
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combined soft X-ray and theoretical investigation discloses the water harvesting behaviour of Mg-MOF-74 at the crystal surface†
Francesco
Tavani‡
 a,
Alessandro
Tofoni‡
a,
Marco
Vandone
b,
Matteo
Busato
a,
Luca
Braglia
cd,
Piero
Torelli
cd,
Maximillian G.
Stanzione
e,
Anthony R.
Armstrong
e,
Russell E.
Morris
e,
Valentina
Colombo
*b and
Paola
D'Angelo
*a
a,
Alessandro
Tofoni‡
a,
Marco
Vandone
b,
Matteo
Busato
a,
Luca
Braglia
cd,
Piero
Torelli
cd,
Maximillian G.
Stanzione
e,
Anthony R.
Armstrong
e,
Russell E.
Morris
e,
Valentina
Colombo
*b and
Paola
D'Angelo
*a
aDipartimento di Chimica, Università degli Studi di Roma La Sapienza, P.le A. Moro 5, I-00185 Rome, Italy. E-mail: p.dangelo@uniroma1.it
bDipartimento di Chimica & UdR INSTM di Milano, Università degli Studi di Milano, Via Golgi 19, 20133 Milan, Italy. E-mail: valentina.colombo@unimi.it
cCNR-Istituto Officina dei Materiali, TASC, 34149 Trieste, Italy
dAREA Science Park, Padriciano 99, I-34149 Trieste, Italy
eSchool of Chemistry, University of St. Andrews, North Haugh, St. Andrews KY16 9ST, UK
First published on 22nd April 2025
Abstract
Metal–organic frameworks (MOFs) are receiving growing interest as transformative materials for real-world atmospheric water harvesting applications. However, obtaining molecular-level details on how surface effects regulate MOF water uptake has proven to be elusive. Here, we present a novel methodology based on ambient pressure soft X-ray absorption spectroscopy (AP-NEXAFS), machine learning-assisted theoretical spectroscopy and molecular dynamics simulations to gain selective insights into the behaviour of water at a MOF crystal surface. We applied our interdisciplinary method to investigate the structural and dynamical properties of water at the surface of the Mg-MOF-74 system, while obtaining complementary information on the water uptake and release from the bulk by synchrotron powder X-ray diffraction. Our investigation pointed out the simultaneous presence of Mg open sites and residual gas-phase water during dehydration, and proved that during water release a high number of surface Mg sites still interact with one or two water molecules. Conversely, when looking at the bulk, a significantly lower number of Mg sites have been found to interact with water molecules in the same experimental conditions. This behaviour suggests that the water adsorption (desorption) process starts from the interior of the material and propagates towards the channel openings. The combined approach based on AP-NEXAFS, PXRD experimental determinations and ML-supported theoretical analyses has been found to be a valuable tool to provide a thorough description of the water harvesting process at both surface and bulk of the crystal.
Introduction
At present, many regions of the world experience water stress due to climate change and society is expected to face growing issues related to water scarcity in the near future.1–5 The limited availability of ground fresh water resources and the energy-intensive infrastructure required to convert seawater into drinking water have motivated significant research efforts to develop materials that may collect water directly from the earth atmosphere, which is estimated to contain about 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 trillion liters of water.6
000 trillion liters of water.6
Metal–organic frameworks (MOFs), a class of porous and crystalline solids composed of metal centers coordinated to organic linkers, have recently emerged as promising candidates for this application owing to their high porosity, unrivaled tunability and suitable water affinity.8–15 MOFs have been proven able to extract atmospheric water in the desert12,16–18 with water yield depending on the kinetics of water uptake and release from the pores,19,20 and this discovery has sparked further interest in understanding what the water capture mechanisms are. Rationalizing how MOFs capture water from the air at the molecular level is in fact crucial to designing more efficient MOF water harvesting systems.21,22 Notably, a recent study8 reported the water harvesting mechanism of MOF-303 ([Al(OH)(PZDC)], where PZDC4− = 1-H-pyrazole-3,5-dicarboxylate) by employing an extensive series of single-crystal X-ray diffraction measurements to locate the water adsorption sites in the MOF with coordinatively saturated metal sites. Such knowledge enabled the multivariate modification of the MOF pores which led to higher water productivity and tunable regeneration temperatures. Previous investigations on how MOFs with open metal sites capture water have evidenced that hydration at low relative pressures results in occupation of the metal sites by water, which is then followed by water clustering into the pores as the relative humidity (RH) increases.11,23–26
Although it is becoming clear that the MOF crystal surface plays a key role in initiating and regulating water uptake into the pores, molecular-level details on how water clustering occurs are still missing. In fact, unveiling the mechanism of how MOF water-binding sites are populated has proven to be experimentally challenging,8 and although bulk probes such as neutron or X-ray diffraction techniques8,20,23,27,28 and diffuse reflectance infrared Fourier transform spectroscopy24,29,30 have been used to investigate MOF water uptake, studies using surface-sensitive techniques have remained very limited.21 Moreover, molecular dynamics (MD) simulations of interfacial processes require comparison with experimental data to propose valid molecular models.21,24,31–33
Soft X-ray absorption spectroscopy (soft-XAS) is at the forefront of surface chemistry studies, offering simultaneous insights into the local structural and electronic environment of a selected photoabsorber with unrivaled sensitivity and specificity.34,35 Appropriately designed reactor cells36 have very recently allowed soft-XAS operando experiments to be performed at atmospheric pressure, a technique referred to as ambient pressure near-edge X-ray fine structure spectroscopy (AP-NEXAFS). Here, soft-XAS is operated in total electron yield (TEY) detection mode which limits the thickness of the probed sample to about 4 nm due to the low electron escape depth.36–39 In this work, for the first time, the AP-NEXAFS technique has been used to gain information on the water harvesting mechanism occurring at the surface of a MOF crystal. The AP-NEXAFS spectroscopic results have been complemented with machine learning (ML)-assisted theoretical calculations, powder X-ray diffraction (PXRD) and MD simulations to provide a unified picture of the water harvesting process. We applied this method to Mg-MOF-74, for which a thorough description of the water harvesting mechanism involving the whole material has been already carried out,40 therefore making this MOF an ideal candidate for a comparison between surface and bulk properties. Moreover, previous experimental and theoretical investigations at the Mg K-edge have been successfully carried out with the AP-NEXAFS technique providing a solid base for the present study.34,39
Results
Synthesis and characterization of Mg-MOF-74
Mg-MOF-74 was synthesized in gram quantities following the previously reported protocol. The thermogravimetric analysis (TGA) and N2 and water sorption profiles (Fig. S1–S3,† respectively) are in agreement with those reported in the literature. Full details on the synthesis and characterization of Mg-MOF-74 are provided in Section 1 of the ESI.†Thermal dehydration of Mg-MOF-74
Fig. 1 shows the crystallographic structure of Mg-MOF-74 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group). This MOF is built from secondary building units (SBUs) joined by H4DOBDC linkers (DOBDC4− = 2,5-dioxido-1,4-benzenedicarboxylate) forming approximately 11 Å wide hexagonal channels41 that run along the crystallographic c-axis.6,42 In each SBU, terminal water molecules are directly bound to the Mg centers which overall result coordinated by the DOBDC linkers in a slightly distorted octahedral geometry (Fig. 1c).
space group). This MOF is built from secondary building units (SBUs) joined by H4DOBDC linkers (DOBDC4− = 2,5-dioxido-1,4-benzenedicarboxylate) forming approximately 11 Å wide hexagonal channels41 that run along the crystallographic c-axis.6,42 In each SBU, terminal water molecules are directly bound to the Mg centers which overall result coordinated by the DOBDC linkers in a slightly distorted octahedral geometry (Fig. 1c).
It is known that Mg-MOF-74 may adsorb water at low relative pressures.43–46Fig. 1d shows the structure of the hydrated Mg-MOF-74 material at 25 °C,7 and highlights the presence of different water adsorption sites in the framework. Five nonequivalent water molecules can be identified in the displayed structural model: the water molecules coordinating the open Mg sites (OW1), those interacting with the Mg-bound water molecules through hydrogen-bonds (OW2 and OW3), those found further away from the framework (OW4), and finally those clustering near the center of the channels (OW5).6 Here, we investigated the Mg-MOF-74 water uptake process and the influence of water desorption on the MOF bulk properties by using synchrotron PXRD.8 In particular, we heated a methanol-exchanged Mg-MOF-74 sample in situ up to 150 °C in a pure He flux (12 standard cubic centimeters per minute (SCCM)) to remove adsorbed impurities. We subsequently cooled the sample down to room temperature (RT) and exposed it to a wet He flux saturated with water in order to fully hydrate the MOF43 (refer to the ESI† for additional details on the hydration protocol). Fig. 1e shows the measured PXRD pattern of the fully hydrated Mg-MOF-74 material and a Le Bail refinement confirms the phase purity of the sample. We then subjected the hydrated Mg-MOF-74 sample to gradual temperature increments from 25 °C to 145 °C in a pure He flux. In situ high-resolution PXRD patterns were collected every 10 °C allowing us to monitor the structural changes occurring upon the release of water from the MOF porous channels (Fig. 2a).8,23 A smooth shift of the peak positions towards lower 2θ values and a gradual reduction of the diffracted intensities are detected in the patterns as a function of temperature due to water release. The in situ PXRD data were analyzed through sequential structureless Le Bail refinements47 and the unit-cell parameters were found to vary during the sample thermal treatment in close connection to the water content evolution, as shown in Fig. 2c and listed in Table S1.† Note that the Mg-MOF-74 sample presents good crystallinity during the thermal treatment up to 145 °C in the essayed experimental conditions.7,23 Between 25 and 100 °C the a and c parameters decrease and increase, respectively, when the amount of water present in the MOF diminishes (the MOF unit-cell change is displayed in Fig. 2b), in agreement with previous investigations.23,40 In this temperature range the percentage variation of the unit-cell parameters is small and amounts to less than ∼1% of their initial values in the hydrated MOF crystal structure. The contraction along the a-axis, which corresponds to a shrinkage of the porous channel radius, may be rationalized as due to a small rearrangement of the MOF framework that minimizes the empty space during water release, and in turn leads to a small elongation of the channels along the c-axis. At temperatures above 100 °C the a and c parameters slightly increase and decrease, respectively (see Fig. 2c), and this effect can be attributed to the progressive departure of the Mg-bound OW1 water from the framework. Note that the observed temperature evolution of the unit-cell parameters nicely matches that found by Rosnes and coworkers40 and it is reasonable to assume that the site occupancies of the individual water molecules evolve as described in this previous work.40 In particular, it has been reported that the OW5 and OW4 water molecules are the first to leave the MOF, followed by the OW3 and OW2 ones.40 At 77 °C most of the water molecules, apart from the Mg-bound OW1 ones, are removed from the MOF framework while above 77 °C the OW1 water content gradually decreases and a small amount of residual water, which may be estimated to be about ∼4% of the initial water content, remains still confined in the MOF between 175 and 200 °C.40 Further, we have collected the TGA traces in the temperature range between 25 and 350 °C on the as prepared and hydrated sample (Fig. S1†) and the desorption of water molecules can be clearly observed as a mass loss at about 100 °C.
 | ||
| Fig. 1 (a) Crystal structure of Mg-MOF-74 viewed down the c-axis, together with the approximate pore channel diameter calculated according to the van der Waals radii of the framework atoms. (b) Side view visualizing the MOF structure along the c-axis. (c) Depiction of the local environment of the Mg sites in Mg-MOF-74, where a single water molecule is directly coordinated to the open Mg site. Atom color code: magnesium (orange), framework oxygen (red), water oxygen (cyan), carbon (gray), hydrogen (white). (d) Crystal structure of hydrated Mg-MOF-74,7 where the five nonequivalent water molecules are highlighted using the color code shown in the inset. (e) Whole powder pattern Le Bail analysis of the synchrotron PXRD pattern collected on the fully hydrated Mg-MOF-74 sample at 25 °C. Experimental, calculated, background and difference traces are depicted in black, red, blue and gray, respectively. The positions of the Bragg reflections are indicated with blue tick marks and the weighted profile residual figure of merit is reported in the plot. | ||
 | ||
| Fig. 2 PXRD monitoring of the Mg-MOF-74 thermal dehydration process. (a) Evolution of the experimental Mg-MOF-74 PXRD patterns (λ = 0.31916 Å) collected during thermal heating from 25 °C (blue) to 145 °C (dark red) in pure He flow (12 SCCM at 1 atm). The inset shows a magnification of the 9.0–15.0 2θ region. (b) Scheme of the Mg-MOF-74 unit-cell highlighting the expansion and contraction along the c- and a-axes, respectively. (c) Temperature evolution of the Mg-MOF-74 unit-cell parameters as determined by the sequential Le Bail refinements (the red and blue dotted lines refer to the a-axis and c-axis, respectively, as indicated by the colored arrows). | ||
To obtain insights into the local structural and electronic properties of the water/MOF interface at the crystal surface, in the second step of our investigation we used the AP-NEXAFS technique to monitor the thermal dehydration of Mg-MOF-74.34,35,39 In particular, we collected AP-NEXAFS spectra at both the Mg and O K-edges on the fully hydrated and dehydrated sample at temperatures between 45 and 200 °C (refer to the ESI† for additional details). Fig. 3a compares the Mg K-edge AP-NEXAFS spectra of the fully hydrated Mg-MOF-74 measured at 45 °C with that of the dehydrated sample collected at 200 °C. A pre-edge peak appears at ∼1306.6 eV when the sample is dehydrated and this is due to a transition allowed by the breaking of the local symmetry around the metal site that induces a strong mixing of the 3s and 3p states. The appearance of the pre-edge structure is associated with the formation of a five-coordinated asymmetric coordination around the Mg metal that goes from an octahedral to a square pyramidal geometry as a consequence of the removal of the water molecule from the apical position.48
 | ||
| Fig. 3 (a) Experimental Mg K-edge AP-NEXAFS spectra of hydrated Mg-MOF-74 collected at 45 °C under wet He flux (blue curve) and of the dehydrated MOF collected at 200 °C under pure He flux (red curve). (b) Theoretical Mg K-edge NEXAFS spectra. The blue curve is calculated from MD simulations of Mg-MOF-74 at a water loading of N = 90 per unit-cell, while the crimson curve is obtained as the NN-weighted average of the NEXAFS theoretical spectrum of the dehydrated MOF and of the NEXAFS spectra calculated from MD simulations for water loadings of N = 18 and N = 36 per unit-cell. (c) Experimental O K-edge AP-NEXAFS spectra collected upon thermal dehydration of Mg-MOF-74 under flowing He (50 SCCM) in the temperature interval between 45 and 200 °C. From top to bottom, the AP-NEXAFS spectra shown in gray were measured at the temperatures of 61, 72 and 88 °C, respectively. | ||
Moreover, inspection of Fig. 3a reveals a red-shift of the main absorption edge onset due to the release of the water molecules. Further, the spectral intensity in the 1314–1317 eV energy range is higher in the spectrum of hydrated Mg-MOF-74 as compared to the dehydrated one. This behaviour has been already observed when the CO2 molecule is desorbed from the Mg-MOF-74 giving rise to an open metal site.48The evolution of the O K-edge AP-NEXAFS spectra recorded during the Mg-MOF-74 thermal dehydration is presented in Fig. 3c. One may clearly observe that in the O K-edge AP-NEXAFS spectrum collected at 45 °C, besides the transitions at 531.0 and 532.3 eV due to the MOF framework, there is the spectral signature of gas-phase water. In particular, there are well separated peaks at 534.0 eV and 536.0 eV which are due to O 1s transitions into the antibonding O–H 4a1 and 2b2 water molecular orbitals, respectively, together with excitations into the water Rydberg orbitals at 537.2, 538.0 and 538.7 eV.49,50 When the MOF is hydrated, the intensities of these transitions are relatively high as they reflect the elevated abundance of gas-phase water molecules confined in the 1-D channels. In a previous work on Zn-MOF-74 it was already suggested that the water molecules inside the 1-D channels had the same IR features as free water molecules in vacuum,51 nevertheless it cannot be excluded that some gaseous water molecules that are not interacting with the MOF framework are detected during the experiment. As temperature is progressively increased leading to water release from the MOF, the intensities of the peaks associated to gas-phase water decrease, finally becoming very weak in the O K-edge AP-NEXAFS spectrum collected at 200 °C (Fig. 3c). The AP-NEXAFS results indicate that after thermal dehydration up to 200 °C, coordinatively unsaturated Mg sites are formed and that most but not all gas-phase water is removed from the Mg-MOF-74 channels.
Computational analysis of water clustering in Mg-MOF-74
In order to obtain molecular-level understanding of the structural and dynamical properties of water clusters in Mg-MOF-74 we performed a computational analysis of the MOF water uptake by means of MD and state-of-the-art ab initio NEXAFS simulations. In particular, the origin and evolution of the Mg K-edge AP-NEXAFS spectral features as a function of increasing water content were investigated by classical MD simulations using the TIP3P water model together with a force field derived from Density Functional Theory (DFT), a combination that has been recently shown to accurately predict the experimental water adsorption isotherms of Mg-MOF-74 (ref. 44) (see the ESI† for details). MD simulations were performed at loadings of N = 18, 36, 54, 72 and 90, where N is the number of water molecules per unit-cell. An octahedral geometry with coordination number (CN) of 1 and a Mg–Owater bond length in the 2.32–2.37 Å range44 were obtained for the Mg first coordination shell, as evidenced by the Mg–Owater radial distribution functions and corresponding running integration numbers shown for each water loading in Fig. S6.†Starting from the microscopic description of the water/MOF systems retrieved from the MD simulations a quantitative analysis of the Mg K-edge AP-NEXAFS spectra was carried out.34,39,52 To this end, converged average Mg K-edge NEXAFS theoretical spectra have been calculated for each water loading increment starting from the configurations provided by 100 MD snapshots. The resulting theoretical NEXAFS spectra calculated for N = 18, 36, 54 and 72 are shown in Fig. S7a, and c–e,† respectively, while Fig. 4a presents the NEXAFS calculations for N = 90. One may note that in all cases there are appreciable differences in the NEXAFS spectra evaluated for each MD snapshot, evidencing the sensitivity of NEXAFS to subtle variations in the local structure of the Mg framework sites.34,39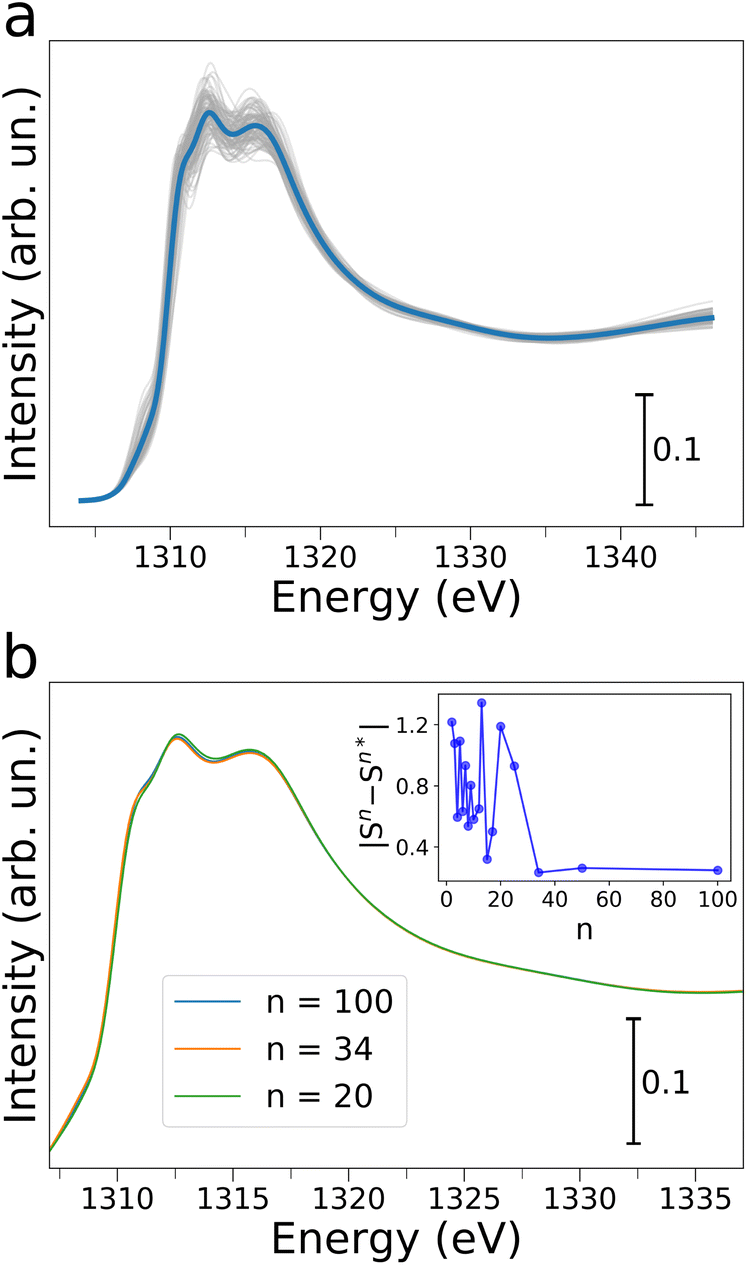 | ||
| Fig. 4 (a) Theoretical Mg K-edge NEXAFS spectra (gray full lines) calculated from 100 MD snapshots of Mg-MOF-74 loaded with 90 water molecules per unit-cell and converged NEXAFS average (light blue) of the 100 simulated curves. (b) Selection of average Mg K-edge NEXAFS theoretical spectra calculated with an increasing number (n) of spectra. The inset displays the total differences between averages of spectra computed with increasing n values. Sn and Sn* are the theoretical averages of n and n* spectra, respectively, with n* immediately preceding the given value of n in the considered sequence. | ||
Fig. 4b shows the Mg K-edge NEXAFS theoretical averages for N = 90 calculated with an increasing number (n) of spectra. One may observe that for n ≥ 34 the theoretical average NEXAFS spectra exhibit very small differences and that they are well-converged (see inset of Fig. 4b). The same observations hold for the theoretical NEXAFS average spectra simulated for N = 18, 36, 54 and 72 (see Fig. S7b and d–f,† respectively), which have all reached convergence for n = 100. We then focused on understanding the water abundance at the MOF surface upon the MOF thermal dehydration. To this end, we employed a supervised ML method, namely an artificial neural network (NN),53–57 to establish relationships between the Mg K-edge NEXAFS experimental spectral features measured at 200 °C during thermal treatment and those of the theoretical NEXAFS spectra calculated at increasing water loading. In particular, we constructed a training set by mixing the theoretical NEXAFS spectra simulated for the dehydrated MOF which exhibits a well-defined pre-edge transition due to the presence of open Mg sites (see Fig. S8†), together with the theoretical NEXAFS spectra calculated from the MD simulations for N = 18 and for N = 36. This spectral mixing procedure was performed using known, appropriate weights wN=0, wN=18, wN=36. An artificial NN was then defined as a nonlinear function f(μi, θ) → {wN=j} (where j = 0, 18, 36) that receives as input a preprocessed and discretized Mg K-edge NEXAFS spectrum and returns the corresponding wN=j value, i.e. the target of the regression algorithm.53,56,57 Three distinct NNs were trained for each weights wN=j, all providing negligible mean squared errors during both training and validation, as shown in Fig. S9.† We then applied the well-trained NNs on the experimental Mg K-edge spectrum measured during the thermal dehydration at 200 °C to estimate the water content at the same temperature by extracting the wN=0, wN=18 and wN=36 values using the regression algorithm. In particular, this procedure yielded wN=0, wN=18 and wN=36 fractional values equal to 0.20(1), 0.58(1) and 0.20(1), respectively, suggesting that at 200 °C in our experimental conditions ∼20% of the MOF surface Mg sites are coordinatively unsaturated, ∼60% are bound to a single water molecule while in ∼20% an additional water molecule interacts with the Mg-bound one. We then employed these NN-extracted values as weights to obtain a theoretical Mg K-edge NEXAFS spectrum capturing the structural and electronic properties of Mg-MOF-74 during the dehydration experiment. The resulting NN-weighted theoretical Mg K-edge NEXAFS spectrum (Fig. 3b, crimson curve) exhibits a pre-edge peak due to the inclusion of open Mg sites in the calculations, as well as a red-shifted main absorption edge and a less intense shoulder in the 1314–1317 eV energy range if compared to those of the theoretical NEXAFS spectrum calculated for N = 90 (Fig. 3b, blue curve). As one may note by comparing Fig. 3a and b there is an excellent agreement between the experimental and theoretical Mg K-edge NEXAFS spectra indicating the reliability of our NN-assisted computational method. Indeed, given the experimental sensitivity of the NEXAFS technique to probing several nanometers below the topmost MOF atomic plane, the detailed structural description of the MOF water adsorption provided by the MD simulations at different hydration levels allowed us to pinpoint the MOF surface composition during real-time conditions. Note that during thermal dehydration in our experimental conditions we highlighted the presence of a higher amount of Mg sites interacting with one or two water molecules and a lower number of open Mg sites at the crystal surface of Mg-MOF-74 if compared to the abundancies of the same Mg sites in the MOF bulk.40 These observations may be attributed both to differences between the MOF surface and bulk behaviour, as well as to significant differences in the experimental procedures we have employed if compared to those of previous work,40 such as differences in sample preparation, activation and heating rate. Further insights into the nature of the interactions established between the water molecules and the MOF framework are gained by dissecting the contribution of increasing water content to the theoretical Mg K-edge NEXAFS spectra. Fig. 5a displays the evolution of MD snapshots of Mg-MOF-74 loaded with N = 18, 36, 54, 72 and 90 water molecules per unit-cell. Each depicted cluster includes the water molecules located within 7 Å of a central Mg site, i.e. the cutoff distance employed in our ab initio NEXAFS analysis.58 As shown in Fig. 5a, when N = 18 each water molecule is directly bound to distinct metal centers and the corresponding theoretical Mg K-edge NEXAFS spectrum (see Fig. 5b) does not possess a pre-edge peak while the intensity of the shoulder in the 1314–1317 eV energy region is rather flattened. In fact, the Mg local coordination environment goes from a square pyramidal to an octahedral one when a water molecule directly binds the given Mg site and, as a consequence, the pre-edge transition is depleted in the NEXAFS simulated spectrum. As an increasing number of water molecules progressively populates the Mg local environment when N = 36, 54 and 72, the intensity of the shoulder located between 1314 and 1317 eV also gradually increases in the corresponding theoretical NEXAFS spectra while this feature remains very similar in the theoretical spectra simulated for N = 72 and N = 90 (Fig. 5b). The theoretical NEXAFS analyses indicates that the pre-edge peak and the intensity of the shoulder transition between 1314 and 1317 eV in the Mg K-edge spectra are sensitive fingerprints of the hydration level around the metal MOF nodes.
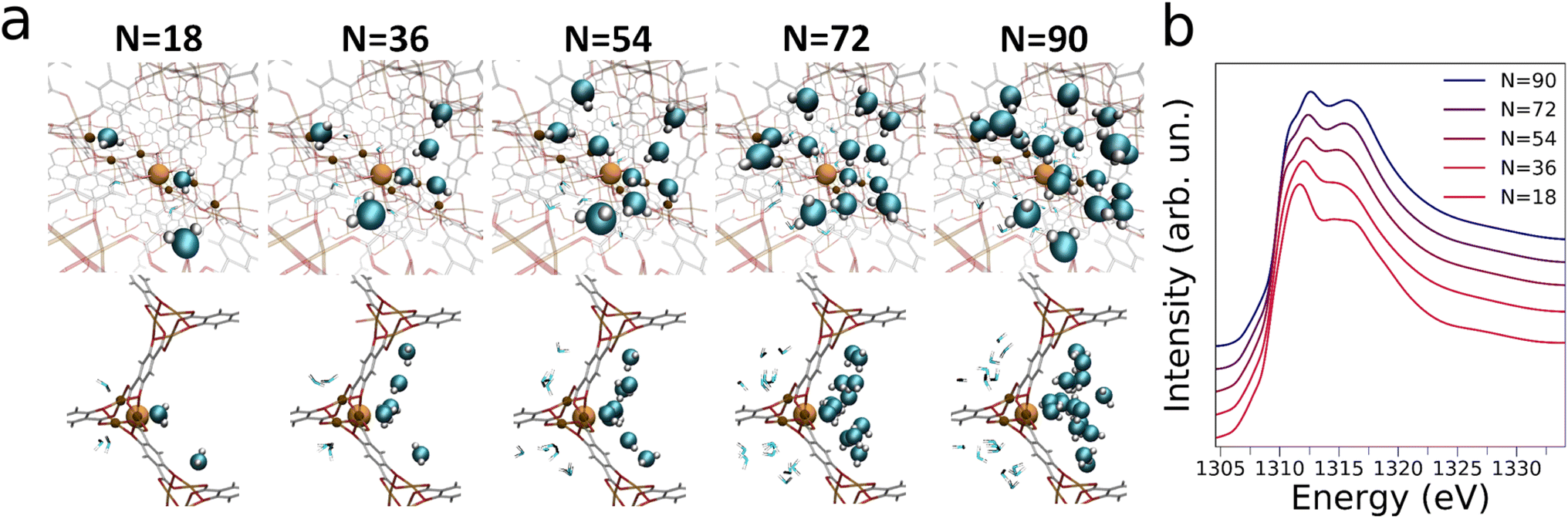 | ||
| Fig. 5 (a) Front (top) and side (bottom) views of MD snapshots of Mg-MOF-74 loaded with N = 18, 36, 54, 72 and 90 water molecules per unit-cell. The clusters include the water molecules located within 7 Å of a central Mg site (full orange sphere). The water molecules found on the same channel as that of the Mg central site are depicted as solid spheres, whereas those found on adjacent channels are portrayed as lines. (b) Theoretical Mg K-edge NEXAFS spectra, calculated as averages of 100 MD snapshots of Mg-MOF-74 loaded with N = 18, 36, 54, 72 and 90 water molecules per unit-cell. | ||
The structural and dynamical behaviour of water in Mg-MOF-74 was further characterized by MD simulations. In particular, the MD analysis evidences that when a single water molecule per Mg node is present in the MOF (a water loading equivalent to N = 18), the water molecules coordinate individual open Mg sites (Fig. 6a and b). These water molecules are well-known to be strongly bound to the Mg atoms, to not diffuse over long distances and may well be considered as part of the framework.59 As additional water molecules populate the framework, 1-D water chains arise between vicinal Mg sites. In particular, each Mg-bound water molecule donates a hydrogen bond to a water molecule contiguous along the c-axis (Fig. S10a†), while acting as hydrogen bond donor and acceptor with two additional water molecules adjacent along the b-axis which are interacting with the π-system of the linker (Fig. S10b†). As the water loading increases, concentric cylindrical shells progressively adsorb onto these 1-D water chains and stretch out into the MOF channels finally populating the channel center more efficiently for N = 90 (Fig. 6d).24,44 Importantly, the MD simulations evidence that the increase of water content slows down the reorientational mobility of the free water molecules adsorbed in the channels, an effect we evaluated by calculating the orientational correlation function of the water O–H bond, C2,OH(t), as function of N. As shown in Fig. 6c, the reorientation rate of the Mg-bound water molecules (N = 18) is faster than those of the free water molecules calculated for increasing water content (N = 36–90), a result consistent with recent theoretical investigations of other MOF systems exhibiting honeycomb-like channels.24,59
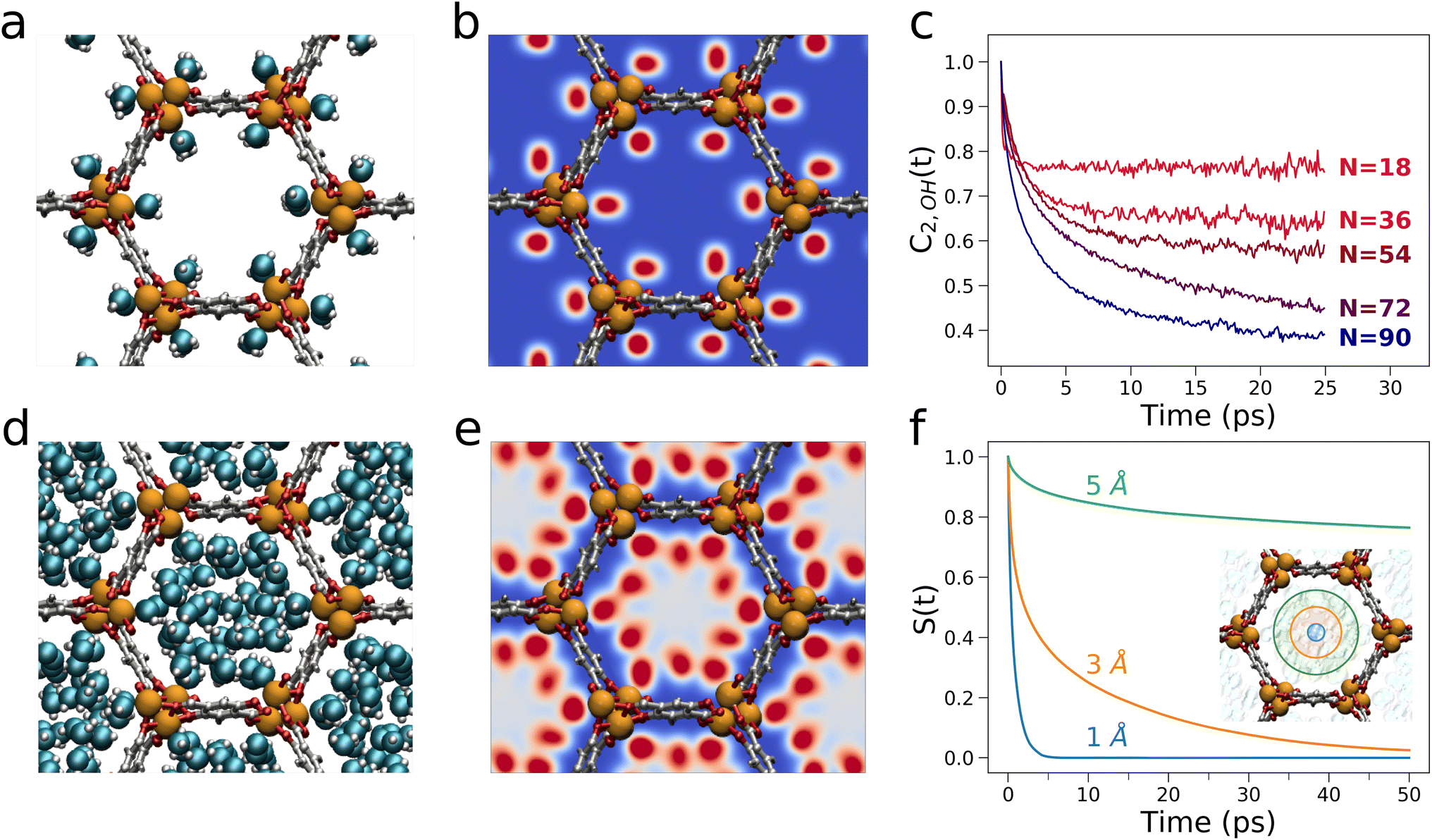 | ||
| Fig. 6 (a) MD snapshot of Mg-MOF-74 with a water loading of N = 18. (b) Density map of water calculated from MD simulations evaluated for a loading of N = 18 (regions with higher water density are depicted using darker red colors). (c) Water orientational correlation functions obtained from MD simulations of Mg-MOF-74 with increasing water loadings. (d) MD snapshot of Mg-MOF-74 with a water loading of N = 90. (e) Density map of water calculated from MD simulations evaluated for a loading of N = 90 (regions with higher water density are depicted using darker red colors). (f) Survival probabilities for water molecules in three cylindrical regions of radii equal to 1 Å, 3 Å and 5 Å inside the porous channels of Mg-MOF-74 loaded with N = 90 water molecules. | ||
As the number of water molecules inside the porous channels increases, also the translational mobility of water confined in Mg-MOF-74 is affected. The water residence probabilities indicate that while the Mg centers are the first occupied sites for N = 18 (Fig. 6b), the framework progressively interacts with adsorbed water molecules for N = 36, 54 and 72 mainly via the DOBDC linker (Fig. S11†),44 with the channel center being populated at higher frequency only for N = 90 (Fig. 6e). Interestingly, while at the highest water loading the average reorientation rate of the free water molecules is reduced, also the mobility of the free water molecules is significantly influenced by the distance from the MOF framework atoms. Fig. 6f displays the survival probability S(t) that water molecules may remain enclosed in three cylindrical regions of radii equal to 1, 3 and 5 Å inside the MOF porous channels for N = 90. One may observe that the S(t) function undergoes a faster decay further away from the framework, and this effect is due to the higher mobility of the water molecules that are more distant from the framework.60
Conclusions
In this work for the first time the AP-NEXAFS technique has been used to study the water harvesting process occurring at the crystal surface of a prototypical MOF and some interesting differences have been detected when looking at the water adsorption and desorption processes occurring at the crystal surface as compared to the bulk. In particular, the combined experimental and theoretical NEXAFS investigations carried out on Mg-MOF-74 pointed out the simultaneous presence of Mg open sites and residual gas-phase water during dehydration, and proved that during water release a high number of surface Mg sites still interact with one or two water molecules. Conversely, when looking at the bulk, a significantly lower number of Mg sites have been found to interact with water molecules in the same experimental conditions.40 This finding may either be due to the presence of defects, which are more common at the crystal surface35 or to the fact that the water adsorption (desorption) process starts from the interior of the material and propagates towards the channel openings.21 According to this picture, the central sections of the MOF channels would be emptied at similar rates leading to a corresponding higher density of unsaturated Mg sites in the MOF bulk, with higher amounts of water being pushed towards and concentrated at the MOF crystal surface. In conclusion, the combined approach based on AP-NEXAFS, PXRD experimental determinations and ML-supported theoretical analyses has been found to be a valuable tool for a thorough description of the water harvesting process at both the crystal surface and bulk. The AP-NEXAFS spectroscopy, that has been used for the first time to unveil the water adsorption behaviour of a MOF, has been proved to be an effective method that can add interesting information on the processes occurring at the MOF crystal surface.Data availability
The data supporting this article have been included as part of the ESI.† The authors have cited additional references within the ESI.†61–72Author contributions
Francesco Tavani: conceptualization, investigation, data curation, writing – original draft. Alessandro Tofoni: conceptualization, investigation, data curation, writing – original draft. Marco Vandone: investigation. Matteo Busato: investigation. Luca Braglia: investigation. Piero Torelli: conceptualization, writing – review & editing. Maximillian G. Stanzione: investigation. Anthony R. Armstrong: conceptualization, writing – review & editing. Russell E. Morris: conceptualization, writing – review & editing. Valentina Colombo: conceptualization, writing – review & editing. Paola D'Angelo: conceptualization, supervision, data curation, investigation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swiss Norwegian beamlines (SNBL, ESRF) and the APE-HE beamline (Elettra) and their staff are acknowledged for provision of beamtime and for invaluable support. The BM31 setup was funded by the Swiss National Science Foundation (grant 206021 189629) and the Research Council of Norway. Part of the calculations were performed on the Galileo100 system of the CINECA supercomputing center (grants IsCa6_MTMOF and IsCb3_MILWHA). The authors acknowledge the European Union-NextGenerationEU under the Italian Ministry of University and Research (MUR), Network 4 Energy Sustainable Transition – NEST project – CUP B53C22004070006 with eCO2 cascade project (CUP B53C22004060006), and the Italian Ministry of University and Research (MUR) for the PRIN project “MOF-MTM: tailoring Metal–Organic Frameworks for the direct Methane To Methanol conversion under mild conditions” number 2022SFC459.Notes and references
- C. J. Vörösmarty, P. Green, J. Salisbury and R. B. Lammers, Science, 2000, 289, 284–288 CrossRef
.
- M. Flörke, C. Schneider and R. McDonald, Nat. Sustain., 2018, 1, 51–58 CrossRef
- B. Malmqvist and S. Rundle, Environ. Conserv., 2002, 29, 134–153 CrossRef
- H. Lin, Y. Yang, Y.-C. Hsu, J. Zhang, C. Welton, I. Afolabi, M. Loo and H.-C. Zhou, Adv. Mater., 2023, 2209073 Search PubMed
- S. Chandrasekara, S. Pathmarajah, J. Obeysekera and M. Vithanage, ACS ES&T Water, 2023, 3, 1454–1462 Search PubMed
-
O. Yaghi, M. J. Kalmutzki and C. S. Diercks, in Water Sorption Applications of MOFs, John Wiley & Sons, Ltd, 2019, ch. 17, pp. 395–427 Search PubMed
- P. D. Dietzel, R. Blom and H. Fjellvåg, Eur. J. Inorg. Chem., 2008, 2008, 3624–3632 CrossRef
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454–459 CrossRef CAS
- Z. Zheng, A. H. Alawadhi, S. Chheda, S. E. Neumann, N. Rampal, S. Liu, H. L. Nguyen, Y.-h. Lin, Z. Rong, J. I. Siepmann, L. Gagliardi, A. Anandkumar, C. Borgs, J. T. Chayes and O. M. Yaghi, J. Am. Chem. Soc., 2023, 145, 28284–28295 CrossRef CAS PubMed
- T. J. Matemb Ma Ntep, M. Wahiduzzaman, E. Laurenz, I. Cornu, G. Mouchaham, I. Dovgaliuk, S. Nandi, K. Knop, C. Jansen, F. Nouar, P. Florian, G. Füldner, G. Maurin, C. Janiak and C. Serre, Adv. Mater., 2023, 2211302 Search PubMed
- A. J. Rieth, S. Yang, E. N. Wang and M. Dincă, ACS Cent. Sci., 2017, 3, 668–672 CrossRef CAS PubMed
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed
- M. L. Valentine, G. Yin, J. J. Oppenheim, M. Dincǎ and W. Xiong, J. Am. Chem. Soc., 2023, 145, 11482–11487 CAS
- M. Logan, S. Langevin and Z. Xia, Sci. Rep., 2020, 10, 1492 CrossRef CAS PubMed
- M. A. Van der Veen, S. Canossa, M. Wahiduzzaman, G. Nenert, D. Frohlich, D. Rega, H. Reinsch, L. Shupletsov, K. Markey, D. E. De Vos, M. Bonn, N. Stock, G. Maurin and E. H. G. Backus, Adv. Mater., 2023, 2210050 Search PubMed
- H. Kim, S. R. Rao, E. A. Kapustin, L. Zhao, S. Yang, O. M. Yaghi and E. N. Wang, Nat. Commun., 2018, 9, 1191 Search PubMed
- A. LaPotin, Y. Zhong, L. Zhang, L. Zhao, A. Leroy, H. Kim, S. R. Rao and E. N. Wang, Joule, 2021, 5, 166–182 CAS
- W. Song, Z. Zheng, A. H. Alawadhi and O. M. Yaghi, Nat. Water, 2023, 1, 626–634 CAS
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS
- A. Fuchs, F. Knechtel, H. Wang, Z. Ji, S. Wuttke, O. M. Yaghi and E. Ploetz, J. Am. Chem. Soc., 2023, 145, 14324–14334 CrossRef CAS
- J. C. Wagner, K. M. Hunter, F. Paesani and W. Xiong, J. Am. Chem. Soc., 2021, 143, 21189–21194 CrossRef CAS PubMed
- X. Liu, X. Wang and F. Kapteijn, Chem. Rev., 2020, 120, 8303–8377 CrossRef CAS PubMed
- P. Dietzel, R. Johnsen, R. Blom and H. Fjellvåg, Chem.–Eur. J., 2008, 14, 2389–2397 CrossRef CAS
- A. J. Rieth, K. M. Hunter, M. Dincă and P. Francesco, Nat. Commun., 2019, 10, 4771 CrossRef PubMed
- S. Li, P. Wu, L. Chen, Y. Tang, Y. Zhang, L. Qin, X. Qin and H. Li, J. Water Process Eng., 2024, 58, 104899 CrossRef
- X. Huang, Q. Qin, Q. Ma and B. Wang, Water, 2022, 14, 3487 CrossRef CAS
- F. Salles, S. Bourrelly, H. Jobic, T. Devic, V. Guillerm, P. Llewellyn, C. Serre, G. Ferey and G. Maurin, J. Phys. Chem. C, 2011, 115, 10764–10776 CrossRef CAS
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS
- K. M. Hunter, J. C. Wagner, M. Kalaj, S. M. Cohen, W. Xiong and F. Paesani, J. Phys. Chem. C, 2021, 125, 12451–12460 CrossRef CAS
- J. M. Olinger and P. R. Griffiths, Anal. Chem., 1988, 60, 2427–2435 CrossRef CAS
- S. Fei, A. Alizadeh, W.-L. Hsu, J.-J. Delaunay and H. Daiguji, J. Phys. Chem. C, 2021, 125, 26755–26769 CrossRef CAS
- A. Lamaire, J. Wieme, S. Vandenhaute, R. Goeminne, S. M. J. Rogge and V. Van Speybroeck, Nat. Commun., 2024, 15, 9997 CrossRef CAS PubMed
- J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678–10679 CrossRef CAS PubMed
- F. Tavani, M. Busato, D. Veclani, L. Braglia, S. Mauri, P. Torelli and P. D'Angelo, ACS Appl. Mater. Interfaces, 2023, 15, 26166–26174 CrossRef CAS PubMed
- L. Braglia, F. Tavani, S. Mauri, R. Edla, D. Krizmancic, A. Tofoni, V. Colombo, P. D'Angelo and P. Torelli, J. Phys. Chem. Lett., 2021, 12, 9182–9187 CrossRef CAS
- C. Castán-Guerrero, D. Krizmancic, V. Bonanni, R. Edla, A. Deluisa, F. Salvador, G. Rossi, G. Panaccione and P. Torelli, Rev. Sci. Instrum., 2018, 89, 054101 CrossRef
- A. Ruosi, C. Raisch, A. Verna, R. Werner, B. A. Davidson, J. Fujii, R. Kleiner and D. Koelle, Phys. Rev. B, 2014, 90, 125120 CrossRef
- F. Bassato, S. Mauri, L. Braglia, A. Y. Petrov, E. Dobovičnik, F. Tavani, A. Tofoni, P. Ferrer, D. Grinter, G. Held, P. D'Angelo and P. Torelli, J. Phys. Chem. Lett., 2024, 15, 8540–8548 CrossRef CAS
- F. Tavani, M. Busato, L. Braglia, S. Mauri, P. Torelli and P. D'Angelo, ACS Appl. Mater. Interfaces, 2022, 14, 38370–38378 CrossRef CAS PubMed
- M. H. Rosnes, B. Pato-Doldán, R. E. Johnsen, A. Mundstock, J. Caro and P. D. Dietzel, Microporous Mesoporous Mater., 2020, 309, 110503 CAS
- J. Zheng, R. S. Vemuri, L. Estevez, P. K. Koech, T. Varga, D. M. Camaioni, T. A. Blake, B. P. McGrail and R. K. Motkuri, J. Am. Chem. Soc., 2017, 139, 10601–10604 CrossRef CAS
- L. J. Wang, H. Deng, H. Furukawa, F. Gándara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2014, 53, 5881–5883 CAS
- D.-A. Yang, H.-Y. Cho, J. Kim, S.-T. Yang and W.-S. Ahn, Energy Environ. Sci., 2012, 5, 6465–6473 RSC
- A. N. Rudenko, S. Bendt and F. J. Keil, J. Phys. Chem. C, 2014, 118, 16218–16227 CrossRef CAS
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC
- A. Le Bail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23, 447–452 CrossRef CAS
- W. S. Drisdell, R. Poloni, T. M. McDonald, J. R. Long, B. Smit, J. B. Neaton, D. Prendergast and J. B. Kortright, J. Am. Chem. Soc., 2013, 135, 18183–18190 CrossRef CAS PubMed
- S. Myneni, Y. Luo, L. A. Näslund, M. Cavalleri, L. Ojamäe, H. Ogasawara, A. Pelmenschikov, P. Wernet, P. Väterlein, C. Heske, Z. Hussain, L. G. M. Pettersson and A. Nilsson, J. Phys. Condens. Matter, 2002, 14, L213 CrossRef CAS
- F. Frati, M. O. J. Y. Hunault and F. M. F. de Groot, Chem. Rev., 2020, 120, 4056–4110 CrossRef CAS
- Y. Li, X. Wang, D. Xu, J. D. Chung, M. Kaviany and B. Huang, J. Phys. Chem. C, 2015, 119, 13021–13031 CrossRef CAS
- A. Caruso, X. Zhu, J. L. Fulton and F. Paesani, J. Phys. Chem. B, 2022, 126, 8266–8278 CrossRef CAS PubMed
- J. Timoshenko, D. Lu, Y. Lin and A. I. Frenkel, J. Phys. Chem. Lett., 2017, 8, 5091–5098 CrossRef CAS
- J. Timoshenko, F. T. Haase, S. Saddeler, M. Rüscher, H. S. Jeon, A. Herzog, U. Hejral, A. Bergmann, S. Schulz and B. Roldan Cuenya, J. Am. Chem. Soc., 2023, 145, 4065–4080 CrossRef CAS
- N. Marcella, Y. Liu, J. Timoshenko, E. Guan, M. Luneau, T. Shirman, A. M. Plonka, J. E. S. van der Hoeven, J. Aizenberg, C. M. Friend and A. I. Frenkel, Phys. Chem. Chem. Phys., 2020, 22, 18902–18910 RSC
- J. Timoshenko, A. Anspoks, A. Cintins, A. Kuzmin, J. Purans and A. I. Frenkel, Phys. Rev. Lett., 2018, 120, 225502 CrossRef CAS PubMed
- J. Timoshenko, C. J. Wrasman, M. Luneau, T. Shirman, M. Cargnello, S. R. Bare, J. Aizenberg, C. M. Friend and A. I. Frenkel, Nano Lett., 2019, 19, 520–529 CrossRef CAS PubMed
- D. Del Giudice, F. Tavani, M. Di Berto Mancini, F. Frateloreto, M. Busato, D. Oliveira De Souza, F. Cenesi, O. Lanzalunga, S. Di Stefano and P. D'Angelo, Chem.–Eur. J., 2022, 28, e202103825 CrossRef CAS
- Z. L. Terranova and F. Paesani, Phys. Chem. Chem. Phys., 2016, 18, 8196–8204 RSC
- A. A. Milischuk and B. M. Ladanyi, J. Chem. Phys., 2011, 135, 174709 CrossRef PubMed
- J. Liu, A. I. Benin, A. M. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 131, 4995–5000 CrossRef CAS PubMed
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- Y. Joly, Phys. Rev. B, 2001, 63, 125120 CrossRef
- O. Bunǎu and Y. Joly, World J. Condens. Matter Phys., 2009, 21, 345501 CrossRef
- Y. Joly, O. Bunău, J. E. Lorenzo, R. M. Galéra, S. Grenier and B. Thompson, J. Phys.: Conf. Ser., 2009, 190, 012007 CrossRef
- A. S. Rosen, J. M. Notestein and R. Q. Snurr, ACS Catal., 2019, 9, 3576–3587 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: MOF synthesis and preliminary characterization, AP-NEXAFS and PXRD in situ experiments, MD simulations, ML-assisted ab initio NEXAFS calculations, Fig. S1–S11 and Table S1. See DOI: https://doi.org/10.1039/d5sc01482d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |