DOI:
10.1039/D4SC08401B
(Edge Article)
Chem. Sci., 2025,
16, 9447-9453
Water-accelerated photooxidation and degradation of lignin linkages mediated by plasmonic catalysts†
Received
11th December 2024
, Accepted 23rd April 2025
First published on 24th April 2025
Abstract
The selective oxidation and degradation of lignin are crucial for realizing its potential as a biofuel or petroleum substitute. Despite the importance of C–C bond cleavage for lignin valorization, this process is significantly challenging. Herein, we present plasmonic gold nanoparticles (Au NPs) as environmentally friendly and reusable photocatalysts for the chemoselective oxidation of the benzylic hydroxyl groups of lignin and subsequent lignin degradation. The oxidation process is driven by the generation of superoxide ions (O2˙−), leading to proton release and initiating lignin photooxidation through a mechanism termed plasmon-driven hydrogen atom abstraction and degradation (p-HAADe). Our results demonstrate the significant suppression of lignin oxidation and degradation in acetonitrile-rich environments, while aqueous conditions notably enhance these processes. Furthermore, two distinct time-dependent regimes are identified, namely, the “oxidation dominant” regime, where lignin oxidation is predominant, and the “degradation dominant” regime, favoring Cα–Cβ bond cleavage. These findings provide crucial insights into optimizing lignin conversion in biofuel applications, highlighting the potential of Au NPs for use in sustainable chemical processes.
Introduction
In conjunction with cellulose and hemicellulose, lignin is a major component of lignocellulose. In recent years, biomass derived from lignocellulose has received growing attention as a renewable energy source with the potential to replace petroleum-based materials.1–3 Natural lignin is a polymer containing abundant aromatic compounds, and it comprises 15–30 wt% of the lignocellulosic biomass.4,5 Furthermore, natural lignin consists of both phenolic and non-phenolic structures. The conversion of natural lignin to low-molecular-weight aromatic compounds is of particular interest because of its potential to yield value-added chemical feedstocks through the cleavage of lignin C–O or C–C bonds.6,7 Previous studies have indicated that the strengths of these bonds in the β-O-4 linkage weakens following hydroxyl group oxidation, rendering it susceptible to cleavage.5,8–10 For example, computational calculations predicted an approximately 14 kcal mol−1 decrease in the Cβ–O bond dissociation enthalpy (BDE) of the β-O-4 linkage upon the oxidation of Cα–OH to Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, thereby facilitating C–O bond cleavage.8,11 Other studies have revealed that Cα–Cβ bond cleavage is possible after the oxidation of the benzylic hydroxyl group.8,12 However, traditional methods for lignin decomposition, such as pyrolysis, acid–base catalysis, organosolvolysis, krafting, and enzymatic decomposition, have numerous drawbacks, including the requirement for high temperatures, low efficiency, and toxic chemical waste generation.13–15 Furthermore, these methods, which generally necessitate harsh conditions, tend to exhibit low product selectivity, thereby complicating the extraction of target compounds.10 To address these challenges, photocatalysis has emerged as a potential technology for lignin decomposition, offering an eco-friendly, highly efficient, and cost-effective process that can be conducted under mild conditions.16,17 To date, research on the photocatalytic decomposition of lignin has explored various types of catalysts, including organic and metal–organic complexes (homogeneous catalysts), in addition to metal chalcogenides and metal oxides (heterogeneous catalysts).6,18–20 Plasmonic nanocrystals, particularly single-component Au nanoparticles (NPs), have attracted considerable attention as visible-light-driven photocatalysts owing to their strong light absorption capability in the visible light region and their excellent catalytic activities at the nanoscale.21–25 Plasmonic Au photocatalysts have been demonstrated to exhibit unique physical behaviors from the perspectives of both kinetics and thermodynamics.26–28 They have also been shown to drive useful chemical reactions such as the CO2 reduction reaction,25,29 C–C bond formation,30,31 bond scission,24 and electrochemical transformations.32,33 Therefore, the use of plasmonic Au photocatalysts, which are eco-friendly and reusable and possess an excellent photostability, is considered to be a potentially effective strategy for transforming lignin derivatives.
O, thereby facilitating C–O bond cleavage.8,11 Other studies have revealed that Cα–Cβ bond cleavage is possible after the oxidation of the benzylic hydroxyl group.8,12 However, traditional methods for lignin decomposition, such as pyrolysis, acid–base catalysis, organosolvolysis, krafting, and enzymatic decomposition, have numerous drawbacks, including the requirement for high temperatures, low efficiency, and toxic chemical waste generation.13–15 Furthermore, these methods, which generally necessitate harsh conditions, tend to exhibit low product selectivity, thereby complicating the extraction of target compounds.10 To address these challenges, photocatalysis has emerged as a potential technology for lignin decomposition, offering an eco-friendly, highly efficient, and cost-effective process that can be conducted under mild conditions.16,17 To date, research on the photocatalytic decomposition of lignin has explored various types of catalysts, including organic and metal–organic complexes (homogeneous catalysts), in addition to metal chalcogenides and metal oxides (heterogeneous catalysts).6,18–20 Plasmonic nanocrystals, particularly single-component Au nanoparticles (NPs), have attracted considerable attention as visible-light-driven photocatalysts owing to their strong light absorption capability in the visible light region and their excellent catalytic activities at the nanoscale.21–25 Plasmonic Au photocatalysts have been demonstrated to exhibit unique physical behaviors from the perspectives of both kinetics and thermodynamics.26–28 They have also been shown to drive useful chemical reactions such as the CO2 reduction reaction,25,29 C–C bond formation,30,31 bond scission,24 and electrochemical transformations.32,33 Therefore, the use of plasmonic Au photocatalysts, which are eco-friendly and reusable and possess an excellent photostability, is considered to be a potentially effective strategy for transforming lignin derivatives.
Thus, in this study, the photodegradation of lignin model compounds (β-O-4 linkages) is investigated using colloidal Au NPs dispersed in water, as outlined in Fig. 1. Furthermore, non-phenolic lignin model compounds were chosen because they require a higher redox potential compared to phenolic lignin, making them more challenging to degrade. This allows us to evaluate the effectiveness of our strategy for achieving efficient lignin degradation. In addition, the effects of the organic solvent (acetonitrile, MeCN) content of the aqueous system on the photooxidation and degradation behaviors are examined. Furthermore, the driving force for the photooxidation of lignin in a water-rich environment is determined.
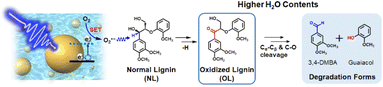 |
| | Fig. 1 Illustration of the stepwise plasmon-driven hydrogen atom abstraction and degradation (p-HAADe) process for the photooxidation of normal lignin (NL) to oxidized lignin (OL), and the subsequent lignin degradation process in a water-rich environment. | |
Results and discussion
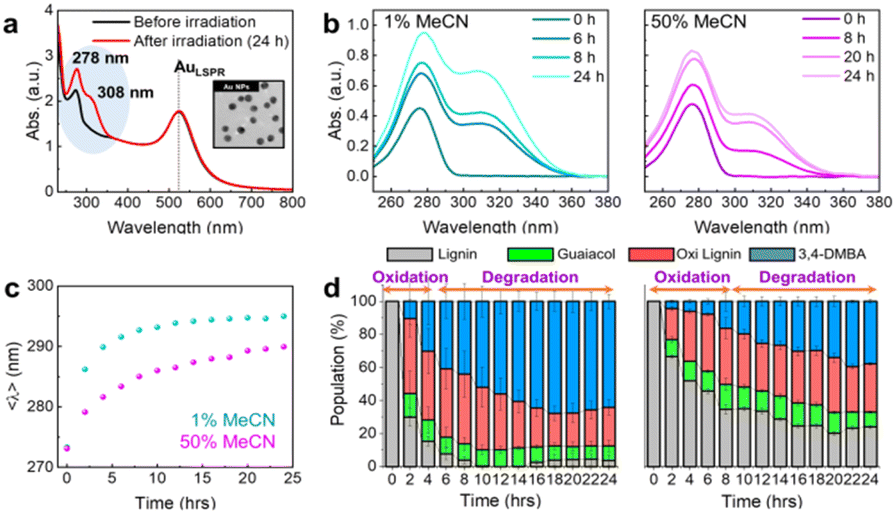
Spherical Au NPs with an average diameter of 13 ± 0.9 nm were employed as plasmonic photocatalysts for the degradation of the synthesized lignin β-O-4 linkage (Fig. S1 and Scheme S1†). Fig. 2a shows the optical characteristics of the reaction mixture before and after the photocatalytic reaction. The intensities of the absorption peaks at 278 and 308 nm increased considerably after irradiation, whereas the localized surface plasmon resonance (LSPR) peak for Au (522 nm) was preserved. The peaks at 278 and 308 nm were therefore attributed to the oxidized form of lignin (Fig. 2a and S2–S4†). A comparative analysis of lignin conversion efficiency was then conducted under various reaction conditions; the results are presented in Table S1.† In the presence of Au NPs and light, lignin photooxidation conversion reached 96.2% after 24 h of light irradiation (Table S1, entry 1 and Fig. S5(a)†). To validate the impact of the Au NPs and light on lignin photooxidation, control experiments were conducted in the absence of light and without the Au NPs (Table S1, entries 2 and 3†). Consequently, a yield of 8% was obtained in the absence of light (Fig. S5(b)†), and no product was detected without the Au NPs (Fig. S5(c)†), demonstrating that both components are required for the reaction to proceed. Furthermore, in the absence of O2, the conversion was <2% (Table S1, entry 4 and Fig. S5(d)†), confirming that O2 is required to facilitate lignin photooxidation.
 |
| | Fig. 2 (a) UV-vis spectra of before (black) and after (red) the photocatalytic lignin photooxidation reaction. The Au LSPR is indicated as a vertical dotted line, showing the stability after irradiation. The inset shows a representative TEM image of the synthesized Au NPs. (b) Transient UV-vis spectra during the photooxidation in 1% MeCN (left) and 50% MeCN (right) in water. (c) Initial moments in the lignin electronic transition as a function of the reaction time (violet: 1% MeCN, magenta: 50% MeCN). (d) Relative populations of normal lignin (NL, gray), oxidized lignin (oxi lignin, OL, red), guaiacol (green), and 3,4-dimethoxybenzaldehyde (3,4-DMBA, light blue) with the corresponding error bars. | |
As presented in Fig. S6, S7(a), and (b),† four species were directly quantified in the ultraviolet-visible (UV-vis) spectra, namely, normal lignin (NL), oxidized lignin (OL), guaiacol, and 3,4-dimethoxybenzaldehyde (3,4-DMBA), wherein the basis spectra demonstrated doublet features. In addition, Fig. 2b displays the changes in the line shapes, which represent the π → π* and n → π* transitions of the electronic structures.15 Notably, the peaks observed at 278 and 308 nm increase significantly in intensity during photooxidation in a water-rich environment containing 1% acetonitrile (MeCN, c.f., a 50% MeCN solution). As shown in Fig. 2c, the initial time-dependent changes reveal a convex trend, with red-shifted electronic transitions of the conjugated bonds being associated with the oxidation reaction (Fig. S7(c)†). The evaluation of the 308/278 nm absorbance ratio as a function of time indicates that the reaction proceeded effectively (Fig. S7(d)†), generating abundant photooxidized and lignin degradation products.
The basis spectra of the four components mentioned above facilitated the development of a protocol for quantifying their relative populations (Fig. S8–S10 and Tables S2–S4†). All basis spectra exhibit two main peaks; therefore, two Gaussian profiles were used to determine the reference parameters of these spectra. As the photocatalytic reaction progressed, the transient signals of the four components gradually changed. These spectra were fit to the sum of four sets of two Gaussian profiles as described by eqn (S1), and ensemble populations were extracted from the integrated area of each Gaussian function. Specific details regarding this protocol are provided in the ESI.† However, it is noted that the fitting algorithm commenced using reference parameters derived from the basis spectra, and various criteria being used to apply the fitting constraints. For instance, considering that the ratio between two Gaussian peaks can be used as a fixed parameter, the initial values for fitting the time-dependent spectra were determined from the fitting results of the previous spectrum. Fitting parameters were also derived from random amplitudes and peak widths within each parameter window, performed over 50 independent fits. The quality of the best fit was verified by comparing the spectra and fits and second derivatives of the spectra (Fig. S8–S10†). The fitting parameters are presented in Tables S2–S4.†Fig. 2d shows the population ensembles as a function of the reaction time under different solvent conditions, i.e., 1 and 50% MeCN (see Fig. S11† for 25% MeCN). Two distinct time regimes are observed. First, the “oxidation dominant” regime exhibits a steep initial rise in the water-rich environment (left plot Fig. 2d) in relation to that observed in the 50% MeCN solution (right plot, Fig. 2d). Second, in the “degradation dominant” regime, the time-dependent variations in the degradation product populations vary significantly between the water-rich and 50% MeCN solutions. This can be accounted for by considering that a water-rich environment may promote lignin degradation by enhancing the photooxidation reaction, which can be inhibited by the presence of MeCN.
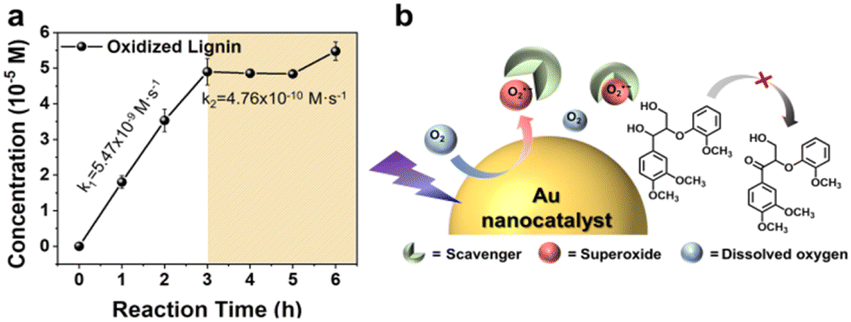
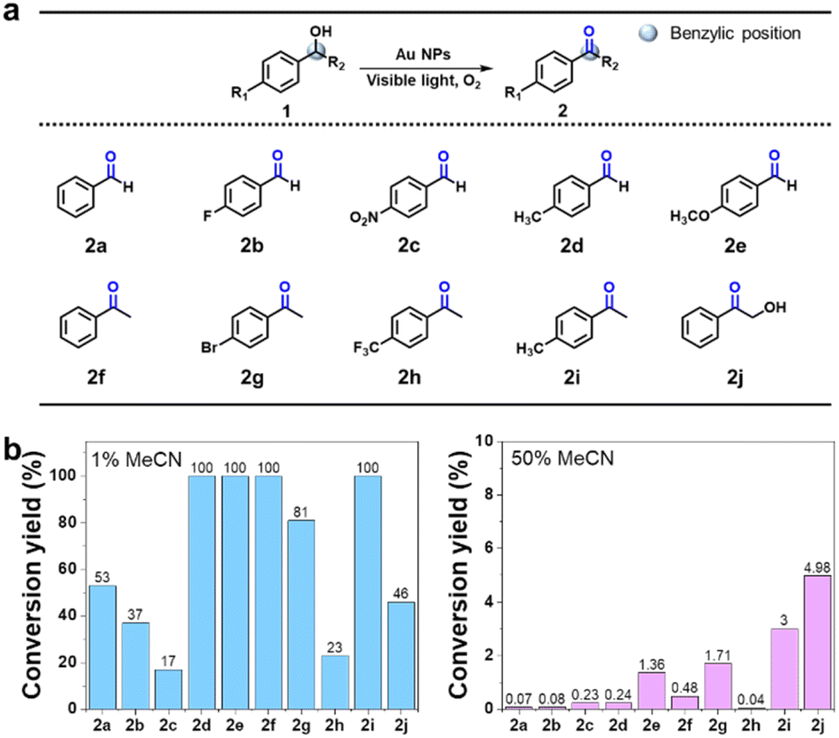
To investigate the photocatalytic reaction mechanism, the increasing levels of the oxidation product were monitored over time from the onset of irradiation. The time-dependent curve representing the oxidation reaction is presented in Fig. 3a, along with the calculated reaction rate. Benzoquinone and hydroquinone were used as scavengers for O2˙− to confirm the generation of OL by these species. Notably, the progression of the photocatalytic reaction led to the gradual formation of OL until the O2˙− scavengers were introduced, at which point the production of OL reached a plateau (Fig. 3a). The initial reaction rate (k1, 5.47 × 10−9 M s−1) decreased upon the addition of the O2˙− scavengers (k2), reaching 4.76 × 10−10 M s−1, which is approximately 10 times lower. A schematic showing the influence of the O2˙− scavengers is given in Fig. 3b. Based on the obtained results, it was proposed that upon visible light excitation, excited electrons from the Au NPs were transferred to the dissolved O2 molecules in the reaction mixture, generating highly reactive O2˙− species. These reactive radicals initially reacted with NL, causing photooxidation. However, the scavengers quickly captured the O2˙− species, thereby hindering the formation of OL. The results of kinetic analyses support this proposed mechanism. Subsequently, a range of simple compounds containing benzylic and aliphatic alcohol moieties (see Fig. 4a and S12†) were evaluated to validate the chemoselective photooxidation reaction. It was found that p-substituted secondary benzylic alcohol compounds underwent oxidation regardless of the presence of electron-donating groups or electron-withdrawing groups as substituents (Fig. 4a and S13–S22†). In contrast, primary alcohols rarely underwent photooxidation (Fig. S23–S26†). Following the photooxidation reaction of benzylic alcohol, a peak corresponding to the CO stretching vibration appeared at ∼1700 cm−1 in the FT-IR spectrum, whereas this peak was not observed for aliphatic alcohols (Fig. S27†). This assessment was in good agreement with the results of lignin photooxidation at the benzylic position, indicating that chemoselective photooxidation had proceeded successfully. This was attributed to the lower dissociation energy of the benzylic C–H bond than those of aliphatic hydrocarbons, leading to facile hydrogen atom abstraction and the formation of relatively stable radical intermediates.34,35 Thus, photooxidation at the benzylic position is favorable. Furthermore, the photooxidation yields were examined relative to the solvent conditions (Fig. 4b). Under water-rich conditions, the conversion yields ranged from 17 to 100% (Fig. 4b, left panel). In contrast, model compounds exposed to MeCN-rich conditions exhibited extremely low conversion yields, typically below a few percent (Fig. 4b, right panel). It was therefore proposed that a higher water content leads to the facile disproportionation of O2˙− with the hydrogen atom at the benzylic position in the β-O-4 linkage (see below eqn (2)), producing the hydroperoxide anion (HO2−). This reactive species promotes deprotonation at the Cα–OH position, generating OL, as detailed below.
| | | AuNPs + hν → eAu− + hAu+ | (1) |
| |  | (3) |
| |  | (4) |
| |  | (5) |
| |  | (6) |
 |
| | Fig. 3 (a) Effect of superoxide ion scavengers on the reaction. (b) Schematic showing of the role of superoxide ion scavengers. | |
 |
| | Fig. 4 (a) Au-catalyzed chemoselective photooxidation of the hydroxyl group at the benzylic position. (b) Chemoselective photooxidation yields determined for the compounds shown in panel (a) under different solvent conditions (left: water-rich, right: MeCN-rich). | |
Conversely, under relatively high MeCN conditions, the disproportionation of O2˙− is suppressed. As a result, hydrogen atom abstraction occurs infrequently, thereby limiting oxidation events.
To establish a structural and energetic basis for interpreting the photooxidation reaction results, quantum mechanical calculations were performed using the Hartree–Fock (HF) and density functional theory (DFT) methods at the HF/6-311+G**, B3LYP/6-311+G**, and B3LYP/6-311G** levels of the Gaussian 16 package.36 A total of 36 different conformational isomers were considered as the initial guesses for the NL and OL species to determine the optimal geometry of the β-O-4-type lignin (Fig. S28 and S29†). The relative single-point energy of a conformational isomer, ΔE, is defined as the energy difference between the isomer (Ei) and the globally optimized “minimum” conformation (Emin; Fig. S30, Tables S5 and S6†). As these calculations are performed for molecules in the absence of a solvent, the resulting optimal geometries may not be realistic. Thus, to address this issue, energies (Ei) in isolated and aqueous environments were refined using the self-consistent reaction field (SCRF) method.37 The geometries of the five key conformations were denoted as “minimum” NL and OL, “flipped” NL and OL, and “twisted” NL, as presented in Fig. S27 and S28.† Due to the energetic similarities between the two conformers, the optimal structure of OL can adopt both the “minimum” and “flipped” conformations (see the Computational details section and Fig. S30 in the ESI†).
Based on the agreement between the HF and DFT methods shown in Fig. S31,† calculations at the B3LYP/6-311G** level of theory were performed to investigate the electrostatic potential and electron density distributions. It was found that, the molecular electrostatic potentials (MEPs) of NL and OL revealed negative and positive electrostatic potentials localized on the oxygen and hydrogen atoms, respectively, because these are neutral compounds (Fig. S32†). The MEP map of NL is consistent with the previously reported DFT results for a similar lignin compound.38 It was deduced that the solvation effect increased the overall polarities of NL and OL, suggesting an enhanced ability for both hydrogen bond donation and acceptance. In addition, the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and HOMO–LUMO gaps were calculated to elucidate the chemical stabilities of NL and OL under different solvation states (Fig. S33, S34 and Table S7†). Interestingly, in both the “minimum” and “flipped” forms, the electron densities transferred from one aromatic ring to another when transitioning from the HOMO to LUMO states. Furthermore, solvation induced a red shift in the OL band gap, whereas NL exhibited a blue shift, as shown in Table S7.†
The cleavage of lignin can be initiated at the Cα–Cβ bond or Cβ–O bond.39–44 Thus, to elucidate the mechanism of selective Cα–Cβ bond cleavage, DFT calculations were performed to determine the energetic gains. As shown in Table S8,† the high BDEs of the Cα–Cβ and Cβ–O linkages (i.e., 50–90 kcal mol−1) typically present a challenge in initiating cleavage.11,45 In the current strategy, the creation of oxygen radicals activates the O–H bond, which promotes Cα–Cβ linkage cleavage. As shown in Fig. 5a, the BDEs of the Cα–H and O–H bonds in neutral NL are relatively high, reaching 83.0 and 91.8 kcal mol−1, respectively.
 |
| | Fig. 5 (a) Bond dissociation energies (BDEs) of the Cα–H and O–H bonds. (b) Orientations of the Cα–O bonds in the planar and non-planar directions. (c) Stabilization energies of radicalized lignin with different structures and orientations. The solvation environments are indicated by different colors. (d) BDEs of the Cα–Cβ bonds across varying configurations. (e) Oxidation–degradation kinetics diagram for β-O-4-type lignin, based on kinetic and structural analyses, showing the molecular electrostatic potential (MEP) map (left), Cα–H and O–H cleavage sites, and stable product conformations in the “oxidation dominant” and “degradation dominant” regimes. | |
Interestingly, the oxygen radical can stabilize the radicalization of lignin when the Cα–O bond is oriented in a planar direction, as shown in Fig. 5b and c. These observations indicate that stabilization could be achieved through conformational preferences. Moreover, lignin with a radical character can effectively benefit from energetic advantages of 4–7 kcal mol−1 (Fig. 5d and Table 1). Thus, based on the stabilization effect, the mechanistic features of the time-dependent photooxidation reaction can be described. Consistent with the UV-vis population analysis, two time-dependent regimes were observed, namely, the “oxidation dominant” regime (early stage, wherein the “minimum” and “flipped” conformational isomers of OL remain interchangeable, with a low energy difference of 0.4 kcal mol−1 between the two isomers), and the “degradation dominant” regime (which indicates energetic advantages for the Cα–Cβ linkage, particularly under aqueous conditions).
Table 1 Stabilization energies of radicalization and Cα–Cβ cleavage for the conformational isomers of lignin at the B3LYP/6-311G** level of theory. The minimum, flipped, and twisted conformations are indicated by subscripts “min,” “flip,” and “twist” labels, respectively. The solvation environment is also indicated
| Radicalization (kcal mol−1) |
Water |
Acetonitrile |
| ΔERadical,min (planar) |
7.5 |
9.2 |
| ΔERadical,flip (planar) |
6.2 |
7.1 |
| ΔERadical,min (non-planar) |
72.4 |
74.2 |
| ΔERadical,flip (non-planar) |
75.5 |
76.4 |
| ΔERadical,twist (non-planar) |
70.0 |
71.0 |
| ΔEC–C (kcal mol−1) |
Water |
Acetonitrile |
| ΔEC–C,min |
−6.7 |
−5.4 |
| ΔEC–C,flip |
−5.8 |
−4.5 |
Conclusions
In conclusion, the photooxidation and degradation of lignin model compounds was demonstrated using a plasmonic Au nanocatalyst in an aqueous environment. Based on the time-dependent spectral changes and structural data, a stepwise mechanism was proposed for the photo-triggered reactions, involving electron transfer, hydrogen abstraction, and degradation. More specifically, electrons in the Au NPs are excited by visible light, and O2˙− is generated through the single-electron transfer process. In a water-rich environment, these reactive oxygen species abstract a hydrogen atom from the benzylic carbon of the NL model compound to form a ketone. The photooxidized state of lignin is stabilized in bulk water by an equilibrium between the “minimum” and “flipped” conformations. Once photooxidation occurs at the benzylic carbon, the Cα–Cβ and C–O bonds in the NL lignin become unstable, leading to their cleavage and the formation of two distinct subunits. This photoreaction system is unique because it employs a single-component catalyst in an aqueous medium, distinguishing it from previous studies that utilize organic-based catalysts in organic media.46–52 Moreover, the chemoselective photooxidation of the benzylic hydroxyl group was achieved in various compounds, contrasting with existing studies that utilized multicomponent heterogeneous catalysts or high temperatures.53–55 Notably, this photooxidation system opens up new possibilities owing to the use of a single-component catalyst without toxic chemicals or harsh reaction conditions. Overall, these findings provide molecular insights into plasmon-driven chemoselective photooxidation in aqueous environments, emphasizing the significance of understanding the molecular basis of selective oxidization. It is anticipated that these findings will shed light on the impact of the photooxidative reaction solvent on the structure, dynamics, and stability of biomass.
Lignin, a biomass-derived material abundantly available from sources such as wood and agricultural residues, has attracted increasing attention as a promising renewable feedstock for the sustainable chemical industry. To enable its practical utilization, it is essential to address key factors such as process stability and reproducibility, along with the reusability and long-term durability of catalytic systems. With continued advancements in these areas, lignin degradation technologies hold significant potential for industrial-scale applications in biorefineries, green chemical synthesis, and energy-related conversion processes.
Data availability
All data can be obtained from the corresponding authors upon request.
Author contributions
Juhee Ha: writing – original draft, formal analysis, data curation. Jiwon Kang: writing – original draft, formal analysis, data curation. Suk Hyun Lim: data curation. Dae Won Cho: visualization, data curation. Kwang-Im Oh: writing – review & editing, supervision, resources, formal analysis. Youngsoo Kim: writing – review & editing, supervision, resources, formal analysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for financial support from the National Research Foundation of Korea (NRF-2022R1A2C1010042, NRF-2022R1A4A5034331, NR-2022-NR072441, and RS-2025-00573677). The authors also thank the Core Research Support Center for Natural Products and Medical Materials (CRCNM) for the technical support related to the TEM measurements. Calculations were performed at the KISTI Supercomputing Center.
Notes and references
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- N. Srisasiwimon, S. Chuangchote, N. Laosiripojana and T. Sagawa, ACS Sustainable Chem. Eng., 2018, 6, 13968–13976 CrossRef CAS.
- E. Subbotina, T. Rukkijakan, M. D. Marquez-Medina, X. Yu, M. Johnsson and J. S. M. Samec, Nat. Chem., 2021, 13, 1118–1125 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- S. Dabral, H. Wotruba, J. G. Hernández and C. Bolm, ACS Sustainable Chem. Eng., 2018, 6, 3242–3254 CrossRef CAS.
- J. Chen, W. Liu, Z. Song, H. Wang and Y. Xie, Bioenergy Res., 2018, 11, 166–173 CrossRef CAS.
- M. Ko, L. T. M. Pham, Y. J. Sa, J. Woo, T. V. T. Nguyen, J. H. Kim, D. Oh, P. Sharma, J. Ryu, T. J. Shin, S. H. Joo, Y. H. Kim and J. W. Jang, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- Y. Cao, N. Wang, X. He, H. R. Li and L. N. He, ACS Sustainable Chem. Eng., 2018, 6, 15032–15039 CrossRef CAS.
- S. Li, Z. J. Li, H. Yu, M. R. Sytu, Y. Wang, D. Beeri, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Energy Lett., 2020, 5, 777–784 CrossRef CAS.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- S. H. Lim, H. Jang, M. J. Kim, K. R. Wee, D. H. Lim, Y. I. Kim and D. W. Cho, J. Org. Chem., 2022, 87, 2289–2300 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. Luo, X. Zhang, J. Lu and J. Zhang, ACS Catal., 2017, 7, 5062–5070 CrossRef CAS.
- Z. Xiang, W. Han, J. Deng, W. Zhu, Y. Zhang and H. Wang, ChemSusChem, 2020, 13, 4199–4213 CrossRef CAS PubMed.
- S. Gazi, W. K. Hung Ng, R. Ganguly, A. M. Putra Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- N. Luo, M. Wang, H. Li, J. Zhang, H. Liu and F. Wang, ACS Catal., 2016, 6, 7716–7721 CrossRef CAS.
- Y. Liu, C. Li, W. Miao, W. Tang, D. Xue, C. Li, B. Zhang, J. Xiao, A. Wang, T. Zhang and C. Wang, ACS Catal., 2019, 9, 4441–4447 CrossRef CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, B. Lassiter, E. A. Carter, P. Nordlander, N. J. Halas, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- Y. Kim, D. Dumett Torres and P. K. Jain, Nano Lett., 2016, 16, 3399–3407 CrossRef CAS PubMed.
- Y. Kim, A. J. Wilson and P. K. Jain, ACS Catal., 2017, 7, 4360–4365 CrossRef CAS.
- Y. Kim, J. G. Smith and P. K. Jain, Nat. Chem., 2018, 10, 763–769 CrossRef CAS PubMed.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- J. Kim, J. Lee, H. Choi, J. Ha, M. Cheon, Y. Seo, Y. Kim and D. Yoo, Nanoscale, 2023, 15, 15950–15955 RSC.
- J. Lee, P. B. Joshi, A. J. Wilson and Y. Kim, J. Phys. Chem. C, 2023, 127, 8096–8103 CrossRef CAS.
- Y. Zhang, W. Guo, Y. Zhang and W. D. Wei, Adv. Mater., 2021, 33, 1–16 Search PubMed.
- E. Contreras, R. Nixon, C. Litts, W. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS PubMed.
- H. Wakefield IV, Q. Jiang and R. S. Klausen, Org. Biomol. Chem., 2022, 20, 1407–1414 RSC.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 720–723 CrossRef.
- Y. Zhang, H. He, K. Dong, M. Fan and S. Zhang, RSC Adv., 2017, 7, 12670–12681 RSC.
- L. Jiang, G. Xu and Y. Fu, ACS Catal., 2022, 12, 9473–9485 CrossRef CAS.
- M.-W. Zheng, Y. Lang, X. Han, L. Jia, H. Zeng and C.-J. Li, Cell Rep. Phys. Sci., 2024, 102009, DOI:10.1016/j.xcrp.2024.102009.
- Y. Kang, X. Yao, Y. Yang, J. Xu, J. Xin, Q. Zhou, M. Li, X. Lu and S. Zhang, Green Chem., 2021, 23, 5524–5534 RSC.
- Y. Li, J. Wen, S. Wu, S. Luo, C. Ma, S. Li, Z. Chen, S. Liu and B. Tian, Org. Lett., 2024, 26, 1218–1223 CrossRef CAS PubMed.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- H. Liu, H. Li, N. Luo and F. Wang, ACS Catal., 2020, 10, 632–643 CrossRef CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- D. W. Cho, R. Parthasarathi, A. S. Pimentel, G. D. Maestas, H. J. Park, U. C. Yoon, D. Dunaway-Mariano, S. Gnanakaran, P. Langan and P. S. Mariano, J. Org. Chem., 2010, 75, 6549–6562 CrossRef CAS PubMed.
- R. DiCosimo and H. C. Szabo, J. Org. Chem., 1988, 53, 1673–1679 CrossRef CAS.
- S. H. Lim, W. S. Lee, Y. I. Kim, Y. Sohn, D. W. Cho, C. Kim, E. Kim, J. A. Latham, D. Dunaway-Mariano and P. S. Mariano, Tetrahedron, 2015, 71, 4236–4247 CrossRef CAS.
- D. W. Cho, J. A. Latham, H. J. Park, U. C. Yoon, P. Langan, D. Dunaway-Mariano and P. S. Mariano, J. Org. Chem., 2011, 76, 2840–2852 CrossRef CAS PubMed.
- P. J. Harvey, H. E. Schoemaker, R. M. Bowen and J. M. Palmer, FEBS Lett., 1985, 183, 13–16 CrossRef CAS.
- S. H. Lim, K. Nahm, C. S. Ra, D. W. Cho, U. C. Yoon, J. A. Latham, D. Dunaway-Mariano and P. S. Mariano, J. Org. Chem., 2013, 78, 9431–9443 CrossRef CAS PubMed.
- V. B. Huynh, Biochem. Biophys. Res. Commun., 1986, 139, 1104–1110 CrossRef CAS PubMed.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- Y. Liu, P. Zhang, B. Tian and J. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 13849–13858 CrossRef CAS PubMed.
- J. L. Dimeglio, A. G. Breuhaus-Alvarez, S. Li and B. M. Bartlett, ACS Catal., 2019, 9, 5732–5741 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jiwon
Kang‡
a,
Suk Hyun
Lim
a,
Dae Won
Cho
a,
Jiwon
Kang‡
a,
Suk Hyun
Lim
a,
Dae Won
Cho