination.20 The current system could overcome this problematic issue to the access of o-fluorinated arylsilanes. Aryl substrates containing the acidic sp2-C–H bonds were successfully silylated at the ipso-sites without the isomerization of the reactive positions (16–20, 59–85%).20 Gratifyingly, substrates with vinyl or alkynyl moieties both generated the corresponding arylsilanes smoothly without reduction of the unsaturated bonds (21–22, 83% and 57%, respectively). Carbonyls such as ester or amide were also viable to form the products in excellent yields (23–24, 95% and 98%, respectively). However, a substrate with a cyclic amide skeleton gave a diminished yield (25, 50%). Reagents with a more reactive ketone or aldehyde group were incompatible in the current system (26 and 27).
Steric hindrance influence on aryl electrophiles was further examined. Ortho-methyl iodobenzene generated the coupling product 28 in a high yield of 88% with an increased catalyst loading (20 mol% of Cu). Sterically encumbered 2,6-dimethyl iodobenzene was also applicable albeit in a decreased yield (29, 55%). π-Extended naphthalenyl and phenanthrenyl rings gave excellent yields (30–31, 90% and 94%, respectively). Finally, the applicability of heteroarenes including indole, furan, and thiophene was all tested to afford the products 32–37 in 74–95% yield. Unfortunately, iodopyridines and iodopyrrole were unsuitable in the current system (38–41). Interestingly, iodopyrazole was successfully reacted to form the coupling product in a good yield of 66% (42). Vinyl iodides were briefly tested, and the products could be obtained smoothly (43 and 44). Finally, scale-up reactions were successfully run even with 50-mmol-scale, furnishing products 6 and 45 in high yields (9.06 g, 80% yield, and 10.5 g, 88% yield, respectively), thus demonstrating the robustness and practicality of the current system. Unfortunately, aryl bromides were not workable in the current protocol, which was probably due to the inability for the Csp2–Br bond activation.
Next, considering the limited halosilane scope especially in terms of trialkyl-substituted reagents in previous reports,12–14 we set out to evaluate the competence of the current system with various silanes (Table 3). Bulkier triethyl chlorosilane was coupled in a moderate yield of 43% with 10 mol% catalyst in 12 h, and a high yield was obtained with an elongated reaction time and a higher catalyst loading (46, 85% isolated yield with 20 mol% of copper in 24 h). n-Butyl dimethyl and isopropyldimethyl chlorosilanes both formed the products in high yields (47 and 48, 79% and 80%, respectively). Surprisingly, the sterically congested tert-butyl dimethyl chlorosilane also participated to furnish product 49 in a moderate yield of 47%. Unfortunately, triisopropyl chlorosilane failed to form the silylation product due to the bulkiness of the silyl reagent (50). Interestingly, aryl chlorosilanes showed superior activities compared with trialkyl silanes. For example, phenyl dimethyl chlorosilane formed a quantitative yield of product 51 (99%) with 10 mol% catalyst in 12 h. Strikingly, the sterically hindered diphenyl methyl chlorosilane and even triphenyl chlorosilane afforded the target products in excellent yields (52 and 53, 99% and 84%, respectively). Vinyl chlorosilane was workable to deliver the product 54 in 99% yield with 10 mol% of copper.
| a Reaction conditions: iodobenzene (0.40 mmol), chlorosilane (0.80 mmol), CuBr·SMe2 (20 mol%), and Zn (1.20 mmol) in the anhydrous fluorobenzene (0.20 mL) at 120 °C for 24 h under nitrogen, isolated yields. b CuBr·SMe2 (10 mol%), 12 h. c 4-Iodo-1,1′-biphenyl used as the reagent. d Chlorosilane (0.40 mmol), aryl iodide (1.60 mmol), Zn (6.0 equiv.). e 1-Fluoro-4-iodobenzene used as the reagent. f Chlorosilane (0.40 mmol), aryl iodide (2.40 mmol), Zn (9.0 equiv.). |
|---|

|
Taking one step further, we were curious about the extension of the system to hydrochlorosilanes, which contain both reactive Si–H and Si–Cl bonds. In fact, the bond dissociation energy of the Si–H bond is much lower than that of the Si–Cl bond (ca. 77 kcal mol−1 and ca. 93 kcal mol−1 for Si–H and Si–Cl bonds, respectively), and hydrochlorosilane reagents via the Si–Cl bond activation are far-less reported.13 Moreover, organometallic reagents are prone to react with the Si–H bonds, forming the Si–C skeletons.21 Intriguingly, the current system enabled the couplings of chlorohydrosilanes and aryl iodides in 72–83% productive yields without difficulty, as exemplified by products 55–57. The use of biphenyl iodide to couple with dimethylchlorosilane was due to the volatility of the product.
Interestingly, silanes with multiple Si–Cl bonds were successfully employed to form the arylation products effectively. For instance, dichlorodimethylsilane reacting with an aryl iodide formed a two-fold arylation product 58 in a fantastic yield (99%) by using an excess amount of aryl iodide. Considering the importance of silole moieties in materials science, bis-arylation of dichlorodibenzosilole was achieved in a moderate yield (59, 58%). Alkyl-substituted dichlorosilanes were also successfully utilized as the coupling partners to furnish the arylation products (60 and 61, 68% and 69%, respectively). Astonishingly, tri-arylation was also demonstrated with trichlorophenylsilane to form product 62 in a high yield of 85%.
Mechanistic studies
After the evaluation of the reaction scope, we performed a series of experiments to reveal the mechanism (Scheme 2). No apparent inhibition was observed with 1,1-diphenyl ethene as the radical scavenger (Scheme 2a, 94% GC yield), indicating that the reaction of the current system was unlikely to occur via radical pathways. To validate the possibility of copper-aryl species involved in the present system, a copper–aryl complex C6F5Cu (Cu-1) was independently prepared and subjected to reaction with trimethyl chlorosilane (Scheme 2b). Astonishingly, the C–Si coupling product 15 was not detected with or without the zinc reductant. Considering the fact that iodopentafluorobenzene (C6F5I) could be used as the reagent in the current silylation protocol (Table 2, product 15) and arylcopper Cu-1 could not react with halosilane in the stoichiometric transformation, copper-aryl species were not likely to be involved in the silylation process and the C–Si bond formation was unlikely to occur on the copper center.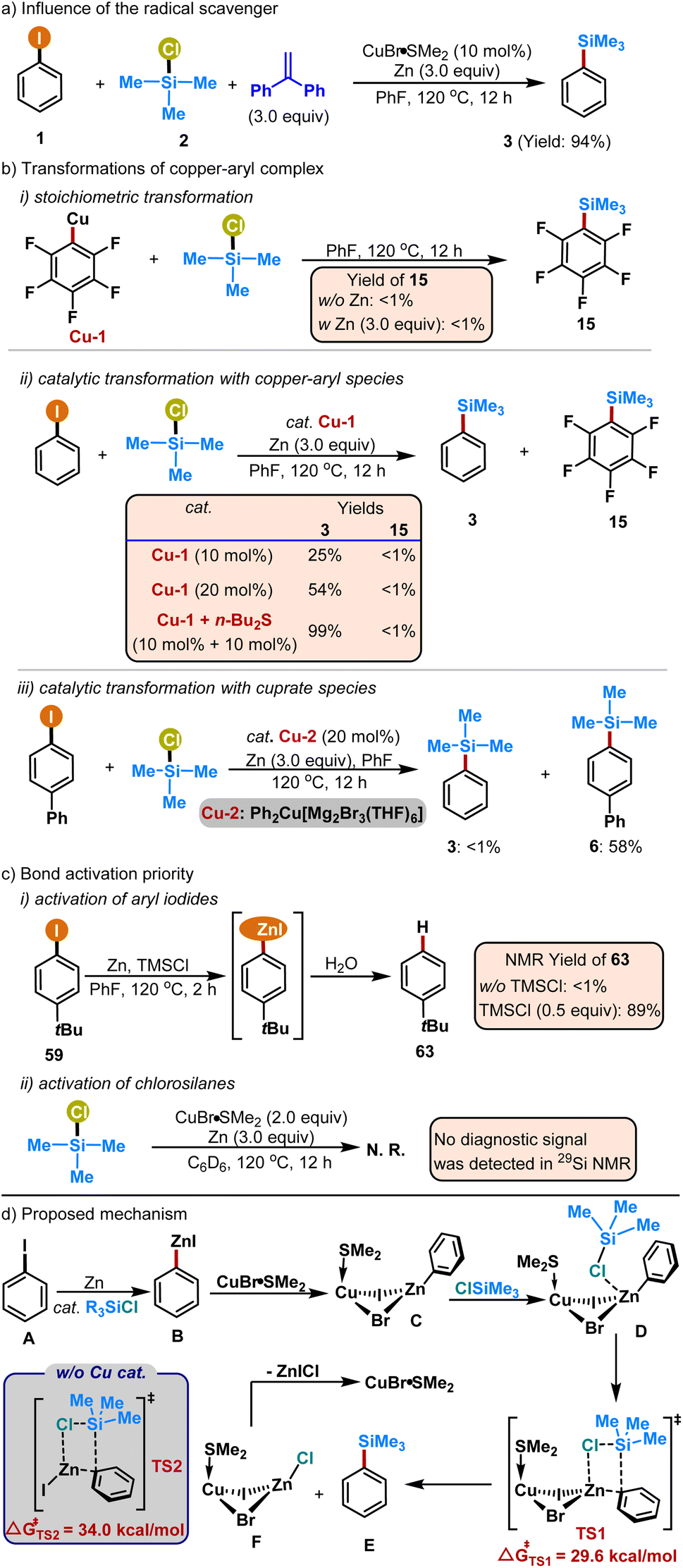 | ||
| Scheme 2 Mechanistic studies and proposed mechanism. | ||
Subsequently, the utilization of Cu-1 as the catalyst was investigated for the model reaction of iodobenzene with trimethyl chlorosilane. The reaction afforded the coupling product 3 in 25% GC yield, and with the increase of the catalyst loading, the yield of the desired product could reach 54% with 20 mol% of the catalyst. As expected, a catalytic amount of sulfide could promote the coupling yield effectively (10 mol% Cu-1 and 10 mol% n-Bu2S, 99% GC yield). Notably, the aryl transfer product 15 from the copper complex Cu-1 was also not detected, indicating that the aryl moiety in the copper-aryl species was unable to couple with halosilanes in the catalytic process, which was in line with the above stoichiometric transformations. Additionally, the employment of cuprate species Cu-2 (20 mol%) as the catalyst only gave a diminished yield of the silylation product (6, 58%), and the aryl transfer product 3 from the cuprate species Cu-2 was also not observed. This experiment further excluded the involvement of cuprate species as the catalytically active species in the current system.
Furthermore, a series of controlled experiments disclosed that the activation priority of iodoarenes was over halosilanes (Scheme 2c), which was in line with their bond dissociation energies.22 Specifically, aryl iodides were activated by the zinc reductant with the addition of chlorosilane to form the arylzinc species,23 while no diagnostic signal was observed when treating the halosilane with zinc and copper salt. Collectively, the above experimental results revealed that the reaction of the current system occurred via a non-radical pathway and the copper-aryl species was not involved in the bond formation process. In fact, it is well documented that copper has the propensity for interaction with halogens,24 and numerous copper–zinc bimetallic adducts containing the Cu–X–Zn moieties (X = halogen) have been reported in the literature.25 Thus, we assumed that the in situ generated arylzinc species B would interact with the copper catalyst forming the corresponding halogen bridging species C, followed by the coordination of halosilanes to form the species D. Subsequently, the aryl group would transfer from species D to generate the silylation product Evia a transition state TS1 (Scheme 2d). Computational studies disclosed that the transition state TS1 requires an energy barrier at 29.6 kcal mol−1 (see the ESI for details†), and this barrier could be overcome since the reactions were conducted at 120 °C. In comparison, the direct reaction of phenylzinc iodide B with trimethyl chlorosilane requires a transition state energy barrier at 34.0 kcal mol−1 (transition state TS2 in Scheme 2d), rendering this pathway highly improbable under the reaction conditions. In addition, the plausible reaction mode was also in line with the additional ligands inhibiting the reaction efficiency (see Table S3 in the ESI†).
Thus, a plausible mechanism is depicted in Scheme 2d based on the literature precedents18–20,24–26 and the above experiments. Iodoarene A was activated by zinc forming arylzinc B by the assistance of halosilanes. The reactive arylzinc species would interact with the copper catalyst to generate the copper–zinc bimetallic species C. Subsequently, the reaction of species C with halosilanes gave the intermediate D, which would afford the silylation product E and copper–zinc adduct Fvia the transition state TS1. Finally, a release of the zinc salt would allow the regeneration of the copper catalyst.
Conclusions
In summary, the present study first demonstrates a practical and efficient copper-catalytic system, which enables the reductive couplings of aryl/vinyl iodides and chlorosilanes with a wide scope and high functionality tolerance, thus offering an efficient and practical method for organosilane synthesis. Furthermore, this work offers a new direction for the utilization of chlorosilanes with copper-catalysis, which may allow us to access the underexplored chemical space of halosilane involved couplings. Further effort would be devoted to broadening the application of copper-catalyzed silylation systems with halosilanes.Data availability
All data for the replication of this work are given in the ESI† or can be obtained from the lead contact upon reasonable request.Author contributions
W. X. organized the project. Q. L. and L. Y. conducted the experiments. H. C. and L. S. performed DFT calculations. W. X., Q. L., L. Y. H. C. and L. S. analysed the data and W. X. wrote the manuscript with the assistance of all authors.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22201034), and the Natural Science Foundation of Shanghai (22ZR1402500) for generous funding.Notes and references
- (a) F. Diederich, and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH, 2004 Search PubMed; (b) J. C. H. Lee, and D. J. Hall, Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase, and M. Oestreich, Wiley-VCH, Weinheim, 2014, pp. 65–132 Search PubMed.
- (a) J. Fotie, C. M. Matherne and J. E. Wroblewski, Chem. Biol. Drug Des., 2023, 102, 235–254 CrossRef CAS PubMed; (b) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed; (c) G. K. Min, D. Hernández and T. Skrydstrup, Acc. Chem. Res., 2013, 46, 457–470 CrossRef CAS PubMed; (d) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- (a) Science of Synthesis, ed. I. Fleming, Thieme, Stuttgart, 2002, ch. 4.4, vol. 4 Search PubMed; (b) The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport, and Y. Apeloig, Wiley, New York, 1998, vol. 2 Search PubMed.
- K. Murakami, K. Hirano, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2008, 47, 5833–5835 CrossRef CAS PubMed.
- (a) A. P. Cinderella, B. Vulovic and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 7741–7744 CrossRef CAS PubMed; (b) B. Vulovic, A. P. Cinderella and D. A. Watson, ACS Catal., 2017, 7, 8113–8117 CrossRef CAS PubMed.
- (a) R. Chandrasekaran, F. Pulikkottil, K. Elama and R. Rasappan, Chem. Sci., 2021, 12, 15719–15726 RSC; (b) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CrossRef CAS PubMed; (c) D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS PubMed.
- (a) C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404–6407 CrossRef CAS PubMed; (b) W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141–12145 CrossRef CAS PubMed.
- (a) I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed; (b) A. A. Toutov, W. B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed; (c) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed; (d) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS PubMed.
- (a) E. McNeill, T. E. Barder and S. L. Buchwald, Org. Lett., 2007, 9, 3785–3788 CrossRef CAS PubMed; (b) H. Yamagishi, H. Saito, J. Shimokawa and H. Yorimitsu, ACS Catal., 2021, 11, 10095–10103 CrossRef CAS; (c) H. Yamagishi, F. Harata, J. Shimokawa and H. Yorimitsu, Org. Lett., 2023, 25, 11–15 CrossRef CAS PubMed.
- (a) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed; (b) W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222–14225 CrossRef CAS PubMed; (c) S. Kamio, M. Nakamoto, T. Yamagishi, M. Oestreich and H. Yoshida, Chem. Commun., 2024, 60, 6379–6382 RSC.
- (a) M. J. James, G. E. Clarke, C. Lee and I. J. S. Fairlamb, J. Chem. Educ., 2022, 99, 2656–2660 CrossRef CAS; (b) J. Rappoport, and I. Marek, The Chemistry of Organolithium Compounds, Wiley, 2004 CrossRef; (c) J. A. Schwindeman, C. J. Woltermann and R. J. Letchford, Chem. Health Saf., 2002, 9, 6–11 CrossRef CAS; (d) P. Slavík, B. R. Trowse, P. O'Brien and D. K. Smith, Nat. Chem., 2023, 15, 319 CrossRef PubMed.
- J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed.
- Z.-Z. Zhao, X. Pang, X.-X. Wei, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2022, 61, e202200215 CrossRef CAS PubMed.
- L.-y. Yang, Y. Qin, Z. Zhao and D. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202407773 CrossRef CAS PubMed.
- (a) L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 18587–18590 CrossRef CAS PubMed; (b) M. Xing, H. Cui and C. Zhang, Org. Lett., 2021, 23, 7645–7649 CrossRef CAS PubMed; (c) J. Duan, Y. Wang, L. Qi, P. Guo, X. Pang and X.-Z. Shu, Org. Lett., 2021, 23, 7855–7859 CrossRef CAS PubMed.
- (a) C. Li, S. Yang and X. Zeng, ACS Catal., 2023, 13, 12062–12073 CrossRef CAS; (b) D. Xing, J. Liu, D. Cai, B. Huang, H. Jiang and L. Huang, Nat. Commun., 2024, 15, 4502 CrossRef CAS PubMed.
- (a) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed; (b) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (c) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (d) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed; (e) H. Zhao, A. K. Ravn, M. C. Haibach, K. M. Engle and C. C. C. Johansson Seechurn, ACS Catal., 2024, 14, 9708–9733 CrossRef CAS PubMed; (f) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (g) I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753–7808 CrossRef CAS; (h) F. Buono, T. Nguyen, B. Qu, H. Wu and N. Haddad, Org. Process Res. Dev., 2021, 25, 1471–1495 CrossRef CAS.
- (a) F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS; (b) M. Eckert, G. Fleischmann, R. Jira, H. M. Bolt, and K. Golka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2006 Search PubMed; (c) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed; (d) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (e) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC; (f) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS PubMed; (g) Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS; (h) H.-Q. Do, R. M. K. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS PubMed.
- (a) Y. Zhou, L. Qiu, J. Li and W. Xie, J. Am. Chem. Soc., 2023, 145, 28146–28155 CrossRef CAS PubMed; (b) J. Xu, X. Zhang, Y. Cao, X. Jian, L. Song and W. Xie, Cell Rep. Phys. Sci., 2024, 287, 102050 CrossRef; (c) J. Zhou, Z. Zhang, Y. Cao and W. Xie, Chem. Sci., 2025, 16, 5109–5117 RSC.
- (a) M. Stratakis, P. G. Wang and A. Streitwieser, J. Org. Chem., 1996, 61, 3145–3150 CrossRef CAS PubMed; (b) H.-Q. Do and O. Daugulis, Org. Lett., 2009, 11, 421–423 CrossRef CAS PubMed.
- (a) N. Hirone, H. Sanjiki, R. Tanaka, T. Hata and H. Urabe, Angew. Chem., Int. Ed., 2010, 49, 7762–7764 CrossRef CAS PubMed; (b) C. Huang, N. Chernyak, A. S. Dudnik and V. Gevorgyan, Adv. Synth. Catal., 2011, 353, 1285–1305 CrossRef CAS PubMed.
- R. Walsh, Acc. Chem. Res., 1981, 14, 246–252 CrossRef CAS.
- (a) A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040–6044 CrossRef CAS PubMed; (b) E. M. Hanada, P. J. McShea and S. A. Blum, Angew. Chem., Int. Ed., 2023, 62, e202307787 CrossRef CAS PubMed.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- G. Kociok-Köhn, M. F. Mahon, K. C. Molloy and A. L. Sudlow, Main Group Met. Chem., 2014, 37, 11–24 Search PubMed.
- L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01304f |
| This journal is © The Royal Society of Chemistry 2025 |