
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor–acceptor type covalent organic frameworks: design, optimization strategies and applications
Haiyang
Liu
a,
Shanshan
Zhu
a,
Yongfeng
Zhi
 *b,
Huijuan
Yue
c and
Xiaoming
Liu
*a
*b,
Huijuan
Yue
c and
Xiaoming
Liu
*a
aCollege of Chemistry, Jilin University, Changchun, 130012, P.R. China. E-mail: xm_liu@jlu.edu.cn
bSchool of Chemistry and Chemical Engineering, Hainan University, Haikou, 570228, China. E-mail: zhiyf@hainanu.edu.cn
cState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun, 130012, P.R. China
First published on 24th June 2025
Abstract
Covalent organic frameworks (COFs) have emerged as one of the hottest research topics in various applications due to their designed structures, adjustable pore sizes, and abundant active sites. However, the high electron–hole recombination rate and short carrier lifetime in non-donor–acceptor type COFs are inevitable issues. The electron donor–acceptor (D–A) type COFs, which are synthesized by introducing donor and acceptor units into the COF skeleton, combined with effective carrier separation, adjustable bandgap, and sensitive photoelectric response, are considered an effective strategy for improving exciton dissociation and optimizing carrier transport. In recent years, D–A type COFs have witnessed exponential expansion in applications spanning photocatalysis, energy storage, and photothermal therapy. Consequently, there exists an imperative necessity to comprehensively summarize and expound upon the challenges and prospects pertaining to the development of D–A type COFs. In this review, we first summarize the common connecting bonds as well as the building blocks for the synthesis of D–A type COFs. Several strategies for optimizing D–A type COFs and their recent progress in photocatalysis, photothermal therapy, and energy storage are then presented. Finally, we delineate the current challenges and impediments of D–A type COFs and offer a forward-looking perspective on the future development of D–A type COFs. This review is poised to encourage researchers with a more profound comprehension of the design strategies and applications of D–A type COFs, thereby inspiring them to conduct more incisive research into the challenges and developmental prospects of D–A type COFs.
 Haiyang Liu | Haiyang Liu obtained his B.Sc degree from Inner Mongolia Minzu University in 2020. Now, he is pursuing his PhD degree in Prof. Xiaoming Liu's group at the Jilin University, China. His research topic is the design, synthesis and application of functional covalent organic frameworks in the field of catalysis. |
 Shanshan Zhu | Shanshan Zhu obtained her B.Sc degree from Xuchang University in 2019 and M.Sc degree from the Zhengzhou University, in 2022. Now, she is pursuing her PhD degree in Prof. Xiaoming Liu's group at the Jilin University, China. Her research focuses on developing novel COFs for catalysis. |
 Yongfeng Zhi | Yongfeng Zhi is an associate professor at School of Chemistry and Chemical Engineering of Hainan University, China. He obtained his PhD degree in chemistry in Prof. Xiaoming Liu's group at Jilin University in 2019. He then joined Prof Qichun Zhang's Group at the Nanyang Technological University as a research fellow. In October 2020, He worked as a postdoctoral researcher under the guidance of Prof. Qing Wang and Prof. Donglin Jiang at National University of Singapore. His research topics mainly focus on synthesis and exploration of covalent organic frameworks (COFs). |
 Huijuan Yue | Huijuan Yue is a full professor at State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry of Jilin University, China. She obtained her PhD degree in Chemistry in 2007 at Jilin University. She worked as a postdoctoral researcher in the lab of Prof. Xiaodong Zou at Stockholm University in 2007–2010. Her current research interests focus on developing dielectric and magnetic nanocomposites for electromagnetic energy absorption. |
 Xiaoming Liu | Xiaoming Liu is a full professor at College of Chemistry of Jilin University, China. He obtained his PhD degree in Chemistry in 2006 at Jilin University. He worked as a postdoctoral researcher in the lab of Prof. Donglin Jiang at Institute for Molecular Science in 2010–2012. His current research interests focus on functional porous organic polymers (POPs) and covalent organic frameworks (COFs) for energy and catalysis. |
1. Introduction
The global energy crisis is progressively becoming a pivotal issue constraining sustainable development. With the continuous consumption of fossil fuels and the sharp rise in global energy demand, the traditional energy system is confronted with formidable challenges.1,2 Realizing green and low-carbon transformation and developing clean energy technologies have become urgent global needs. Covalent organic framework materials (COFs) are porous materials composed of organic molecules connected by covalent bonds, with high structural tunability and excellent physical chemical properties, which can overcome the limitations of traditional inorganic materials (e.g., smaller specific surface area, insufficiently rich functional and structural diversity, etc.).3–6 Due to their excellent chemical stability, pore structure, adjustable specific surface area, and diverse functionalization possibilities, COFs have been widely used in various fields such as gas storage,7,8 catalysis,9–13 separation,14–17 luminescent materials,18 wastewater treatment,19 solvent permeation,20 and sensors.21–24 Therefore, COF materials, as a new type of functional material, provide broad application prospects for addressing energy crises and promoting carbon neutrality. Although COFs have shown great potential in materials science, traditional COFs often have certain limitations in electronic and optical properties, especially in optoelectronic device applications, where efficient electronic transmission and light response performance are difficult to achieve. This limitation is mainly due to the insufficient band structure and electronic coupling effect of organic monomers in the intrinsic structure of COFs, which urgently need further optimization.To address this issue, COFs with donor–acceptor (D–A) structures have emerged. D–A type COFs are materials constructed through carefully designed interactions between donor and acceptor units.25,26 This design strategy endows D–A type COFs with a unique electronic structure and functional performance, which can significantly improve the performance of materials in applications such as optoelectronics and energy storage. Donor units are usually aromatic amines or thioaromatic compounds with higher electron density, while acceptor units are aromatic carbonyl or nitro compounds with lower electron density. Under the action of these units, D–A type COFs can not only achieve electronic structure control in the microstructure of the material, but also effectively transfer electrons inside the material, as well as enhance its light-absorbing ability, thereby significantly improving its photoelectric performance.27 Therefore, D–A type COFs have shown wide potential for applications in the field of optoelectronics, especially in photocatalysis, solar cells, and photothermal therapy. For example, under the action of an intramolecular built-in electric field, the photocatalytic hydrogen evolution rate of TeTpb COF with D–A structure composed of truxenone and 1,3,5-tris (p-formyl-phenyl) benzene-based materials reached 21.6 mmol g−1 h−1.28 Wang et al. incorporated the D–A type BABT-COF (benzotrithiophene as the D unit, tris (4-aminophenyl) amine as the A unit) as an efficient additive into the hole transport layer, and the assembled perovskite solar cell achieved a power conversion efficiency of 18.56%.29 In addition, the CTCS-COF hydrogel could achieve the rapid separation of electron–hole pairs under the interaction of D–A (curcumin as the D unit, porphyrins as the A unit), resulting in a rapid on-wound photothermal sterilization effect.30 These studies on D–A type COFs have shown encouraging results, giving us great expectations for the future development of D–A type COFs. In summary, the design concept and multifunctionality of D–A type COFs have demonstrated excellent performance in optoelectronic and energy storage applications, and with the continuous improvement of synthesis methods and structural design, their future application prospects in multiple technical fields are becoming increasingly broad.
Although there are a handful of reviews on D–A type COFs that cover many important research advances,31,32 most of them focus on the connection of material bonds, basic characteristics, and some application areas. Summaries on energy storage or how to improve the performance of D–A type COFs by optimizing their electronic structure are not available. In addition, D–A-type COFs are a very hot field, and the number of articles on D–A-type COFs and the speed of their development have exploded in recent years. Therefore, there is an urgent need to summarize the advantages of D–A-type COFs, construction strategies, and recent advances in photocatalysis, photothermal therapy, and energy storage applications. Hence, this review aims to further supplement and improve the construction modules of D–A type COFs and their research in optical and electrical applications, and introduce several strategies for further optimizing the performance of D–A type COFs (Fig. 1). Additionally, the review will highlight the latest research developments, particularly the emergence of novel structures and innovative breakthroughs in enhancing the application performance of D–A type COFs in optoelectronic devices. We believe that in the future, with the continuous development of synthesis technology and in-depth theoretical research, D–A type COFs are expected to play a more important role in fields such as photocatalytic reactions, optoelectronic devices, energy storage and conversion.
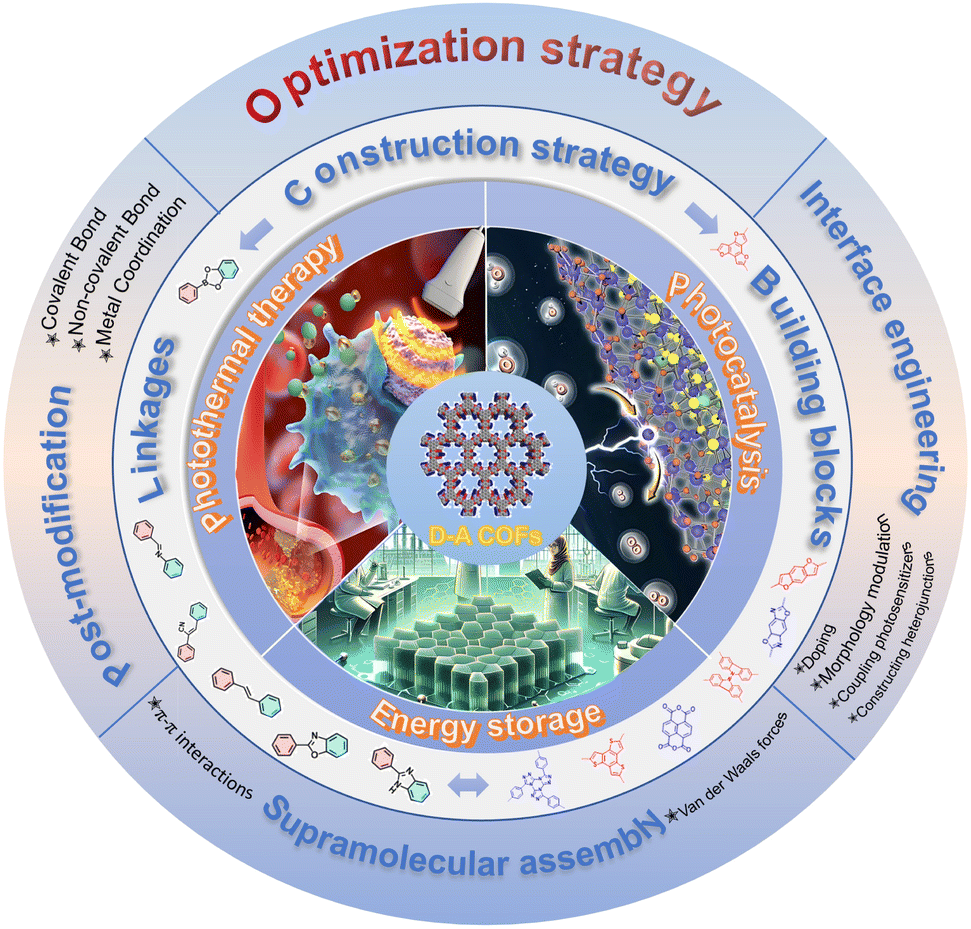 | ||
| Fig. 1 Schematic diagram of the construction strategy, optimization strategy, and application of D–A type COFs. | ||
2. Definition and advantages of D–A type COFs
D–A type COF is a covalent organic framework material formed by alternating donor and acceptor units. In contrast to conventional COFs, D–A type COFs introduce donor acceptor structures in molecular design, which can endow the material with electronic properties, optical properties, and chemical activity that are different from general COFs. Donor units are typically electron rich aromatic groups, such as nitrogen-containing aromatic hydrocarbons or pyridine groups. These functional groups have strong electron donating ability. The acceptor unit is generally an aromatic group containing electron-withdrawing groups, such as ketone groups, semi carbazide groups, etc. These functional groups have strong electron attraction ability. This D–A structure engenders an asymmetric electronic structure of the material, endowing it with special optoelectronic properties such as high light absorption capacity and enhanced electron hole separation efficiency. These characteristics make D–A type COFs exhibit great advantages in photocatalysis, photothermal therapy, and energy storage. Below, we will introduce these advantages in detail:(1) Advantages of D–A type COFs in photocatalysis.
Efficient electron transport: the donor acceptor structure of D–A type COFs can effectively separate excited electrons and holes, improving the efficiency of photocatalytic reactions. This structural design optimizes the migration path of electrons, allowing electrons and holes to remain separated for a long time and reducing recombination.
Wide spectral absorption: the electronic structure characteristics of D–A type COFs can adjust the band structure of the material, thereby achieving absorption of visible light and even near-infrared light. This gives D–A type COFs significant advantages in photocatalytic water splitting, H2O2 generation and organic conversion reactions.33–35
Contains built-in electric field: due to the presence of electronic push–pull structures in the molecule, D–A type COFs will form an intrinsic electric field at the microscale. This built-in electric field can promote the migration of electrons from the donor region to the acceptor region in the excited state, while suppressing the recombination of electrons and holes.
(2) Advantages of D–A type COFs in photothermal therapy.
Enhanced light absorption capability: D–A type COF materials can absorb a wide range of light, especially in the near-infrared region, which is crucial for deep tumor treatment. The efficient light absorption ability enables it to generate a large amount of heat during the photothermal conversion process, effectively killing tumor cells.
High photothermal conversion efficiency: the conjugated system and electronic transition behavior of D–A type COFs enable excited state electrons to rapidly convert into thermal energy through non radiative transitions, resulting in high photothermal conversion efficiency.
Good biocompatibility: by designing the surface functional groups of D–A type COFs reasonably, its biocompatibility can be improved, the toxicity to normal cells can be reduced, and the selectivity of therapeutic effects can be enhanced.
(3) Advantages of D–A type COFs in energy storage.
Enhancement of ion migration ability: in the D–A structure, under external voltage, the electron conduction path and ion diffusion path are often highly coupled, which can achieve synergistic migration of electrons and ions, helping to improve the overall dynamic and rate performance.
Good conductivity: by adjusting the donor and acceptor units, D–A type COFs can optimize the material's conductivity, thereby improving the energy density and power density of batteries or supercapacitors.
Multi-electron redox mechanism: by designing multiple reversible electron donor or acceptor units, D–A type COFs can realize multi-electron transfer reactions and significantly increase the number of electrons that can be stored per unit mass, thus enhancing the specific capacity.
Long cycle life: due to its excellent structural stability and compressive strength, D–A type COFs can maintain high electrochemical performance during long-term charge and discharge processes, exhibiting excellent cycle stability.
As a result, D–A type COFs demonstrate excellent performance in various fields such as photocatalysis, photothermal therapy, and energy storage through their unique donor acceptor structure. Their efficient electronic transmission, wide spectral absorption, adjustable pore structure, and good stability make them a candidate for future high-performance materials. These advantages make D–A type COFs have broad application prospects in fields such as sustainable energy, medical treatment, and intelligent energy storage.
3. Construction strategy of D–A type COFs
D–A type COFs are constructed when donor and acceptor units establish a highly ordered network through covalent bonds. The characteristic of D–A type COFs is that the D and A building units are alternately arranged on the plane to enhance the π–π conjugation effect of the interlayer stacking structure. Owing to the predesign ability of COF structures, the selection of building units and bond connections that make up D–A type COF materials is crucial. The types of D–A units and bonding junctions can affect not only the stability and crystallinity of COFs, but also the in-plane transport mechanisms of carriers. In this section, we provide an overview of the key connections and common donor and acceptor units used for designing D–A type COFs.3.1 Linkages
The intricacy of synthesis, extent of conjugation, and stability of COFs are mainly influenced by the choice of connection method. Through suitable monomer connections, these monomers undergo reversible condensation reactions to form extended frameworks with specific geometric shapes, which are covalently linked together.2 At present, various reactive linking groups have been harnessed to construct COFs, and the reactions used to synthesize COFs can be divided into seven categories based on the types of covalent bonds they form. They are: (1) B–O (such as boronic ester,36 borosilicate,37etc.); (2) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (such as imine,38 hydrazone,39 azone);40 (3) CC (such as cyanovinylidene,41 alkene,42 and benzofuran);43 (4) CNaromatic (such as triazine,44 quinoline,45 phenazine,46 imidazole,47 oxazole,48etc.); (5) C–O (such as dioxane49 and ester50); (6) C–N (such as β-ketoenamine,51 amide,52 hydrazine,53 quinoxaline,54etc.); and (7) others (such as boron zinc,55 silicate,56 and azo57). However, not all of the aforementioned connections have been used to form the D–A type COF. Generally speaking, the formation of D–A arrays through condensation between donor and acceptor monomers is crucial for typical D–A type COFs. So far, the connection types of D–A type COFs have been reported as shown in Fig. 2, including but not limited to classic bond linkages such as boronic ester, imine, and ethylene connections.
N (such as imine,38 hydrazone,39 azone);40 (3) CC (such as cyanovinylidene,41 alkene,42 and benzofuran);43 (4) CNaromatic (such as triazine,44 quinoline,45 phenazine,46 imidazole,47 oxazole,48etc.); (5) C–O (such as dioxane49 and ester50); (6) C–N (such as β-ketoenamine,51 amide,52 hydrazine,53 quinoxaline,54etc.); and (7) others (such as boron zinc,55 silicate,56 and azo57). However, not all of the aforementioned connections have been used to form the D–A type COF. Generally speaking, the formation of D–A arrays through condensation between donor and acceptor monomers is crucial for typical D–A type COFs. So far, the connection types of D–A type COFs have been reported as shown in Fig. 2, including but not limited to classic bond linkages such as boronic ester, imine, and ethylene connections.
 | ||
| Fig. 2 Formation of linkage bonds for D–A type COFs. | ||
D–A type COFs linked with boronic esters: Boric acid ester connection represents the initially reported type of connection in COFs and exhibits significant thermal stability. The significant crystallinity observed in COFs connected by boronic acid esters can be attributed to the highly reversible copolymerization reaction between boronic acid and catechol derivatives. However, COFs with boronic ester connections characterized by non-conjugated bonds exhibit limited carrier mobility due to their dependence on the stacking of conjugated units to form channels. To enhance photocatalytic activity, incorporating donor and acceptor units into these COFs can effectively improve carrier mobility. Jiang et al. synthesized the first D–A type COF connected by boronic acid ester, which utilized triphenylamine as the electron donor and benzothiazole as the electron acceptor (Fig. 3a).58 The D–A type COF generates unidirectional columnar arrays and periodicity of D-on-D and A-on-A in an extended and layered crystal framework with AA stacking. This bipolar independent pathway enables the transport of electrons and holes, while the vertically arranged heterojunction with a wide D–A interface significantly improves the photoconductivity. However, their low stability towards hydrolysis and oxidation processes limits their use as heterogeneous catalysts.
 | ||
| Fig. 3 (a) Schematic representation of 2D D–A COFs with self-sorted and periodic electron donor–accepter ordering and bicontinuous conducting channels. Reproduced from ref. 58 with permission from John Wiley and Sons, copyright 2012. (b) Synthetic Routes to CQ-COFs from imine-linked COFs. Reproduced from ref. 62 with permission from American Chemical Society, copyright 2022. | ||
D–A type COF linked with imine: The COF connected by imine is synthesized through the Schiff base reaction of aromatic amines and aldehydes. This synthesis method overcomes the problem of easy hydrolysis of boron ester linked COFs, while endowing imine linked COFs with excellent chemical stability (although their formation process is easily reversible).59 However, the reversibility of imine bonds also makes D–A type COFs connected by imines excellent in crystallinity and specific surface area, making them a widely studied object. The development of imine-based COFs not only significantly improves the chemical stability and applicability of materials, but also greatly expands the research space for new structures, topological structures, synthesis methods, and functions.60 This is due to the widespread presence and easy availability of amines and aldehydes. Therefore, imine bonds have become the most common connecting bonds in the synthesis of D–A type COFs and other materials. Recently, Li et al. introduced three receptor units containing different heteroatoms (O, S, and Se) and synthesized corresponding photoactive imine linked COFs (Py-BO-COF, Py-BT-COF, and Py-BSE-COF) with different D–A structures.61 Among them, Py-BT-COF had the best exciton separation ability and interface carrier migration ability, and its hydrogen evolution rate could reach up to 10.0 mmol g−1 h−1. In terms of stability, the chemical stability of imine-based COFs in acidic and alkaline media is higher than that of their boron counterparts. However, nucleophilic reagents can affect their integrity and trigger their disassembly, which is not advisable. Therefore, some efforts have been made to increase this stability. For example, an interesting strategy is to convert imines into other more stable functional groups, such as thiazole or quinoline, while maintaining their crystallinity and porosity. For example, Wang and his team utilized an intramolecular Povarov reaction to synthesize imine linked COFs and cyclize them into quinoline linked CQ COFs (Fig. 3b).62 This CQ COF had excellent chemical stability against strong acids, strong bases, and redox reagents.
D–A type COF linked with ethylene: COFs connected by imines are widely used in the research of D–A type COFs due to their excellent crystallinity and ease of synthesis. However, due to the local conjugation properties of imine bonds, the carrier transport performance of such COFs is somewhat limited. Compared with COFs connected with imines, COFs connected with CC typically have stronger conjugation and exhibit higher stability to acids or bases.63 These materials have a complete conjugated system, which enhances their light absorption ability and electron delocalization. In addition, they also retain the inherent characteristics of COFs, such as high specific surface area and excellent crystallinity. The Knoevenagel condensation between aldehydes and benzyl cyanides has been widely used for the preparation of COFs with cyanovinylidene linkages. For example, Zhu and his colleagues successfully prepared fully conjugated Bpy-DAN-COF with the D–A structure using K2CO3 as a base catalyst.64 The bipyridine unit of the acceptor facilitates the formation of a molecular specific electron transport pathway with the donor benzothiophene unit, exhibiting excellent charge separation and transfer efficiency. Specifically, the BTT-BpyDAN-COF exhibited a high hydrogen evolution rate of 10.1 mmol g−1 h−1 and an excellent apparent quantum efficiency of 4.83% under visible light irradiation. Due to the presence of cyanide substituents on the double bond, the stability of ethylene linked COFs synthesized through Knoevenagel condensation is compromised. To overcome this issue, Aldol condensation can be used to prepare unsubstituted CC-linked COFs. Guo et al. synthesized two types of ethylene linked COFs (TMT-BT-COF and TMT-TT-COF) using benzoic anhydride as a catalyst and aldol condensation reaction (using thienothiophene and benzothiazole groups as the donor and electron-deficient triazine units as the acceptor).65 Both types of COFs with D–A type vinyl connections exhibited high charge transfer efficiency and significant catalytic activity. As a result, both of them demonstrate remarkably catalytic activity in the oxidation of styrene to benzaldehyde with molecular oxygen, with an exceptionally high conversion rate (≥92%) and selectivity (≥90%).
Therefore, the connecting bonds of D–A type COFs play a crucial role in enhancing material properties, and different types of connecting bonds provide unique performance and advantages for D–A type COFs. In the future, by combining new chemical bonds and self-assembly strategies, D–A type COFs featuring higher efficiency, stability, and specific functions can be developed, promoting their applications in energy, catalysis, and drug delivery fields.
3.2 Building blocks
The common donor and acceptor units reported in the literature are shown in Fig. 4. Building blocks play a crucial role in the formation of D–A type COFs. They not only determine the structure and topological characteristics of COFs, but also directly affect their electronic properties, stability, and functionality. In this section, we primarily introduce the common building blocks used to form D–A type COFs, including triazine, thiazolo[5,4-d] thiazole, benzothiadiazole, diketopyrrolopyrrole, naphthalene diimides, pyrene, thiophene and benzothiophene unit.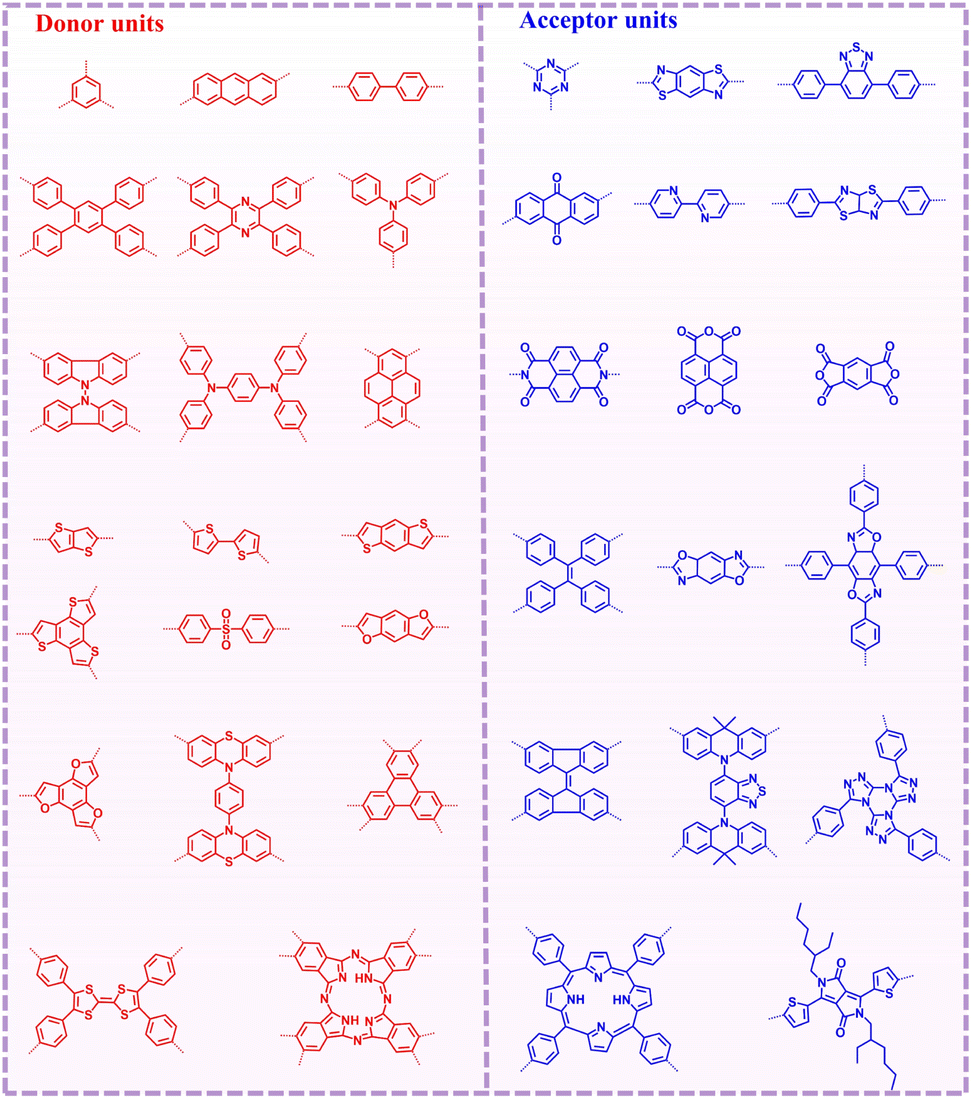 | ||
| Fig. 4 Partial construction units for D–A type COFs. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μmol g−1 h−1.
000 μmol g−1 h−1.
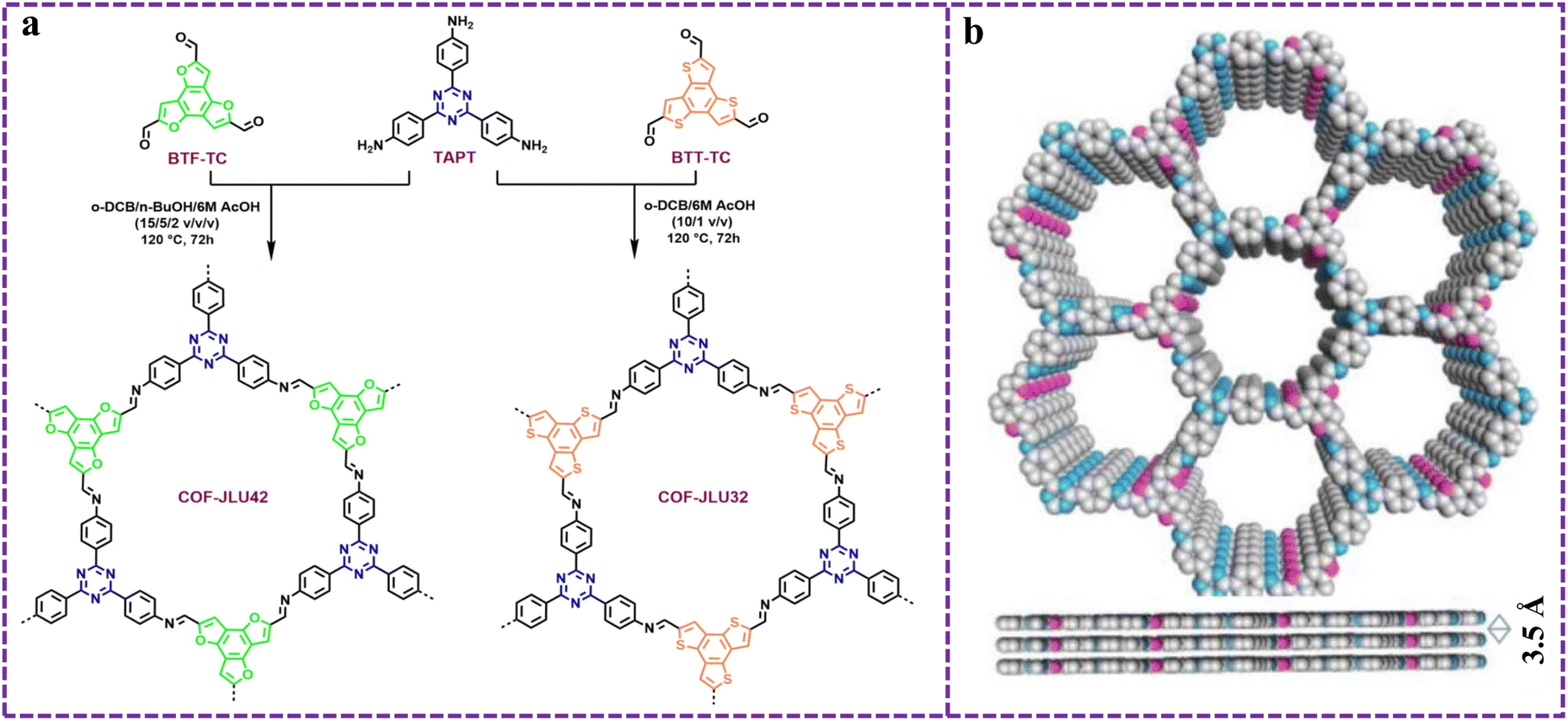 | ||
| Fig. 5 (a) Synthetic conditions and chemical structures of COF-JLU32 and COF-JLU42, and (b) top and side views of the AA stacking structure of COF-JLU42. Reproduced from ref. 76 with permission from Royal Society of Chemistry, copyright 2024. | ||
As is well known, the optical properties and photocatalytic performance of COFs can be adjusted by changing the monomers or making simple modifications.77,78 However, the key factors affecting the optical, electrical, and photocatalytic performance of COFs still need further exploration. Tang et al. synthesized a series of [3 + 3] COFs by the condensation reaction of 1,3,5-tris (4-formylphenyl) benzotriazine units with different substituents (–H, –OH, and –CF3) on the aromatic ring.79 Research findings have indicated that the existence of –OH substituents could narrow the bandgap and improve conductivity, while COFs with Tz skeletons can improve charge separation efficiency due to the interaction of D–A. Therefore, among these COFs, OH-TFP-TTA with both Tz skeleton and –OH functional group exhibited the highest photocatalytic activity in a visible light induced reduction dehalogenation reaction. Isomers of triazine based COFs with different imine bond directions exhibited significant differences in photocatalytic activity due to differences in imine polarization properties.80 Among them, the bipyridine unit acted as an electron donor and acceptor in both reverse and forward imine cases, respectively, and these interactions ultimately led to Re-f-COF isomers as effective photocatalysts for CO2 reduction. Furthermore, density functional theory can be used to predict the optoelectronic properties of the designed COFs. Jiang et al. calculated and evaluated the band alignment and charge transfer characteristics of potential D–A COFs.81 By comparing the charge difference between the ground state and excited state, the effect of D–A action on the charge separation degree and exciton binding energy of different COFs was elucidated. Guided by the above results, corresponding D–A type COFs with different HOMO–LUMO orbitals were synthesized (Fig. 7a), among which TPT-OMe-COF had the best photocatalytic hydrogen production activity, and the activity trends of other COFs were correlated with the activity trends calculated for exciton binding energy. The long reaction time and environmental sensitivity of traditional solvothermal methods limit their practical applications. In view of this, Tz based TzPm-COF with a D–A structure could rapidly generate high crystallinity through the microwave-assisted method.82 TzPm-COF had a low exciton binding energy and could effectively generate O2˙−, ultimately driving the photooxidation amination reaction with high recyclability.
 | ||
| Fig. 6 Synthesis of PyTz-COF under solvothermal conditions. Reproduced from ref. 86 with permission from John Wiley and Sons, copyright 2020. | ||
Tz units have abundant heteroatoms with lone pair electrons, which can undergo coordination interactions and improve the crystallinity of COFs, which makes D–A type COFs consisting of the Tz unit have a wide range of applications. Liang and his team designed and prepared a D–A type COF (TPDA-TZDA) based on the Tz unit for enhancing power conversion efficiency in perovskite solar cells.88 Utilizing the coordination effect generated by the interaction between N atoms on the Tz unit and Pb2+, which effectively increased the grain size and crystallinity, suppressed the defects in the perovskite, and promoted the charge transport within the films. The multi-component COF1-AO based on Tz could also have a certain affinity for uranium in seawater, and the extraction efficiency of uranium could reach a record high of 2.45 mg−1 g per day.89 In addition, Tong and his team prepared two different types of D–A structured COFs containing Tz units (COF-TD1 and COF-TD2), which exhibited excellent degradation activity for various antibiotics.90 Under the effect of electronic push–pull, COF-TD1 had good degradation performance in complex water environments and real sunlight, which had certain practical application ability.
 | ||
| Fig. 7 (a) Construction of D–A COFs composed of amine monomers and functionalized terephthalaldehydes, as well as the corresponding HOMO–LUMO orbitals. Reproduced from ref. 81 with permission from Springer Nature, copyright 2023. (b) Synthetic routes of Py-XTP-BT-COFs under solvothermal conditions, and AA stacking mode of Py-ClTP-BT-COF. Reproduced from ref. 100 with permission from John Wiley and Sons, copyright 2020. | ||
Among many different bond connections, constructing fully conjugated COFs can achieve ideal light absorption and charge separation.77,101 Combining the electron-donating pyrene unit with the electron-deficient BT unit enabled the synthesis of a fully conjugated Py-BSZ-COF.102 The introduction of the BT unit induced a redshift in visible light absorption and a rapid separation of photogenerated charges. The Py-BSZ-COF could mediate the photocatalytic oxidative amine coupling of thioamides and the cyclization to 1,2,4-thiadiazoles with high yields and recoverability. The work demonstrated the great potential of fully conjugated COFs with a D–A structure in light-driven organic synthesis. Wang et al. reported two sp2-c conjugated COFs with BT units (HDU-107 and HDU-108).103 Among them, the conjugation of the cyano-functional group of HDU-108 provided a concentrated electronegative active center, which made the COF material active site-rich and enhanced the photocatalytic reduction of Cr(VI).
Benzobisthiazole is a derivative of BT, which can serve as a long linker for BT units and has a stronger electron-deficient ability. Zhao and his team fabricated three D–A type COFs (BTH-1, 2, 3) with benzobisthiazole as a building unit through Knoevenagel condensation.104 The integration of the cyanoethylene bond and the BTH unit provided an electron-deficient π-conjugated symmetric structure, and the semiconducting properties of the π-conjugated COFs could be modulated by changing the electronic properties of the other donor units. BTH-3 with benzotrithiophene as the electron-donor unit exhibited the highest photocatalytic hydrogen precipitation ability under visible light irradiation.
Similarly, Chen et al. reported a D–A type COF (Tz-COF) linked with benzobisthiazole, which accelerated the exciton dissociation by controlling the D–A interaction and thus prolonged the lifetime of photogenerated carriers.94 The optimized Tz-COF-3 had an apparent quantum yield of 6.9% at 420 nm.
Due to the extended conjugated structure of the DPP units and the ability to contribute to efficient carrier transport, COFs composed of DPP show promising applications in organic optoelectronics. As shown in Fig. 8a, Liu and his partners constructed two 2D COFs (DPP-TBB-COF and DPP-TPP-COF) with high crystallinity and AA stacking structure via imine bonding.109 The ordered intermolecular filling structure and the D–A interaction gave the COFs a wide light absorption range as well as suitable HOMO/LUMO energy levels, thus the COFs possessed multifunctional photovoltaic properties (photothermal conversion, supercapacitor performance, and bipolar semiconductor behavior). Lee and his team loaded coplanar organic small molecule DPP units into 2D COFs,110 resulting in stronger conjugation effects within the layered network to enhance light absorption, while a porous polyvinyl alcohol network was used to enhance the hydrophilicity of the overall COFs. Therefore, the prepared layered COFs (COFHS) achieve a high light hot water evaporation rate of 2.5 kg m−2 h−1 and a good ability to purify polluted water under one solar irradiation. Similarly, the DPP2-HHTP-COF coupled with borate ester had excellent photovoltaic properties with conductivity values up to 10−6 S cm−1.111
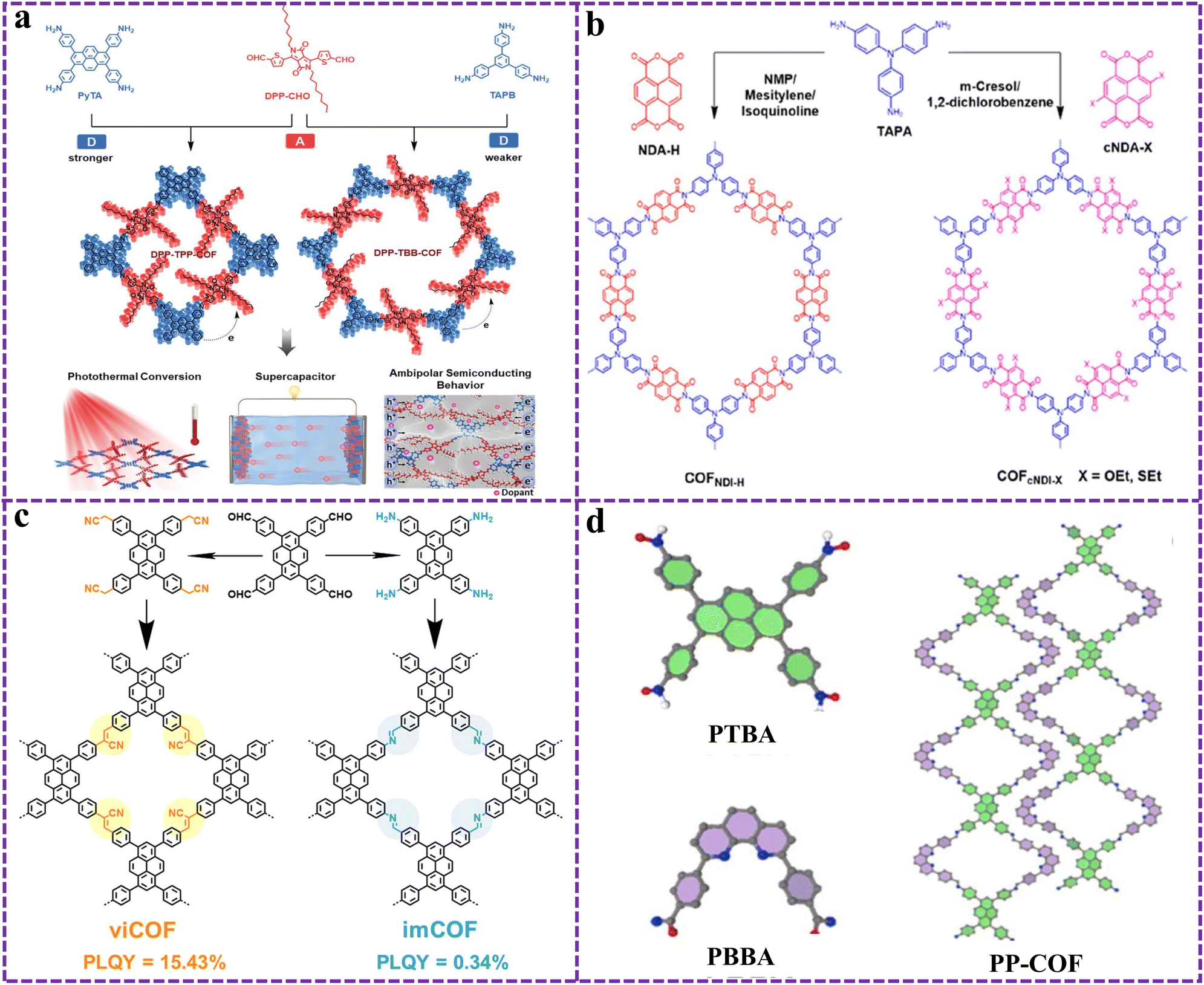 | ||
| Fig. 8 (a) Schematic diagram of the synthesis of a D–A type COF based on 2D DPP, aiming to achieve multifunctional photoelectric conversion properties. Reproduced from ref. 109 with permission from John Wiley and Sons, copyright 2024. (b) Chemical structures of the COFNDI-H, COFcNDI-OEt, and COFcNDI-SEt. Reproduced from ref. 117 with permission from American Chemical Society, copyright 2023. (c) Schematic representation of the syntheses of imCOF and viCOF. Reproduced from ref. 127 with permission from John Wiley and Sons, copyright 2023. (d) Construction of PP-COF. Reproduced from ref. 130 with permission from Royal Society of Chemistry, copyright 2024. | ||
The bandgap and carrier separation ability are important indicators for evaluating the photoactivity of COFs. Generally, it can be achieved by inserting different substituents on the photoactive unit or changing the interlayer spacing. In view of this, Zhan and his partners obtained NDI COFs (COFcNDI-OEt and COFcNDI-SEt) with customizable bandgaps by inserting different substituents without changing the global structure, and compared them with COFNDI-H without substituents (Fig. 8b).117 The insertion of substituents narrowed the bandgap and interlayer distance of COFs, making it easier for charge delocalization. Moreover, ultrafast spectroscopic data were used to perform kinetic analyses to estimate the lifetime of charge recombination. Overall, current studies remain limited in terms of the correlation between photophysical properties and structural features (e.g., interlayer distances).
In addition to the imine linked Py-based COFs generated by the Schiff base reaction mentioned above, the formation of ethylene linked sp2 conjugated COFs through Knoevenagel condensation is also a common method for generating Py-based D–A COFs. Jiang et al. synthesized the first crystalline sp2 c-COF based on Py groups.124 The band gap of sp2 c-COF was 1.9 eV, the aperture was 2 nm, and it had a paramagnetic carbon structure with high spin density. Subsequently, a series of Py-based sp2 conjugated COFs emerged. Recently, Zhang and his partners prepared two ethylene linked g–COF–DMDP-1 and g–COF–DMDP-2 with a high crystallinity honeycomb structure using 5,10-dimethyl-4,9-diazepene as the key monomer.125 One of the COFs had a specific surface area of 1238 m2 g−1. Due to its high hydrophilicity and nitrogen content, g–COF–DMDP-1 produced a high H2O2 yield of 3820 μmol g−1 h−1 in pure water. The 2D D–A type COF with sp2 carbon connections containing Py and tetra styrene units synthesized by the Knoevenagel condensation method exhibited high thermal stability, with a decomposition temperature of 509 °C.126 Moreover, the connection of different bonds in COFs can affect the exciton relaxation mode and fluorescence intensity. As shown in Fig. 8c, Han and his collaborators found a significant difference in solid-state photoluminescence quantum yield between vinyl and imino-based Py-COFs, with values of 15.43% and 0.34%, respectively.127
Py-based COFs are capable of forming 3D and 1D COFs in addition to the common 2D structures. Initially, Wang et al. synthesized 3D COFs (Py-COF) based on a Py-based [4 + 4] imine linkage via the solvothermal method.128 The 3D COF had a dual interpenetrating layer topology and was the first reported fluorescent 3D COF that could be used for explosive detection. Similarly, the two ffc topological structures of D–A type 3D COFs (TFP-Py and TFP-BF) reported by Mahdy exhibit excellent photocatalytic degradation of organic pollutants and hydrogen production performance.129 In addition to 3D COF, 1D COFs (PP-COF) consisting of pyrene with aniline and phenanthroline with aldehyde groups had been reported recently, and the building units and model diagram of PP-COF are shown in Fig. 8d.130 Compared with 2D PyTTA-COF, the prepared PP-COF maintains good crystallinity even after 24 h of immersion in different organic solvents as well as strong acids and bases. Moreover, the good photo-responsive properties and excellent light absorption ability of PP-COF could promote the photocatalytic indole functionalization and oxidized thiol coupling reaction.
Bicarbazole is usually composed of two carbazole units connected by covalent bonds. Its structure not only retains the conjugation and electron donating properties of carbazole, but also brings a series of enhancement effects due to molecular extensibility and symmetry. Kaskel and his colleagues reported two novel equi network chemically stable COFs based on carbazole units (COF-DUT-175 and DUT-176).136 The reversible protonation of imine N in COF can lead to changes in the electronic structure of the conjugated π-system, resulting in characteristic pH responses over a wide pH range without COF decomposition. Both types of COFs are expected to become one of the best materials for indoor humidity control applications. Recently, two D–A type COFs based on carbazole were first used for piezoelectric photocatalytic synthesis of H2O2.137 Vinyl modified Cz-COF achieved record high H2O2 production (9212 μmol g−1 h−1) from air and pure water through piezoelectric photocatalysis. The efficient production rate of H2O2 is due to the synergistic effect between the polarization electric field induced by ultrasound and multiple charge transfer channels separated in space.
Thiophene based COF materials are commonly used as photoactive units due to their good dispersibility, developed pore size, and strong light absorption ability. For example, D–A type COF-S materials prepared from thiophene units were used as photo-initiators to synthesize polymethyl methacrylate (PMMA) through free radical polymerization of methyl methacrylate (MMA) under visible light.140 Similarly, the DTT-ABBO-COF prepared by Chen and his partners had good stability, high activity and recyclability and could be used as an environmentally friendly multiphase catalyst for α-oxidation amination.141 In addition, TTF-PDI-COF,142 ThTz COF,143 TTDA-COF,144 and JLNU-309 (ref. 145) were all D–A type COFs formed by thiophene as a photoactive unit. However, most COFs are crystalline agglomerated powders, which hinder the exposure of surface catalytic active sites and have poor hydrophilicity. As shown in Fig. 9a, Li et al. used a simple electrochemical strategy to exfoliate COFs in water, resulting in exfoliated COFs with more accessible active sites and promoting the dissociation of photoexcited electrons.146 The specific surface area of Exfo-TTB-PT increased from 762 m2 g−1 to 1056 m2 g−1 after exfoliation, and the hydrogen production also reached a maximum of 27.24 mmol h−1 g−1.
 | ||
| Fig. 9 (a) Schematic diagram of the COF electrochemical stripping device, and water contact angle before and after peeling of two types of COFs. Reproduced from ref. 146 with permission from Royal Society of Chemistry, copyright 2024. (b) The synthetic route of thiophene-enriched BUCT–COF–11, and schematic diagram of devices. Reproduced from ref. 147 with permission from John Wiley and Sons, copyright 2024. (c) The model compound of COF-Ph and the isomeric COFs (COF-α and COF-β) dangling with thiophene units, and schematic illustration of the oxygen reduction process on isomeric COF catalysts. Reproduced from ref. 148 with permission from John Wiley and Sons, copyright 2024. | ||
Furthermore, the introduction of the thiophene structure can significantly improve the conductivity of COF materials and enhance catalytic activity by optimizing the electronic structure. For example, a thiophene-rich fully conjugated 3D COF (BUCT–COF–11) with excellent metal-free ORR activity and semiconductor properties was reported by Wang et al. (Fig. 9b).147 The introduction of the thiophene ring significantly promoted charge transfer and improved electrocatalytic efficiency. The peak power density of the fuel cell composed of BUCT–COF–11 as the cathode was as high as 493 mW cm−2. This was the first study which reported that the cathode catalyst of fuel cells was an intrinsic COF material, which broadened the application scope of COFs.
As is well known, the catalytic activity of COFs mainly depends on the molecular structure design. Usually, when the building unit is composed of different elements, different bonding directions and angles, it can lead to completely different molecular configurations, stacking methods, and unsaturated states, even for isomers. At present, it is difficult to further explore the relationship between molecular structure and catalytic activity by controlling a single factor. Based on the above problems, as shown in Fig. 9c, Long and his team employed a thiophene positional isomerization strategy to modulate the molecular geometry, and directionally designed and developed 2-thiophene-containing COF-α and 3-thiophene-containing COF-β heterogeneous COF electrocatalysts (COF-α and COF-β).148 The pentacyclic carbon near the sulfur atom (thiophene α-position) could be accurately identified as the active site for the catalytic reaction of COFs by a combination of in situ Raman spectroscopy and DFT calculations. The ORR reaction mechanism diagram of the COF is shown in Fig. 9c (on the right). Due to the localized charge effect and having more exposed active sites, COF-β exhibited higher ORR performance than COF-α, which had non-localized charge around the dangling thiophene unit.
In general, there exist two methods for the synthesis of D–A type COFs with benzothiophene groups. One is by post-modified bromine functionalization of vinyl-conjugated COFs, followed by further conversion to the thiophene-linked conjugated system 2D COF-S by thiolation cyclization.151 The high π-conjugation extended the light absorption edge of COF-S from 480 nm to 800 nm, while the intrinsic D–A structure promotes the separation of photogenerated electrons–holes. The second approach is to utilize benzothiophene-containing aldehyde or amine monomers that can synthesize imine-linked benzothiophene-based COFs under mild conditions. For example, Baek and his research group designed and synthesized four benzothiophene-based COFs with aldehyde groups and investigated the effect of conjugation degree on photocatalytic performance.152 Due to the different degrees of conjugation in the four COFs, the bandgap of their π–π* leaps becomes narrower with the extension of conjugation, and the wavelength of absorption becomes longer. Moreover, the electron affinity increases with the increase of the number of aromatic rings, and the stabilization of the structure increases when the COFs get extra electrons. Similarly, JLNU-310,311,153 BTT-TAPT-COF,154 and DADP-COF155 were all benzothiophene-based COFs synthesized by the Schiff base reaction. Compared to 2D COFs, fully conjugated 3D COFs have abundant open channels and isolated bits, which overcome the interlayer charge transfer bottleneck of 2D COFs and provide more charge transfer pathways. Recently, Wang and his partners synthesized a series of fully conjugated three-dimensional COFs (BUCT–COF–20, 21, 22 and 23) by combining benzothiophene units with saddle shapes and biphenyls with different substituents.156 The non-conjugated 3D COFs containing sp3 carbon-based tetrahedral linkages had much lower hydrogen production rates than the four conjugated 3D COF, confirming that the extended π-conjugated structure could provide efficient charge transport channels and enhanced photocatalytic performance. The maximum hydrogen evolution rate of the fully conjugated BUCT–COF–20 was 40.36 mmol g−1. Importantly, this represented the first report of intrinsically metal-free 3D COFs unitized for photocatalytic hydrogen evolution, and the achievement greatly expanded the application scope of 3D COFs.
4. Optimization strategy of D–A type COFs
In D–A type COFs, the different electron affinities between D and A promote charge transfer from D to A in the excited state. Ultimately, the electrons are favorably concentrated on the acceptor unit, which can effectively promote exciton dissociation. However, there are several strategies that can further improve the performance of D–A type COFs, such as post-modification, supramolecular assembly, and interface engineering, which have achieved some remarkable results until now.4.1 Post-modification
4.1.1.1 Covalent bond modification. Nucleophilic substitution reaction: takes advantage of the reactive sites existing in the COF structure, such as aryl halides.157,158 These sites are capable of reacting with nucleophilic reagents. For example, organic molecules with nucleophilic groups like amino groups and hydroxyl groups can react with the halogenated groups on COFs.159 Through this nucleophilic substitution reaction, novel functional groups or molecular fragments can be introduced into the COF framework.
Click chemistry: it refers to a class of chemical reactions that are distinguished by high efficiency and high selectivity, which is often employed for the post-modification of COFs.160 Among them, the copper-catalyzed azide–alkyne cycloaddition reaction stands out as one of the most prevalently used click chemical reactions.161 By introducing azide groups or alkyne groups onto COFs and then reacting with molecules containing corresponding complementary groups, the functionalization of COFs can be achieved rapidly and efficiently. In this way, a diverse range of functional molecules or groups can be introduced onto the surface or within the framework of COFs.
Condensation reaction: taking advantage of groups such as aldehyde groups and amino groups contained in the COF structure, new functional groups or structural units can be introduced through condensation reactions. For instance, aldehyde groups and amino groups can undergo a Schiff base reaction to form imine bonds.162,163 Through this approach, functional molecules containing amino groups or aldehyde groups can be linked to COFs, thus achieving the modification of COFs. In addition, common post-modification reactions include the Povarov reaction,164,165 nitrogen-containing Diels–Alder reaction,166 and oxidative cyclization reaction.167
4.1.1.2 Metal coordination modification. Direct coordination: the solution that contains metal ions is blended with the COF material. Certain coordination sites within the COF structure, such as nitrogen-containing heterocyclic groups including pyridine168,169 and imidazole,170 as well as oxygen-containing groups like carboxyl groups and hydroxyl groups, can directly coordinate with metal ions.171 Consequently, the metal ions are bonded to the COFs, forming metal–COF complexes, which endow COFs with new properties, such as catalytic activity.172
Ligand exchange: firstly, COFs are made to coordinate with a ligand to form a stable COF–ligand complex. Subsequently, this complex is immersed in a solution that contains the target metal ions. Through the ligand exchange reaction, the target metal ions substitute the original ligand and coordinate with the COFs, thereby achieving the metal coordination modification of COFs.173–175 This method can more precisely control the coordination environment and the loading amount of metal ions.
 | ||
| Fig. 10 (a) Post-synthetic annulation of NJU-319Fe. Reproduced from ref. 176 with permission from American Chemical Society, copyright 2023. (b) Schematic diagram of the confinement of uniformly dispersed POM clusters within the pores of COFs through covalent bonds. Reproduced from ref. 183 with permission from American Chemical Society, copyright 2022. (c) Schematics of the synthesis of COF-284-NH2. Reproduced from ref. 159 with permission from American Chemical Society, copyright 2023. (d) Scheme of the synthesis of the thiazole-linked COF (THZ-DMTD) through post-synthetic modification of TTT-DMTD COF. Reproduced from ref. 185 with permission from John Wiley and Sons, copyright 2023. | ||
Some COFs will have poor hydrophilicity and dispersion, which makes it difficult to apply them in liquid-phase reactions. The hydrophilicity of COFs can be well changed by post-modification to solve the above problems. For instance, in order to be able to design a bifunctional material that can fulfill the need of integrating hydrogen supply and proton conduction, Liu et al. improved the hydrophilicity and carrier separation of the material itself by converting the cyano-group on the synthesized D–A-type PyBT-COF into a carboxyl group.177 The water contact angle of PyBT-COF-COOH was much smaller than that of PyBT-COF, which enabled stronger intermolecular interactions with water and easier dispersion in water. As a result, the post-modified PyBT–COF–COOH exhibited a strong photocatalytic hydrogen precipitation rate as well as proton conductivity. Fluorescent COF (TMT-TA) had good crystallinity and stability, but poor dispersibility in water. In view of this, Yan and his team also introduced carboxyl groups into TMT-TA through a post-modification strategy, and the obtained TMT-TA-COOH not only had good dispersibility in water, but also had high sensitivity for the quantitative identification of 5-HIAA.178 In addition, the application of TMT-TA-COOH in fingerprinting had some potential due to the interaction of residual amino acid bonds and fatty compounds in fingerprints with the carboxyl groups on the surface of TMT-TA-COOH. In the same way, the hydrophilicity of triazinyl COFs could be improved by in situ growth of FeOOH clusters on TaTz COFs.179 The post-modified super-hydrophilic TaTz-FeOOH photocatalysts exhibited effective photo-oxidation of various organic pollutants.
The most common technique for building functional COFs is the post-modification strategy. After modification, some functional groups can be introduced into the COF framework, such as coordinating some metals or connecting organic small molecules with catalytic sites, thereby enhancing the photocatalytic performance of COFs. Initially, Jiang and his partners utilized click reactions to anchor fullerenes covalently in the nanochannels of COFs, transforming them into ordered D–A structured framework materials.180 Baldwin et al. synthesized a series of post-modified COFs by embedding alkyl, alcohol, or aryl groups into the phenol portion of a COF.181 The grafting of these functional units resulted in COFs that had a good affinity and sensitivity for a number of volatile gases, and also a high selectivity for toluene and isopropanol. Embedding metals atomically into the framework of COFs as catalytic active sites is also a commonly used post-modification strategy. Ye and his team embedded single atom Co into the bipyridine unit of D–A type sp2 c-COFdpy as a catalytic active site for photocatalytic CO2 reduction.182 The structural advantage of this CC connection allowed carriers to easily reach the Co site through an electronic cascade, resulting in a photocatalytic CO production rate of 17.93 mmol g−1 and a selectivity of 81.4% for sp2 c-COFdpy-Co. Compared with supramolecular interactions such as van der Waals force, hydrogen bonding force, Coulomb force, etc., metal clusters stabilized in pores through covalent bonds are undoubtedly the most stable. TCOF-MnMo6 was a modified COF with a bex topology synthesized by covalently connecting polyoxometalates (POMs) uniformly dispersed in D–A type COFs (Fig. 10b).183 Specifically, covalent bonds provided efficient electron transfer, and highly dispersed MnMo6 clusters serve as catalytic active sites for CO2 reduction. The CO yield of TCOF-MnMo6 was 37.25 μmol g−1 h−1, which is much higher than that of ECOF-MnMo6. Post-modified functionalized COFs can exhibit better activity in photocatalytic reactions, indicating that the post-modification strategy is an effective and simple functionalization method. Recently, Yaghi et al. synthesized two COFs (COF-284 and COF-285) that were stable under extreme conditions through aldehyde alcohol ring trimerization.159 In Fig. 10c, subsequent post-synthetic modifications of primary amines were carried out to reveal the ability of these two COFs to absorb CO2 in flue gas and air. Overall, the problem with introducing metal or small molecule strategies is that while they can introduce excellent active sites for COFs, they also reduce their crystallinity, and it is also difficult to ensure that the post-modified groups can be uniformly distributed on the COF backbone.
Converting COFs with poor chemical stability linked to imines into COFs with high chemical stability and conjugation linked to thiazoles is also a common post-modification strategy. At the earliest, Yaghi and his team converted the imine bond into two COFs with thiazole and oxazole bonds by linker substitution and oxidative cyclization, and this derived COF material exhibited higher chemical stability.184 Byon and his partners compared the stability of COF electrodes in organic lithium batteries with imine connections and those converted to thiazole connections.167 It was shown that the structural stability of the organic electrodes was enhanced by the π-conjugation and excellent crystallinity of the thiazole moiety, providing extended recyclability and a fast-charging process. The AZO-1 COF electrode connected with thiazole exhibits excellent cycling stability and a power density of approximately 2800 Wk g−1. In contrast, AZO-2 COF connected with imine and AZO-3 COF connected with β-ketoamine were prone to decomposition in azo reactions due to chemical instability and non-coupling, resulting in poor cycling performance. Furthermore, sulfur assisted chemical conversion methods could be used to convert TTT-DMTD COFs containing high-density redox sites into robust thiazole linked D–A type THZ-DMTD COFs (Fig. 10d).185 Briefly, the reaction of singlet sulfur with the aromatic imine from COF oxidizes the imine to thiamine, followed by further oxidative cyclization of the thiamine to form a thiazole ring. The capacity and stability of Li–S batteries were enhanced while ensuring crystallinity. In addition, this post-sulfurization process can not only maintain crystallinity, but also expand the π-conjugation degree of the COF itself, which is beneficial for light collection. Chen et al. similarly utilized singlet sulfur to convert imine-conjugated D–A type 4PE-N COFs into thiazole-conjugated 4PE-N-S COFs.186 This strategy extends the conjugated structure of the COFs in both the x and y directions, which leads to higher electron transport and light absorption. It is worth mentioning that the thiazole-linked 4PE-N-S COFs still have good crystallinity when immersed in common organic solvents or strong acids and bases for 3 days. In contrast, the structures of 4PE-N and 4PE-TT COFs were disrupted when immersed under the same conditions.
4.2 Supramolecular assembly
Supramolecules usually refer to two or more molecules that are bonded together through secondary bonds between molecules to form a multimolecular system at the molecular level. Generally, they are automatically bonded into orderly and organized polymeric systems by non-chemically established weak forces such as hydrogen bonding, coulombic interactions of charges, van der Waals forces, and π–π interactions (formation of stacks of off-domain π-bonds).187,188 Unlike traditional chemistry which mainly studies covalent bonds, supramolecular chemistry studies weak and reversible non-covalent interactions between molecules, which can be used to significantly accelerate the reaction rate or allow highly selective reactions. As an example, ultra-thin bithiazole-based COF nanosheets (COF-Bta-NSs) with a thickness of about 1.95 nm were prepared in high yield by Zhu et al.189 The COFs were assembled with acetylcholinesterase (AChE) through strong supramolecular interactions, and electrochemical biosensors with excellent performance were constructed, which could be used for the efficient detection of organophosphorus pesticides. The AChE molecules were confirmed to be immobilized on the surface of COF-Bta-NSs through dense hydrogen bonding interactions by various spectroscopic means, which improved the sensitivity and detection durability of the sensor. In addition to this, the heteroatom-hydrogen bonding connection between the COF and the metal coordination played a decisive role in the interfacial transfer in supramolecular systems.190 Specifically, the photogenerated electrons in the COF were transferred to the metal coordination compounds for CO2 reduction through the bridge of heteroatoms and hydrogen bonds, and the stronger this hydrogen bonding effect, the higher the photocatalytic CO2 to CO conversion rate. In supramolecular systems, this heteroatom–hydrogen bond bridging electron transfer pathway confers high photocatalytic performance and provides a way to rationally design efficient supramolecular photosystems.The π–π supramolecular interactions formed by the stacking of off-domain π-bonds are capable of enhancing the visible light trapping and facilitating charge transfer of D–A type COFs. TT-Por(M)-COFs, for example, had π-stacked column arrays and ordered D–A heterojunction structures that exhibited enhanced intra- and inter-layer charge separation and migration, enabling the efficient photoreduction of CO2.139 The vertically oriented D–A separation column of the COF was capable of forming a plate-like interface of periodically ordered heterojunctions, leading to the generation of supramolecular π–π interactions. This will effectively separate the photogenerated carriers and facilitate their migration along the column to the surface, realizing rapid carrier migration and separation. The transient absorption spectra and theoretical computational analyses indicated that the long-term charge retention induced by the intra-layer ordered D–A structure and π-stacked column arrays were the reason for the excellent photoreduction of CO2.
In general, z-direction modulation of 2D COFs relies on spontaneous π–π interactions to form 3D structures, which is difficult to realize. Despite the importance of π–π stacking in the formation, property modulation and stability of COFs, only a few have been proposed to improve the vertical stacking behavior of 2D COF layers. Inspired by the structural modulation mode of graphite intercalation compounds, Lin et al. utilized a supramolecular self-assembly system to embed guest molecules between the covalent layers of the COF structure through a bottom-up strategy, thereby generating D–A intercalation COFs capable of modulating the z-direction of the lamellar heterostructure (Fig. 11a).119 Specifically, stabilized intercalated COFs with alternating D–A stacking arrangements were constructed using electron-deficient PDI as the acceptor unit, electron-rich perylene as the donor unit and intercalating agent in the vertical direction, and p-phenylenediamine as the polymerizing agent in the lateral direction. This strategy overcame the dispersive interactions between the large π–π surfaces as well as the electrostatic D–A pairs between most of the COFs with insufficient attractive forces. TTF-intercalated COF@C was also an intercalated COF synthesized by supramolecular assembly, which used triazine as the acceptor unit and electron-rich thiofulvalene as the donor unit and intercalating agent. This intercalated COF was able to efficiently convert the light energy into heat energy with a photothermal conversion efficiency of up to 38.1%.191 Moreover, this intercalated COF could be encapsulated by cancer cell membranes with good biocompatibility and tumor-targeting ability, and can effectively inhibit tumor growth under laser irradiation. Overall, a stable D–A alternate stacking arrangement is a necessary prerequisite for the successful construction of D–A intercalated COFs, and this novel assembly method is expected to be able to provide new nanotechnology applications in the future.
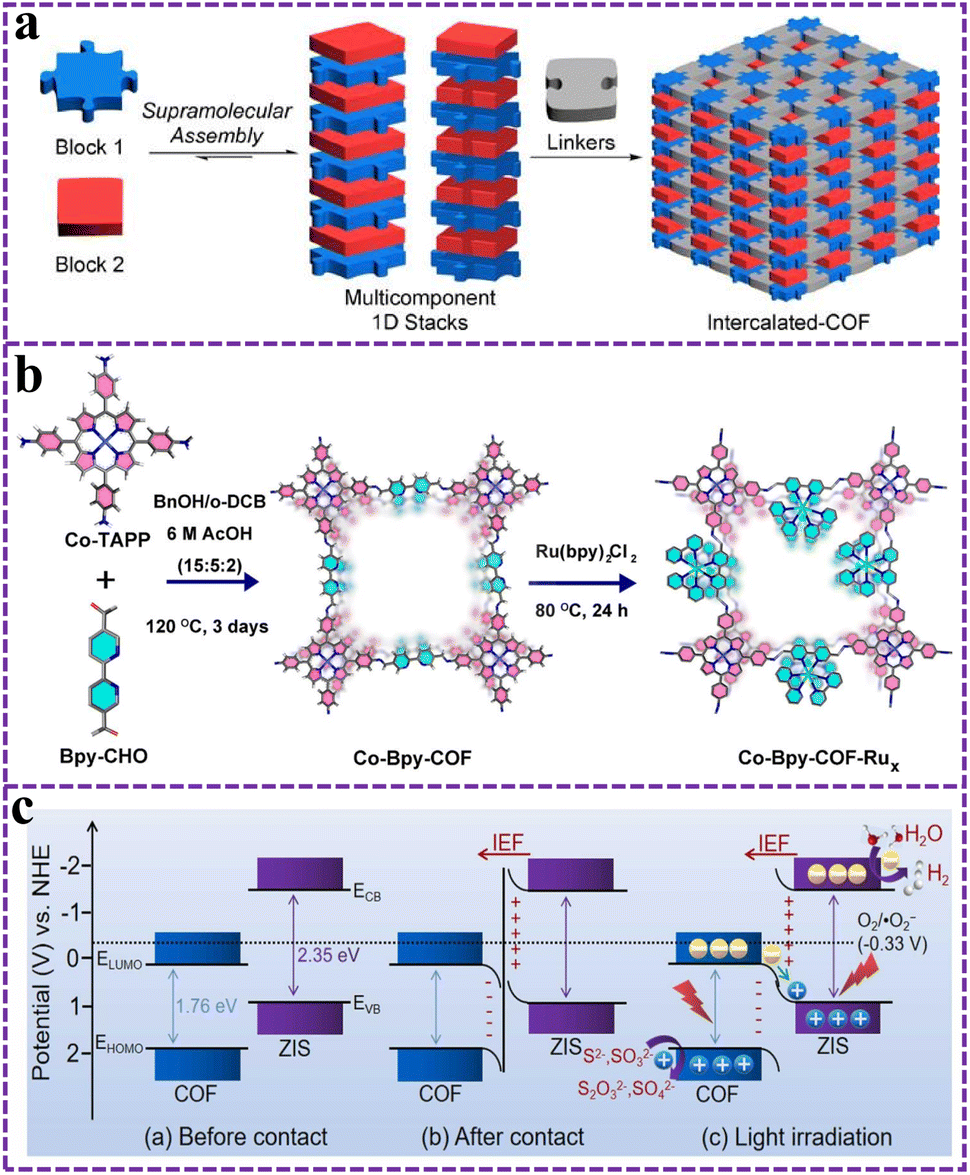 | ||
| Fig. 11 (a) Schematic illustration of supramolecular multicomponent 1D stacks toward intercalated-COFs. Reproduced from ref. 119 with permission from American Chemical Society, copyright 2020. (b) Schematic illustration of the construction of Co-Bpy-COF and its post-synthetic modification to Co-Bpy–COF–Rux. Reproduced from ref. 201 with permission from American Chemical Society, copyright 2023. (c) Charge-transfer mechanism of the S-scheme COF-ZIS heterostructure before contact, after contact in the dark and light irradiation. Reproduced from ref. 205 with permission from Elsevier, copyright 2024. | ||
4.3 Interface engineering
The design and construction of interfacial engineering of COF catalysts are the key factors to improve their catalytic performance and stability. Currently, the common optimization of COF interfacial engineering mainly includes doping,192,193 morphology modulation,194 coupling photosensitizers195 and constructing heterojunctions.196,197 This optimization of interfacial engineering can produce a 1 + 1 > 2 effect with the interaction between D–A type COFs, which can substantially improve the performance and application ability of COFs. Both metal doping and non-metal doping are effective methods to enhance COFs in terms of energy band structure, band gap, properties and other parameters. This facilitates charge carrier separation, optical trapping range expansion, energy band structure optimization and substrate adsorption/desorption. In order to explore new ways with modulating exciton dissociation in COFs to promote photocatalysis, Li and his team doped the metal Ni into fluorenylketone-based D–A type COFs by doped (Ni–COF-SCAU-1).198 The addition of Ni significantly modified the exciton effect and promoted the surface hydrogen precipitation reaction under photoexcitation. The results showed that the hydrogen precipitation rate of Ni–COF-SCAU-1 reached 198 mmol g−1 h−1 at 420 nm under visible light and the AQE was as high as 43.2%, which was much higher than that of COF-SCAU-1. This metal-doping strategy opens up a new pathway for exciton modulation in COFs. Wang and his partners constructed a copper-porphyrin doped D–A type COF (CTF-BTA2OH-Por(Cu)) for multiple efficient sterilization.199 The Cu-porphyrin doping enhanced the ROS generation and could destroy proteins for sterilization in the absence of light. Non-metallic doping extends the conjugated system, facilitates enhanced light absorption and charge separation and transport rather than recombination. COF-IsoQ-Tp was a controllable N-doped D–A type COF based on bottom-up molecular precursor selection.200 The N-doped array of ordered photocatalytic sites functioned within the porous β-ketoamine carbon framework in the reticular COF-IsoQ-Tp. Compared with the homostructured COF-Naph-Tp without N doping, COF-IsoQ-Tp provided excellent peak photoelectric activity (J = −16 μA cm−2) and its photocatalytic hydrogen evolution rate was also three orders of magnitude higher.Coupling a photosensitizer on a D–A-type COF enables a long-lived triple excited state photosensitive donor to extend the excited state lifetime of the active site and also generates an internal driving force with a well-defined direction like a built-in electric field to enhance the photovoltaic efficiency. In Fig. 11b, Huang et al. used a photosensitizer (Ru(bpy)3Cl2) with a good excited donor state characterization for photovoltaic CO2 reduction by integrating it into Co-bpy-COF.201 The problem that the excited state in the active site is easily dissipated by thermal release, luminescence and electron transfer to nearby substances resulting in short excited state lifetime has been solved. Specifically, the powerful donor photosensitizer Ru(bpy)3Cl2 with a long-lived triplet excited state could transfer excitation electrons to the Co-bpy-COF, lowering the energy barrier for photo-electrocatalytic CO2 reduction. Notably, only trace amounts of H2 and CO production were detected on the Co-bpy-COF in the absence of Ru(bpy)3Cl2. Strategies for coupling photosensitizers can be constructed not only by coupling to D–A COFs through bonding interactions, but also by loading. Chen and his team constructed magnetic Fe3O4@D–A COFs as photoactive materials, which were used as photocurrent–polarity switching factors by loading ZnSe quantum dots onto the COFs.202 ZnSe quantum dots as photosensitive materials were matched with D–A type COF energy levels to change the polarity of the photocurrent, which were used as mir-138 sensors with high selectivity and sensitivity. However, the system of this strategy is more complicated and it is not possible to determine the real active site or the magnitude of the role played by each player, so in-depth exploratory mechanistic studies are lacking.
It is well known that improving the separation rate of electron holes in D–A type COFs is an important factor to enhance their photocatalytic performance.203 And the formation of a heterojunction can enable the transfer of photogenerated carriers from the COF to the surface of another semiconductor, thus realizing the effective separation of photogenerated electrons and holes. In addition, heterojunctions may not only inherit the properties of individual components, but may also exhibit new properties, which may synergistically contribute to the enhancement of photoactivity. The more common types of heterojunctions studied are mainly type-II heterojunctions, Z-scheme heterojunctions, and S-scheme heterojunctions. For type-II heterojunction, Ding et al. recently obtained heterojunction COF materials with excellent photocatalytic nitrogen fixation performance by compositing a D–A type COF with g-C3N4 with N vacancies. The optimized COF composite showed an NH2 production efficiency of 646 μmol h−1 g−1 at 420 nm without adding a sacrificial agent.
But the type II heterojunction still has fundamental problems at present, such as the efficiency of the photogenerated electron hole separation is at the cost of reducing the reduction and oxidation ability of the two semiconductor photocatalysts. In addition, the presence of photogenerated electrons and holes in the original photocatalysts inhibits the interfacial transfer of electron holes in the other catalysts due to electrostatic interactions. Comparatively, the formation of Z-scheme as well as S-scheme heterojunctions can well avoid the above problems. However, due to some defects and irrationality in the mechanism of the Z-scheme heterojunction when it was initially set up, the charge transfer mechanism of the Z-scheme heterojunction has been gradually replaced by that of the S-scheme heterojunction.204 Wang and his partners formed core–shell structural complexes with S-scheme heterojunctions by growing ZnIn2S4 nanosheets in situ on a porphyrin-based COF with a D–A structure.205 Specifically, the COF and ZnIn2S4 form a built-in electric field due to the difference in Fermi energy levels when they are in contact. Under the effect of the built-in electric field and energy band bending, the electrons and holes with strong reducing and oxidizing ability were retained (Fig. 11c). The PHE efficiency of the COF-ZIS composite was 2711 μmol h−1 g−1 with an AQY of up to 2.45% at 400 nm when Pt was the co-catalyst. However, the biggest disadvantage of heterojunctions is that most of the heterojunctions formed by the compositing of two materials are independent and not in contact with each other, and the number of heterojunctions formed is very small. Therefore, precise control and improvement of the number of heterojunctions formed is the way to greatly improve the photocatalytic efficiency. In addition, lattice mismatches, defects at phase interfaces, and electronic energy band shifts may occur at heterojunctions during long-term cycling tests.
5. Application of D–A type COFs
The previous sections emphasized the advantages of D–A type COFs, including high crystallinity, porosity, and stability. In addition, the selection of D and A molecular units, the choice of connecting bonds, the regulation of crystallinity and conjugation, and the functionalization of active centers give D–A type COFs different properties. This can meet the needs of many applications such as photocatalysis, photothermal therapy and energy storage (Table 1).| Application | Photocatalyst | Linkage | Lightsource | Conditiona | Performance | Ref. |
|---|---|---|---|---|---|---|
| a AA: ascorbic acid; TEOA: triethanolamine. | ||||||
| H2 evolution | PyPz-COF | Imine | ≥420 nm | H2O/AA | 7542 μmol g−1 h−1 | 118 |
| Py-DNII-COF | Imine | ≥420 nm | H2O/NMP/AA | 625 μmol g−1 h−1 | 206 | |
| COF-F | Imine | AM 1.5 | H2O/AA | 10580 μmol g−1 h−1 |
207 | |
| Py-FTP-BT-COF | Imine | >420 nm | H2O/AA | 177.50 μmol h−1 | 100 | |
| sp2c-Py-BT COF | Vinylene | >420 nm | H2O | 17.2 μmol g−1 h−1 | 210 | |
| BTT-BpyDAN-COF | Vinylene | >420 nm | H2O/TEOA | 10100 μmol g−1 h−1 |
64 | |
| COF-JLU45 | Vinylene | >420 nm | H2O/AA | 272500 μmol g−1 h−1 |
209 | |
| COF-alkene | Vinylene | >420 nm | H2O/TEOA | 2330 μmol g−1 h−1 | 211 | |
| DCNA-1 | Imine | >420 nm | H2O/TEOA | 27900 μmol g−1 h−1 |
212 | |
| HIAM-0015 | Imine | >420 nm | H2O/AA | 17990 μmol g−1 h−1 |
213 | |
| COF-JLU35 | Vinylene | >420 nm | H2O/AA | 70800 μmol g−1 h−1 |
214 | |
| HBT-COF | Imine | ≥420 nm | H2O/AA | 19.00 μmol h−1 | 91 | |
| TtaTfa-COF | Imine | >420 nm | H2O/TEOA | 20700 μmol g−1 h−1 |
215 | |
| BTT-BPy-COF | Vinylene | >420 nm | H2O/AA | 15800 μmol g−1 h−1 |
216 | |
| MAC-FA1/S-COF | Imine | >420 nm | H2O/AA | 100000 μmol g−1 h−1 |
218 | |
| H2O2 production | TAPD-(Me)2 COF | Imine | 420–700 nm | H2O/ethanol/O2 | 234.52 μmol g−1 h−1 | 223 |
| TP-DPBD30-COF | Hydrazone | >300 nm | H2O/air | 9200 μmol g−1 h−1 | 224 | |
| TT-DTDA-COF | Imine | >420 nm | H2O/O2 | 1302 μmol g−1 h−1 | 225 | |
| TAPT-TFPA COFs@Pd IC | Imine | AM 1.5 | H2O/ethanol/O2 | 2143 μmol g−1 h−1 | 226 | |
| COF-JLU90 | Imine | AM 1.5 | H2O/O2 | 9800 μmol g−1 h−1 | 221 | |
| COF-N32 | Imine | >420 nm | H2O/O2 | 605 μmol g−1 h−1 | 227 | |
| PB-COF | Imide | AM 1.5 | H2O/O2 | 2044 μmol g−1 h−1 | 228 | |
| JUC-675 | Imine | >420 nm | CH3CN/benzylamine | 22800 μmol g−1 h−1 |
229 | |
| COF-TPT-Azo | Azobenzene | >420 nm | H2O/O2 | 1498 μmol g−1 h−1 | 230 | |
| COF-1 | Imine | >420 nm | Seawater | 6930 μmol g−1 h−1 | 231 | |
| EBA-COF | Vinylene | >420 nm | H2O/ethanol/O2 | 1830 μmol g−1 h−1 | 74 | |
| TTF-BT-COF | Imine | AM 1.5 | H2O/O2 | 276000 μmol g−1 h−1 |
232 | |
| TDB-COF | Imine | AM 1.5 | H2O/O2 | 723.5 μmol g−1 h−1 | 233 | |
| ECUT-COF-50 | Imine | ≥400 nm | H2O/air | 4742 μmol g−1 h−1 | 234 | |
| Organic conversion | Bpy-sp2c-BTT-COF | Vinylene | LED | — | Thioether oxidation (93%) | 235 |
| En-COF-P | Ketoenamine | LED | — | Thioether oxidation (99%) | 160 | |
| TpAQ-COF | Imine | LED | — | Oxidation of amines (92%) | 236 | |
| COF-NUST-36 | Imine | LED | — | Oxidation of amines (98%) | 237 | |
| TP-PB-COF | Imine | LED | — | Indole derivative C-3 thiocyanation (97%) | 238 | |
| COF-JLU24 | Imine | LED | — | Indole derivative C-3 thiocyanation (98%) | 122 | |
5.1 Photocatalysis
In addition to the effect of substituents, recent studies have shown that covalent bonding between different photoactive units is a key factor in determining the photocatalytic activity of COFs.208 In contrast, sp2-linked fully conjugated COFs can enhance charge-carrier mobility and promote charge transfer, which is highly desirable for photocatalytic applications.209 The imine-Py-BT COFs linked by imine bonds and the cyanoethylene-linked sp2c-Py-BT COFs then had obvious differences in the photocatalytic activity of water splitting (Fig. 12a).210 The sp2c-Py-BT COF with the D–A energy band structure could directly decompose H2O into H2 and O2 under visible light irradiation, whereas the imine-Py-BT COF was not able to catalyze the total water cleavage reaction because of the insufficient OER capacity. Experiments and DFT calculations had showed that cyanoethylene bonding could reduce the exciton binding energy of the COF (Fig. 12b), which was more conducive to promoting charge separation and the dissociation of photo-generated electrons. Similarly, the cyanoethylene-linked COFs constructed from benzotrithiophene as the donor unit exhibited a good photocatalytic hydrogen evolution rate (10.1 mmol g−1 h−1) and AQY (4.83%).64 This was mainly attributed to the fact that the bipyridine unit favored the formation of an intermolecular electron transfer pathway with benzotrithiophene, thus promoting charge separation and transfer efficiency. Yu and his team constructed three COFs with different connecting bonds but the same structure, and similarly found that the photocatalytic hydrogen precipitation performance of the COFs connected by cyanoethylene bonds was much better than that of the imine- and imide-connected COFs, and reached an AQE of 6.7% at 420 nm.211
 | ||
| Fig. 12 (a) Synthetic routes and chemical structures and (b) exciton dissociation rates of sp2c-Py-BT COF and imine-Py-BT COF. Reproduced from ref. 179 with permission from John Wiley and Sons, copyright 2024. (c) The chemical backbones of the isomeric COFs DCNA and DNCA, and (d) comparison of the photocatalytic HER rates of all DCNA_AC and DNCA_AC COFs studied in this project. Reproduced from ref. 212 with permission from Springer Nature, copyright 2022. (e) Syntheses of COF-JLU35 and COF-JLU36 through Knoevenagel and Schiff-base condensation reactions, respectively, and (f) comparison of mass normalized HER performance between COF-JLU35, COF-JLU36 and other COF materials. Reproduced from ref. 214 with permission from American Chemical Society, copyright 2023. | ||
In addition to the aforementioned building block connections and bond types that can influence photophysical properties, the structural isomers of the bonds are equally important for photocatalytic performance studies. Previous work on imine-linked COFs had typically been random in the choice of which was the amino-functionalized monomer as well as whether it was the aldehyde-functionalized monomer, or the monomer that was cheaper or easier to prepare had been chosen. However, this decision may significantly affect the properties of the final material as it predetermines the orientation of the CN bond to the different linkers. In response to the stoichiometry proposed above, as shown in Fig. 12c, Thomas and his team designed and synthesized imine-bonded D–A-type COFs with differently oriented isomers, (D–CN–A (DCNA) and D–NC–A (DNCA)), respectively, in order to study and compare the effects that their structures had on the photovoltaic properties.212 Although the change in CN bond orientation did not alter the topology of the COFs, the DCNA-1 COFs were more ordered than DNCA-1, had more than twice the specific surface area of DNCA-1, and had a higher charge transfer efficiency. As a result, all DCNA COFs exhibited stronger photocatalytic hydrogen precipitation performance than the corresponding DNCA COFs (Fig. 12d). Similarly, a series of D–A-type COFs with different imine bond orientations based on BT units also exhibit markedly different photocatalytic hydrogen precipitation abilities.213 HIAM-0015 with a stronger electron-deficient capacity showed the highest hydrogen precipitation rate, which was about 15 times higher than that of its isomer HIAM-0015v. This strategy provides new ideas for the design and synthesis of COFs, and it is believed that this concept of bond isomerization can be extended to synthesize a wider range of connecting bonds for COFs in addition to imine bonds. Liu and his team constructed two photoactive three component D–π–A materials by introducing electron-deficient triazine and electron-rich benzothiophene groups into the framework (Fig. 12e).214 This fully π-conjugated bond effectively broadened the visible light capture range of COFs and improved charge transfer and separation efficiency. Among them, COF-JLU35 had a hydrogen evolution rate of 70.8 ± 1.9 mmol g−1 h−1 under visible light, which is superior to that of the previously reported COF materials (Fig. 12f).
Subtle structural modifications can significantly modulate the band structure and interfacial properties. For instance, the photocatalytic hydrogen precipitation performance could be improved by changing the hydrophilicity of D–A type COFs. Jiang and his partners reported and compared the photoelectric properties of HBT-COF containing hydroxyl groups and BT-COF without hydroxyl groups.91 It was found that introducing hydroxyl groups into BT based COFs and polymers improved the hydrophilicity and energy level structure of COF, and enhanced the effective photo-induced charge transfer and segregation. The HER performance of HBT-COF was 5 times higher than that of BT-COF. Furthermore, protonation of the imine bond was also able to improve the hydrophilicity of the D–A-type COF, which enhanced the photocatalytic hydrogen precipitation performance of TtaTfa-COF (20.7 mmol g−1 h−1).215 This post-protonation modification can be applied not only in imine-linked COFs, but also in fully conjugated ethylene-linked COFs. Recently, Wang and his team prepared a protonated sp2 bonded BTT-BPy-PCOF using bipyridine as a postmodified protonation site.216 This protonation modification improved the charge separation efficiency and increased the hydrophilicity in the COF pore. The HER rate of the protonated BTT-BPy-PCOF was as high as 15.8 mmol g−1 h−1, which was about 6 times higher than that of the unmodified BTT-BPh-COF. Importantly, this post-modified protonation is universal.
In addition to the modulation of D–A COFs described above, compositing with other materials is also an effective way to prepare highly active D–A type COF photocatalysts. Bimetallic Pt-based nanoclusters were loaded onto D–A COFs for photocatalytic hydrogen production.217 The interaction between COFs and metal nanoclusters promoted the adsorption and activation of H2O molecules and accelerated the release of hydrogen. Under visible light, PtCo2@COF exhibited the highest photocatalytic activity with a turnover frequency of 486 min−1. Similarly, metal–organic rings with a photosensitizing effect (MAC-FA1) and coral-like S-COF were constructed with direct Z-scheme photocatalysts for the hydrogen evolution reaction through supramolecular interactions.218 The optimized 4% MAC-FA1/S–COF exhibited a hydrogen evolution rate of 100 mmol g−1 h−1, which was much higher than that of S-COF alone, and was one of the best COF-based photocatalytic hydrogen evolution catalysts at that time.
 | ||
| Fig. 13 (a) Schematic of manufacturing H2O2 solution via continuous photocatalysis in a flow reactor and flow reactor performance for continuous manufacture of pure H2O2 solution. Reproduced from ref. 224 with permission from Springer Nature, copyright 2024. (b) Schematic illustration of the octupolar structure and photosynthesis of H2O2 using COF-N31, COF-N32 and COF-N33. Reproduced from ref. 227 with permission from Springer Nature, copyright 2023. (c) Illustration of the synthesis and topology representation of 3D COFs, and photocatalytic H2O2 production performance of COF-1 and COF-2 in seawater. Reproduced from ref. 231 with permission from John Wiley and Sons, copyright 2024. | ||
The magnitude of intramolecular polarity in COF has a significant effect on exciton formation and dissociation. Tong and his team found that the effect of the magnitude of intramolecular polarity on the photocatalytic performance also follows the volcano model by varying the number of phenyl groups on the triazine unit.227Fig. 13b shows that the polarity of D–A type COFs had a significant impact on the photocatalytic production of H2O2. When the polarity is weak, it will limit exciton dissociation, while when the polarity is too strong, it will lead to a weak π-conjugation effect, suppress exciton formation, and thus reduce the photostability of the COF. Therefore, the use of an appropriate amount of phenyl could increase the intramolecular polarity of the COFs themselves and thus regulated the degree of exciton dissociation to efficiently generate H2O2. The carbonyl group in polyimide COFs has the property of absorbing and storing electrons, which is an ideal active site for photoreduction of O2 to H2O2.228 Therefore, the H2O2 yield of PB-COF with an electron-rich carbonyl active center was 4.22 times higher than that of PT-COF with a relatively electron-deficient carbonyl active center. The generation of value-added organic chemicals along with the generation of H2O2 is an advantage of photocatalysis, and importantly, the substrate of the value-added chemicals also acts as a sacrificial agent to inhibit the electron–hole complexation rate. In view of this, Fang et al. synthesized three COFs based on the D–A structure of the BT unit for simultaneous benzylamine oxidation and H2O2 generation.229 As expected, the addition of the sacrificial agent benzylamine allowed the COF to have more e− to generate O2˙−, which captured protons to form ˙OOH intermediates and subsequently captured protons and electrons for conversion to H2O2. In the structure of a COF, even one atom difference can have a great impact on its photoelectric properties as well as the generation of H2O2. COF-TPT-Azo was formed through the insertion exchange of COF-TPT-TPA using a linker, and only one N atom was different between the two.230 The NN bond in this novel azobenzene-conjugated COF-TPT-Azo was the active site for photocatalysis, exhibiting a narrower band gap and charge transfer ability, and its photocatalytic H2O2 yield under alkaline conditions was 7.9 times higher than that of the CN-conjugated COF-TPT-TPA. Moreover, two 3D COFs (COF-1 and COF-2) reported by Wang et al. could reduce the energy loss for filling the triplet excited state and lower the jump energy barrier from the ground state to the singlet excited state, resulting in high selectivity and efficiency of the two COFs in the photocatalytic production of H2O2 from natural seawater (Fig. 13c).231
As shown in Fig. 14, total H2O2 photosynthesis can be achieved by both the oxygen reduction reaction (ORR) and water oxidation reaction (WOR). Mi and his partners reported that the ORR and WOR could occur simultaneously in EBA-COF and BTEA-COF, with H2O2 yields up to 1830 μmol g−1 h−1.74 In contrast to the two-electron ORR pathway, the WOR consists of three competing pathways, including a one-electron transfer pathway for ˙OH generation, a two-electron transfer pathway for H2O2 generation, and a four-electron transfer pathway for photo-induced oxygen precipitation.239 Therefore, it is crucial to enhance the 2e− WOR pathway rather than the 4e− WOR to achieve simultaneous H2O2 production from the WOR/ORR reaction. Lan and his team reported COFs with integrated redox centers, named TTF-BT-COF.240 Tetrathiafulvalene units were used as oxidation sites for the 2e− WOR and BT units as reduction sites for the ORR, this structure could generate a large amount of H2O2 under light irradiation. As a result, TTF-BT-COF could achieve H2O2 yields up to 276000 μmol h−1g−1 without sacrificial agents and could produce high concentrations of H2O2 (∼18.7 wt%) in prolonged batch experiments, which had a great potential for practical applications. The modification of the thioether group can narrow the band gap of TDB-COF and promote the photogenerated mobility.233 Most importantly, it can effectively modulate the energy band structure for the 2e− WOR and accelerate the 2e− ORR process to generate H2O2. Luo and his team synthesized two three-component COFs with D–A–π–D and D–A–π–A structures for the ORR and WOR to efficiently generate H2O2 from air and water.234 It was found that ECUT–COF–50 with the D–A–π–D structure had a higher O2 adsorption capacity and H2O2 generation rate compared to ECUT–COF–51 with the D–A–π–A structure. The CC in both COFs could serve as the active site for both the ORR and WOR. Currently, significant progress has been made in the study of H2O2 production from COF-based photocatalysts.241 However, this research is still at an early stage with low solar conversion efficiency. Therefore, finding more available monomers to construct COFs with efficient solar energy conversion is an important direction for future development of catalytic generation of H2O2.
 | ||
| Fig. 14 Mechanism diagram of photocatalytic production of H2O2. | ||
C bond connection had stronger photoreduction and electron transfer abilities than the imine bond connection to COF, and could generate more O2˙− and 1O2 for selective thioether oxidation reaction under green light driving. Lang and his partners designed and synthesized two isomers with different thiazole bond orientations, COFs-Tz-1 and COF-TZ-2.243 COF-TZ-2 exhibited a higher photocatalytic activity for selective oxidation of sulfides due to its higher dipole moment giving it a better in-plane electron transfer and charge separation ability. En–COF–P was a new type of COFs linked by β-ketoamine, formed by polymerization of amino alkyne clicks, which was a novel approach to synthesize COFs.160 As shown in Fig. 15a, En–COF–P exhibited a strong visible light absorption range and was able to drive thioether oxidation reactions with up to 99% conversion and 99% selectivity while Im–COF–B polymerized by Schiff base has only a 12% conversion rate. This work expands the types of chemical bonds in COFs.
 | ||
| Fig. 15 (a) Schematic diagram of the synthesis and photocatalytic performance of En–COF–P. Reproduced from ref. 160 with permission from American Chemical Society, copyright 2024. (b) Schematic diagram of two methods for constructing NQ-COFA1. Reproduced from ref. 244 with permission from Royal Society of Chemistry, copyright 2023. (c) The design and preparation of COF-980, and its NIR-II fluorescence imaging-guided in vivo phototherapy. Reproduced from ref. 256 with permission from John Wiley and Sons, copyright 2024. (d) Schematic illustration of 2D COF fabrication, in vivo tumor therapy. Reproduced from ref. 258 with permission from American Chemical Society, copyright 2019. | ||
The formation of benzimidazole derivatives by photosynthetic cyclization is also a common organic transformation reaction. In Fig. 15b, NQ-COFA1 could be formed through a multi-component one pot reaction and two-step post modification, which converts imine bonds in COFs to quinoline linked bonds through cyclization.244 This one-pot strategy was simpler and had a higher specific surface area as well as crystallinity than the two-step synthetic post-modification approach. NQ-COFA1 was an efficient O2˙− mediated photocatalyst for the synthesis reaction of cyclized imidazole derivatives. Subsequently, Xiang et al. synthesized ionic NQ-COFD4-Me and achieved a yield of 91–98% in photocatalytic benzimidazole synthesis.245 Similarly, TPT-COF with excellent carrier separation and migration rate exhibited better catalytic performance for the photocatalytic reaction of benzimidazole than TTT-COF, with a yield of 96% and high stability.246 The next organic conversion reaction is benzylamine coupling reaction. The main reactive oxygen species of the anthraquinone compound TpAQ-COF was O2˙−, which could selectively oxidize amines under O2 and green light, and had good recyclability.236 Zhang and his partners doped COFs with S and Se atoms and found that doping Se atoms in semimetals could reduce the bandgap and exciton binding energy.237 Therefore, COF-NUST-36 doped with Se atoms had a high yield of 98% and a selectivity of 97.5% for amine oxidation. Dong and his team designed and synthesized COFs based on the binding energies of COFs with different amine-based monomer linkers by DFT calculations.247 It was found that the bipyridine-N site of the D–A structured BTT-BPy-COF exhibited enhanced oxygen adsorption and activation, as well as electron transfer ability, which facilitated photocatalytic benzylamine coupling with conversion and selectivity up to 99%.
D–A COFs have been used as photocatalysts for an increasing number of organic small molecule transduction reactions in recent years.248 By modulating the D–A interactions within the COFs, the optimized TP-PB COFs had narrower band gaps as well as stronger photocurrent responses, and could be used as highly efficient photocatalysts for indole derivative C-3 thiocyanation.238 In addition, COF-JLU24 was used for photocatalytic C-3 functionalization of indole, and its catalytic activity was superior to that of the metal free photocatalyst g-C3N4.122 The MeO-TBT-COF containing methoxy on the surface showed the highest photocatalytic activity and recoverability in the C-3 thiocyanation reaction conversion of indole derivatives.249 Moreover, under visible light, BF COFs with the D–A structure could promote the C–S bond formation reaction between β-ketoesters and NH4SCN, efficiently generating multi-substituted olefins with a yield of up to 98%.250 The D–A type COF (COF-JLU33) constructed from triazine units and benzothiophene units was used to carry out the photosynthesis of α-trifluoromethyl ketones in heterogeneous photocatalytic systems.251 Similarly, D–A type PTBC-Por-COF based on porphyrin could effectively generate O2˙− in heterogeneous photocatalytic systems, and served as an efficient photocatalyst for sulfide oxidation and 2-bromoacetophenone reduction dehalogenation.252 In addition, 3D TAPB-ETTBC COFs with oxidation and reduction active centers could be used as photocatalysts for various organic reactions.253
5.2 Photothermal therapy
D–A type COF materials can not only be applied in the fields of photocatalysis and electrocatalysis, but also have important significance in the field of biomedicine. Photothermal therapy (PDT) is a novel non-invasive tumor treatment method that uses photosensitizers to convert light energy into heat energy under light irradiation, killing tumor cells while reducing drug resistance and side effects.254,255 In addition, the D–A structure can induce photo-generated charge transfer between donor and acceptor units, thereby suppressing fluorescence and enhancing photothermal efficiency. Zhang and his team constructed a D–A type COF (COF-980) capable of second near-infrared (NIR-II) phototherapy and fluorescence bioimaging (Fig. 15c).256 The COF-980 had good photostability, chemical stability, and excellent reactive oxygen species (ROS) generation efficiency, and was capable of triggering a large amount of intracellular ROS under laser light to kill cancer cells. Furthermore, COF-980 delivers clear in vivo images at longer emission wavelengths and could guide efficient photodynamic therapy for 4T1 tumors with minimal side effects. At present, the preparation of monodisperse nanoscale COFs is difficult, and such nanoscale D–A type COFs can facilitate the diffusion of reactive oxygen species. Xie and his team were able to control the size of the COFs (around 400 nm) with uniform spherical morphology and colloidal stability by adjusting the amount of acetic acid catalyst.257 This nanoscale DPPN COF had the highest photothermal conversion ability under 808 nm laser irradiation, which had significant anti-cancer effects. The synergistic application of photodynamic therapy (PTT) and photothermal therapy (PDT) can achieve better therapeutic results. As shown in Fig. 15d, porphyrin-based COFs (Por-COFs) with a D–A structure could act as photosensitizers enabling the combined action of PTT and PDT to significantly eliminate tumors at 635 nm laser light.258 Furthermore, another D–A type porphyrin-based HPCOF showed good photo-imaging ability and photothermal conversion efficiency.259 This HPCOF can be used as both a photothermal agent and a photosensitizer, enabling the combined treatment of tumors with PDT and PTT.In addition to that, D–A type COFs can be a good carrier for transporting targeted drugs. The D–A structure in COF-PDA-FA can provide multiple channels and pathways for carrier transport, which gives it excellent photothermal properties.260 And the large specific surface area and porosity in COF-PDA-FA make it a good carrier for targeting and delivering glucose oxidase to tumor cells. Finally, this COF-PDA-FA with high IR absorption could induce endogenous mitochondria and apoptosis in combination with the generated ROS, which ultimately led to the removal of cancer cells with insufficient energy supply. Sun et al. in situ loaded heteropolymer blue into a COF by a one-pot method, and the resulting HPB@COF had good pH-responsive release characteristics, biocompatibility, and high tumor inhibition efficiency.261 Under irradiation with an 808 nm near-infrared laser, HPB@COF could make the tumor temperature rise at high temperature to kill the tumor. In fact, although there are many studies of COFs for photothermal tumor therapy, there is still a long way to go before real clinical cancer treatment. This is because the photothermal conversion efficiency and photodynamic therapy efficiency of COFs have not reached the expected results. In addition, the dispersive stability of COF materials in a short time and at a low concentration in water as well as the lack of selectivity to tumor cells still limits the rapid development of practical applications.
5.3 Energy storage
000 charge discharge cycles in acidic electrolytes (Fig. 16b). Similarly, DPP-TBB-COF composed of DPP and 1,3,5-triphenylbenzene could promote charge transfer under the action of D–A, with a superior specific capacitance of 384 F g−1 at a current density of 0.3 A g−1 and good durability.109
 | ||
| Fig. 16 (a) Synthesis of PAE-M1M2PcF8 (M1, M2 = Ni, Cu, Zn) with dioxin linkages. Reproduced from ref. 265 with permission from John Wiley and Sons, copyright 2022. (b) The cycling stability of PAE-NiNiPcF8 for MSC at 50 mV s−1 (inset: CV curves at 1st, 5000th, and 10000th cycle). Reproduced from ref. 109 with permission from John Wiley and Sons, copyright 2022. (c) The GCD curves of Ni-TAPP/rGO at different current densities from 0.5 to 10 A g−1. Reproduced from ref. 267 with permission from Royal Society of Chemistry, copyright 2024. (d) Covalent TCNQ immobilization in COF through a [2 + 2] cycloaddition reaction accompanied by a ring opening of the strained intermediate. Reproduced from ref. 269 with permission from American Chemical Society, copyright 2024. | ||
Variations in surface charge density and pore size induced by different electron acceptor units are important for charge storage. Van Der Voort et al. investigated the charge storage mechanism of D–A type COFs consisting of TTFs in ionic liquid electrolyte-based supercapacitors.266 The changes in charge density affect the buildup of ionic liquids in the vicinity of the pores, and the electronic polarity of the pores could influence the confinement process of the ionic liquids in the pores. TTF-Por COF had a wider EDLC window in ionic liquid electrolytes, and the energy density of the EDLC device was 58 Wh kg−1, which was almost 6 times higher than that of organic electrolytes. In addition, TTF–COF–based devices retained about 75–88% capacitance after 100 hours of use in ionic liquid electrolytes. Coupling organic compounds with multiple electron channels and redox reversibility to graphene sheets is expected to overcome the bottleneck of low energy density in organic-based supercapacitors. Ni-TAP/rGO and Ni-TAPP/rGO were D–A type COFs composed of thiofulvalene units and graphene connected by covalent bonding.267 The formation of a heterojunction at the interface promotes the efficient utilization of COFs and charge transfer, and electrons could be rapidly transferred from the highly conductive graphene oxide to the vertically grown COFs. In Fig. 16c, the capacitance of the Ni-TAPP/rGO electrode at 0.5 A g−1 current was 367.5 F g−1. The energy density and power density of the Ni-TAPP/rGO electrode were as high as 51.0 Wh kg−1 and 1.78 kW kg−1, respectively. For using D–A type COFs as supercapacitor electrodes, it is important to introduce different donor and acceptor units or further achieve functionalization, for example, enhancing hydrophilicity to improve electrolyte wettability, or introducing active functional groups to enhance pseudocapacitive behavior.
Hindering the decomposition of perovskite materials and improving the stability of the materials are of great importance for improving PCE. Doping of COFTPDA-TZDA in the FAPbI3 layer was able to improve the charge transport within the perovskite membrane and increase the grain size of the perovskite membrane.88 Importantly, it was able to suppress the perovskite defects through the N-atom coordination effect, thus enhancing the stability of PSCs. After storage for 480 h at approximately 60% relative humidity, the PCE of COFTPDA-TZDA treated PSCs was as high as 23.51% and had high cycling stability (Fig. 17a). Similarly, doping DA-COFs into the FAPbI3 layer of PSCs could produce the same effect, and the highest power conversion efficiency of the constructed PSCs was 23.19%, with good humidity stability.270 In addition, this strategy of doping D–A type COFs can also be applied to inverted PSCs. For example, Yu et al. used two novel COFTPA and COFICZ to modify inverted PSCs, and this strategy reduced carrier complex losses and improved the stability of the perovskite membrane.271 The corresponding device structure diagram and PCE performance diagram are shown in Fig. 17b. The energy level matching resulted in a p-type doped interface which effectively enhanced the charge extraction and transport in PSCs, and the maximum PCE of inverted PSCs treated with COFs was 25.68%.
 | ||
| Fig. 17 (a) J–V characteristics of the PSCs without and with COFTPDA-TZDA under standard illumination conditions. Reproduced from ref. 88 with permission from CCS Chemistry, copyright 2024. (b) Device structure of the inverted PSC and J–V curves of the COF-modulated inverted PSC devices. Reproduced from ref. 271 with permission from John Wiley and Sons, copyright 2024. (c) Cycling performance of the pouch cell at 0.2 A g−1 for 100 cycles. Reproduced from ref. 272 with permission from John Wiley and Sons, copyright 2024. (d) Galvanostatic cycling of Li–Li symmetric cells at a current density of 5 mA cm−2. Reproduced from ref. 274 with permission from John Wiley and Sons, copyright 2023. (e) Long-term cycles of full cells based on Li@2C-TA-CTF, Li@3C-TF-CTF, and Li@4C-TA0.5TF0.5-CTF at 3C. Reproduced from ref. 134 with permission from John Wiley and Sons, copyright 2024. (f) Schematic illustration of the synthesis of VCOF-1@CNT and the inverse vulcanization of VCOF-1@CNT with elemental sulfur. Reproduced from ref. 275 with permission from Elsevier, copyright 2024. (g) Schematic of the COF-TMT-BT‖Zn(CF3SO3)2‖Zn energy storage system. Reproduced from ref. 278 with permission from John Wiley and Sons, copyright 2023. | ||
The COF materials are renowned for their strong porosity and sustainability, and have enormous potential as cathodes for aqueous ZIBs.276 Tao and his team designed NT-COF with a D–A structure as a cathode to develop photo-responsive aqueous organozinc batteries.277 The ordered stacking of D and A fragments establishes a reliable pathway for charge transport and transfer, which results in an effective coupling of intramolecular charge separation and reversible redox chemistry. The NT-COF cathode assembled ZIB battery had a high discharge voltage platform of about 1.4 V, and a discharge capacity of 214 mA h g−1. However, the low stability of COF materials in aqueous solutions limits the long-term cycling of ZIBs. Therefore, COF-TMT-BT formed by aldol condensation had irreversible CC bonds, making it insoluble in water-based electrolytes and enhancing cycling stability.278 The BT unit in COF-TMT-BT acted as an electrochemically active group and the assembled ZIBs are shown in Fig. 17g. The specific capacity of the ZIBs was 283.5 mA h g−1 with a power density of 23.2 kW kg−1 at a current density of 0.1 A g−1, and still had good specific capacity values after 800 cycles.
Overall, in electrochemical energy storage, the precise arrangement of organic units in COF structures can determine their effectiveness, and these structures can be carefully tailored to meet the requirements of specific applications. In addition, structural stability in COFs is important for the long-lasting operation of energy devices. Therefore, it is possible to develop stable structures and suitable charge density distributions by fine-tuning the pore structure and density of D–A type COFs, which will improve ion-selective transport in energy storage and conversion.
6. Conclusion and prospect
This work started with a comprehensive review of the conformational relationships in D–A type (COFs), focusing on those built from common building blocks and bond connections. Understanding these relationships is fundamental as it reveals the underlying structural features governing D–A type COFs' properties. Next, it explores strategic approaches for optimizing D–A type COFs. Supramolecular assembly, for instance, arranges molecular components via non-covalent interactions to achieve a more favorable structure and enhanced properties. Post-modification synthesis tailors the chemical functionality of pre-synthesized COFs, enabling broader applications. Interfacial engineering, moreover, manipulates COFs' interfaces to enhance their performance. And then this review also summarizes recent progress in photocatalysis, photothermal therapy, and energy storage involving D–A structured COFs. In photocatalysis, they can drive reactions under light due to the favorable charge separation. In photothermal therapy, they convert light to heat for potential cancer treatment, and in energy storage, they may contribute to better devices. In addition, D–A type COFs are a novel material with high research and application value in photovoltaics. Their unique electronic structure allows precise electron control, crucial for charge transfer in photovoltaic devices. Their wide-range light absorption maximizes solar energy use, and high specific surface area offers more reaction sites. Embedding functional groups, choosing bonding modes, and combining donors and acceptors endow them with high structural and optoelectronic tunability. Nevertheless, despite great potential, D–A type COF material research faces challenges like large-scale synthesis, stability under harsh conditions, and performance optimization for specific applications (Fig. 18).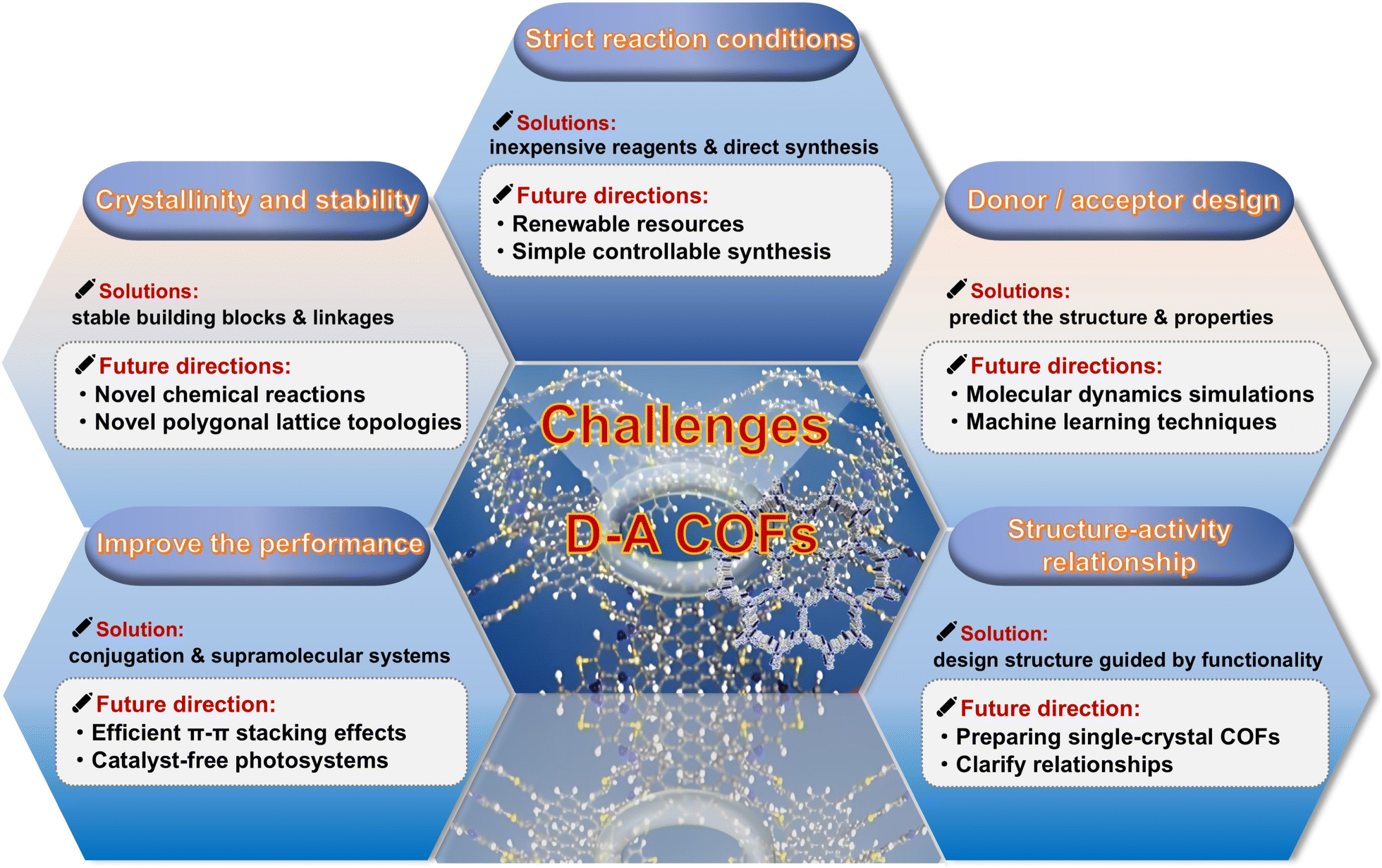 | ||
| Fig. 18 Current challenges and future prospects of D–A type COFs. | ||
(1) D–A COFs must maintain stability after functionalization in the environment for sustainable and regenerative long-term operation. Therefore, crystallinity and stability of the pore structure must be key to the rational design of D–A COF-based materials for the target application. Employing stable linkers and establishing strong covalent bonds can enhance the durability of COFs, and the environmental conditions (pH, temperature, and presence of solvents) of D–A-type COFs need to be considered and stable building blocks and linkers should be selected accordingly. Furthermore, exploring new irregular polygonal lattice topologies and improving their stability is an important aspect. This is because it enables the growth of the polymer backbone in an anisotropic tiling manner and its structural ordering as compared to the conventional ortho polygonal scheme. In addition, most of the formation of D–A type COFs is specific to the molecular precursor. In other words, only some building blocks with suitable functional groups are able to react with each other to form crystal networks. Therefore, novel chemical reactions involving the use of dynamic covalent bonds should be vigorously explored.
(2) From an economic perspective, the synthesis of D–A COFs generally requires high reaction temperatures, long reaction times, or complex solvent conditions, resulting in poorly controllable synthesis processes as well as high costs. Meanwhile, the use of transition metal catalysts in the cross-coupling reaction introduces trace metals, which affect the objective evaluation of COF catalysts. In addition, some reported D–A-based COFs require high inputs due to multi-step synthesis and harsh reaction conditions. Therefore, the use of inexpensive raw materials and direct synthesis routes can effectively reduce the overall cost of the catalysts. Future research should be devoted to optimizing the synthesis process of D–A type COFs and developing simpler and controllable synthesis methods. For example, the use of microwave radiation and acoustic chemistry can accelerate the reaction rate and improve the yield, but there are fewer related studies. In addition, exploring self-assembly strategies to promote the formation of ordered structures is also an important research direction. In addition, the synthesis of D–A type COFs by utilizing renewable resources (e.g., biomass) or using inexpensive precursors will help to reduce the production cost and promote their realization in large-scale industrial applications.
(3) In D–A type COFs, the selection and arrangement of donor and acceptor units directly affect the electronic structure and their efficiency in photovoltaic and catalytic reactions. However, most of the previous donor and acceptor units are random or empirical and lack a good theoretical basis. In the future, machine learning techniques and molecular dynamics simulations can be used to fine-tune the design of the electronic structure of the D–A interface and predict the electronic structure and physicochemical properties of the material. This will help accelerate the discovery of more exciting and high-performance D–A type COF materials.
(4) The poor electrical conductivity and the lack of active sites for highly selective photocatalytic energy conversion of D–A type COFs have been the limiting factors for the substantial improvement of their performance. Previous researchers have typically used noble metals such as Pt and Pd as co-catalysts to enhance the surface reaction kinetics. However, their limited availability and the heavy metal contamination problem associated with transition metal oxides have hindered their large-scale and widespread application. In the future, efficient π–π stacking effects can be designed, macromolecular conjugation systems can be employed or structures with long-range electron migration can be constructed, which in turn can enhance the electrical conductivity and active sites of D–A type COFs. In addition, it is crucial to vigorously explore low-cost, environmentally friendly, and efficient co-catalysts or develop co-catalyst-free photosystems.
(5) While research in bond chemistry has greatly broadened the structural types of D–A type COFs, a comprehensive understanding of the intrinsic relationship between the selected covalent bonds and the resulting material properties remains a significant challenge. In many cases, covalent bonds act primarily to connect organic units into a well-defined network rather than as a direct source of material functionality. The exact effect of covalent linkages on the overall properties of the resulting materials remains unclear. Due to the complexity of the interactions between organic units as well as between neighboring layers, conducting systematic studies of single-crystal COFs remains a great challenge. Therefore, there is a great need for a generalized method capable of preparing single-crystal COFs to help reveal the mechanism of covalent linkages in these materials.
We are confident that these challenges will be addressed as researchers explore D–A type COFs in depth. In the future, the electronic structure design of D–A type COFs will be optimized across multiple dimensions, particularly through the precise modulation of donor and acceptor units, fine-tuning the energy band structure, and enhancing electron transfer efficiency. These advancements will significantly enhance their performance in applications such as photovoltaics, catalysis, gas separation, and storage.
Data availability
The type of this manuscript is a review and related data are cited from others. Therefore, there are no raw experimental or computational data associated with this article.Author contributions
Conceptualization: Haiyang Liu, Yongfeng Zhi, and Xiaoming Liu; writing–original draft: Haiyang Liu, Shanshan Zhu, and Huijuan Yue; writing–review and editing: Yongfeng Zhi and Xiaoming Liu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project of the National Natural Science Foundation of China (No. 52373210, 52073119), Natural Science Foundation of Jilin Province (No. 20230101029JC), Hainan Provincial Natural Science Foundation of China (224QN181), and State Key Laboratory of Inorganic Synthesis and Preparative Chemistry (No. 2024-7).Notes and references
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 Search PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 Search PubMed.
- E. Asayesh-Ardakani, M. Rahmani, A. Hosseinian, S.-B. Ghaffari and M.-H. Sarrafzadeh, Coord. Chem. Rev., 2024, 518, 216087 CrossRef CAS.
- X. Kang, E. R. Stephens, B. M. Spector-Watts, Z. Li, Y. Liu, L. Liu and Y. Cui, Chem. Sci., 2022, 13, 9811–9832 RSC.
- Y. Liu, X. Liu, A. Su, C. Gong, S. Chen, L. Xia, C. Zhang, X. Tao, Y. Li, Y. Li, T. Sun, M. Bu, W. Shao, J. Zhao, X. Li, Y. Peng, P. Guo, Y. Han and Y. Zhu, Chem. Soc. Rev., 2024, 53, 502–544 RSC.
- Y. Qian and H.-L. Jiang, Acc. Chem. Res., 2024, 57, 1214–1226 CrossRef CAS PubMed.
- C. Kang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, K. Chai, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2022, 144, 20363–20371 CrossRef CAS.
- Y. Kumar, I. Ahmad, A. Rawat, R. K. Pandey, P. Mohanty and R. Pandey, ACS Appl. Mater. Interfaces, 2024, 16, 11605–11616 CrossRef CAS.
- Y. Liu, L. Li, Z. Sang, H. Tan, N. Ye, C. Sun, Z. Sun, M. Luo and S. Guo, Nat. Synth., 2024, 4, 134–141 CrossRef.
- J. Qiu, K. Meng, Y. Zhang, B. Cheng, J. Zhang, L. Wang and J. Yu, Adv. Mater., 2024, 36, 2400288 CrossRef CAS PubMed.
- Z. Zhang, Q. Zhang, Y. Hou, J. Li, S. Zhu, H. Xia, H. Yue and X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202411546 CrossRef CAS PubMed.
- Z. Zhao, W. Chen, G. Zhang and Y. Chen, J. Mater. Chem. A, 2023, 11, 26052–26062 RSC.
- R. Xue, Y.-S. Liu, M.-Y. Wang, H. Guo, W. Yang and G.-Y. Yang, Mater. Horiz., 2023, 10, 4710–4723 RSC.
- S. Wang, Y. Yang, X. Liang, Y. Ren, H. Ma, Z. Zhu, J. Wang, S. Zeng, S. Song, X. Wang, Y. Han, G. He and Z. Jiang, Adv. Funct. Mater., 2023, 33, 2300386 CrossRef CAS.
- K. Xu, Y. Zheng, J. Zhou, Y. Zhao, X. Pang, L. Cheng, H. Wang, X. Zhang, R. Zhang and Z. Jiang, Adv. Funct. Mater., 2024, 2417383 Search PubMed.
- Q. Wang, Y. Guo, Y. Long, Y. Liu, Z. Wang, Y. Liu and X. Zhang, J. Membr. Sci., 2024, 701, 122766 CrossRef CAS.
- J. Chang, H. Li, J. Zhao, X. Guan, C. Li, G. Yu, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, Chem. Sci., 2021, 12, 8452–8457 RSC.
- C. Krishnaraj, A. M. Kaczmarek, H. S. Jena, K. Leus, N. Chaoui, J. Schmidt, R. Van Deun and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2019, 11, 27343–27352 CrossRef CAS.
- T. Skorjanc, D. Shetty, F. Gándara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Chem. Sci., 2020, 11, 845–850 RSC.
- A. Giri, G. Shreeraj, T. K. Dutta and A. Patra, Angew. Chem., Int. Ed., 2023, 62, e202219083 CrossRef CAS PubMed.
- S. Wang, Y. Fu, F. Wang, X. Wang, Y. Yang, M. Wang, J. Wang, E. Lin, H. Ma, Y. Chen, P. Cheng and Z. Zhang, J. Am. Chem. Soc., 2024, 146, 33509–33517 CrossRef CAS PubMed.
- S. Wang, Y. Han, V. A. Reddy, M. C.-Y. Ang, G. Sánchez-Velázquez, J. M. Saju, Y. Cao, D. T. Khong, P. K. Jayapal, R. Cheerlavancha, S. I. Loh, G. P. Singh, D. Urano, S. Rajani, B. Marelli and M. S. Strano, Nat. Commun., 2024, 15, 9300 CrossRef CAS PubMed.
- W. Zhang, Q. Sun, Y. Zhu, J. Sun, Z. Wu and N. Tian, ACS Sens., 2024, 9, 3262–3271 CrossRef CAS.
- N. P. Bradshaw, Z. Hirani, L. Kuo, S. Li, N. X. Williams, V. K. Sangwan, L. E. Chaney, A. M. Evans, W. R. Dichtel and M. C. Hersam, Adv. Mater., 2023, 35, 2303673 CrossRef CAS.
- G. A. Leith, A. A. Berseneva, A. Mathur, K. C. Park and N. B. Shustova, Trends. Chem., 2020, 2, 367–382 CrossRef CAS.
- Y. Liu, X. Jiang, L. Chen, Y. Cui, Q.-Y. Li, X. Zhao, X. Han, Y.-C. Zheng and X.-J. Wang, J. Mater. Chem. A, 2023, 11, 1208–1215 RSC.
- L. Wang and W. Zhu, Adv. Sci., 2023, 11, 2307227 CrossRef.
- L. Hao, R. Shen, C. Qin, N. Li, H. Hu, G. Liang and X. Li, Sci. China Mater., 2024, 67, 504–513 CrossRef CAS.
- N. Wang, S.-Q. Ye, M. Li, H. Pan and G.-W. Wang, Sol. RRL, 2023, 8, 202300790 Search PubMed.
- P. Li, B. Li, C. Wang, X. Zhao, Y. Zheng, S. Wu, J. Shen, Y. Zhang and X. Liu, Composites, Part B, 2023, 252, 110506 CrossRef CAS.
- Y. Xia, W. Zhang, S. Yang, L. Wang and G. Yu, Adv. Mater., 2023, 35, 2301190 CrossRef CAS.
- J. Zhao, J. Ren, G. Zhang, Z. Zhao, S. Liu, W. Zhang and L. Chen, Chem. Eur. J., 2021, 27, 10781–10797 CrossRef CAS PubMed.
- M. Lv, X. Ren, R. Cao, Z. Chang, X. Chang, F. Bai and Y. Li, Polymers, 2022, 14, 4893 CrossRef CAS PubMed.
- J. Liu, S. Tang, J. Wang, P. Zhang, Z. Wang, L. Wang, R. Wen and J. Huang, J. Environ. Chem. Eng., 2024, 12, 114802 CrossRef CAS.
- X. Wang, H. Li, S. Zhou, J. Ning, H. Wei, X. Li, S. Wang, L. Hao and D. Cao, Adv. Funct. Mater., 2025, 2424035 CrossRef.
- A. I. B. Adrien, P. Côté, N. W. Ockwig, A. J. M. Michael O'Keeffe and O. M. Yaghi, Science, 2005, 310, 1120411 Search PubMed.
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Côté and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS PubMed.
- J. R. H. Fernando, J. Uribe-Romo, H. Furukawa, C. Klöck and a. O. M. Y. Michael O'Keeffe, J. Am. Chem. Soc., 2008, 131, 4570–4571 Search PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 Search PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed.
- Y. Su, Y. Wan, H. Xu, K.-i. Otake, X. Tang, L. Huang, S. Kitagawa and C. Gu, J. Am. Chem. Soc., 2020, 142, 13316–13321 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef.
- P.-L. Wang, S.-Y. Ding, Z.-C. Zhang, Z.-P. Wang and W. Wang, J. Am. Chem. Soc., 2019, 141, 18004–18008 Search PubMed.
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef CAS PubMed.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed.
- C. Zhao, H. Lyu, Z. Ji, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 14450–14454 Search PubMed.
- X. Guan, Y. Qian, X. Zhang and H. L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202306135 CrossRef.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519–15522 Search PubMed.
- H. L. Nguyen, C. Gropp, N. Hanikel, A. Möckel, A. Lund and O. M. Yaghi, ACS Cent. Sci., 2022, 8, 926–932 CrossRef CAS PubMed.
- V. A. Kuehl, P. H. H. Duong, D. Sadrieva, S. A. Amin, Y. She, K. D. Li-Oakey, J. L. Yarger, B. A. Parkinson and J. O. Hoberg, ACS Appl. Mater. Interfaces, 2021, 13, 37494–37499 CrossRef CAS PubMed.
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823 RSC.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed.
- Z.-B. Zhou, P.-J. Tian, J. Yao, Y. Lu, Q.-Y. Qi and X. Zhao, Nat. Commun., 2022, 13, 2180 Search PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Nat. Rev. Chem., 2022, 6, 881–898 CrossRef CAS.
- Z. Song, Y. Xie, X. Song, J. Tang, J. Wang, B. Z. Tang and Z. Li, J. Mater. Chem. C, 2024, 12, 19094–19102 RSC.
- X.-R. Ren, B. Bai, Q. Zhang, Q. Hao, Y. Guo, L.-J. Wan and D. Wang, J. Am. Chem. Soc., 2022, 144, 2488–2494 CrossRef CAS PubMed.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475 CrossRef.
- C.-C. Gu, C.-Q. Ni, R.-J. Wu, L. Deng, J. Zou, H. Li, C.-Y. Tong, F.-H. Xu, B.-C. Weng and R.-L. Zhu, J. Colloid Interface Sci., 2024, 658, 450–458 CrossRef CAS PubMed.
- J. Wang, S. Qiao, M. Yang and Z. Guo, Small, 2024, 21, 2409292 CrossRef.
- A. Hayat, H. Ali, Z. Ajmal, A. Alshammari, M. M. Alghamdi, A. A. El-Zahhar, N. Almuqati, M. Sohail, A. M. Abu-Dief, S. Khan, Y. Al-Hadeethi, M. Z. Ansari and Y. Orooji, Prog. Mater. Sci., 2025, 147, 101352 Search PubMed.
- P. Xiong, S. Zhang, R. Wang, L. Zhang, Q. Ma, X. Ren, Y. Gao, Z. Wang, Z. Guo and C. Zhang, Energy Environ. Sci., 2023, 16, 3181–3213 RSC.
- R. Luo, H. Lv, Q. Liao, N. Wang, J. Yang, Y. Li, K. Xi, X. Wu, H. Ju and J. Lei, Nat. Commun., 2021, 12, 6808 CrossRef CAS PubMed.
- X. Li, J. Zhou, W. Yin, B. Xie, Z. Liu and J.-J. Liu, J. Catal., 2024, 437, 115640 Search PubMed.
- K. Verma, Mohit and K. R. J. Thomas, Langmuir, 2024, 40, 24148–24161 CrossRef CAS PubMed.
- Y. Song, A. Li, P. Li, L. He, D. Xu, F. Wu, F. Zhai, Y. Wu, K. Hu, S. Wang and M. V. Sheridan, Chem. Mater., 2022, 34, 2771–2778 CrossRef CAS.
- W. Wang, F. Meng, Y. Bai, Y. Lu, Q. Yang, J. Feng, Q. Su, H. Ren and Q. Wu, ChemSusChem, 2024, 17, e202301916 CrossRef CAS PubMed.
- L. Wang, J. Chakraborty, K. S. Rawat, M. Deng, J. Sun, Y. Wang, V. V. Speybroeck and P. Van Der Voort, Sep. Purif. Technol., 2025, 359, 130368 Search PubMed.
- L. Zhai, Z. Xie, C.-X. Cui, X. Yang, Q. Xu, X. Ke, M. Liu, L.-B. Qu, X. Chen and L. Mi, Chem. Mater., 2022, 34, 5232–5240 CrossRef CAS.
- X. Ma, J. Kang, W. Cao, Y. Wu, C. Pang, S. Li, Z. Yi, Y. Xiong, C. Li, M. Wang, Z. Xu and J. Li, J. Colloid Interface Sci., 2024, 659, 665–675 Search PubMed.
- C. Yang, Z. Zhang, J. Li, Y. Hou, Q. Zhang, Z. Li, H. Yue and X. Liu, Green Chem., 2024, 26, 2605–2612 RSC.
- D. N. Ampong, E. Effah, E. A. Tsiwah, A. Kumar, E. Agyekum, E. N. A. Doku, O. Issaka, F. O. Agyemang, K. Mensah-Darkwa and R. K. Gupta, Coord. Chem. Rev., 2024, 519, 216121 CrossRef CAS.
- A. Rodríguez-Camargo, K. Endo and B. V. Lotsch, Angew. Chem., Int. Ed., 2024, 63, e202413096 CrossRef.
- H. Liu, C. Li, H. Li, Y. Ren, J. Chen, J. Tang and Q. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 20354–20365 CrossRef CAS PubMed.
- D. H. Streater, E. R. Kennehan, D. Wang, C. Fiankor, L. Chen, C. Yang, B. Li, D. Liu, F. Ibrahim, I. Hermans, K. L. Kohlstedt, L. Luo, J. Zhang and J. Huang, J. Am. Chem. Soc., 2024, 146, 4489–4499 CrossRef CAS.
- Y. Qian, Y. Han, X. Zhang, G. Yang, G. Zhang and H.-L. Jiang, Nat. Commun., 2023, 14, 3083 CrossRef CAS PubMed.
- A. Dey, S. Chakraborty, A. Singh, F. A. Rahimi, S. Biswas, T. Mandal and T. K. Maji, Angew. Chem., Int. Ed., 2024, 63, e202403093 CrossRef CAS PubMed.
- L. Qin, C. Ma, J. Zhang and T. Zhou, Adv. Funct. Mater., 2024, 34, 2315157 CrossRef.
- D.-M. Xue, Y.-J. Zhang, J.-W. Chen, H. Yang, R.-J. Xie, S.-C. Qi, Y.-Q. Bu, F. Liu, H.-H. Zhang and J. Lalevée, Chem. Eng. J., 2024, 502, 157874 Search PubMed.
- H. Yu, J. Zhang, X. Yan, C. Wu, X. Zhu, B. Li, T. Li, Q. Guo, J. Gao, M. Hu and J. Yang, J. Mater. Chem. A, 2022, 10, 11010–11018 RSC.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2020, 60, 1869–1874 CrossRef PubMed.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- L. Yao, S. Liu, L. Li, B. Ge, W. Jiao, S. Zong, X. Song, D. Zhou and Z. Liang, CCS Chem., 2024, 6, 1721–1730 CrossRef CAS.
- M. Hao, Y. Xie, X. Liu, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, JACS Au, 2023, 3, 239–251 CrossRef CAS.
- Y. Hou, F. Liu, B. Zhang and M. Tong, Environ. Sci. Technol., 2022, 56, 16303–16314 CrossRef CAS PubMed.
- C. Lin, X. Liu, B. Yu, C. Han, L. Gong, C. Wang, Y. Gao, Y. Bian and J. Jiang, ACS Appl. Mater. Interfaces, 2021, 13, 27041–27048 CrossRef CAS PubMed.
- J.-H. Dou, Z.-A. Yu, J. Zhang, Y.-Q. Zheng, Z.-F. Yao, Z. Tu, X. Wang, S. Huang, C. Liu, J. Sun, Y. Yi, X. Cao, Y. Gao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2019, 141, 6561–6568 CrossRef CAS PubMed.
- M.-Y. Yang, S.-B. Zhang, M. Zhang, Z.-H. Li, Y.-F. Liu, X. Liao, M. Lu, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2024, 146, 3396–3404 CrossRef CAS PubMed.
- F. Liu, Y. He, X. Liu, Z. Wang, H.-L. Liu, X. Zhu, C.-C. Hou, Y. Weng, Q. Zhang and Y. Chen, ACS Catal., 2022, 12, 9494–9502 CrossRef CAS.
- J. Xu, W. Liu, L. Jiang, X. Jing, L. L. Liu and Z. Li, Small, 2023, 19, 202304989 Search PubMed.
- J. Zhang, W. Zhou, J. Zhao, L. Xu, X. Jiang, Z. Li, Y. Peng and G. Li, Small, 2024, 202408324 Search PubMed.
- Z. Zhao, Y. Zheng, C. Wang, S. Zhang, J. Song, Y. Li, S. Ma, P. Cheng, Z. Zhang and Y. Chen, ACS Catal., 2021, 11, 2098–2107 CrossRef CAS.
- X. Zhong, H. Chen, M. Wang, S. Gan, Q. He, W. Chen and F. He, Macromolecules, 2019, 52, 2393–2401 CrossRef CAS.
- B. McDearmon, Z. A. Page, M. L. Chabinyc and C. J. Hawker, J. Mater. Chem. C, 2018, 6, 3564–3572 RSC.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- Z. Wang, P. Yeary, Y. Fan and W. Lin, Chem. Sci., 2024, 15, 4920–4925 Search PubMed.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- C. Wang, W. Lu, W. Song, Z. Zhang, C. Xie, Y. Li and J. Wang, Appl. Catal., B, 2025, 361, 124583 CrossRef CAS.
- Y. Wang, W. Hao, H. Liu, R. Chen, Q. Pan, Z. Li and Y. Zhao, Nat. Commun., 2022, 13, 100 CrossRef CAS PubMed.
- B. Mishra, S. Dutta, U. Pal, S. Rana, S. K. Mishra, T. Saha-Dasgupta and P. Pachfule, Small, 2025 DOI:10.1002/smll.202411199.
- Z. Chen, J. Zhang, F. Yu, Y. He, J. Kang, S. Xiao, F. Song, C. Zhang, R. Liang and J. Qiu, Sep. Purif. Technol., 2025, 359, 130376 CrossRef CAS.
- Y. Fang, Y. Liu, H. Huang, J. Sun, J. Hong, F. Zhang, X. Wei, W. Gao, M. Shao, Y. Guo, Q. Tang and Y. Liu, Nat. Commun., 2024, 15, 4856 CrossRef CAS.
- B. Gole, V. Stepanenko, S. Rager, M. Grüne, D. D. Medina, T. Bein, F. Würthner and F. Beuerle, Angew. Chem., Int. Ed., 2017, 57, 846–850 CrossRef PubMed.
- L. Luo, C. Li, Y. Wang, P. Chen, Z. Zhou, T. Chen, K. Wu, S. Y. Ding, L. Tan, J. Wang, X. Shao and Z. Liu, Small, 2024, 20, 202402993 Search PubMed.
- Z. Huang, Y. H. Luo, W. Y. Geng, Y. Wan, S. Li and C. S. Lee, Small Methods, 2021, 5, 2100036 CrossRef CAS PubMed.
- S. Rager, A. C. Jakowetz, B. Gole, F. Beuerle, D. D. Medina and T. Bein, Chem. Mater., 2019, 31, 2707–2712 CrossRef CAS PubMed.
- Y. Zhang, G. Wu, H. Liu, R. Tian, Y. Li, D. Wang, R. Chen, J. Zhao, S. Liu, Z. Li and Y. Zhao, Mater. Chem. Front., 2021, 5, 6575–6581 RSC.
- M. Martínez-Fernández, E. Martínez-Periñán, S. Royuela, J. I. Martínez, F. Zamora, E. Lorenzo and J. L. Segura, Appl. Mater. Today, 2022, 26, 101384 CrossRef.
- J. Lv, Y. X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M. D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS.
- S. Royuela, E. Martínez-Periñán, M. P. Arrieta, J. I. Martínez, M. M. Ramos, F. Zamora, E. Lorenzo and J. L. Segura, Chem. Commun., 2020, 56, 1267–1270 RSC.
- C. Weng, X. Li, Z. Yang, H. Long, C. Lu, L. Dong, S. Zhao and L. Tan, Chem. Commun., 2022, 58, 6809–6812 RSC.
- Y. Luo, Z. Chang, J. Pei, Z. Guo and H. Zhan, Nano Lett., 2023, 23, 9266–9271 CrossRef PubMed.
- F. D. Wang, L. J. Yang, X. X. Wang, Y. Rong, L. B. Yang, C. X. Zhang, F. Y. Yan and Q. L. Wang, Small, 2023, 19, 202207421 Search PubMed.
- H. Li, P. Shao, S. Chen, G. Li, X. Feng, X. Chen, H.-J. Zhang, J. Lin and Y.-B. Jiang, J. Am. Chem. Soc., 2020, 142, 3712–3717 CrossRef CAS.
- N. Liu, S. Xie, Y. Huang, J. Lu, H. Shi, S. Xu, G. Zhang and X. Chen, Adv. Energy Mater., 2024, 14, 202402395 Search PubMed.
- G.-B. Wang, H.-P. Xu, K.-H. Xie, J.-L. Kan, J. Fan, Y.-J. Wang, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2023, 11, 4007–4012 RSC.
- Z. Li, S. Han, C. Li, P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC.
- G. Zhang, M. Zhao, L. Su, H. Yu, C. Wang, D. Sun and Y. Ding, ACS Appl. Mater. Interfaces, 2023, 15, 20310–20316 CrossRef CAS.
- E. Jin, Q. X. Mizue Asada, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- X. Chi, Z. Zhang, M. Li, Y. Jiao, X. Li, F. Meng, B. Xue, D. Wu and F. Zhang, Angew. Chem., Int. Ed., 2024, 202418895 Search PubMed.
- W. Li, L. Gao, H. Tang, J. Qin, Y. Jiao, L. Wang, S. Xian, Q. Zhou and Z. Wang, Appl. Mater. Today, 2024, 37, 102124 CrossRef.
- Y. Wang, Y. Z. Cheng, K. M. Wu, D. H. Yang, X. F. Liu, X. Ding and B. H. Han, Angew. Chem., Int. Ed., 2023, 62, e202310794 CrossRef CAS.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS.
- J. H. Wang, A. E. Hassan, A. M. Elewa and A. F. M. El-Mahdy, J. Mater. Chem. A, 2024, 12, 14005–14021 RSC.
- L. Wang, L. Wang, Q. Zhao, X. Ji, M. Zhao, Y. Zhang and M. Lai, J. Mater. Chem. A, 2024, 12, 28424–28436 RSC.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- K. Verma, M. A. Addicoat and K. R. Justin Thomas, ACS Appl. Polym. Mater., 2024, 6, 3909–3917 CrossRef CAS.
- H. He, R. Shen, Y. Yan, D. Chen, Z. Liu, L. Hao, X. Zhang, P. Zhang and X. Li, Chem. Sci., 2024, 15, 20002–20012 RSC.
- X. M. Lu, H. Wang, Y. Sun, Y. Xu, W. Sun, Y. Wu, Y. Zhang, C. Yang and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202409436 CrossRef CAS.
- H. Li, J. Fan, M. Ran, R. A. Borse, S.-X. Lin and D. Yuan, Angew. Chem., Int. Ed., 2025, 64, e202500937 CrossRef CAS.
- L. Gilmanova, V. Bon, L. Shupletsov, D. Pohl, M. Rauche, E. Brunner and S. Kaskel, J. Am. Chem. Soc., 2021, 143, 18368–18373 CrossRef CAS.
- Z. Li, Z. Dong, Z. Zhang, B. Wei, C. Meng, W. Zhai, Y. Wang, X. Cao, B. Han and Y. Liu, Angew. Chem., Int. Ed., 2025, 64, e202420218 CrossRef CAS.
- Q. Wu, M. J. Mao, Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Small, 2020, 17, 202004933 Search PubMed.
- S. S. Zhao, J. Liang, D.-H. Si, M.-J. Mao, Y. B. Huang and R. Cao, Appl. Catal., B, 2023, 333, 122782 CrossRef CAS.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- H. Liu, X. Yan, W. Chen, Z. Xie, S. Li, W. Chen, T. Zhang, G. Xing and L. Chen, Sci. China Chem., 2021, 64, 827–833 CrossRef CAS.
- H. Xu, S. Xia, C. Li, Y. Li, W. Xing, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202405476 CrossRef CAS PubMed.
- R.-C. Chen, C.-M. Wu, L.-H. Chung, J. Hu, M.-C. Tang, Z. Lin, X. Yang, Z. Xu and J. He, Chem. Mater., 2024, 36, 9928–9938 CrossRef CAS.
- C. Jin, Y. Pang, R. Wang, S. Wu, H. He, L. Gao, J. Liu, L. Chen, Y. Li, X. Yan and B. Wang, Appl. Catal., A, 2024, 685, 119906 CrossRef CAS.
- Y. Yao, Y. Han, S. Qi, D. Sun, J. Lang, B. Hu, Y. Ma and C. Liu, Int. J. Hydrogen Energy, 2024, 63, 184–192 CrossRef CAS.
- T. Wang, R. Zhang, P. Zhai, M. Li, X. Liu and C. Li, J. Mater. Chem. A, 2024, 12, 1292–1299 RSC.
- R. Bao, Z. Xiang, Z. Qiao, Y. Yang, Y. Zhang, D. Cao and S. Wang, Angew. Chem., Int. Ed., 2022, 62, e202216751 CrossRef.
- Q. Wang, C. Wang, K. Zheng, B. Wang, Z. Wang, C. Zhang and X. Long, Angew. Chem., Int. Ed., 2024, 63, e202320037 CrossRef CAS PubMed.
- H. Liu, X. Zheng, J. Xu, X. Jia, M. Chao, D. Wang and Y. Zhao, ACS Appl. Mater. Interfaces, 2023, 15, 16794–16800 CrossRef CAS PubMed.
- S. Chang, C. Li, H. Li, L. Zhu and Q. Fang, Chem. Res. Chin. Univ., 2022, 38, 396–401 CrossRef CAS.
- Y. Zhao, S. Li, J. Han, G. Fu, L. Liang, M. Chen, X. Wang and T. Zhang, ACS Mat. Lett., 2024, 6, 3985–3992 CrossRef CAS.
- J. P. Jeon, Y. J. Kim, S. H. Joo, H. J. Noh, S. K. Kwak and J. B. Baek, Angew. Chem., Int. Ed., 2023, 62, e202217416 CrossRef CAS PubMed.
- Y. Ma, Y. Yao, S. Qi, C. You, Y. Wu, C. Liu, B. Hu and K. Cui, Sep. Purif. Technol., 2025, 356, 129972 CrossRef CAS.
- F. Huang, Y. Wang, X. Dong and X. Lang, J. Mater. Chem. A, 2024, 12, 7036–7046 RSC.
- Y. Ran, M. Yang, J. Li, J. Song, Y. Wang, Z. Li and L. Yuan, Sep. Purif. Technol., 2025, 355, 129603 CrossRef CAS.
- Y. Wang, Z. Qiao, H. Li, R. Zhang, Z. Xiang, D. Cao and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202404726 CrossRef CAS.
- Q. Rong, X. Chen, Q. Cheng, Z. Huang and S. He, ACS Sustainable Chem. Eng., 2024, 12, 13306–13315 CrossRef CAS.
- H. Ma, B. Liu, B. Li, L. Zhang, Y.-G. Li, H.-Q. Tan, H.-Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 Search PubMed.
- X. Han, Z. Zhou, K. Wang, Z. Zheng, S. E. Neumann, H. Zhang, T. Ma and O. M. Yaghi, J. Am. Chem. Soc., 2023, 146, 89–94 CrossRef PubMed.
- Z. Gu, J. Tian, Y. Li, H. Li, Y. Chai and X. Chi, ACS Mat. Lett., 2024, 6, 5292–5299 CrossRef CAS.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Angew. Chem., Int. Ed., 2016, 56, 346–349 CrossRef.
- H.-Y. Zhang, Y. Yang, C.-C. Li, H.-L. Tang, F.-M. Zhang, G.-L. Zhang and H. Yan, J. Mater. Chem. A, 2021, 9, 16743–16750 RSC.
- M. Kaur, S. Kumar, S. A. Younis, M. Yusuf, J. Lee, S. Weon, K.-H. Kim and A. K. Malik, Chem. Eng. J., 2021, 423, 130230 CrossRef CAS.
- B.-J. Yao, W.-X. Wu, L.-G. Ding and Y.-B. Dong, J. Org. Chem., 2021, 86, 3024–3032 CrossRef CAS PubMed.
- X.-T. Li, J. Zou, T.-H. Wang, H.-C. Ma, G.-J. Chen and Y.-B. Dong, J. Am. Chem. Soc., 2020, 142, 6521–6526 Search PubMed.
- H. Lyu, H. Li, N. Hanikel, K. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 12989–12995 CrossRef CAS PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 2003735 CrossRef CAS.
- P. Liu, K. Cai, D.-J. Tao and T. Zhao, Appl. Catal., B, 2024, 341, 123317 CrossRef CAS.
- Z.-Z. Wu, T. Xia, Y. Liang, Y.-P. Li, Z.-Y. Sui, L.-J. Feng, D.-X. Wu, X.-L. Tian and Q. Chen, Rare Met., 2024, 43, 3096–3106 CrossRef CAS.
- G. Matthys, A. Laemont, N. De Geyter, R. Morent, R. Lavendomme and P. Van Der Voort, Small, 2024, 20 Search PubMed.
- J. Huang, X. Liu, W. Zhang, Z. Liu, H. Zhong, B. Shao, Q. Liang, Y. Liu, W. Zhang and Q. He, Chem. Eng. J., 2021, 404, 127136 CrossRef CAS.
- Y. Hu, G. Liu, T. Song, X. Hu, B. Long and G.-J. Deng, Appl. Catal., B, 2025, 361, 124587 CrossRef CAS.
- Y. Wang, Y. Zheng, S. han, F. Chen and J. Hu, Appl. Surf. Sci., 2025, 698, 163089 CrossRef CAS.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- Y. Liang, T. Xia, Z. Wu, Y. Yang, Y. Li, Z. Sui, C. Li, R. Fan, X. Tian and Q. Chen, Mater.Today Chem., 2022, 24, 100777 CrossRef CAS.
- P. Dong, X. Xu, R. Luo, S. Yuan, J. Zhou and J. Lei, J. Am. Chem. Soc., 2023, 145, 15473–15481 CrossRef CAS PubMed.
- S. Yang, W. Liu, Y. Zhang, X. Jia, J. Sun, C. Zhang and M. Liu, J. Mater. Chem. A, 2024, 12, 28161–28169 RSC.
- X. Quan and B. Yan, ACS Sustainable Chem. Eng., 2023, 11, 7466–7474 CrossRef CAS.
- Y. Wang, Y. Deng, H. Xia, R. Zhang, J. Liu, H. Zhang, Y. Sun, Z. Zhang and X. Lu, Small Methods, 2023, 8, 2300163 CrossRef PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- L. D. Tran, D. C. Moore, B. C. Patra, J. Choi, C. M. Hampton, M. E. Loveday, D. D. Bhagwandin, I. Renggli, C. Muratore, L. F. Drummy, D. Zhao, N. R. Glavin and L. A. Baldwin, Adv. Funct. Mater., 2024, 34, 202402208 Search PubMed.
- Y. Xiang, W. Dong, P. Wang, S. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. Jin, Y. Weng, H. Chen and J. Ye, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- M. Lu, M. Zhang, J. Liu, T.-Y. Yu, J.-N. Chang, L.-J. Shang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 9099–9103 CrossRef CAS PubMed.
- R. Yan, B. Mishra, M. Traxler, J. Roeser, N. Chaoui, B. Kumbhakar, J. Schmidt, S. Li, A. Thomas and P. Pachfule, Angew. Chem., Int. Ed., 2023, 62, e202302276 CrossRef CAS.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. Van Hecke, R. Morent, N. De Geyter, K. Leus, H. Chen and P. Van Der Voort, Green Chem., 2023, 25, 3069–3076 RSC.
- K. Dey, S. Mohata and R. Banerjee, ACS Nano, 2021, 15, 12723–12740 CrossRef CAS.
- Y. Zhang, X. Guan, Z. Meng and H.-L. Jiang, J. Am. Chem. Soc., 2025, 147, 3776–3785 CrossRef CAS PubMed.
- W. Wei, S. Zhou, D. D. Ma, Q. Li, M. Ran, X. Li, X. T. Wu and Q. L. Zhu, Adv. Funct. Mater., 2023, 33, 2302917 CrossRef CAS.
- Y. He, Y. Zhao, X. Wang, Z. Liu, Y. Yu and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202307160 CrossRef PubMed.
- X. Zhang, Z. Wen, L. Pan, R. Wei, W. Pan, Y. Li, F. Lu, Y. Gao, N. Li and B. Tang, Mater. Today Nano, 2023, 24, 100418 CrossRef CAS.
- Z. Tang, J. Chen, Y. Xu, Z. Li, L. Sheng, Y. Hu, X. Wang, J. Wang, Y. Tang, X. He and H. Xu, Chem. Mater., 2024, 36, 4437–4443 CrossRef CAS.
- Y. Ma, Y. Han, Y. Yao, T. Zhou, D. Sun, C. Liu, G. Che, B. Hu, V. Valtchev and Q. Fang, Chem. Sci., 2024, 15, 12488–12495 RSC.
- L. Ye, Z. Xia, Q. Xu, Y. Yang, X. Xu, H. Jin and S. Wang, Chem. Commun., 2023, 59, 9872–9875 RSC.
- Q. An, S. Su, W. Hu, Y. Wang, T. Liang, X. Li and C. Li, Nanoscale, 2023, 15, 19815–19819 RSC.
- Y. Wang, Z. Hu, W. Wang, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao, G. Yu and Y.-N. Liu, Chem. Sci., 2021, 12, 16065–16073 RSC.
- M. Zhang, M. Lu, Z. L. Lang, J. Liu, M. Liu, J. N. Chang, L. Y. Li, L. J. Shang, M. Wang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 6500–6506 CrossRef CAS PubMed.
- R. Shen, X. Li, C. Qin, P. Zhang and X. Li, Adv. Energy Mater., 2023, 13, 2203695 CrossRef CAS.
- Z.-W. Zhou, C.-X. Liu, C.-X. Cai, Y.-J. Wei, K. Li, X.-Q. Yu, Y.-H. Liu and N. Wang, J. Environ. Chem. Eng., 2024, 12, 113533 CrossRef CAS.
- L. Cognigni, T. Gobbato, E. Benazzi, L. Paoloni, B. D. Vizio, R. Bonetto, F. Rigodanza, A. Bonetto, S. Agnoli, M. Bonchio and P. Costa, ChemSusChem, 2024, e202401977 Search PubMed.
- Q.-J. Wu, D.-H. Si, S. Ye, Y.-L. Dong, R. Cao and Y.-B. Huang, J. Am. Chem. Soc., 2023, 145, 19856–19865 CrossRef CAS PubMed.
- K. Xiao, R. Zhu, C. Du, X. Zhang and J. Chen, Sens. Actuators, B, 2023, 380, 133403 CrossRef CAS.
- S. Ge, K. Wei, W. Peng, R. Huang, E. Akinlabi, H. Xia, M. W. Shahzad, X. Zhang, B. B. Xu and J. Jiang, Chem. Soc. Rev., 2024, 53, 11259–11302 Search PubMed.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- C. Cui, X. Xu, X. Zhao, N. Xi, M. Li, X. Wang, Y. Sang, X. Yu, H. Liu and J. Wang, Nano Energy, 2024, 126, 109632 CrossRef CAS.
- I. M. A. Mekhemer, M. M. Elsenety, A. M. Elewa, K. D. G. Huynh, M. M. Samy, M. G. Mohamed, D. M. Dorrah, D. C. K. Hoang, A. F. Musa, S.-W. Kuo and H.-H. Chou, J. Mater. Chem. A, 2024, 12, 10790–10798 RSC.
- M. Wang, Z. Wang, M. Shan, J. Wang, Z. Qiu, J. Song and Z. Li, Chem. Mater., 2023, 35, 5368–5377 CrossRef CAS.
- Y. Yang, N. Luo, S. Lin, H. Yao and Y. Cai, ACS Catal., 2022, 12, 10718–10726 CrossRef CAS.
- S. Ma, Z. Li, Y. Hou, J. Li, Z. Zhang, T. Deng, G. Wu, R. Wang, S. w. Yang and X. Liu, Angew. Chem., Int. Ed., 2025, e202501869 CAS.
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2023, 36, 202305313 Search PubMed.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Adv. Sci., 2020, 7, 1902988 CrossRef CAS PubMed.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- C. Q. Han, J. X. Guo, S. Sun, Z. Y. Wang, L. Wang and X. Y. Liu, Small, 2024, 20, 202405887 Search PubMed.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 Search PubMed.
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- L. Dai, A. Dong, X. Meng, H. Liu, Y. Li, P. Li and B. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300224 CrossRef CAS PubMed.
- Y. Liu, Y. Shi, H. Wang and S. Zhang, Nano Res., 2024, 17, 5835–5844 CrossRef CAS.
- X. A. Li, Z. Z. Liang, Y. C. Zhou, J. F. Huang, X. L. Wang, L. M. Xiao and J. M. Liu, Aggregate, 2023, 5, e442 CrossRef.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- S. Feng, L. Wang, L. Tian, Y. Liu, K. Hu, H. Xu, H. Wang and J. Hua, Chem. Sci., 2024, 15, 11972–11980 RSC.
- Z. Zhang, Y. Hou, S. Zhu, L. Yang, Y. Wang, H. Yue, H. Xia, G. Wu, S. w. Yang and X. Liu, Angew. Chem., Int. Ed., 2025, e202505286 CAS.
- M. Liu, P. He, H. Gong, Z. Zhao, Y. Li, K. Zhou, Y. Lin, J. Li, Z. Bao, Q. Yang, Y. Yang, Q. Ren and Z. Zhang, Chem. Eng. J., 2024, 482, 148922 CrossRef CAS.
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed.
- Y. Chen, R. Liu, Y. Guo, G. Wu, T. C. Sum, S. W. Yang and D. Jiang, Nat. Synth., 2024, 3, 998–1010 CrossRef CAS.
- Y. Wang, H. Zhao, P. Li, J. Zhang, X. Sun, R. Zhang, Y. Guo, Y. Dong and Y. Zhu, Chem. Eng. J., 2024, 491, 151825 CrossRef CAS.
- Y. Liu, L. Li, H. Tan, N. Ye, Y. Gu, S. Zhao, S. Zhang, M. Luo and S. Guo, J. Am. Chem. Soc., 2023, 145, 19877–19884 CrossRef CAS PubMed.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14, 4344 CrossRef CAS PubMed.
- W. Chi, B. Liu, Y. Dong, J. Zhang, X. Sun, C. Pan, H. Zhao, Y. Ling and Y. Zhu, Appl. Catal., B, 2024, 355, 124077 CrossRef CAS.
- J.-C. Liu, C. Tuo, W.-Y. Xiao, M.-Y. Qi, Y. Yusran, Z.-T. Wang, H. Li, C.-S. Guo, J.-L. Song, S.-L. Qiu, Y.-J. Xu and Q. Fang, Angew. Chem., Int. Ed., 2024, e202416240 Search PubMed.
- H. H. Sun, Z. B. Zhou, Y. Fu, Q. Y. Qi, Z. X. Wang, S. Xu and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202409250 CrossRef CAS PubMed.
- Y. Xie, F. Mao, Q. Rong, X. Liu, M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Adv. Funct. Mater., 2024, 2411077 CrossRef CAS.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- Z. Zhou, M. Sun, Y. Zhu, P. Li, Y. Zhang, M. Wang and Y. Shen, Appl. Catal., B, 2023, 334, 122862 CrossRef CAS.
- L. Guo, L. Gong, Y. Yang, Z. Huang, X. Liu and F. Luo, Angew. Chem., Int. Ed., 2024, e202414658 Search PubMed.
- F. Huang, F. Zhang, Y. Wang, K. Zhang and X. Lang, Appl. Catal., B, 2025, 361, 124628 CrossRef CAS.
- K. Xiong, F. Zhang, Y. Wang, B. Zeng and X. Lang, J. Colloid Interface Sci., 2023, 643, 340–349 CrossRef CAS PubMed.
- Z. Gu, Z. Shan, Y. Wang, J. Wang, T. Liu, X. Li, Z. Yu, J. Su and G. Zhang, Chin. Chem. Lett., 2024, 35, 108356 CrossRef CAS.
- F. Tao, W. Zhou, Z. Li, X. Jiang, L. Wang, Z. Yu, J. Zhang and H. Zhou, ACS Mat. Lett., 2024, 6, 1120–1129 CrossRef CAS.
- X. Hu, X. Zeng, Y. Liu, J. Lu and X. Zhang, Nanoscale, 2020, 12, 16008–16027 RSC.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- S. Suleman, Y. Zhang, Y. Qian, J. Zhang, Z. Lin, Ö. Metin, Z. Meng and H. L. Jiang, Angew. Chem., Int. Ed., 2023, 63, e202314988 CrossRef PubMed.
- Y. Wang, F. Zhang, F. Huang, X. Dong, B. Zeng, X.-K. Gu and X. Lang, Appl. Catal., B, 2024, 354, 124103 CrossRef CAS.
- H. Pang, D. Huang, Y. Zhu, X. Zhao and Y. Xiang, Chem. Sci., 2023, 14, 1543–1550 RSC.
- D. Huang, Y. Zhang, H. Pang, X. Hu and Y. Xiang, J. Mater. Chem. A, 2024, 12, 18512–18518 RSC.
- Y. Pang, Y. Li, X. Gu, D. Wang, H. He, Y. Gou, B. Yuan, L. Chen and B. Wang, J. Catal., 2024, 433, 115497 CrossRef.
- J. Kou, G. Wang, H. Guo, L. Li, J. Fang, J. Ma and Z. Dong, Appl. Catal., B, 2024, 352, 124020 CrossRef.
- A. Chakraborty, M. Roy, A. Alam, D. Adhikari and P. Pachfule, Green Chem., 2024, 26, 9619–9651 RSC.
- K. Cai, W. Wang, J. Zhang, L. Chen, L. Wang, X. Zhu, Z. Yu, Z. Wu and H. Zhou, J. Mater. Chem. A, 2022, 10, 7165–7172 RSC.
- W.-K. An, S.-J. Zheng, X. Xu, L.-J. Liu, J.-S. Ren, L. Fan, Z.-K. Yang, Y. Ren and C. Xu, Appl. Catal., B, 2022, 316, 121630 CrossRef CAS.
- Z. Li, J.-a. Wang, S. Ma, Z. Zhang, Y. Zhi, F. Zhang, H. Xia, G. Henkelman and X. Liu, Appl. Catal., B, 2022, 310, 121335 CrossRef CAS.
- H. Shan, D. Cai, X. Zhang, Q. Zhu, P. Qin and J. Baeyens, Chem. Eng. J., 2022, 432, 134288 CrossRef CAS.
- D. Zhu, Y. Chen, Y. Zhu, C.-Y. Liu, Q. Yan, X. Wu, K. Ling, X. Zhang, P. M. Ajayan, T. P. Senftle and R. Verduzco, Macromolecules, 2024, 57, 1038–1049 CrossRef CAS.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- Q. Zhang, D.-H. Qu, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2022, 144, 2022–2033 CrossRef CAS.
- X. Zhang, Y. Dou, S. Liu, P. Chen, Y. Wen, J. Li, Y. Sun and R. Zhang, Adv. Heanlthc. Mater., 2024, 13, 2303842 CrossRef CAS PubMed.
- R. Xia, X. Zheng, C. Li, X. Yuan, J. Wang, Z. Xie and X. Jing, ACS Nano, 2021, 15, 7638–7648 CrossRef CAS PubMed.
- K. Wang, Z. Zhang, L. Lin, J. Chen, K. Hao, H. Tian and X. Chen, Chem. Mater., 2019, 31, 3313–3323 CrossRef CAS.
- Y. Liu, K. Yang, J. Wang, Y. Tian, B. Song and R. Zhang, Mater. Today Bio, 2024, 25, 100981 CrossRef CAS.
- S. Song, D. Wang, K. Zhao, Y. Wu, P. Zhang, J. Liu, G. Yang, P. Gong and Z. Liu, Chem. Eng. J., 2022, 442, 135963 CrossRef CAS.
- W. Wang, Y. Song, J. Chen, Y. Yang, J. Wang, Y. Song, J. Ni, M. Tang, J. Zhao, Y. Sun, T. Sun and J. Peng, J. Mater. Chem. B, 2022, 10, 1128–1135 RSC.
- H. Zhang, Y. Geng, J. Huang, Z. Wang, K. Du and H. Li, Energy Environ. Sci., 2023, 16, 889–951 RSC.
- P. Dubey, V. Shrivastav, T. Boruah, G. Zoppellaro, R. Zbořil, A. Bakandritsos and S. Sundriyal, Adv. Energy Mater., 2024, 14, 2400521 CrossRef CAS.
- M. Chafiq, A. Chaouiki and Y. G. Ko, Energy Storage Mater., 2023, 63, 103014 CrossRef.
- K. J. Nana Li, F. Rodríguez-Hernández, H. Mao, S. Han, X. Fu, J. Zhang, C. Yang, C. Ke and X. Zhuang, Adv. Sci., 2022, 9, 2104898 CrossRef PubMed.
- A. Chatterjee, J. Sun, K. S. Rawat, V. Van Speybroeck and P. Van Der Voort, Small, 2023, 19, 202303189 Search PubMed.
- A. Zhang, P. Ran, X. Han, S. Ke, A. Qiu, Z. Zhang, Y. Lv, M. Ding and J.-L. Zuo, J. Mater. Chem. A, 2024, 12, 22037–22044 RSC.
- S. Wang, T. Wu, J. Guo, R. Zhao, Y. Hua and Y. Zhao, ACS Cent. Sci., 2024, 10, 1383–1395 CrossRef CAS PubMed.
- E. Zhou, X. Zhang, L. Zhu, D. Yuan and Y. Wang, Adv. Funct. Mater., 2023, 33, 2213667 CrossRef CAS.
- Z. Li, Z. Zhang, R. Nie, C. Li, Q. Sun, W. Shi, W. Chu, Y. Long, H. Li and X. Liu, Adv. Funct. Mater., 2022, 32, 2112553 CrossRef CAS.
- S. Yang, J. He, Z. Chen, H. Luo, J. Wei, X. Wei, H. Li, J. Chen, W. Zhang, J. Wang, S. Wang and G. Yu, Adv. Mater., 2024, 36, 2408686 CrossRef CAS PubMed.
- C. Li, A. Yu, W. Zhao, G. Long, Q. Zhang, S. Mei and C. J. Yao, Angew. Chem., Int. Ed., 2024, 63, e202409421 CrossRef CAS PubMed.
- Y. Yang, C. Zhang, G. Zhao, Q. An, Z. y. Mei, Y. Sun, Q. Xu, X. Wang and H. Guo, Adv. Energy Mater., 2023, 13, 2300725 CrossRef CAS.
- G. Zhao, H. Ma, C. Zhang, Y. Yang, S. Yu, H. Zhu, Y. Sun and H. Guo, Nano-Micro Lett., 2024, 17, 21 CrossRef PubMed.
- X. Yu, Y. Zhao, Q. Bao, W. Wang, Y. Li, J. Xiao, Z. Sui, X. Tian and Q. Chen, J. Colloid Interface Sci., 2024, 675, 970–979 CrossRef CAS PubMed.
- D. Ma, H. Zhao, F. Cao, H. Zhao, J. Li, L. Wang and K. Liu, Chem. Sci., 2022, 13, 2385–2390 RSC.
- S. Wang, C. Zhu, J. Ji, M. Li, L. Zhao, F. Cai and Z. Tao, Small Methods, 2024, 2400557 CrossRef CAS.
- H. Peng, S. Huang, V. Montes-García, D. Pakulski, H. Guo, F. Richard, X. Zhuang, P. Samorì and A. Ciesielski, Angew. Chem., Int. Ed., 2023, 62, e202216136 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |